The Engine of Amplification: How DNA Polymerase Drives PCR Success in Biomedical Research
This article provides a comprehensive analysis of the critical function of DNA polymerase in the Polymerase Chain Reaction (PCR), tailored for researchers, scientists, and drug development professionals.
The Engine of Amplification: How DNA Polymerase Drives PCR Success in Biomedical Research
Abstract
This article provides a comprehensive analysis of the critical function of DNA polymerase in the Polymerase Chain Reaction (PCR), tailored for researchers, scientists, and drug development professionals. It explores the fundamental biochemical properties of DNA polymerases—including thermostability, fidelity, processivity, and specificity—and their direct impact on PCR efficacy. The scope extends from foundational principles and methodological applications for various biomedical uses to advanced troubleshooting, optimization strategies, and the rigorous validation and comparative analysis required for robust assay development in clinical and research settings.
The Core Machinery: Understanding DNA Polymerase Biochemistry and PCR Mechanics
Core Principles of the Polymerase Chain Reaction
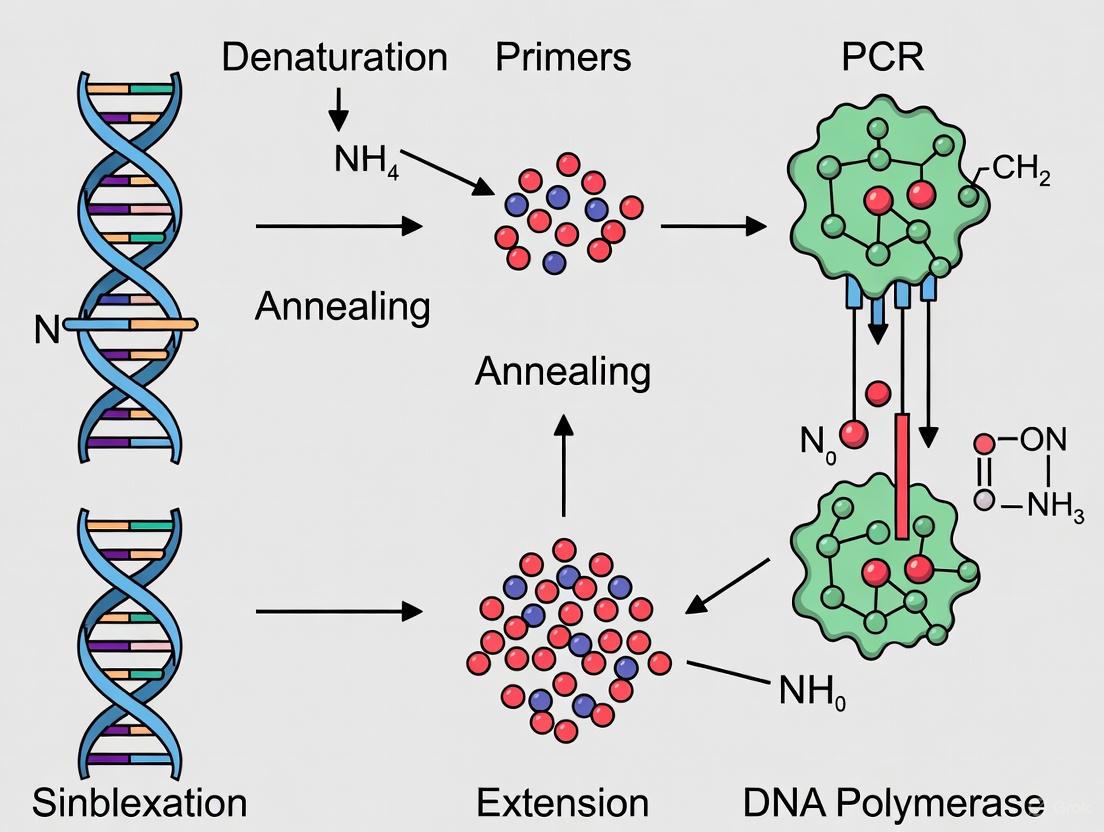
The Polymerase Chain Reaction (PCR) is a foundational in vitro technique that enables the exponential amplification of a specific DNA sequence, revolutionizing molecular biology, diagnostics, and drug development [1] [2]. This process allows researchers to generate millions to billions of copies of a particular DNA segment from a minimal starting amount, facilitating analysis and manipulation [3].
The core PCR process is a repetitive cycle comprising three fundamental steps, each dependent on precise temperature control [1] [3]:
- Denaturation: The double-stranded DNA template is heated to 94–98°C, disrupting the hydrogen bonds between complementary bases to yield single-stranded DNA molecules.
- Annealing: The temperature is lowered to 50–65°C, allowing short, single-stranded DNA primers to hybridize to their complementary sequences on either side of the target DNA segment.
- Extension: The temperature is raised to 72°C, the optimal temperature for a thermostable DNA polymerase to synthesize a new DNA strand by adding nucleotides to the 3' end of each primer.
This cycle is typically repeated 25-40 times, leading to an exponential accumulation of the target amplicon [1] [4]. The following diagram illustrates this cyclical process and the exponential growth of DNA products.
DNA Polymerase: The Engine of PCR
The DNA polymerase enzyme is the core component driving the PCR process, and its properties are critical for successful amplification. The discovery of Taq DNA polymerase from Thermus aquaticus was a pivotal advancement, as this thermostable enzyme can withstand the repeated high temperatures of the denaturation step without being inactivated [1] [3]. DNA polymerases for PCR are characterized by four key properties: thermostability, specificity, fidelity, and processivity [5].
Key Characteristics of DNA Polymerases
- Thermostability: This refers to the enzyme's ability to retain activity after multiple exposures to high temperatures (≥95°C). While Taq polymerase is sufficiently thermostable for many applications, enzymes from hyperthermophilic archaea like Pyrococcus furiosus (Pfu polymerase) exhibit even greater stability, with a half-life at 95°C that is approximately 20 times longer than that of Taq [5].
- Specificity: Specificity ensures that amplification is limited to the intended target sequence. Nonspecific amplification can occur if the polymerase synthesizes DNA from misprimed targets or primer-dimers. Hot-start PCR methods were developed to mitigate this; the polymerase is kept in an inactive state by antibodies, chemical modifications, or aptamers during reaction setup at room temperature, and is activated only after the first high-temperature denaturation step, thereby dramatically improving specificity [5] [6].
- Fidelity: Fidelity is the accuracy of DNA replication, defined by the enzyme's error rate (number of misincorporated nucleotides per base synthesized). DNA polymerases with proofreading activity possess a 3'→5' exonuclease domain that can recognize and excise mismatched nucleotides. High-fidelity polymerases are essential for applications like cloning, sequencing, and mutagenesis [5] [7]. Fidelity is often expressed relative to Taq polymerase.
- Processivity: Processivity is the number of nucleotides a polymerase can incorporate in a single binding event. A highly processive enzyme can synthesize long DNA fragments more efficiently and is better at amplifying difficult templates, such as those with complex secondary structures or high GC content [5].
Comparative Analysis of DNA Polymerase Properties
Table 1: Key Characteristics of Common DNA Polymerases
| Polymerase | Source Organism | Proofreading (3'→5' Exo) | Relative Fidelity (vs. Taq) | Typical Application |
|---|---|---|---|---|
| Taq | Thermus aquaticus | No | 1X | Routine PCR, genotyping [5] [1] |
| Pfu | Pyrococcus furiosus | Yes | ~7-10X | High-fidelity cloning, protein expression [5] |
| PrimeSTAR GXL | Engineered | Yes | 6.5X | Long-range, high-fidelity PCR [7] |
| PrimeSTAR Max | Engineered | Yes | 29X | Ultra-high-fidelity applications [7] |
| KOD | Thermococcus kodakarensis | Yes | ~10X | High-speed, high-fidelity amplification [5] |
Table 2: End-Product Properties and Cloning Compatibility
| Polymerase Type | Terminal Structure | Recommended Cloning Method |
|---|---|---|
| Standard Taq | 3' dA Overhang | TA Cloning [7] |
| Proofreading (e.g., Pfu, PrimeSTAR) | Blunt End | Blunt-End Cloning [7] |
| Any Blunt-End Polymerase | A-Tailed (after treatment) | TA Cloning (post A-tailing) [7] |
The following diagram provides a logical framework for selecting the appropriate DNA polymerase based on the primary goal of the experiment.
Essential PCR Methodologies and Protocols
Basic PCR Protocol
A standard PCR reaction includes the following components [3]:
- Template DNA: 10–500 ng of genomic DNA or plasmid.
- Primers: 0.1–1.0 µM each of forward and reverse primer.
- Taq DNA Polymerase: 0.5–2.5 units per 50 µL reaction.
- dNTPs: 200 µM each of dATP, dCTP, dGTP, and dTTP.
- Reaction Buffer: Provides optimal pH and MgCl₂ (typically 1.5–2.0 mM final concentration).
A sample thermal cycling program is [3]:
- Initial Denaturation: 94°C for 2 minutes.
- Amplification (25–35 cycles):
- Denature: 94°C for 30 seconds.
- Anneal: 55°C (or 5°C below primer Tm) for 30 seconds.
- Extend: 72°C for 1 minute per kilobase of product.
- Final Extension: 72°C for 5–10 minutes.
Specialized PCR Techniques
- Quantitative PCR (qPCR): This technique allows for the detection and quantification of amplified DNA in real-time using fluorescent reporters. Two primary detection chemistries are used [8] [9]:
- DNA-binding dyes (e.g., SYBR Green I): These dyes fluoresce brightly when bound to double-stranded DNA, providing a simple and cost-effective method. However, they can bind to any dsDNA, including nonspecific products and primer-dimers [9].
- Hydrolysis probes (e.g., TaqMan probes): These are sequence-specific oligonucleotides labeled with a fluorescent reporter and a quencher. The 5'→3' exonuclease activity of Taq polymerase cleaves the probe during amplification, separating the reporter from the quencher and generating a fluorescent signal. This method increases specificity and enables multiplexing [8] [9].
- Reverse Transcription PCR (RT-PCR): This method is used to amplify RNA sequences. It involves a two-step process: RNA is first reverse-transcribed into complementary DNA (cDNA) using a reverse transcriptase enzyme, followed by standard PCR amplification of the cDNA [1] [8].
- Nested PCR: This technique increases specificity and yield by using two sets of primers in two successive PCR runs. The second set of primers binds within the first PCR product, thereby reducing background from nonspecific amplification in the initial reaction [4] [7].
- Touchdown PCR: This strategy enhances specificity by starting with an annealing temperature higher than the primer's calculated Tm and gradually decreasing it in subsequent cycles. This ensures that amplification in the initial cycles is highly stringent, favoring the correct target [7].
Advanced PCR Technologies and Applications
PCR technology has evolved significantly, leading to sophisticated methods that enhance sensitivity, quantification, and throughput.
- Digital PCR (dPCR): This technique provides absolute quantification of nucleic acids without the need for a standard curve. The reaction mixture is partitioned into thousands of individual droplets or microchambers, so that each contains either zero or one (or a few) target molecules. After PCR amplification, the fraction of positive reactions is counted to determine the original concentration of the target with high precision [4] [10].
- Multiplex PCR: This allows for the simultaneous amplification of multiple targets in a single reaction by using multiple pairs of primers. Critical success factors include designing primers with similar annealing temperatures and ensuring they do not interact with each other to form primer-dimers [4] [7].
- Color Cycle Multiplex Amplification (CCMA): An advanced qPCR method that significantly increases multiplexing capacity. Instead of assigning one color per target, CCMA uses programmable delays to create a unique sequence, or "color cycle," for each target. With four fluorescence colors, this method can theoretically discriminate up to 136 distinct DNA targets in a single tube [10].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for PCR
| Reagent/Material | Function | Technical Considerations |
|---|---|---|
| Hot-Start DNA Polymerase | Inhibits polymerase activity at room temperature to prevent nonspecific amplification and primer-dimer formation. | Available as antibody-mediated, chemically modified, or aptamer-based. Crucial for high-throughput setups [5] [6]. |
| Proofreading High-Fidelity Polymerase | Accurately replicates template DNA with low error rates for applications requiring high sequence fidelity. | Contains 3'→5' exonuclease activity. Essential for cloning, sequencing, and mutagenesis [5] [7]. |
| dNTP Mix | The building blocks (dATP, dCTP, dGTP, dTTP) for de novo DNA synthesis by the polymerase. | Used at 200 µM each in a standard reaction. Quality is critical for efficient amplification [3]. |
| Optimized Buffer Systems | Provides a stable chemical environment (pH, ionic strength) and co-factors (like Mg²⁺) for maximal polymerase activity. | MgCl₂ concentration is a critical variable and must be optimized; it is chelated by dNTPs [6] [3]. |
| PCR Additives (DMSO, Betaine) | Assist in amplifying difficult templates (e.g., GC-rich sequences) by reducing secondary structure and lowering DNA melting temperature. | Often used empirically at concentrations of 1–10% (v/v). Can inhibit some polymerases, so compatibility must be checked [6] [3]. |
The polymerase chain reaction (PCR) stands as a cornerstone technique in molecular biology, and its efficacy is fundamentally dictated by the properties of the DNA polymerase enzyme employed. This in-depth technical guide examines the four critical characteristics of DNA polymerases—thermostability, fidelity, processivity, and specificity—within the context of their collective function in PCR research and drug development. We explore the molecular mechanisms underpinning each property, provide structured quantitative comparisons of enzymes from various microbial origins, and detail experimental methodologies for their assessment. Furthermore, this whitepaper visualizes the interrelationships between these properties and presents a curated toolkit of research reagents, equipping scientists with the knowledge to select optimal enzymes for specific applications, from routine amplification to high-fidelity cloning and sensitive diagnostic assays.
The function of DNA polymerase in PCR research extends far beyond simple DNA synthesis; it is the core engine that drives the amplification of specific nucleic acid sequences. The selection of an appropriate DNA polymerase is a critical determinant of experimental success, influencing everything from amplicon yield and accuracy to the ability to amplify challenging templates. Early PCR protocols relied on the Klenow fragment of E. coli DNA polymerase I, which was heat-labile and required replenishment after each denaturation cycle [11]. A transformative advancement came with the introduction of thermostable DNA polymerases derived from thermophilic microorganisms, which withstand the repeated high-temperature denaturation steps intrinsic to PCR [5] [11].
Among these, Taq DNA polymerase, isolated from the bacterium Thermus aquaticus, became the benchmark due to its thermostability and optimal activity temperature around 75–80°C [11]. However, native Taq polymerase has limitations, including a lack of proofreading activity and susceptibility to nonspecific amplification. These challenges prompted the exploration of enzymes from other bacterial and archaeal sources, such as Pyrococcus furiosus (Pfu polymerase), and the development of engineered polymerases through directed evolution and protein fusion technologies [5] [12] [13]. This evolution has yielded modern PCR enzymes with enhanced properties, making them versatile tools for a broad range of biological applications, including cloning, sequencing, diagnostics, and gene expression analysis [5]. This guide delves into the four key characteristics—thermostability, fidelity, processivity, and specificity—that define a DNA polymerase's performance and utility in a research setting.
The Four Key Characteristics of DNA Polymerases
Thermostability
Definition and Molecular Basis Thermostability refers to the ability of a DNA polymerase to retain its folded structure and catalytic activity at the high temperatures required for PCR, particularly during the denaturation step (typically 94–98°C). This property is an inherent characteristic of enzymes derived from thermophilic and hyperthermophilic organisms that thrive in hydrothermal vents and hot springs [5] [14]. Their structural stability is achieved through various molecular mechanisms, including reinforced hydrophobic cores, increased ionic interactions, and compact packing [5].
Comparative Analysis of Thermostable Polymerases While Taq polymerase (from Thermus aquaticus) is thermostable, its half-life diminishes significantly above 90°C (approximately 40 minutes at 95°C) [11]. In contrast, archaeal DNA polymerases like Pfu (from Pyrococcus furiosus) exhibit superior thermostability; Pfu is about 20 times more stable than Taq at 95°C [5]. This enhanced stability is crucial for applications involving prolonged high-temperature incubations, such as amplifying DNA with extensive secondary structures or GC-rich regions.
Table 1: Thermostability and Optimal Conditions for Common DNA Polymerases
| Polymerase | Organismal Origin | Origin Type | Optimal Extension Temperature | Half-Life at High Temperature | Key Feature |
|---|---|---|---|---|---|
| Taq | Thermus aquaticus | Bacterium | 72–80°C [11] | ~40 min at 95°C [11] | First widely adopted thermostable polymerase |
| Tth | Thermus thermophilus | Bacterium | 74°C [12] | Similar to Taq [12] | Possesses reverse transcriptase activity |
| Pfu | Pyrococcus furiosus | Archaeon | 75°C [5] [12] | ~2 hours at 95°C [5] | High fidelity due to proofreading |
| KOD | Thermococcus kodakarensis | Archaeon | 75°C [12] | Extremely high [5] | High processivity and speed (~120 bp/sec) [12] |
| 9°Nm | Thermococcus sp. | Archaeon | 75–85°C [14] | Stable at 100°C [14] | Extremely thermostable, used in demanding applications |
Practical Implications for PCR Highly thermostable polymerases are less likely to denature irreversibly over multiple PCR cycles, ensuring consistent performance and enabling the amplification of long or complex templates [5] [14]. However, a notable limitation of some hyperthermostable archaeal polymerases (e.g., Pfu) is their inability to amplify uracil-containing templates, as they possess a uracil-binding pocket as part of a DNA repair mechanism [5]. This can be a critical factor in techniques like bisulfite sequencing or PCR carryover prevention protocols that utilize uracil.
Fidelity
Definition and Mechanism of Proofreading Fidelity is the accuracy of DNA synthesis, defined as the inverse of the error rate (number of misincorporated nucleotides per total nucleotides polymerized) [5]. High-fidelity DNA polymerases are essential for applications where sequence accuracy is paramount, such as cloning, mutagenesis, and next-generation sequencing library preparation.
The primary mechanism for high fidelity is the 3′→5′ exonuclease activity, or proofreading activity [5] [15]. This activity resides in a separate enzyme domain from the polymerase activity. When a mismatched nucleotide is incorporated, it causes a temporary stall in DNA synthesis. This delay allows the mispaired nucleotide to be translocated to the exonuclease active site, where it is excised before polymerization resumes with the correct nucleotide [5].
Table 2: Fidelity and Error Rates of Selected DNA Polymerases
| Polymerase | Proofreading (3′→5′ Exo) | Fidelity (Relative to Taq) | Error Rate (per base per duplication) | Impact on 3 kb Amplicon after 30 Cycles (% error-free molecules) [16] |
|---|---|---|---|---|
| Taq | No | 1x | 1.5–8.0 × 10⁻⁵ [12] | ~0% (Every molecule contains ~2 errors) |
| OneTaq | Yes (Low) | 2x [15] | - | - |
| Pfu | Yes | ~10x [5] | 1.3 × 10⁻⁶ [12] | 74.8% [16] |
| KOD | Yes | 12–12.5x [12] [17] | 1.2–3.5 × 10⁻⁶ [12] | - |
| Phusion | Yes | 39–50x [15] [17] | ~2.8 × 10⁻⁷ [12] | 96.04% (GC Buffer) [16] |
| Q5 | Yes | ~280x [15] [17] | - | - |
Measuring Fidelity Fidelity is typically measured using several established assays [5] [17]:
- Colony Screening (lacZα/lacI Assay): A PCR-amplified fragment of the lacZ or lacI gene is cloned. Errors during PCR that inactivate the gene result in a color change (e.g., from blue to white colonies on X-Gal plates), allowing for the calculation of error frequency [5] [17].
- Sanger Sequencing: Cloned PCR products are individually sequenced to identify mutations introduced during amplification.
- Next-Generation Sequencing (NGS): PCR amplicons are directly sequenced en masse using NGS platforms, providing a highly detailed and comprehensive view of polymerase error rates and spectra (including insertions and deletions) [5].
It is critical to note that fidelity comparisons between polymerases are only valid when the same measurement method and cycling parameters are used, as error rates can be influenced by amplicon length and cycle number [5].
Processivity
Definition Processivity is defined as the number of nucleotides a DNA polymerase incorporates into a growing DNA strand per single binding event with the template [5]. A highly processive enzyme remains attached to the template and continuously synthesizes DNA, while a low-processivity enzyme dissociates frequently.
Factors Influencing Processivity The intrinsic processivity of a polymerase is governed by its affinity for the DNA template. Engineering efforts have successfully enhanced this property by fusing the polymerase domain to other DNA-binding domains, such as the Sso7d protein from Sulfolobus solfataricus or the helix-hairpin-helix (HhH) motif of a topoisomerase [5] [12]. These fusion domains act as a "sliding clamp," increasing the enzyme's grip on the DNA and thereby boosting processivity by 2- to 5-fold [5].
Applications of High-Processivity Polymerases Polymerases with high processivity are particularly beneficial for:
- Long-Range PCR: Amplifying DNA fragments >10 kb requires a polymerase that does not dissociate frequently [15].
- Challenging Templates: Efficiently amplifying through GC-rich regions, secondary structures, and sequences with stable hairpins [5].
- Inhibitor-Rich Samples: Tolerating common PCR inhibitors found in blood (heparin), plant tissues (xylan, humic acid), and forensic samples, as the stronger DNA binding helps the enzyme overcome the inhibitory effects [5].
Specificity
Definition and the Problem of Nonspecific Amplification Specificity in PCR refers to the selective amplification of only the intended target sequence. A primary challenge to specificity is nonspecific amplification, which includes the extension of misprimed targets and the formation of primer-dimers. These undesirable products compete for reaction components, reducing the yield and sensitivity of the target amplicon [5].
Hot-Start Technology The most effective solution to enhance specificity is hot-start PCR [5] [13]. This technique involves inhibiting the DNA polymerase's activity during reaction setup at room temperature, where primers can anneal nonspecifically. Full activity is restored only after the first high-temperature denaturation step.
- Mechanism: The most common method uses antibodies or aptamers that bind to and inhibit the polymerase at lower temperatures. During the initial denaturation (e.g., >90°C), the inhibitor is degraded or denatured, releasing active polymerase [5].
- Benefits: Hot-start polymerases significantly reduce primer-dimer formation and nonspecific background, increase target yield, allow for room-temperature reaction setup for high-throughput applications, and improve reproducibility [5].
Table 3: Comparison of Specificity-Enhancing Methods
| Method | Mechanism | Advantages | Disadvantages |
|---|---|---|---|
| Manual Hot-Start | Polymerase is added after the reaction reaches high temperature. | Simple in concept. | Laborious, increases contamination risk, poor reproducibility. |
| Antibody-Based | An inhibitor antibody is bound to the polymerase and inactivated by heat. | Highly effective, convenient, suitable for high-throughput. | Slightly higher cost. |
| Chemical Modification | The polymerase is chemically blocked; the block is removed by heat. | Effective inhibition. | Requires longer initial activation step. |
| Physical Separation | A wax barrier separates polymerase from other components until melting. | Effective. | Extra setup step required. |
Visualizing DNA Polymerase Characteristics and Workflows
The following diagrams illustrate the core concepts and experimental workflows related to DNA polymerase characteristics.
Interplay of DNA Polymerase Characteristics in PCR Success
This diagram summarizes how the four key characteristics of DNA polymerases contribute to successful PCR outcomes and the technologies that enhance them.
Experimental Workflow for Assessing Polymerase Fidelity
This diagram outlines a standard colony-based assay (e.g., lacZ/lacI) for measuring the error rate of a DNA polymerase.
The Scientist's Toolkit: Essential Reagents and Methodologies
Research Reagent Solutions
Table 4: Key Reagents for PCR Optimization and Their Functions
| Reagent | Function in PCR | Example Use Case |
|---|---|---|
| Hot-Start DNA Polymerase | Inhibits enzyme activity during setup to prevent nonspecific amplification and primer-dimer formation. [5] | Essential for all high-specificity applications, including high-throughput and multiplex PCR. |
| High-Fidelity Polymerase Mix | A blend of a polymerase with proofreading activity and one without, offering a balance of high accuracy and robust amplification. [12] | Long-range PCR and cloning where high yield and fidelity are both required. |
| PCR Enhancers (DMSO, Betaine) | Reduce secondary structure formation in GC-rich templates and improve primer annealing specificity. [13] | Amplification of difficult templates with high GC content or stable secondary structures. |
| dNTP Mix | The essential building blocks (dATP, dCTP, dGTP, dTTP) for DNA strand synthesis by the polymerase. [18] | A fundamental component of every PCR reaction; quality and concentration affect yield and fidelity. |
| MgCl₂ Solution | Acts as a cofactor for DNA polymerase activity; concentration is critical for enzyme efficiency and specificity. [18] | Requires optimization for each primer-template system; affects primer annealing and strand dissociation. |
| Optimized Reaction Buffers | Provide the optimal pH and ionic strength (e.g., KCl) for polymerase activity and stability. [18] [13] | Specific buffers (HF, GC) are often tailored to different polymerases and template types. |
Detailed Experimental Protocol: Validating Hot-Start Specificity
Objective: To demonstrate the effectiveness of a hot-start DNA polymerase in reducing nonspecific amplification compared to a non-hot-start enzyme.
Materials:
- Test DNA template (e.g., human genomic DNA)
- Forward and reverse primers for a specific target (e.g., 2 kb fragment)
- Hot-start DNA polymerase and corresponding buffer
- Standard (non-hot-start) DNA polymerase and corresponding buffer
- dNTP mix
- Sterile water
- Thermal cycler
- Agarose gel electrophoresis equipment
Methodology:
- Reaction Setup: Prepare two identical 50 μL PCR mixtures containing template, primers, dNTPs, and buffer. Add the hot-start polymerase to one tube and the standard polymerase to the other.
- Room-Temperature Challenge: Instead of immediately placing the tubes in the thermal cycler, incubate both reaction mixtures at room temperature (25°C) for a set period (e.g., 0 hours, 24 hours, 72 hours). [5]
- PCR Amplification: After the challenge period, transfer all tubes to a pre-heated thermal cycler and run the following protocol:
- Initial Denaturation/Activation: 94°C for 2 minutes
- 35 Cycles:
- Denaturation: 94°C for 30 seconds
- Annealing: 55–60°C for 30 seconds
- Extension: 72°C for 2 minutes
- Final Extension: 72°C for 5 minutes
- Analysis: Analyze the PCR products using agarose gel electrophoresis.
Expected Outcomes: The reactions with the hot-start polymerase will show a strong, specific band of the expected size (2 kb) even after prolonged room-temperature incubation, with little to no nonspecific background. The reactions with the standard polymerase will show significant nonspecific amplification (smearing or multiple bands) and primer-dimer formation, which will worsen with longer room-temperature incubation [5]. This experiment visually validates the critical importance of hot-start technology for reaction specificity and robustness.
The intricate interplay between thermostability, fidelity, processivity, and specificity defines the functional capacity of a DNA polymerase in PCR research. Moving beyond the one-size-fits-all approach of early PCR, modern molecular biology demands a strategic selection of enzymes based on these core characteristics. The advent of engineered polymerases and sophisticated reagent systems has empowered researchers to tackle increasingly complex challenges, from amplifying long, GC-rich genomic segments for cloning to detecting minute quantities of pathogen DNA with utmost accuracy in diagnostic assays. A deep understanding of these principles, as outlined in this guide, enables scientists to not only optimize existing protocols but also to innovate new applications, thereby driving progress in genetic research, drug development, and clinical diagnostics. The continued evolution of DNA polymerase technology promises to further expand the boundaries of what is possible with PCR.
The polymerase chain reaction (PCR) stands as one of the most transformative technologies in molecular biology, enabling the exponential amplification of specific DNA sequences from minimal starting material. At the heart of this technique lies DNA polymerase, the enzyme responsible for synthesizing new DNA strands during each amplification cycle. The evolution of PCR from a cumbersome manual process to an automated, high-fidelity technique has been driven largely by advances in our understanding and engineering of DNA polymerases [1] [19]. Early PCR methodologies utilized the Klenow fragment of E. coli DNA Polymerase I, which required manual replenishment after each denaturation cycle due to heat sensitivity [20]. This limitation was overcome with the discovery and implementation of thermostable DNA polymerases from extremophilic organisms, revolutionizing molecular biology, biomedical research, and drug development workflows [21] [22].
The ongoing refinement of DNA polymerases for specialized applications represents an active frontier in biotechnology research. This whitepaper examines the characteristics of natural DNA polymerases, with particular focus on the widely utilized Taq and Pfu enzymes, and explores how engineering approaches are expanding their functionality for cutting-edge research applications, including the incorporation of non-standard nucleotides and improved performance under challenging conditions [23] [24].
DNA Polymerase Fundamentals: Structure and Function
Conserved Architecture and Mechanism
DNA polymerases share a conserved structural architecture often described as resembling a "right hand," consisting of palm, fingers, and thumb domains [22] [20]. The palm domain contains the catalytic site where phosphoryl transfer occurs, while the fingers domain is involved in nucleotide binding and recognition. The thumb domain interacts with duplex DNA, maintaining the correct positioning of the primer-template complex [20]. These enzymes catalyze template-directed DNA synthesis in a metal-dependent reaction where the 3′-OH of the primer acts as a nucleophile attacking the α-phosphate of the incoming deoxynucleoside triphosphate (dNTP), forming a new phosphodiester bond and releasing pyrophosphate [23].
Key Performance Characteristics
Several critical properties define the utility of DNA polymerases for PCR applications:
- Fidelity: The accuracy of DNA sequence replication, largely determined by the enzyme's intrinsic selectivity and any proofreading activity [5]. DNA polymerases with 3′→5′ exonuclease activity can excise misincorporated nucleotides, significantly reducing error rates [20].
- Processivity: The number of nucleotides incorporated per enzyme-binding event [5]. Highly processive polymerases remain bound to the template for extended synthesis periods, improving efficiency especially for long templates.
- Thermostability: The ability to retain structure and function at elevated temperatures essential for PCR's repeated denaturation steps [21] [5].
- Specificity: The enzyme's ability to selectively amplify target sequences without generating nonspecific products, often enhanced through "hot-start" modifications that inhibit polymerase activity until elevated temperatures are reached [5].
Natural DNA Polymerases: From Taq to Pfu
Taq DNA Polymerase
Taq DNA polymerase, isolated from the thermophilic bacterium Thermus aquaticus found in hot springs, became the foundational enzyme for modern PCR due to its thermostability [21] [22]. With an optimum temperature of 75–80°C, Taq polymerase can replicate 1,000 base pairs in less than 10 seconds at 72°C [21]. This high processivity and rapid extension speed made it ideal for routine PCR applications. However, Taq polymerase lacks 3′→5′ proofreading activity, resulting in a relatively high error rate of approximately 1 in 9,000 nucleotides [21]. This limitation makes it less suitable for applications requiring high accuracy, such as cloning and sequencing. Additionally, Taq polymerase adds a single adenine overhang to the 3′ ends of PCR products, facilitating TA cloning strategies but producing amplicons that are not blunt-ended [20].
Pfu DNA Polymerase
Pfu DNA polymerase, derived from the hyperthermophilic archaeon Pyrococcus furiosus, offers superior thermostability and proofreading capabilities compared to Taq [21]. Its 3′→5′ exonuclease activity enables correction of misincorporated nucleotides, achieving an error rate of approximately 1 in 1.3 million base pairs—about 150 times more accurate than Taq [21]. This high fidelity makes Pfu preferred for applications where sequence accuracy is critical. The trade-off for this accuracy is slower synthesis speed, requiring 1–2 minutes to amplify 1 kb of DNA compared to Taq's less than 10 seconds [21]. Additionally, Pfu generates blunt-ended PCR products due to its proofreading activity, requiring different cloning strategies than Taq's TA cloning [20].
Comparative Analysis of Natural Polymerases
Table 1: Characteristics of Natural DNA Polymerases Used in PCR
| Characteristic | Taq DNA Polymerase | Pfu DNA Polymerase | KOD DNA Polymerase |
|---|---|---|---|
| Source Organism | Thermus aquaticus (bacterium) | Pyrococcus furiosus (archaeon) | Thermococcus kodakaraensis (archaeon) |
| Optimal Temperature | 75–80°C | ~90°C | ~85°C |
| Proofreading Activity | No 3′→5′ exonuclease | 3′→5′ exonuclease present | 3′→5′ exonuclease present |
| Error Rate | ~1 in 9,000 nucleotides | ~1 in 1.3 million bp | High (comparable to Pfu) |
| Extension Speed | <10 sec for 1 kb | 1–2 min for 1 kb | High speed |
| PCR Product Ends | 3′ A-overhang | Blunt ends | Blunt ends |
| Fidelity Relative to Taq | 1x | ~10-50x | ~10-50x |
Table 2: Performance Characteristics of Selected Commercial Polymerases
| Polymerase Name | Max Target Length (kb) | Extension Speed (kb/min) | Proofreading | Primary Applications |
|---|---|---|---|---|
| HGS Diamond Taq | 2 | 60x | No | PCR, qPCR, multiplex assays |
| Precision DNA Polymerase | 6 | 1 | Yes | Whole genome sequencing, site-directed mutagenesis |
| TaqFast DNA Polymerase | 12 | 4-6 | Yes | Fast PCR, high-throughput PCR |
| TransTaq HiFi DNA Polymerase | 15 | 1-2 | Yes | Complex templates, GC/AT-rich templates |
| Hot Diamond Taq | 15 | N/A | No | AT and GC rich regions, difficult templates |
| PFU DNA Polymerase | 6 | 20 | Yes | High-fidelity amplification, blunt-end cloning |
Engineering Advanced DNA Polymerases
Directed Evolution and Rational Design
The limitations of naturally occurring DNA polymerases have prompted extensive engineering efforts to enhance their functionality for specialized applications. Two primary approaches have emerged: directed evolution, which screens large libraries of polymerase variants for desired traits, and rational design based on structural knowledge [23]. Compartmentalized self-replication (CSR) represents a powerful directed evolution method where single polymerase clones are captured in water-in-oil emulsions, enabling PCR amplification within the emulsion and linking genotype to phenotype [23]. More recently, droplet-based optical polymerase sorting (DrOPS) has allowed high-throughput screening by encapsulating single cells carrying polymerase variants with activity assay reagents [23].
Rational design efforts have focused on understanding structure-function relationships in DNA polymerases. Studies of polymerase structures, such as the Klenow fragment of E. coli DNA polymerase I and Klentaq (the large fragment of T. aquaticus DNA polymerase), have identified conserved motifs critical for function [22]. Motif A (residues 605-617 in Taq) in the palm domain contains catalytic residues like Asp610 and Glu615, while Motif B (residues 659-671) forms the O-helix in the fingers domain that contacts incoming nucleotides [22]. Strategic modifications of these regions have yielded polymerases with altered properties.
Engineering for Novel Functions
Engineering efforts have produced DNA polymerases with capabilities beyond those found in nature:
- Incorporation of Non-standard Nucleotides: Engineered polymerases can replicate DNA containing unnatural base pairs (UBPs) that expand the genetic alphabet, enabling applications in synthetic biology and biotechnology [22] [24]. For example, the Benner laboratory developed an artificially expanded genetic information system (AEGIS) with additional base pairs like P:Z and B:S, while the Romesberg laboratory created polymerases that replicate hydrophobic base pairs with orthogonal packing interactions [24].
- Enhanced Processivity: Fusion of DNA polymerases with DNA-binding domains, such as Sso7d, has dramatically improved processivity, enabling amplification of longer templates and more efficient replication through challenging sequences [5].
- Resistance to Inhibitors: Engineering has produced polymerases that function efficiently in the presence of common PCR inhibitors found in blood, plant tissues, and environmental samples, facilitating amplification from crude samples without extensive purification [5].
- Improved Thermostability: Polymerases from hyperthermophilic organisms have been engineered for even greater stability, allowing PCR under more stringent conditions and enabling applications like PCR of damaged DNA from ancient samples [23].
Table 3: Engineered DNA Polymerase Functions and Applications
| Engineering Approach | Polymerase Example | Enhanced Function | Research Applications |
|---|---|---|---|
| Chimeragenesis | Taq-Pfu chimeras | Proofreading with thermostability | High-fidelity PCR |
| Point Mutations | KlenTaq variants | Hot-start capability | Specific amplification, high-throughput setups |
| Directed Evolution | ZP Klentaq | Unnatural base pair incorporation | Synthetic biology, expanded genetic alphabets |
| Domain Fusion | Sso7d-fused polymerases | Increased processivity | Long-range PCR, difficult templates |
| CSR Evolution | Tth Pol I variants | Activity in challenging conditions | Amplification from crude samples |
Experimental Approaches for Polymerase Analysis
Fidelity Assessment Methods
Several established methods enable quantitative assessment of DNA polymerase fidelity:
- Colony Screening Assay: This classical approach involves PCR amplification of a reporter gene (e.g., lacZα), cloning into an appropriate vector, and determining the mutation frequency by blue/white colony screening. White colonies indicate mutations that inactivate the lacZα gene, allowing calculation of error rates based on the proportion of white colonies [5].
- Sanger Sequencing: Cloned PCR products are sequenced to identify mutations introduced during amplification, providing detailed information about error types and distribution [5].
- Next-Generation Sequencing: PCR amplicons are directly sequenced using NGS platforms, enabling comprehensive analysis of error rates and spectra across the entire amplified product without cloning bias [5].
Processivity and Activity Assays
Processivity measurements typically employ primer extension assays with limiting enzyme concentrations and time-course sampling. Reaction products are separated by denaturing polyacrylamide gel electrophoresis to determine the average number of nucleotides incorporated per binding event [5]. Thermostability is assessed by measuring polymerase half-life at elevated temperatures, often through residual activity testing after incubation at 95°C or higher for varying durations [20].
Diagram 1: DNA polymerase engineering workflows combining rational design and directed evolution approaches.
Research Reagent Solutions
Table 4: Essential Research Reagents for DNA Polymerase Studies
| Reagent Category | Specific Examples | Research Function | Application Notes |
|---|---|---|---|
| DNA Polymerases | Taq, Pfu, KOD, engineered variants | DNA strand elongation | Selection depends on required fidelity, speed, and product characteristics |
| Primers | Target-specific oligonucleotides | Define amplification region | Design critical for specificity; length (20-25 nt) and Tm optimization required |
| Nucleotides | dNTPs (dATP, dCTP, dGTP, dTTP) | DNA synthesis building blocks | Balanced concentrations essential for fidelity; modified nucleotides for specialized applications |
| Buffer Components | MgCl₂, Tris-HCl, (NH₄)₂SO₄, KCl | Optimal enzymatic environment | Mg²⁺ concentration critical for fidelity and efficiency; varies by polymerase |
| Hot-Start Additives | Antibodies, aptamers, chemical inhibitors | Enhance specificity | Inhibit polymerase activity at room temperature; activated during initial denaturation |
| Fidelity Enhancers | Betaine, DMSO, glycerol | Improve amplification efficiency | Assist with GC-rich templates and complex secondary structures |
| Detection Systems | SYBR Green, TaqMan probes, ethidium bromide | Amplicon quantification/visualization | Selection depends on quantitative needs and equipment availability |
Applications in Research and Drug Development
The evolution of DNA polymerases has enabled diverse applications across biomedical research and pharmaceutical development:
- Diagnostic Assays: PCR-based pathogen detection, including SARS-CoV-2 identification during the COVID-19 pandemic, relies on robust DNA polymerases for sensitive and specific amplification [1]. Reverse transcription PCR (RT-PCR) combines reverse transcriptase with DNA polymerase to detect RNA viruses.
- Gene Expression Analysis: Quantitative real-time PCR (qPCR) utilizes DNA polymerases with consistent performance characteristics to measure gene expression levels across experimental conditions, with applications in biomarker identification and drug response assessment [25].
- Cloning and Mutagenesis: High-fidelity polymerases like Pfu and engineered variants are essential for accurate gene cloning, site-directed mutagenesis, and protein engineering campaigns in drug development pipelines [21] [5].
- Synthetic Biology: Engineered polymerases capable of incorporating unnatural nucleotides enable the creation of novel genetic systems with expanded functionality, including the production of proteins containing non-canonical amino acids [22] [24].
- Forensic and Ancient DNA Analysis: Polymerases with enhanced processivity and damage tolerance facilitate amplification of degraded or damaged DNA from forensic samples and ancient specimens [23].
Diagram 2: Standard PCR workflow with temperature-dependent stages and polymerase function.
The journey from Taq to Pfu DNA polymerase represents more than just a switch between enzymes—it exemplifies the ongoing evolution of PCR technology to meet increasingly sophisticated research demands. Natural DNA polymerases from thermophilic and hyperthermophilic organisms provide distinct advantages and limitations that make them suitable for different applications, with Taq offering speed and simplicity, while Pfu provides superior accuracy at the cost of slower synthesis.
The future of DNA polymerase development lies in engineered variants that combine the most desirable properties while adding novel functionalities. Directed evolution and rational design approaches continue to yield polymerases with enhanced fidelity, processivity, and ability to incorporate modified nucleotides. These advances support emerging applications in synthetic biology, next-generation sequencing, diagnostics, and drug development.
As PCR remains foundational to molecular biology and biomedical research, the ongoing refinement of DNA polymerases will continue to expand experimental possibilities, enabling researchers to address increasingly complex biological questions and develop novel therapeutic interventions. The integration of computational modeling, high-throughput screening, and structural biology will further accelerate the development of tailored polymerase solutions for specialized research needs.
The Polymerase Chain Reaction (PCR) is a fundamental, indispensable technique in molecular biology that allows for the exponential amplification of specific DNA sequences. Introduced in 1985 by Kary Mullis, for which he was later awarded the Nobel Prize in Chemistry, PCR has become a cornerstone of modern biomedical research, clinical diagnostics, and drug development [1]. At its core, the PCR process is a repetitive, temperature-driven cycle that relies on the precise activity of a unique enzyme: thermostable DNA polymerase. This guide deconstructs the three essential steps of the PCR cycle—denaturation, annealing, and extension—framing them within the critical context of DNA polymerase function. The characteristics of this enzyme, including its thermostability, fidelity, and processivity, are not merely background details; they are the very factors that dictate the efficiency, specificity, and success of the entire amplification process [5]. Understanding this interplay is crucial for researchers and drug development professionals aiming to optimize protocols, develop novel assays, or accurately interpret genetic data.
The Central Role of DNA Polymerase in PCR
DNA polymerase is the enzymatic engine of the PCR reaction, responsible for synthesizing new DNA strands complementary to the template. Its properties directly determine the quality and quantity of the amplification product. Key characteristics include [5]:
- Thermostability: The enzyme must withstand repeated exposure to high temperatures (≥95°C) during the denaturation step without significant loss of activity. Taq DNA polymerase, isolated from the thermophilic bacterium Thermus aquaticus, was a revolutionary discovery that eliminated the need to add fresh enzyme after each cycle [1] [19].
- Fidelity: This refers to the accuracy of DNA replication. High-fidelity polymerases possess 3′→5′ exonuclease (proofreading) activity, which allows them to correct misincorporated nucleotides, reducing error rates. This is paramount in applications like cloning and mutagenesis [5] [26].
- Processivity: Defined as the number of nucleotides added per enzyme-binding event, high processivity enables the efficient amplification of long targets and templates with complex secondary structures [5].
- Specificity: "Hot-start" polymerases are engineered to be inactive at room temperature, preventing non-specific amplification and primer-dimer formation during reaction setup. Activation occurs only after the initial high-temperature denaturation step [5].
Table 1: Key Characteristics of DNA Polymerases in PCR
| Characteristic | Impact on PCR Performance | Example Enzymes |
|---|---|---|
| Thermostability | Determines ability to withstand denaturation temperatures; essential for automated thermal cycling. | Taq, Pfu |
| Fidelity (Proofreading) | Governs replication accuracy; critical for cloning and sequencing. Low-fidelity enzymes (e.g., Taq) are sufficient for routine detection. | Pfu (High), Taq (Low) |
| Processivity | Affects speed and efficiency, especially for long amplicons or GC-rich templates. | Engineered Taq variants |
| Specificity (Hot-Start) | Reduces non-specific amplification and primer-dimers by inhibiting activity at low temperatures. | Antibody-inhibited Taq |
The PCR Cycle: A Three-Step Thermal Process
A standard PCR cycle consists of three discrete temperature-dependent steps, repeated 25–40 times. This process is automated using a thermal cycler [26] [27].
Step 1: Denaturation
- Purpose: To separate the double-stranded DNA template into single strands, providing access for the primers.
- Typical Temperature: 94–98°C for 20–30 seconds [28] [27].
- Mechanism: High thermal energy breaks the hydrogen bonds between complementary base pairs, yielding two single-stranded DNA molecules.
- DNA Polymerase Context: This step also serves to activate hot-start DNA polymerases by dissociating inhibitory antibodies or compounds. The thermostability of the polymerase is critical here, as it must remain functional through dozens of these high-temperature exposures [5]. For highly complex DNA (e.g., genomic DNA) or GC-rich sequences (>65%), a longer initial denaturation of 1–3 minutes may be required [28].
Step 2: Annealing
- Purpose: To allow the forward and reverse primers to bind (anneal) to their complementary sequences on the single-stranded template DNA, flanking the target region.
- Typical Temperature: 50–65°C for 20–40 seconds [28] [27].
- Mechanism: The reaction temperature is lowered to a point that stabilizes hydrogen bonding between the primer and its exact complementary sequence. The optimal temperature is typically 3–5°C below the melting temperature (Tm) of the primers [28] [19].
- DNA Polymerase Context: The annealing temperature is a critical determinant of specificity. If too low, primers may bind imperfectly, leading to non-specific amplification. If too high, primer binding may not occur at all. The buffer provided with the DNA polymerase, particularly its magnesium ion (Mg²⁺) concentration, also strongly influences annealing efficiency and specificity [29] [28] [27].
Step 3: Extension
- Purpose: For DNA polymerase to synthesize a new DNA strand by extending the primer in the 5′ to 3′ direction.
- Typical Temperature: 72°C for 20–60 seconds per kilobase of target DNA [28] [26].
- Mechanism: The DNA polymerase recognizes the 3′-OH end of the annealed primer and sequentially adds free deoxynucleoside triphosphates (dNTPs) that are complementary to the template strand.
- DNA Polymerase Context: This is the step where the enzyme's core function is executed. The processivity of the polymerase dictates the synthesis rate (e.g., Taq is "fast," while Pfu is "slow") [26]. The extension temperature is set to the optimum for the specific enzyme being used (e.g., 72°C for Taq) to ensure maximal activity and accuracy [19]. The duration of this step must be optimized based on both the length of the amplicon and the synthesis speed of the polymerase [28].
Diagram 1: PCR Thermal Cycling Process. The three core steps are repeated 25-40 times, with the DNA product doubling in theory each cycle [26] [19].
Advanced PCR Protocol and Optimization
A robust PCR protocol requires careful preparation and optimization of each component. Below is a detailed methodology for a standard reaction using Taq DNA polymerase.
Standard PCR Reaction Setup
Table 2: Components of a Standard 50 μL PCR Reaction [29]
| Component | Final Concentration/Amount | Function & Critical Notes |
|---|---|---|
| PCR-Grade Water | To 50 μL | Nuclease-free to prevent degradation of DNA and reagents. |
| 10X Reaction Buffer | 1X | Provides optimal pH (8.0-9.5) and ionic conditions (e.g., KCl) for polymerase activity [26]. |
| MgCl₂ | 1.5 - 2.5 mM | Essential cofactor for DNA polymerase. Concentration must be optimized; it affects primer annealing and enzyme activity [29] [27]. |
| dNTP Mix | 200 μM each | Building blocks (dATP, dCTP, dGTP, dTTP) for new DNA synthesis. Unbalanced concentrations can induce errors. |
| Forward Primer | 0.1 - 1.0 μM | Defines the start of the target sequence on one strand. |
| Reverse Primer | 0.1 - 1.0 μM | Defines the start of the target sequence on the complementary strand. |
| Template DNA | 1 - 100 ng (genomic) | The DNA containing the target sequence to be amplified. Quality is critical; contaminants can inhibit the reaction. |
| Taq DNA Polymerase | 1.0 - 2.5 Units | The enzyme that catalyzes DNA synthesis. Thermostable and lacking proofreading activity. |
Experimental Protocol [29] [28]:
- Mix Preparation: Combine all components in a sterile, thin-walled PCR tube on ice, adding the DNA polymerase last to prevent premature activity.
- Thermal Cycling: Load the tube into a thermal cycler and run the program outlined in Table 3.
- Post-Amplification: After cycling, a final hold at 4-15°C allows for short-term storage. The amplified DNA (amplicon) is typically analyzed by agarose gel electrophoresis.
Table 3: Typical Thermal Cycler Parameters for a Three-Step PCR
| Step | Temperature | Time | Notes |
|---|---|---|---|
| Initial Denaturation | 94-98°C | 1-3 min | Ensures complete separation of complex DNA; activates hot-start polymerases. |
| Cycling (25-40x) | |||
| › Denaturation | 94-98°C | 20-30 sec | |
| › Annealing | 50-65°C | 20-40 sec | Critical optimization point. |
| › Extension | 72°C | 30-60 sec/kb | Time depends on polymerase speed and amplicon length. |
| Final Extension | 72°C | 5-15 min | Ensures all nascent strands are fully synthesized. |
| Final Hold | 4-15°C | ∞ | For short-term product storage. |
Critical Optimization Strategies
- Annealing Temperature Optimization: Use a thermal cycler with a gradient block to test a range of temperatures (e.g., 45-65°C) in a single run. The optimal temperature yields the highest amount of specific product with minimal background [28].
- MgCl₂ Titration: Perform a series of reactions with MgCl₂ concentrations varying from 1.0 mM to 3.0 mM in 0.5 mM increments. The correct concentration maximizes yield and specificity [29].
- Cycle Number Determination: Too few cycles (<25) yield low product; too many (>45) increase non-specific products and errors. For low-copy-number targets (e.g., <10 copies), up to 40 cycles may be needed [28].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for PCR
| Reagent / Solution | Critical Function | Technical & Research Context |
|---|---|---|
| Hot-Start DNA Polymerase | Inhibits polymerase activity at room temperature, drastically reducing non-specific amplification and primer-dimer formation. | Essential for high-throughput and highly specific assays. Available via antibody-based inhibition, chemical modification, or aptamer binding [5]. |
| High-Fidelity Polymerase Blends | Provides superior replication accuracy through 3′→5′ proofreading exonuclease activity. | Critical for cloning, sequencing, and mutagenesis. Often a blend of enzymes (e.g., Taq and Pfu) to balance speed and fidelity [5] [26]. |
| PCR Enhancers/Additives | Compounds like DMSO, glycerol, or betaine that aid in denaturing difficult templates (e.g., GC-rich sequences). | Vital for amplifying challenging targets. They lower the melting temperature of DNA, facilitating strand separation during denaturation [28]. |
| dNTPs | The fundamental nucleotides (dATP, dCTP, dGTP, dTTP) required for DNA strand synthesis. | Quality and balance are crucial. Ultrapure dNTPs prevent reaction failure. Unbalanced mixes can lead to misincorporation errors. |
| Optimized Buffer Systems | Provide the optimal salt (K⁺) and pH environment for the specific DNA polymerase used. | Specialized buffers can enable universal annealing temperatures, simplifying multiplex PCR assay design [28]. |
Evolution of PCR: qPCR and dPCR
The fundamental PCR principle has evolved into powerful quantitative and absolute quantification techniques.
- Quantitative PCR (qPCR) / Real-Time PCR: This method allows for the quantification of DNA as it is amplified. It uses fluorescent dyes (e.g., SYBR Green) or sequence-specific probes (e.g., TaqMan) to monitor product accumulation in real-time during the exponential phase of amplification. The cycle threshold (Ct) value is used for quantification, making it a gold standard for gene expression analysis (via RT-qPCR) and viral load detection [1] [30].
- Digital PCR (dPCR): A more recent advancement, dPCR partitions a PCR reaction into thousands of nanoscale reactions. This allows for absolute quantification of target DNA without a standard curve by counting the positive and negative partitions. dPCR offers superior precision for detecting small fold-changes and is less susceptible to PCR inhibitors, making it valuable for detecting rare mutations and copy number variations [26] [31].
Diagram 2: DNA Polymerase Functional Domains. The 5'→3' polymerase domain catalyzes DNA synthesis, while the 3'→5' exonuclease domain in high-fidelity enzymes enables proofreading by excising mismatched nucleotides [5].
The deconstruction of the PCR cycle into its three core steps—denaturation, annealing, and extension—reveals an elegant and powerful biochemical process. However, its efficacy is entirely dependent on the sophisticated properties of DNA polymerase. From the thermostability that enables automated thermal cycling to the fidelity that ensures data integrity in genetic analysis, this enzyme is the functional heart of PCR. For researchers and drug developers, a deep understanding of this relationship is not merely academic. It is a practical necessity for designing robust assays, troubleshooting failed experiments, and pushing the boundaries of sensitivity and accuracy in applications ranging from basic gene cloning to the cutting-edge fields of quantitative and digital PCR. As PCR continues to be a foundational tool, the interplay between enzyme characteristics and cycling parameters will remain a central tenet of successful molecular research.
The DNA polymerase enzyme is the fundamental molecular machine responsible for synthesizing new DNA strands, a process that is the very bedrock of genetic inheritance, molecular biology, and modern biotechnology. Its core function—the template-directed addition of nucleotides in a strict 5' to 3' direction—is the central theme of this technical guide. Within the context of Polymerase Chain Reaction (PCR) research, understanding this activity transcends basic enzymology; it is critical for optimizing assay specificity, fidelity, and efficiency in applications ranging from diagnostic test development to drug discovery [4] [32]. This whitepaper provides an in-depth analysis of the 5' to 3' polymerization mechanism, its kinetic characterization, and its direct implications for PCR-based research and development.
The Fundamental Mechanism of 5' to 3' Synthesis
Chemical Basis and Directionality
DNA polymerase catalyzes the synthesis of DNA molecules from nucleoside triphosphates, the molecular precursors of DNA. The enzyme facilitates a nucleophilic attack where the 3' hydroxyl (3'-OH) group of the last nucleotide in the growing primer strand attacks the alpha phosphate of the incoming deoxynucleoside triphosphate (dNTP). This results in the formation of a phosphodiester bond with the release of a pyrophosphate molecule (PPi) [33]. This chemical reaction dictates an inherent directionality: new nucleotides can only be added to the 3' hydroxyl end, causing the chain to grow in the 5' to 3' direction, meaning the 5' phosphate of the incoming nucleotide is linked to the 3' hydroxyl of the existing chain [33]. Consequently, as the DNA polymerase moves along the template strand in a 3' to 5' direction, it synthesizes a new, complementary strand in the antiparallel 5' to 3' direction.
Structural Basis for Fidelity and Processivity
The known DNA polymerases exhibit a highly conserved structure, often described as resembling a right hand with thumb, finger, and palm domains [33]. Each domain plays a distinct role in the 5' to 3' polymerization activity:
- Palm domain: Serves as the catalytic engine, housing residues that coordinate metal ions (typically Mg²⁺) to catalyze the phosphoryl transfer reaction [33].
- Finger domain: Binds the incoming dNTP and undergoes a conformational change to enclose the substrate, ensuring proper base pairing with the template. This "closed" state is crucial for nucleotide selection [34] [33].
- Thumb domain: Interacts with the newly synthesized DNA duplex, playing a key role in processivity—the number of nucleotides added per enzyme-binding event [33].
This structured arrangement allows the polymerase to rapidly and accurately copy DNA templates. Processive DNA polymerases can add hundreds to thousands of nucleotides per binding event, a capability dramatically enhanced in replication complexes through interaction with sliding DNA clamp proteins [33].
Diagram of 5' to 3' Polymerization Activity
Kinetic Analysis of the Polymerization Pathway
A minimal reaction pathway for DNA polymerases involves several elementary steps that precede and follow the chemical bond formation. Kinetic studies using techniques like chemical quench-flow and fluorescence-based assays have been essential in delineating these steps and identifying potential rate-limiting checkpoints, especially for understanding fidelity [34].
The Reaction Pathway and Conformational Changes
The pathway begins with the binding of the DNA polymerase (E) to the DNA primer-template (DNA), forming a binary complex (E•DNA). The correct incoming dNTP then binds to form a ternary complex (E•DNA•dNTP). A critical, often rate-limiting, step is a conformational change in the polymerase (from an "open" to a "closed" state) that correctly positions the dNTP in the active site (E*•DNA•dNTP). This is followed by the chemical step of phosphodiester bond formation. Finally, another conformational change and the release of pyrophosphate occur before the polymerase translocates to the next position [34].
Experimental Kinetic Approaches
Several kinetic experiment designs are used to dissect this pathway:
- Single-Turnover Kinetics: The DNA polymerase is present in excess over the DNA substrate. This setup ensures all DNA is bound, allowing measurement of the maximum rate of nucleotide incorporation (k~pol~) and the apparent dissociation constant (K~D~) for the dNTP, reporting on steps up to and including chemistry [34].
- Burst Kinetics: When enzyme and DNA concentrations are similar, a biphasic reaction is observed. A rapid "burst" of product formation corresponds to the first round of catalysis from pre-formed E•DNA complexes, followed by a slower steady-state phase limited by product release and enzyme turnover. This is diagnostic for a rate-limiting step after chemistry [34].
- Pulse-Chase and Pulse-Quench Experiments: These experiments can distinguish between a slow chemical step and a slow conformational step preceding it. A difference in product yield between a chase (drives intermediates to product) and a quench (instantly stops the reaction) indicates accumulation of a pre-chemical intermediate [34].
Table 1: Key Kinetic Parameters for DNA Polymerase 5' to 3' Elongation
| Parameter | Description | Experimental Method | Typical Values for Wild-Type Polymerases |
|---|---|---|---|
| k~pol~ | Maximum rate of nucleotide incorporation (s⁻¹) | Single-Turnover Kinetics | 10 to >100 s⁻¹ [34] |
| K~D, dNTP~ | Apparent dissociation constant for dNTP (μM) | Single-Turnover Kinetics | Varies with polymerase and template sequence [34] |
| k~cat~ | Steady-state turnover number (s⁻¹) | Steady-State Kinetics | Often limited by product dissociation (k~off~) [34] |
| Processivity | Average nucleotides added per binding event | Single-Molecule/Synchronized Assays | Varies widely; enhanced by sliding clamps [33] |
| Fidelity | Error rate (incorrect nucleotides per base pair) | Specific Assays (e.g., reversion) | ~10⁻⁴ to 10⁻⁶; can reach ~10⁻⁷ with proofreading [33] |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Studying and Utilizing 5' to 3' Polymerization
| Reagent / Material | Function in Polymerization Research/Application |
|---|---|
| Defined Oligonucleotides | Serve as primer and template substrates for kinetic studies and PCR assay development [34]. |
| dNTPs (dATP, dTTP, dCTP, dGTP) | The fundamental building blocks incorporated during 5' to 3' chain elongation [33]. |
| Thermostable DNA Polymerases (e.g., Taq, KOD, Pfu) | Essential for PCR; withstand high denaturation temperatures. Vary in fidelity, speed, and processivity [4] [35]. |
| α-³²P-dNTP or Fluorescently-Labeled dNTPs | Enable detection and quantification of synthesized DNA products in gel-based and real-time assays [34]. |
| MgCl₂ / Mg²⁺ | Critical divalent cation cofactor for catalytic activity in nearly all DNA polymerases [33]. |
| Phosphorothioate dNTPs | Used in mechanistic studies; the sulfur substitution creates a chiral center and can be used to probe the chemical step (elemental effect) [34]. |
| Proofreading Polymerases (e.g., Pfu, Q5) | Possess 3'→5' exonuclease activity to excise misincorporated nucleotides, significantly enhancing replication fidelity [36]. |
Implications for PCR Research and Diagnostic Applications
The precise 5' to 3' activity of DNA polymerase is the linchpin of PCR technology. Its performance directly dictates the success and accuracy of countless applications in research and drug development.
Fidelity and Error Correction in PCR
The intrinsic fidelity of a DNA polymerase is a paramount concern, especially in applications like sequencing and mutation detection. Fidelity is achieved through a multi-step mechanism. Initial selectivity occurs at the nucleotide incorporation step, where the enzyme's active site has a strong kinetic preference for correctly base-paired dNTPs. For polymerases possessing 3'→5' exonuclease (proofreading) activity, a second layer of fidelity exists. If a mismatch is incorporated, the DNA is transferred from the polymerase active site to the exonuclease domain, where the incorrect nucleotide is excised before synthesis resumes [33] [36]. The absence of this activity in polymerases like Taq can limit the maximum reliable amplicon size and increase error rates, a critical consideration for experimental design [36].
Technological Advancements Driven by Polymerase Engineering
The core understanding of 5' to 3' synthesis has fueled the development of advanced PCR methodologies. Quantitative PCR (qPCR) relies on the predictable accumulation of DNA product during the exponential phase of amplification to quantify template abundance [4] [37]. Digital PCR (dPCR) takes this further by partitioning a sample into thousands of individual reactions, allowing for absolute quantification of nucleic acids based on Poisson statistics, pushing sensitivity to the single-molecule level [4]. Furthermore, the drive for point-of-care diagnostics has led to the engineering of novel polymerase formulations and the development of innovative platforms like microfluidic PCR and photonic PCR, which leverage rapid thermal cycling to accelerate the 5' to 3' polymerization process [4].
Diagram of Proofreading Mechanism
The 5' to 3' polymerization activity of DNA polymerase is a masterpiece of evolutionary molecular engineering. Its unwavering directionality, governed by the chemistry of phosphodiester bond formation, is executed with remarkable speed and accuracy by a conserved structural framework. For the research scientist and drug developer, a deep understanding of this mechanism—from the kinetic intricacies of nucleotide selection to the functional consequences of proofreading—is not merely academic. It is a practical necessity for designing robust PCR assays, interpreting complex data, and leveraging next-generation technologies like dPCR and ultrafast microfluidic systems that continue to push the boundaries of molecular diagnostics and therapeutic discovery. As PCR solidifies its role as an indispensable platform in precision medicine, the fundamental principles of how DNA polymerase synthesizes DNA strands remain the foundation upon which innovation is built.
Precision Tools: Selecting and Applying DNA Polymerases in Research and Diagnostics
The function of DNA polymerase in PCR research extends far beyond simple DNA synthesis; it is the core determinant of success in diverse molecular biology applications. This in-depth technical guide provides researchers and drug development professionals with a structured framework for selecting the appropriate DNA polymerase based on the specific requirements of cloning, quantitative PCR (qPCR), and sequencing. By examining key enzyme properties—including fidelity, processivity, thermostability, and specificity—we establish clear guidelines to optimize experimental outcomes, ensure data integrity, and streamline workflows in biomedical research.
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology that amplifies specific DNA sequences through repeated cycles of denaturation, annealing, and extension [38]. At the heart of this process is DNA polymerase, a thermostable enzyme that synthesizes new DNA strands complementary to the target template [39]. Since the introduction of Taq DNA polymerase, which revolutionized PCR by enabling workflow automation, significant advancements have been made in engineering DNA polymerases with enhanced properties for specialized applications [5] [39]. The function of DNA polymerase in PCR research is multifaceted, impacting not only the success of amplification but also the accuracy, yield, and suitability of the products for downstream applications. This guide examines how different polymerase characteristics must be matched to the specific demands of cloning, qPCR, and sequencing to achieve optimal results.
Critical DNA Polymerase Properties for PCR Success
Selecting the optimal DNA polymerase requires a thorough understanding of four key biochemical properties that directly impact PCR performance and outcomes.
Fidelity: Accuracy in DNA Replication
Fidelity refers to the accuracy of DNA replication, measured as the error rate of misincorporated nucleotides per total bases synthesized [5]. High-fidelity DNA polymerases possess 3′→5′ exonuclease (proofreading) activity, which allows them to detect and excise mismatched nucleotides during synthesis [5]. This capability is quantified relative to Taq DNA polymerase; for example, Q5 High-Fidelity DNA Polymerase demonstrates 280 times higher fidelity than standard Taq polymerase [40]. Fidelity is paramount in applications like cloning and sequencing where sequence accuracy affects functional analysis.
Processivity: Efficiency of DNA Synthesis
Processivity defines the number of nucleotides a polymerase can incorporate per single binding event to the DNA template [5] [41]. Enzymes with high processivity exhibit greater efficiency in amplifying long templates, GC-rich sequences, and targets with complex secondary structures [5]. They also demonstrate enhanced resistance to common PCR inhibitors found in blood or plant tissues [5]. Processivity directly impacts amplification yield and the ability to generate long amplicons, making it crucial for applications such as long-range PCR and whole genome amplification.
Thermostability: Withstanding High Temperatures
Thermostability indicates an enzyme's ability to retain activity after prolonged exposure to high temperatures required for DNA denaturation (typically >90°C) [5]. While Taq polymerase has a half-life of approximately 40 minutes at 95°C, enzymes from hyperthermophilic archaea like Pfu DNA polymerase show approximately 20 times greater thermal stability [5]. This property is essential for efficient amplification of templates with high GC content and for preventing enzyme depletion during extended cycling protocols.
Specificity: Precision in Target Amplification
Specificity refers to the enzyme's ability to amplify only the intended target sequence without generating nonspecific products [5]. Hot-start DNA polymerases enhance specificity through antibody-based or chemical inhibition that prevents enzymatic activity during reaction setup until the initial high-temperature denaturation step [5]. This mechanism prevents primer-dimer formation and mispriming, which is particularly critical in multiplex PCR and low-copy number amplification [41].
Application-Specific DNA Polymerase Selection
Polymerases for Cloning Applications
DNA cloning requires high-fidelity amplification to ensure accurate sequence representation in vectors. Proofreading polymerases are essential to minimize mutations that could compromise downstream functional analyses.
Table 1: DNA Polymerase Selection for Cloning Applications
| Polymerase Type | Key Features | Recommended Use Cases | Resulting Ends | Example Products |
|---|---|---|---|---|
| High-Fidelity | 3′→5′ exonuclease activity, high accuracy (50-280x Taq) | PCR cloning, sequencing, site-directed mutagenesis | Blunt | Q5 High-Fidelity, Phusion High-Fidelity [40] |
| Standard Taq | No proofreading, terminal transferase activity | TA cloning of short inserts (≤5 kb) | 3′ dA overhangs | Taq DNA Polymerase [42] |
Experimental Protocol: PCR Cloning with Blunt-End Ligations
- Amplify Insert: Perform PCR using a high-fidelity DNA polymerase (e.g., Q5 or Phusion) with the following reaction conditions [40] [42]:
- 1X Reaction Buffer
- 0.2 mM each dNTP
- 0.5 µM forward and reverse primers
- 10-50 ng template DNA
- 1 unit DNA polymerase
- Nuclease-free water to 50 µL
- Thermal Cycling:
- Initial denaturation: 98°C for 30 seconds
- 35 cycles: [98°C for 10 seconds, 55-72°C for 20 seconds, 72°C for 30 seconds/kb]
- Final extension: 72°C for 2 minutes
- Hold at 4°C
- Purify PCR Product: Use a PCR purification kit to remove enzymes, salts, and unused dNTPs [42].
- Ligate into Vector: Incubate purified blunt-end PCR product with a linearized blunt-end vector using T4 DNA ligase at room temperature for 2 hours or 16°C overnight [42].
- Transform: Introduce ligation product into competent E. coli cells via heat shock or electroporation.
DNA Cloning Workflow
Polymerases for qPCR and Real-Time Applications
qPCR requires polymerases that combine rapid cycling capability with compatibility with fluorescent detection systems. The getPCR method exemplifies a specialized qPCR application for quantifying genome editing efficiency [43].
Table 2: DNA Polymerase Selection for qPCR Applications
| Polymerase Property | Importance in qPCR | Optimal Characteristics |
|---|---|---|
| Speed | Enables fast cycling for high-throughput applications | Fast extension rate (2-4 seconds/kb) |
| dUTP Tolerance | Prevents carryover contamination in diagnostic assays | Able to incorporate dUTP for UDG carryover prevention systems [44] |
| Fluorescent Probe Compatibility | Enables real-time detection of amplification | Works with intercalating dyes (SYBR Green) and hydrolysis probes (TaqMan) |
| Inhibition Resistance | Ensures reliable results from complex samples | Maintains activity in presence of blood components or other inhibitors [5] |
Experimental Protocol: getPCR for Genome Editing Efficiency Quantifications The getPCR (genome editing test PCR) method utilizes the sensitivity of Taq DNA polymerase to 3′ end mismatches to selectively amplify wild-type but not indel-modified sequences [43].
Primer Design:
- Design "watching primers" that span the nuclease cutting site with the 3′ end placed to discriminate against indel mutations.
- Use 4 cumulative watching bases (sum of forward and reverse primer watching bases) for optimal specificity [43].
- Select adenine as the 3′ terminal base for maximum mismatch discrimination [43].
Reaction Setup:
- Prepare two parallel qPCR reactions for each sample:
- Test reaction: With watching primers spanning the cut site
- Control reaction: With primers amplifying an unedited reference region
- Use a standard qPCR master mix containing:
- 1X Reaction Buffer
- 0.2 mM each dNTP
- 0.1-1 µM watching primers and control primers
- Hot-start Taq DNA polymerase
- Intercalating fluorescent dye (e.g., SYBR Green)
- 10-100 ng genomic DNA
- Nuclease-free water to 20 µL
- Prepare two parallel qPCR reactions for each sample:
qPCR Cycling Conditions:
- Initial activation: 95°C for 2 minutes
- 40 cycles: [95°C for 15 seconds, 60°C for 20 seconds, 72°C for 30 seconds]
- Fluorescence acquisition at the end of each extension step
Data Analysis:
- Calculate the difference in quantification cycles (∆Cq) between test and control reactions
- Determine the percentage of wild-type DNA using the ∆∆Cq method: % wild-type = 2^(-∆Cq) × 100
- Calculate editing efficiency: % editing = 100 - % wild-type [43]
Polymerases for Sequencing Applications
DNA sequencing applications demand the highest fidelity amplification to ensure accurate base representation. Polymerases with proofreading capabilities are essential to prevent amplification errors that could be misinterpreted as genetic variants.
High-Fidelity DNA Polymerases for Next-Generation Sequencing (NGS) Library Preparation NGS library preparation requires precise amplification with minimal introduction of errors that could compromise variant calling.
Amplify Target Regions:
- Use high-fidelity polymerases with >100x fidelity of Taq (e.g., Q5 High-Fidelity DNA Polymerase) [40].
- Reaction conditions should maintain high fidelity:
- Minimal cycle number (25-30 cycles) to reduce mutation accumulation
- Balanced dNTP concentrations (0.2 mM each)
- Sufficient template DNA (10-100 ng) to minimize stochastic effects
Purify and Quantify Amplicons:
- Clean up PCR products using bead-based purification systems
- Quantify using fluorometric methods to ensure equal representation of samples
Library Validation:
- Assess amplicon size distribution using microfluidic analyzers
- Pool libraries at equimolar concentrations for multiplexed sequencing
Polymerase Properties Guide Application Selection
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for DNA Polymerase Applications
| Reagent | Function | Application Notes |
|---|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion) | Amplifies DNA with minimal errors | Essential for cloning and sequencing; produces blunt-ended amplicons [40] |
| Hot-Start Taq DNA Polymerase | Reduces nonspecific amplification by requiring heat activation | Ideal for qPCR, multiplex PCR, and amplification from low template amounts [5] |
| dNTP Mix | Building blocks for DNA synthesis | Use balanced concentrations (0.2 mM each) for optimal fidelity [44] |
| MgCl₂ Solution | Cofactor for DNA polymerase activity | Concentration must be optimized (typically 1.5-3.5 mM); affects enzyme fidelity and yield [44] |
| PCR Buffer | Provides optimal ionic and pH conditions | Specific to each polymerase; may include stabilizers and enhancers [44] |
| TOPO Cloning Vectors | Enables rapid ligation without traditional ligase | Contains covalently bound topoisomerase I for 5-minute ligation times [42] |
Matching DNA polymerase properties to application requirements is fundamental to successful PCR research. Cloning demands high-fidelity polymerases to maintain sequence integrity, qPCR benefits from fast, specific enzymes compatible with detection chemistries, and sequencing requires the utmost accuracy to preserve base-level resolution. By understanding the relationship between enzyme characteristics—fidelity, processivity, thermostability, and specificity—and application needs, researchers can make informed decisions that optimize experimental outcomes, reduce costly failures, and accelerate scientific discovery in biomedical research and drug development.
Within the framework of modern molecular biology research, the polymerase chain reaction (PCR) stands as a foundational technique, and the function of DNA polymerase is its central biochemical engine. The discovery and utilization of Taq DNA polymerase, a thermostable enzyme isolated from Thermus aquaticus, revolutionized PCR by eliminating the need to add fresh enzyme after each denaturation cycle, thereby enabling automation [1] [45]. This protocol deep dive provides an in-depth technical guide to the standard setup of a PCR using Taq DNA polymerase, framed within the broader context of how this enzyme's specific properties dictate the reaction's success. For researchers and drug development professionals, a meticulous understanding of this interaction between enzyme, reagents, and cycling parameters is critical for generating robust, reproducible data in applications ranging from gene cloning to diagnostic assay development.
Taq DNA polymerase is a thermostable enzyme with a half-life of approximately 40 minutes at 95°C, exhibiting a nucleotide incorporation rate of about 60 bases per second at 70°C [44]. It possesses 5'→3' DNA polymerase activity and 5'→3' exonuclease activity, but lacks 3'→5' proofreading capability, which has direct implications for the fidelity of the synthesized amplicons [46]. The enzyme's functional state involves a critical interaction with the DNA template: it first binds to the primer-template junction, forming a binary complex, before engaging with dNTPs to catalyze the step-wise elongation of the DNA strand [47]. The following workflow delineates the core procedural and biochemical logic of a standard PCR experiment.
Reaction Component Considerations
The success of a PCR experiment is contingent upon the precise optimization of each reaction component. The following table provides a detailed overview of these critical components, their functions, and their recommended concentrations or amounts in a standard 50 µL reaction.
Table 1: Essential Components for a Standard 50 µL PCR Reaction with Taq DNA Polymerase
| Component | Function & Role of DNA Polymerase | Recommended Quantity/Concentration | Key Considerations |
|---|---|---|---|
| Template DNA | Provides the target sequence for amplification. DNA polymerase binds to its single-stranded form after denaturation [47]. | 1–1000 ng (e.g., 0.1–1 ng plasmid DNA; 5–50 ng genomic DNA) [44] [45]. | High purity is critical; contaminants like phenol or EDTA can inhibit Taq polymerase [48]. |
| Taq DNA Polymerase | Enzyme that synthesizes new DNA strands by incorporating dNTPs complementary to the template [44] [47]. | 1–2.5 units per 50 µL reaction [44] [45]. | Hot-start versions are recommended to minimize nonspecific amplification at room temperature [48]. |
| Primers | Short oligonucleotides that define the start and end of the target sequence. Provide the 3'-OH group for Taq polymerase to initiate synthesis [44]. | 0.1–1 µM each primer (typically 20–50 pmol per reaction) [44] [45]. | Must be designed with Tm within 55–70°C and avoid self-complementarity [44] [45]. |
| dNTPs | Building blocks (dATP, dCTP, dGTP, dTTP) for new DNA synthesis. Taq polymerase incorporates them into the growing strand [44]. | 200 µM of each dNTP (e.g., 50 µM each of dATP, dCTP, dGTP, dTTP) [45]. | Unbalanced concentrations increase PCR error rate; excess dNTPs can chelate Mg²⁺ [44] [48]. |
| Magnesium Ions (Mg²⁺) | Essential cofactor for Taq polymerase activity. Stabilizes the DNA duplex and catalyzes phosphodiester bond formation [44] [49]. | 1.5–2.0 mM (as MgCl₂ or MgSO₄); requires optimization [44] [45]. | Critical optimization parameter; excess Mg²⁺ reduces fidelity and increases nonspecific binding [48] [49]. |
| PCR Buffer | Provides optimal ionic environment (e.g., KCl at ~50 mM) and pH for Taq polymerase activity and primer-template binding [49] [45]. | 1X concentration (e.g., 5 µL of 10X buffer in a 50 µL reaction) [46] [45]. | Often supplied with the enzyme. May contain MgCl₂; if not, it must be added separately [46]. |
The Scientist's Toolkit: Research Reagent Solutions
A meticulously prepared toolkit is fundamental for experimental reproducibility. Below is a list of essential materials and their specific functions in a standard Taq-based PCR.
Table 2: Research Reagent Solutions for Standard PCR
| Item | Function/Justification |
|---|---|
| Hot-Start Taq DNA Polymerase | Reduces nonspecific amplification and primer-dimer formation by remaining inactive until a high-temperature activation step [48]. |
| Nuclease-Free Water | Serves as the reaction solvent; ensures no contaminating nucleases degrade primers, template, or enzyme. |
| 10X PCR Reaction Buffer | Typically supplied with the enzyme, it contains salts like KCl and Tris-HCl to maintain optimal pH and ionic strength for polymerase activity and primer annealing [46]. |
| 25 mM MgCl₂ Solution | Separate Mg²⁺ source for fine-tuning this critical cofactor's concentration, especially if not pre-included in the 10X buffer [49] [45]. |
| 10 mM dNTP Mix | A prepared, pH-balanced equimolar mixture of all four dNTPs (dATP, dCTP, dGTP, dTTP) to ensure balanced nucleotide incorporation [45]. |
| Target-Specific Primer Aliquots | High-quality, HPLC-purified oligonucleotides resuspended in nuclease-free water or TE buffer to a defined concentration (e.g., 20 µM) [44] [48]. |
| Control Template DNA | A well-characterized DNA sample (e.g., plasmid, genomic DNA) to serve as a positive control for the PCR reaction. |
| PCR Tubes/Plates | Thin-walled polypropylene tubes or plates designed for efficient heat transfer in thermal cyclers. |
Detailed Experimental Protocol
Step-by-Step Laboratory Methodology
This procedure is adapted from established protocols [46] [45] and must be performed using sterile techniques and nuclease-free reagents.
Preparation and Calculation:
- Wear gloves to prevent contamination from nucleases on the skin.
- Thaw all reagents (except Taq polymerase) on ice and mix thoroughly by gentle vortexing before use. Centrifuge briefly to collect contents at the bottom of the tube.
- Calculate the required volumes for all components based on a final 50 µL reaction volume. It is highly recommended to prepare a Master Mix for multiple reactions to minimize pipetting error and ensure consistency. Prepare enough for n+1 reactions.
Master Mix Assembly (on ice):
- In a sterile 1.5 mL microcentrifuge tube, combine the reagents in the following order for a single 50 µL reaction:
- Nuclease-Free Water (Q.S. to 50 µL)
- 10X PCR Buffer: 5 µL
- 10 mM dNTP Mix: 1 µL
- 25 mM MgCl₂ (if needed): Variable (e.g., 1.5-3 µL for 0.75-1.5 mM final concentration; requires optimization)
- 20 µM Forward Primer: 1.25 µL (0.5 µM final)
- 20 µM Reverse Primer: 1.25 µL (0.5 µM final)
- Taq DNA Polymerase (e.g., 5 U/µL): 0.5 µL (2.5 Units)
- Mix the Master Mix gently by pipetting up and down 20-25 times. Do not vortex after adding the enzyme.
- In a sterile 1.5 mL microcentrifuge tube, combine the reagents in the following order for a single 50 µL reaction:
Aliquoting and Adding Template:
- Dispense the appropriate volume of Master Mix into each PCR tube or well.
- Add the calculated volume of template DNA to each respective tube. Include a negative control containing nuclease-free water instead of template DNA.
- Cap the tubes securely. If using a thermal cycler without a heated lid, add a drop of mineral oil (~50 µL) to each tube to prevent evaporation [46].
Thermal Cycling:
- Place the tubes in the pre-heated thermal cycler and start the programmed run. A standard cycling protocol is detailed in Section 3.2.
Post-Amplification Analysis:
- After cycling, hold the reactions at 4°C.
- Analyze the PCR products by loading 5-10 µL of the reaction onto an ethidium bromide-stained agarose gel for electrophoresis and UV visualization to confirm amplification specificity and yield.
Thermal Cycling Parameters
The cycling parameters are designed to exploit the biochemical properties of Taq DNA polymerase. The following table outlines a standard three-step protocol.
Table 3: Standard Thermal Cycling Parameters for a Three-Step PCR Protocol
| Step | Temperature | Duration | Function & Biochemical Rationale |
|---|---|---|---|
| Initial Denaturation | 94–95°C | 1–3 minutes | Fully denatures complex DNA (e.g., gDNA) into single strands and may activate hot-start Taq polymerase. Essential for a uniform start [28] [49]. |
| Denaturation | 94–95°C | 15–30 seconds | Separates the newly formed double-stranded DNA products in each cycle, providing single-stranded templates for the next round. |
| Annealing | 45–65°C* | 15–60 seconds | Allows primers to bind (anneal) to their complementary sequences on the single-stranded DNA templates. Temperature is primer-specific and critical for specificity [28]. |
| Extension | 72°C | 1 minute per kb | The optimal temperature for Taq polymerase activity. The enzyme synthesizes new DNA strands from the 3' end of the primers [28] [46]. |
| Final Extension | 72°C | 5–10 minutes | Ensures all amplicons are fully extended. For cloning, a 30-minute incubation is recommended to add 3'-dA overhangs [28]. |
| Hold | 4°C | ∞ | For short-term storage of the product. |
*The annealing temperature (Ta) must be optimized. A good starting point is 3–5°C below the calculated Tm of the primers [28].
The molecular interplay between Taq polymerase, DNA, and nucleotides during thermal cycling can be visualized as a series of conformational changes and catalytic steps that drive the amplification process.
Troubleshooting and Optimization Guide
Even with a standardized protocol, optimization is often required for specific templates or primer sets. The table below addresses common issues and provides evidence-based solutions.
Table 4: Common PCR Problems and Optimization Strategies
| Observation | Potential Cause | Recommended Optimization Strategy |
|---|---|---|
| No or Low Yield | Insufficient template, low primer Ta, insufficient Mg²⁺, low number of cycles [48]. | - Verify template quality/quantity (104–107 molecules recommended) [49].- Increase Mg²⁺ concentration in 0.5 mM increments [45].- Lower Ta in 2–3°C increments [28].- Increase cycle number to 35–40 for low-copy targets [28]. |
| Nonspecific Bands/Smearing | Excess enzyme, low primer Ta, excess Mg²⁺, high primer concentration, high cycle number [44] [48]. | - Use a hot-start enzyme [48].- Increase Ta in 2–3°C increments [28] [48].- Titrate Mg²⁺ downward [48].- Reduce primer concentration (optimize 0.1–1 µM) [44]. |
| Primer-Dimer Formation | Primer 3'-end complementarity, excess primers, low Ta [48] [45]. | - Redesign primers to avoid 3' complementarity [45].- Use higher Ta and shorter annealing times [49].- Optimize primer concentration. |
| Poor Fidelity (Mutation Incorporation) | Lack of proofreading in Taq, excess Mg²⁺, unbalanced dNTPs, high cycle number [48]. | - Use polymerases with proofreading activity for cloning.- Optimize Mg²⁺ and use balanced dNTP concentrations [44] [48].- Reduce cycle number and increase template input. |
The DNA polymerase enzyme serves as the core engine of the polymerase chain reaction (PCR), catalyzing the template-directed synthesis of new DNA strands. In advanced PCR applications, the inherent properties of the DNA polymerase—including its thermostability, processivity, and fidelity—directly determine experimental success [50]. While basic PCR targets straightforward DNA sequences, research and diagnostic applications often require techniques that push the boundaries of conventional amplification. These advanced methods, including long-range PCR, high-GC content amplification, and multiplex PCR, present unique challenges that are primarily addressed through polymerase selection and reaction optimization. This guide examines these sophisticated applications within the broader context of DNA polymerase function, providing researchers with current methodologies to overcome common technical barriers in molecular biology research and drug development.
The versatility of PCR has found use in diverse areas of scientific research, including diagnostic medicine, agriculture, and forensics [50]. However, amplifying complex or long DNA segments remains challenging due to the biochemical limitations of early DNA polymerases. The development of engineered enzyme blends and specialized reagents has since enabled researchers to tackle increasingly difficult templates, from extensive genomic loci to structurally complex GC-rich regions [50].
Long-Range PCR: Amplifying Extended DNA Fragments
Long-range PCR involves the amplification of DNA fragments typically exceeding 5 kilobase pairs (kb), with some applications extending to 20 kb or more [50]. This technique is crucial for applications such as genome mapping, full-length gene cloning, and detection of large structural variations.
DNA Polymerase Requirements for Long Amplicons
The success of long-range PCR depends critically on using DNA polymerases with enhanced processivity and proofreading capability. Standard Taq DNA polymerase is insufficient for long amplifications due to its relatively low replication fidelity and inability to efficiently synthesize extended fragments [50]. Instead, researchers employ either:
- High-fidelity polymerases: Engineered enzymes with superior proofreading (3'→5' exonuclease) activity to correct misincorporated bases during amplification [50].
- Polymerase blends: Optimized mixtures that combine the processivity of one enzyme with the proofreading ability of another, providing both efficiency and accuracy for long templates [50].
Key Optimization Strategies for Long-Range PCR
Successful long-range PCR requires careful optimization of several reaction parameters beyond polymerase selection:
- Extension Time: Increase extension time significantly compared to standard PCR—typically 1 minute per 1,000 base pairs for fragments up to 3-4 kb, with progressively longer times for larger amplicons [51].
- Template Quality: Use high-quality, intact DNA template to avoid sheared fragments that cannot serve as viable templates for long amplification [51].
- Stabilizing Additives: Incorporate PCR enhancers such as betaine, DMSO, or glycerol to reduce secondary structure and improve polymerase processivity [50].
- Magnesium Concentration: Optimize Mg²⁺ concentration in 0.5 mM increments up to 4 mM, as insufficient Mg²⁺ will yield no product while excess may cause non-specific amplification [51].
Table 1: Essential Components and Their Optimization for Long-Range PCR
| Component | Standard PCR | Long-Range PCR | Optimization Strategy |
|---|---|---|---|
| DNA Polymerase | Standard Taq | High-fidelity/blended enzymes | Use polymerase with proofreading activity |
| Extension Time | 1 min/kb | 1-3 min/kb (increasing with length) | Scale time with product length |
| Cycle Number | 25-35 | 30-40 | Increase for low template concentration |
| Template DNA | 1pg-1μg | 10ng-1μg (high quality) | Ensure intact, high-molecular-weight DNA |
| Additives | Often omitted | Betaine, DMSO, glycerol | Test combinations for specific template |
Overcoming GC-Rich Content Challenges
GC-rich DNA sequences (approximately >60% GC content) present significant amplification challenges due to their high thermal stability and propensity to form secondary structures [52]. These regions are common in promoter elements and regulatory sequences of biologically significant genes, making their amplification essential for many research applications.
Biochemical Challenges of GC-Rich Amplification
The difficulties associated with GC-rich templates stem from fundamental biochemical properties:
- Thermal and Structural Stability: GC-rich sequences exhibit higher melting temperatures due to three hydrogen bonds between G-C base pairs compared to two in A-T pairs, requiring higher denaturation temperatures [53].
- Secondary Structure Formation: GC-rich regions readily form stable hairpin loops and intramolecular structures that impede polymerase progression and primer annealing [52].
- Incomplete Denaturation: At standard denaturation temperatures (94-95°C), GC-rich regions may not fully denature, leading to polymerase stalling and amplification failure [53].
Specialized Solutions for GC-Rich Templates
Multiple strategies have been developed to overcome the challenges of GC-rich amplification:
- Specialized Polymerases and Buffers: Commercial systems like OneTaq GC Buffer (NEB) and AccuPrime GC-Rich DNA Polymerase (ThermoFisher) are specifically engineered for high-GC templates [53]. These systems may incorporate polymerase enzymes from extremophilic organisms that naturally replicate GC-rich genomes.
- PCR Additives: Chemical additives play a crucial role in facilitating GC-rich amplification:
- Betaine: Reduces DNA melting temperature by disrupting base stacking, effectively equalizing the stability of GC and AT base pairs [50].
- DMSO: Interferes with hydrogen bonding and disrupts secondary structures that impede polymerase progression [52].
- Glycerol and formamide: Lower template denaturation temperature and reduce secondary structure formation [50].
- Temperature Modifications: Increasing denaturation temperature to 98°C or using a touchdown PCR approach can improve complete strand separation for problematic templates [53].
- Modified Nucleotides: Incorporating 7-deaza-dGTP, a dGTP analog that disrupts Hoogsteen base pairing, can significantly improve amplification of refractory GC-rich regions [53].
Table 2: PCR Additives for Challenging Templates and Their Mechanisms
| Additive | Final Concentration | Primary Mechanism | Application |
|---|---|---|---|
| Betaine | 1-1.5 M | Reduces base stacking energy; equalizes Tm of AT and GC pairs | GC-rich templates |
| DMSO | 5-10% | Disrupts hydrogen bonding; prevents secondary structures | GC-rich, long amplicons |
| Formamide | 1-5% | Lowers DNA melting temperature; denatures secondary structures | GC-rich templates |
| Glycerol | 5-15% | Stabilizes enzymes; lowers DNA denaturation temperature | Long-range PCR |
| 7-deaza-dGTP | 150 μM (with 50 μM dGTP) | Reduces hydrogen bonding; disrupts secondary structures | Extremely GC-rich regions |
Diagram: Strategic approaches to overcome GC-rich amplification challenges
Multiplex PCR: Simultaneous Multi-Target Amplification
Multiplex PCR enables the simultaneous amplification of multiple distinct DNA targets in a single reaction, significantly enhancing throughput and efficiency for diagnostic and research applications [54]. This technique is particularly valuable when sample material is limited or when comprehensive pathogen detection is required.
Design Principles for Robust Multiplex Assays
Developing an effective multiplex PCR assay requires careful consideration of several factors:
- Primer Compatibility: All primer pairs must function efficiently under identical thermal cycling conditions with minimal cross-dimer formation or non-specific priming [54]. Primer pairs should have closely matched melting temperatures (typically within 5°C) [51].
- Amplicon Differentiation: Multiplex products must be distinguishable by size (gel electrophoresis), fluorescent label (real-time PCR), or melting temperature (melting curve analysis) [54].
- Reaction Balancing: Optimize primer concentrations to prevent amplification bias, where some targets amplify more efficiently than others due to competition for limited reagents [54].
- Detection Systems: Advanced multiplex systems use fluorophore-labeled probes (TaqMan) or DNA-binding dyes with high-resolution melt analysis to distinguish multiple targets, sometimes detecting up to 20 or more targets simultaneously [54].
Advanced Applications of Multiplex PCR
Multiplex PCR has evolved beyond basic research to become a cornerstone of modern molecular diagnostics:
- Infectious Disease Panels: Comprehensive panels can simultaneously detect numerous bacterial, viral, and fungal pathogens from respiratory, bloodstream, or gastrointestinal samples [55]. During the SARS-CoV-2 pandemic, multiplex RT-PCR enabled differentiation between SARS-CoV-2, influenza A/B, and other respiratory pathogens with similar symptoms [54].
- Antimicrobial Resistance Detection: Multiplex assays can identify not only pathogens but also associated antibiotic resistance genes, guiding appropriate antimicrobial therapy and supporting antimicrobial stewardship programs [55].
- Genetic Analysis: Applications include short tandem repeat (STR) profiling for forensics, detection of genetic deletions (e.g., Duchenne muscular dystrophy), and identification of copy number variations [54].
Diagram: Comprehensive workflow for multiplex PCR assay development and analysis
Research Reagent Solutions
Selecting appropriate reagents is critical for success in advanced PCR applications. The following table outlines essential solutions for challenging PCR scenarios:
Table 3: Research Reagent Solutions for Advanced PCR Applications
| Reagent Category | Specific Examples | Function in Advanced PCR |
|---|---|---|
| Specialized Polymerases | OneTaq (NEB), Q5 (NEB), AccuPrime GC-Rich | Enhanced processivity, proofreading, GC-rich amplification |
| PCR Enhancers | Betaine, DMSO, GC Enhancer solutions | Disrupt secondary structures, improve polymerase efficiency |
| Optimized Buffer Systems | GC buffers, long-range buffers, proprietary mixes | Provide ideal chemical environment for challenging templates |
| Detection chemistries | TaqMan probes, SYBR Green, EvaGreen | Enable real-time monitoring, multiplexing, melt curve analysis |
| dNTP Formulations | dNTPs with 7-deaza-dGTP, high-fidelity mixes | Reduce secondary structure, improve incorporation accuracy |
Integrated Experimental Protocols
Comprehensive Protocol for GC-Rich Long-Range PCR
This integrated protocol combines approaches for amplifying long, GC-rich DNA fragments:
Reaction Setup:
- 1X specialized PCR buffer (commercial GC or long-range formulation)
- 1.5-2.0 mM Mg²⁺ (optimize empirically)
- 200 μM each dNTP
- 0.2-0.5 μM each forward and reverse primer
- 1M betaine
- 5% DMSO
- 1.25 units specialized polymerase blend (e.g., OneTaq or similar)
- 10-100 ng high-quality genomic DNA template
- Nuclease-free water to 50 μL final volume
Thermal Cycling Parameters:
- Initial denaturation: 95°C for 2 minutes
- 35 cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: Temperature gradient from 65-72°C for 30 seconds (determine optimal)
- Extension: 68°C for 1 minute per 1,000 bp (extend for longer fragments)
- Final extension: 68°C for 5-10 minutes
- Hold at 4°C
Post-Amplification Analysis:
Development and Validation Protocol for Multiplex PCR
This protocol outlines the systematic development of a multiplex assay for pathogen detection:
Primer and Probe Design:
- Design primers targeting conserved regions of each pathogen genome
- Ensure amplicon sizes are distinct (if using size-based detection) or have different Tm values (if using melt analysis)
- Verify specificity using BLAST against non-target sequences
- Label probes with distinct fluorophores for real-time detection
Initial Optimization:
- Test each primer pair individually in singleplex reactions
- Determine optimal annealing temperature using gradient PCR
- Titrate primer concentrations to achieve balanced amplification
- Optimize Mg²⁺ concentration (typically 1.5-3.0 mM)
Multiplex Reaction Assembly:
- 1X master mix containing buffer, dNTPs, Mg²⁺
- Optimized concentrations of all primer pairs (typically 0.1-0.3 μM each)
- Sequence-specific probes if using probe-based detection
- 1.25-2.5 units DNA polymerase
- 5-10 μL template DNA
- Nuclease-free water to final volume
Amplification and Analysis:
Advanced PCR applications continue to expand the utility of this fundamental molecular biology technique in research and clinical diagnostics. The success of long-range PCR, GC-rich amplification, and multiplexing hinges on understanding the functional properties of DNA polymerase enzymes and their interaction with reaction components. Through strategic selection of specialized polymerases, optimization of reaction conditions, and implementation of appropriate additives, researchers can overcome the challenges associated with complex templates. The continued refinement of these approaches supports ongoing advances in genomics, pathogen detection, and personalized medicine, demonstrating the enduring importance of PCR as a cornerstone technology in the life sciences.
Within the broader context of a thesis on the function of DNA polymerase in PCR research, the significance of reaction specificity cannot be overstated. The DNA polymerase enzyme is the core catalytic engine of the Polymerase Chain Reaction (PCR), responsible for template-directed synthesis of new DNA strands [25]. However, a fundamental challenge in conventional PCR is that many DNA polymerases, including the commonly used Taq DNA polymerase, retain significant enzymatic activity at the non-stringent temperatures encountered during reaction setup (room temperature or on ice) [57] [58]. This residual activity facilitates the extension of misprimed sequences and primer-dimers, leading to nonspecific amplification that competes with the target amplicon, reduces yield, and compromises assay sensitivity and reliability [57] [59]. Hot-Start PCR represents a refined methodological solution to this problem, engineered specifically to control and constrain DNA polymerase activity until high stringency conditions are achieved, thereby enhancing the precision and performance of PCR-based research and diagnostics [60].
The Problem of Nonspecific Amplification in Conventional PCR
In conventional PCR, the reaction mixture containing all essential components—DNA polymerase, primers, nucleotides, and template—is assembled at room temperature or on ice. Under these low-stringency conditions, primers can bind to non-target sequences with partial complementarity (mispriming) or to each other (primer-dimer formation) [59] [58]. The DNA polymerase, being partially active even at these sub-optimal temperatures, can extend these incorrectly bound primers, synthesizing undesired DNA products [57].
The negative impacts of this nonspecific amplification are multifold:
- Reduced Target Yield: Reaction components (nucleotides, enzymes, primers) are consumed by the synthesis of nonspecific products, leaving fewer resources for the amplification of the desired target [59].
- Decreased Sensitivity: For targets present in low copy numbers, such as in pathogen detection or single-gene analysis, the competition from nonspecific amplification can drastically reduce, or even prevent, the detection of the specific product [57] [60].
- Unreliable Results: The co-amplification of multiple products can lead to complex and uninterpretable results, compromising downstream analysis like cloning, sequencing, or clinical diagnosis [57] [25].
While preparing the reaction on ice can lower polymerase activity, it is an insufficient measure to completely prevent nonspecific synthesis [57]. Hot-Start PCR addresses this limitation by incorporating a mechanism that fundamentally blocks DNA polymerase activity until the first high-temperature denaturation step begins.
Core Mechanisms of Hot-Start PCR
Hot-Start PCR technologies employ diverse strategies to inhibit DNA polymerase activity during reaction setup. The inhibition is reversibly relieved by the initial heat denaturation step of the thermal cycler (typically >90°C), allowing amplification to proceed only under stringent conditions. The main mechanisms are summarized below and detailed in Table 1.
Table 1: Comparison of Major Hot-Start PCR Mechanisms
| Mechanism | Key Feature | Activation Trigger | Pros | Cons |
|---|---|---|---|---|
| Antibody-Based [57] [58] | A monoclonal antibody binds the polymerase's active site. | Heat denaturation (≥90°C) degrades the antibody. | Short activation time; full restoration of native enzyme activity [57]. | Animal-origin antibodies; higher exogenous protein content [57]. |
| Chemical Modification [57] [58] | Polymerase is covalently modified with inert chemical groups. | Prolonged heating (e.g., 10-12 min at 95°C) removes groups. | Highly stringent inhibition; animal-origin free [57]. | Longer activation time; can impair long amplicon (>3kb) yield [57]. |
| Affibody/Aptamer-Based [57] [58] | Peptide (Affibody) or oligonucleotide (Aptamer) binds polymerase. | Heat denaturation dissociates the binder. | Short activation time; animal-origin free (Aptamer) [57]. | Can be less stringent; bench stability may be reduced [57]. |
| Physical Separation [58] [61] | Polymerase is physically separated via wax barriers or within bacterial cells. | Wax melts or cell membranes are disrupted by heat. | Simple, cost-effective (e.g., EcoliTaq method) [61]. | Can be less convenient for high-throughput setups. |
| Modified dNTPs [62] | dNTPs are blocked with a thermolabile group at the 3'-O position. | Initial denaturation cleaves the protecting group. | Directly blocks the substrate; can be used with any polymerase [62]. | May require optimization of Mg²⁺ concentration [62]. |
| Modified Primers [59] | Primers contain thermolabile groups (e.g., OXP) at the 3' end. | Heat converts the modified primer to a functional form. | High specificity; applicable to any polymerase [59]. | Requires specialized primer synthesis. |
The logical workflow of how these mechanisms collectively enhance PCR specificity is illustrated below.
Experimental Protocols for Key Hot-Start Methods
Protocol: Antibody-Based Hot-Start PCR
This protocol uses a Taq DNA polymerase complexed with a neutralizing monoclonal antibody [57] [58].
Reaction Setup (on ice):
- Prepare a master mix containing:
- 1X PCR Buffer (with MgCl₂, typically final [MgCl₂] = 1.5-2.5 mM)
- Forward and Reverse Primers (0.1-1.0 µM each, final concentration)
- dNTP Mix (200 µM each dNTP, final concentration)
- Antibody-Hot Start Taq DNA Polymerase (1.0-1.5 units per 50 µL reaction)
- Template DNA (variable, e.g., 10-100 ng genomic DNA)
- Nuclease-free water to volume.
- Mix gently by pipetting. Do not vortex.
- Prepare a master mix containing:
Thermal Cycling:
- Initial Denaturation/Activation: 95°C for 2-5 minutes. This step denatures the antibody and activates the polymerase.
- Amplification (25-40 cycles):
- Denature: 95°C for 15-30 seconds.
- Anneal: Primer-specific temperature (50-65°C) for 15-30 seconds.
- Extend: 72°C for 1 minute per kb of amplicon length.
- Final Extension: 72°C for 5-10 minutes.
- Hold: 4°C.
Product Analysis:
- Analyze 5-10 µL of the PCR product by agarose gel electrophoresis and visualize with an intercalating dye [25].
Protocol: dNTP-Mediated Hot-Start PCR
This protocol utilizes dNTPs modified with a thermolabile protecting group (e.g., CleanAmp dNTPs) [62].
Reagent Preparation:
- Thaw the Hot-Start dNTP mix on ice or at room temperature. Vortex and pulse centrifuge.
- If using a concentrated stock, dilute with water or buffer (pH 8-10.5) to a working concentration.
Reaction Setup (on ice):
- Prepare a master mix containing:
- 1X PCR Buffer (pH 8-9, may require optimization)
- MgCl₂ (2.5 mM final concentration is a starting point; note: for every 0.2 mM increase in Hot Start dNTPs, add at least 1.0 mM extra MgCl₂)
- Forward and Reverse Primers (concentration as optimized)
- Hot-Start dNTP Mix (400 µM final concentration of each dNTP is recommended)
- Standard Taq DNA Polymerase (1.25 units per 25 µL reaction)
- Nuclease-free water.
- Mix gently by pipetting.
- Aliquot the master mix into tubes.
- Add template DNA (e.g., 5 µL per 20 µL master mix for a 25 µL total reaction).
- Prepare a master mix containing:
Thermal Cycling:
- Initial Denaturation/Activation: 95°C for 5-10 minutes. This step removes the protecting groups from the dNTPs.
- Amplification (30-40 cycles):
- Denature: 95°C for 30 seconds.
- Anneal: 55-65°C for 30 seconds.
- Extend: 72°C for 1 minute/kb.
- Final Extension: 72°C for 7 minutes.
- Hold: 4°C.
Product Analysis:
- Analyze a 10 µL aliquot by agarose gel electrophoresis [62].
Protocol: Novel EcoliTaq Direct Hot-Start PCR
This cost-effective protocol uses whole E. coli cells expressing Taq polymerase, providing a physical hot-start barrier [61].
EcoliTaq Preparation:
- Culture E. coli expressing recombinant Taq polymerase.
- Harvest, wash, and resuspend cells to a final OD₆₀₀ of 0.8.
Specialized Buffer Preparation:
- Prepare a high-pH Tricine-based buffer (pH 8.6) containing 2% Tween 20 and 0.4 M trehalose to overcome PCR inhibitors in direct amplification from blood.
Reaction Setup:
- Prepare a master mix containing:
- 1X Tricine Buffer (with 2% Tween 20, 0.4 M trehalose, pH 8.6)
- Primers and dNTPs (standard concentrations)
- EcoliTaq suspension (e.g., a 1:2 dilution of the OD 0.8 stock).
- Template (e.g., 1 µL of whole blood directly, without DNA purification).
- Mix gently.
- Prepare a master mix containing:
Thermal Cycling:
- Initial Denaturation/Activation: 95°C for 5-10 minutes. This lyses the E. coli cells and releases the active Taq polymerase.
- Amplification: Follow standard cycling parameters for the target amplicon.
- Analysis: Proceed with standard gel electrophoresis.
Quantitative Benefits and Research Applications
The implementation of Hot-Start technology provides measurable improvements in PCR performance. The following table synthesizes key quantitative and qualitative benefits as demonstrated across multiple studies.
Table 2: Documented Benefits and Applications of Hot-Start PCR
| Benefit / Application | Key Finding / Description | Significance |
|---|---|---|
| Specificity Enhancement | Near-complete elimination of primer-dimer artifacts and nonspecific bands in complex genomic DNA amplifications [57] [59]. | Enables cleaner results for cloning, sequencing, and diagnostic applications. |
| Sensitivity Increase | Reliable detection of low-copy-number targets (e.g., down to 200 CFU/mL of S. typhimurium in whole blood) [61]. | Critical for pathogen detection, viral load monitoring, and single-cell genomics. |
| Target Yield Improvement | Consistent increase in the yield of the desired amplicon due to reduced competition for reagents [57] [60]. | Provides higher-quality product for downstream applications like NGS library prep. |
| Application: Direct PCR | Successful amplification directly from crude samples (whole blood) using specialized buffers with EcoliTaq [61]. | Bypasses time-consuming DNA extraction steps, accelerating high-throughput screening. |
| Application: Allele-Specific PCR | 100% concordance with commercial kits in HLA-B27 genotyping of 110 clinical samples using EcoliTaq [61]. | Ensures high fidelity in SNP detection and genotyping for pharmacogenomics and disease association studies. |
| Automation Compatibility | Reaction mixes are stable at room temperature, making them suitable for automated liquid-handling platforms [57]. | Facilitates reproducible, high-throughput screening in clinical and drug discovery settings. |
The Scientist's Toolkit: Essential Research Reagents
Selecting the appropriate reagents is fundamental to designing a successful Hot-Start PCR experiment. The following table catalogues key solutions and their functions.
Table 3: Essential Reagents for Hot-Start PCR Research
| Reagent / Material | Function / Description | Example Use-Case |
|---|---|---|
| Antibody-Hot Start Polymerase | Polymerase inhibited by antibody binding; activated by heat. Examples: Platinum Taq, DreamTaq HS [57] [58]. | Standard high-specificity PCR and qPCR where rapid activation is needed. |
| Chemically Modified Polymerase | Polymerase inactivated by covalent modification; requires prolonged heating. Example: AmpliTaq Gold [57]. | Applications requiring very stringent inhibition during setup. |
| Hot-Start dNTP Mix | dNTPs blocked with thermolabile groups; activate upon heating. Example: CleanAmp dNTPs [62]. | Provides hot-start capability with any standard DNA polymerase. |
| OXP-Modified Primers | Primers with thermolabile 4-oxo-1-pentyl groups at 3' terminus; converted to native form by heat [59]. | Extreme specificity requirements; useful with any polymerase. |
| EcoliTaq Cells | E. coli cells expressing Taq polymerase; physical separation provides hot-start [61]. | Cost-effective, direct PCR from inhibitory samples like whole blood. |
| High-pH Enhancer Buffer | Buffer containing additives (e.g., Tween 20, trehalose) to counteract PCR inhibitors [61]. | Direct PCR from challenging samples (blood, plant tissue, soil). |
| MgCl₂ Solution | Essential co-factor for DNA polymerase activity; concentration must be optimized and may need adjustment with Hot-Start dNTPs [58] [62]. | A critical optimization parameter for all PCR reactions. |
Hot-Start PCR is a pivotal advancement in the ongoing evolution of DNA polymerase functionality in PCR research. By strategically imposing an initial, reversible inhibition on the polymerase—through methods as diverse as antibodies, chemical modification, substrate blocking, or physical separation—the technique effectively suppresses the nonspecific amplification that has long plagued conventional PCR. The resultant enhancements in specificity, sensitivity, and yield are quantitatively demonstrable and have profound implications. For researchers and drug development professionals, the adoption of Hot-Start protocols translates to more reliable genotyping, more sensitive pathogen detection, more efficient cloning, and more robust performance in automated, high-throughput diagnostic pipelines. As PCR continues to be a cornerstone technique in genetics and molecular biology, the strategic implementation of Hot-Start mechanisms ensures that the fundamental enzyme, the DNA polymerase, performs its catalytic role with maximal precision and efficacy.
The Role of DNA Polymerase in Reverse Transcription PCR (RT-PCR) for Infectious Disease Diagnostics
The polymerase chain reaction (PCR) stands as a cornerstone of molecular biology, and its integration with reverse transcription (RT) has been pivotal for infectious disease diagnostics. While reverse transcriptase enzymes are traditionally credited for the initial RNA-to-DNA conversion, the DNA polymerase enzyme is the fundamental engine driving the amplification and detection of genetic material in RT-PCR. Within the context of PCR research, the function of DNA polymerase has evolved from a simple thermostable enzyme amplifying DNA to a more sophisticated component capable of enhancing the speed, sensitivity, and specificity of RNA pathogen detection. This technical guide explores the critical role of DNA polymerase in RT-PCR, detailing its mechanisms, the innovations shaping its use, and the practical methodologies for developing robust diagnostic assays for infectious diseases, including SARS-CoV-2, influenza, and other viral pathogens.
Fundamental Principles of RT-PCR and the Central Role of DNA Polymerase
RT-PCR is a two-enzyme process designed to detect and quantify RNA targets. The process begins with the reverse transcriptase enzyme synthesizing a complementary DNA (cDNA) strand from an RNA template. This cDNA then serves as the template for the DNA polymerase enzyme in the subsequent PCR amplification [63] [64]. DNA polymerase's role is to catalyze the synthesis of new DNA strands complementary to the cDNA template, exponentially amplifying the target sequence for detection and quantification.
The performance of DNA polymerase is critical to the success of the entire assay. Key characteristics include:
- Thermostability: Essential for withstanding the high denaturation temperatures (typically 95°C) of repeated PCR cycles without significant loss of activity [65].
- Processivity: Refers to the number of nucleotides a DNA polymerase can add per binding event. Higher processivity leads to more efficient amplification, especially for longer targets.
- Fidelity: The accuracy with which the enzyme copies the template sequence. High-fidelity polymerases are crucial for minimizing mutations during amplification.
- 5'→3' Nuclease Activity: This activity, exhibited by enzymes like Taq DNA polymerase, is fundamental to hydrolysis probe (e.g., TaqMan) assays. It enables the enzyme to cleave a fluorescently-labeled probe during amplification, generating a detectable signal [64] [65].
RT-PCR can be performed in one-step or two-step formats, each with distinct implications for workflow and DNA polymerase use, as outlined in the table below.
Table 1: Comparison of One-Step and Two-Step RT-PCR Protocols
| Feature | One-Step RT-PCR | Two-Step RT-PCR |
|---|---|---|
| Procedure | Reverse transcription and PCR amplification occur in a single tube [63] [64]. | Reverse transcription and PCR amplification are performed in separate tubes [63] [64]. |
| Advantages | Faster setup, reduced pipetting steps, lower risk of contamination, suitable for high-throughput [63]. | Flexible; cDNA product can be stored and used for multiple PCR assays; reactions can be individually optimized [63]. |
| Disadvantages | Compromised reaction conditions for both enzymes; less sensitive; detection of fewer targets per sample [63]. | Higher risk of contamination due to more handling; more time-consuming [63]. |
| Role of DNA Polymerase | Works in a pre-mixed buffer that is a compromise for both RT and PCR. Must be compatible with reverse transcriptase. | Operates in an optimally buffered environment specifically for DNA amplification. |
Advanced Innovations in DNA Polymerase for RT-PCR
Recent biotechnology advances have led to significant innovations in DNA polymerase engineering, enhancing their utility in diagnostics.
Engineered DNA Polymerases with Reverse Transcriptase Activity
A groundbreaking innovation is the engineering of novel DNA polymerase variants capable of catalyzing both the reverse transcription and PCR amplification steps. This eliminates the need for a separate viral reverse transcriptase, truly streamlining the one-step RT-PCR process [65]. For instance, novel variants of Thermus aquaticus (Taq) DNA polymerase I have been developed by combining different mutation pools (e.g., L459M, S515R, I638F, M747K). These single-enzyme systems are highly thermostable and compatible with both dye- and probe-based detection methods, enabling quantitative multiplex RT-PCR for targets like SARS-CoV-2 without the need for Mn²⁺ additives or enzyme fusions [65].
Digital PCR and Enhanced Quantification
Digital PCR (dPCR) is another advancement that leverages DNA polymerase in a novel way. In dPCR, the PCR reaction mixture is partitioned into thousands of individual nanoreactions. DNA polymerase then amplifies the target in each partition. A key advantage of dPCR is that it provides absolute quantification of the nucleic acid target without the need for a standard curve, a requirement in traditional real-time RT-PCR [66] [67]. Studies have shown that dPCR offers superior accuracy and precision, particularly for quantifying intermediate viral loads of pathogens like influenza, RSV, and SARS-CoV-2, thereby enhancing diagnostic accuracy and the understanding of co-infection dynamics [67].
Experimental Protocols and Methodologies
This section details a standard two-step RT-PCR protocol and a novel single-enzyme assay, highlighting the critical role of DNA polymerase at each stage.
Detailed Protocol: Two-Step RT-qPCR for Viral Detection
Step 1: Reverse Transcription (cDNA Synthesis)
- RNA Template Preparation: Extract and purify total RNA from the clinical sample (e.g., nasopharyngeal swab). RNA quality and concentration should be accurately determined [68].
- Primer Annealing: Prepare a mixture containing the extracted RNA and a primer. Common choices include:
- Oligo(dT) primers: Bind to the poly-A tail of mRNA, suitable for eukaryotic mRNA [63].
- Random hexamers: Anneal to all types of RNA, useful for transcripts with secondary structure or when analyzing non-polyadenylated RNA (e.g., viral RNA) [63].
- Sequence-specific primers: Provide the highest specificity by binding to a known sequence within the target RNA [63].
- cDNA Synthesis: Add a reverse transcriptase (e.g., M-MLV or AMV), dNTPs, and reaction buffer. Incubate at a defined temperature (e.g., 37–50°C) for 30–60 minutes to synthesize the first-strand cDNA [68].
Step 2: Quantitative PCR (qPCR) Amplification
- Reaction Setup: Prepare a master mix containing:
- DNA Polymerase: A thermostable DNA polymerase (e.g., Taq polymerase) [65].
- PCR Buffer: Provides optimal pH and salt conditions for polymerase activity.
- Primers: Forward and reverse primers specific to the target cDNA sequence.
- dNTPs: Nucleotides for DNA synthesis.
- Probe: A TaqMan hydrolysis probe labeled with a fluorophore and quencher [64].
- cDNA Template: Add the synthesized cDNA from Step 1.
- Thermal Cycling: Run the reaction in a real-time PCR instrument with the following typical steps [69]:
- Initial Denaturation: 95°C for 2-5 minutes to activate the DNA polymerase and denature the cDNA.
- Amplification (40-50 cycles):
- Denature: 95°C for 15-30 seconds.
- Anneal/Extend: 60°C for 30-60 seconds. During this step, the DNA polymerase amplifies the target and cleaves the probe, releasing fluorescence.
- Data Analysis: The cycle threshold (Ct) at which fluorescence exceeds the background is determined. The Ct value is inversely proportional to the starting amount of the target RNA [66].
Protocol: Single-Enzyme Multiplex RT-PCR Using Engineered Taq Variants
This protocol, derived from recent research, demonstrates a simplified workflow [65].
- Template and Assay Design: Design primer and TaqMan probe sets for multiple RNA targets (e.g., different viral genes), each with a distinct fluorescent dye.
- Reaction Assembly: In a single tube, combine:
- RNA template (can be from unprocessed samples).
- An engineered Taq DNA polymerase variant with RT activity (e.g., RT-Taq).
- dNTPs.
- A multiplexed mix of gene-specific primers and TaqMan probes.
- Thermal Cycling: Perform a single, unified thermal cycling protocol that accommodates both the reverse transcription and the PCR amplification steps. No separate incubation for cDNA synthesis is needed.
- Detection: Monitor the fluorescence of each dye in real-time to quantify all RNA targets simultaneously in the same well.
The following workflow diagram contrasts the steps of traditional two-step RT-PCR with the novel single-enzyme approach.
The Scientist's Toolkit: Essential Reagents for RT-PCR
The following table catalogs the key reagents required for RT-PCR experiments, with a focus on the role of DNA polymerase and associated components.
Table 2: Essential Research Reagents for RT-PCR Assays
| Reagent | Function | Examples & Notes |
|---|---|---|
| DNA Polymerase | Amplifies cDNA template; in probe-based assays, provides 5' nuclease activity for cleavage [64] [65]. | Taq DNA Polymerase, engineered Taq variants (e.g., RT-Taq) [65]. |
| Reverse Transcriptase | Synthesizes cDNA from an RNA template. | M-MLV RT, AMV RT. Not required for single-enzyme systems [68]. |
| Primers | Short DNA sequences that define the start and end points of amplification. | Sequence-specific primers; design to span exon-exon junctions to avoid genomic DNA amplification [63]. |
| Fluorescent Probes/Dyes | Enable real-time detection of amplification products. | TaqMan Probes: Require 5' nuclease activity of DNA polymerase [64]. SYBR Green I: Binds dsDNA; requires post-run melt curve analysis to verify specificity [64]. |
| dNTPs | The building blocks (A, dT, G, C) for synthesizing new DNA strands. | Quality and concentration are critical for efficient amplification. |
| Reaction Buffer | Provides optimal chemical environment (pH, salts) for enzyme activity and fidelity. | Often includes MgCl₂, a essential cofactor for DNA polymerase. |
| Standard Curves | Essential for accurate quantification in real-time RT-PCR. | Serial dilutions of standards with known concentration; however, they contribute to inter-assay variability [66]. |
Data Presentation and Analysis in RT-PCR
Accurate quantification in RT-PCR relies on understanding key parameters derived from the amplification plot and standard curve. The DNA polymerase's efficiency directly influences these metrics.
Table 3: Key Quantitative Parameters in RT-qPCR Analysis
| Parameter | Description | Impact & Ideal Range |
|---|---|---|
| Cycle Threshold (Ct) | The cycle number at which fluorescence crosses a predefined threshold [66]. | Inversely proportional to the starting quantity of the target. A lower Ct indicates a higher initial target concentration. |
| Amplification Efficiency | The rate at which the target is amplified per cycle, calculated from the standard curve slope [66]. | Ideal: 90–110% (Slope ≈ -3.1 to -3.6). Lower efficiency suggests poor reaction optimization; higher indicates non-specific amplification [66] [69]. |
| Standard Curve | A plot of Ct values against the logarithm of known template concentrations used for quantification [66]. | Essential for relative quantification. Variability in standard curves between experiments is a major source of error, necessitating their inclusion in each run for reliable results [66]. |
| Linear Dynamic Range | The range of template concentrations over which the assay maintains a linear relationship between Ct and log concentration. | A wider range allows for accurate quantification of both high and low abundance targets. |
Recent studies highlight the variability inherent in RT-qPCR. For example, an analysis of 30 independent standard curves for SARS-CoV-2 N2 gene detection showed an efficiency of 90.97% with a high coefficient of variation (4.38–4.99%), underscoring the necessity of including a standard curve in every experiment for reliable quantification [66]. In contrast, digital RT-PCR (dRT-PCR) eliminates this need by providing absolute quantification through Poisson statistics, demonstrating superior accuracy for respiratory virus quantification, particularly at medium and high viral loads [67].
DNA polymerase is far more than a simple PCR component; it is the central driver of performance, accuracy, and innovation in RT-PCR for infectious disease diagnostics. From its fundamental role in catalyzing DNA amplification in traditional assays to the capabilities of newly engineered variants that unify reverse transcription and amplification, DNA polymerase continues to be a focal point of PCR research. As diagnostics move towards greater simplicity, multiplexing, and precision—exemplified by single-enzyme systems and digital PCR—the evolution of DNA polymerase will remain critical. These advancements promise to deliver more robust, accessible, and reliable diagnostic tools, ultimately enhancing pandemic preparedness and global public health responses.
Solving the Puzzle: A Strategic Guide to PCR Troubleshooting and Optimization
The polymerase chain reaction (PCR) stands as a cornerstone technique in molecular biology, enabling the amplification of specific DNA sequences with profound implications for basic research, clinical diagnostics, and drug development [1]. The core engine of this process is the DNA polymerase, a thermostable enzyme that catalyzes the template-directed synthesis of new DNA strands. The functional properties of DNA polymerase—including its fidelity, processivity, thermostability, and specificity—are therefore directly instrumental to the success or failure of any amplification experiment [5] [70]. Even with optimized protocols, researchers frequently encounter technical challenges that can compromise data integrity. This guide provides an in-depth analysis of three common PCR problems—no product, non-specific bands, and low yield—framed within the context of DNA polymerase function, and offers detailed, actionable troubleshooting methodologies.
The Function of DNA Polymerase in PCR
A comprehensive understanding of DNA polymerase mechanics is prerequisite to effective PCR troubleshooting.
Polymerase Fidelity: Fidelity refers to the accuracy of DNA replication. Enzymes like Taq polymerase lack proofreading activity (3'→5' exonuclease), leading to an error rate of approximately 1 in 6,000 bases [70]. High-fidelity polymerases (e.g., Pfu, Q5) possess proofreading capabilities, which excise misincorporated nucleotides, reducing error rates to as low as 1 in 1.68 million bases (280x higher than Taq) [5] [70]. Low fidelity can manifest not only as sequence errors but also in failed reactions if mismatches prevent further extension.
Processivity: This property denotes the number of nucleotides a polymerase can incorporate per single binding event. A highly processive enzyme remains attached to the template through challenging secondary structures or long amplicons, thereby enhancing efficiency and yield [5]. Engineered chimeric polymerases fused to DNA-binding domains exhibit significantly improved processivity, enabling faster amplification of long or complex targets [70].
Thermostability: Essential for PCR, thermostability allows the enzyme to withstand repeated denaturation temperatures (94–98°C). While Taq polymerase is stable, hyperthermostable enzymes derived from archaea (e.g., Pfu from Pyrococcus furiosus) retain activity even better under prolonged high-temperature conditions, which is crucial for denaturing GC-rich templates [5].
Specificity and Hot-Start Activation: DNA polymerases can exhibit residual activity at room temperature, leading to the extension of misprimed sequences or primer-dimers during reaction setup. Hot-start polymerases are inhibited by antibodies, aptamers, or chemical modifications until a high-temperature activation step (e.g., >90°C) is applied. This mechanism dramatically improves specificity by preventing off-target amplification before cycling begins [5] [71].
The interplay of these properties dictates PCR performance. The following sections dissect common amplification failures through this lens, providing a mechanistic understanding for each problem.
Problem 1: No Amplification Product
The complete absence of a desired product post-amplification is a frequent challenge, often stemming from factors that directly inhibit the core function of the DNA polymerase.
Systematic Troubleshooting Methodology
A logical, step-by-step protocol is the most efficient path to resolution.
Verify Reaction Integrity: Confirm that all essential components were added to the reaction mix. Always include a positive control (a known template and primer pair that has previously worked) to validate all reagents and equipment [72].
Assess Template DNA Quality and Quantity:
- Quantity: For standard PCR with 25-30 cycles, approximately 10^4 copies of the template DNA are typically sufficient [73]. For human genomic DNA, 30-100 ng is standard, though 10 ng may suffice for high-copy number genes [73].
- Quality: Analyze template integrity by agarose gel electrophoresis. Degraded DNA appears as a smear. Use spectrophotometry (A260/A280 ratio) to check for contaminants like phenol or EDTA, which chelate essential Mg²⁺ ions [74] [48]. If quality is poor, repurify the template via ethanol precipitation, dialysis, or commercial cleanup kits [75].
Optimize Primer Design and Annealing Temperature:
- Primer Design: Ensure primers are 15-30 nucleotides long with a GC content of 40-60% [73]. The melting temperatures (Tm) for forward and reverse primers should be within 5°C of each other [73]. Avoid complementarity, especially at the 3' ends, to prevent primer-dimer formation [74] [75].
- Annealing Temperature: If the calculated Tm does not yield a product, perform a temperature gradient PCR, testing a range from 5°C below to 5°C above the calculated Tm [75]. The optimal annealing temperature is typically 3-5°C below the primer Tm [48].
Check for PCR Inhibitors: Common inhibitors include heparin, hemoglobin, phenol, humic acids, and high salt concentrations [1] [72]. They can obstruct the polymerase's active center or chelate Mg²⁺ [74]. Mitigate this by diluting the template 10- to 100-fold or through additional purification [72]. Using polymerases with high processivity can also improve tolerance to inhibitors [5].
Evaluate Polymerase and Cycling Conditions: Confirm that a sufficient amount of active enzyme is used. Increase the number of PCR cycles to 35-40 for low-abundance targets [72]. Ensure the thermal cycler is correctly calibrated, as a malfunctioning block can prevent denaturation or extension [75].
Research Reagent Solutions
The following reagents are essential for diagnosing and resolving a "no product" outcome.
| Reagent/Category | Specific Examples | Function & Application Note |
|---|---|---|
| High-Processivity DNA Polymerase | OneTaq DNA Polymerase (NEB), Platinum II Taq (Thermo Fisher) | Enhanced affinity for template; superior for amplifying long targets or templates with secondary structures [75] [48]. |
| DNA Cleanup Kit | Monarch PCR & DNA Cleanup Kit (NEB) | Removes common PCR inhibitors (salts, organics) and purifies template DNA [75]. |
| dNTP Mix | Prepared balanced solutions (e.g., 10 mM each) | Prevents misincorporation and polymerase stalling due to unbalanced nucleotide concentrations [75]. |
| Mg²⁺ Solution | MgCl₂ or MgSO₄ (25-50 mM stocks) | Serves as an essential cofactor for polymerase activity. Concentration must be optimized [75] [48]. |
| Template Repair Mix | PreCR Repair Mix (NEB) | Repairs damaged template DNA (e.g., nicked or oxidized bases) that can halt polymerase progression [75]. |
The logical relationship between potential causes and their solutions is mapped in the workflow below.
Problem 2: Non-Specific Amplification
The appearance of multiple, unintended bands on a gel indicates non-specific amplification, a problem primarily linked to the specificity of the DNA polymerase and reaction stringency.
Systematic Troubleshooting Methodology
Employ Hot-Start Polymerase: This is the most critical step. Use a hot-start enzyme inhibited by an antibody, aptamer, or chemical modification. This prevents polymerase activity during reaction setup at low temperatures, eliminating the extension of misprimed templates [5] [71]. For manual setup, assemble reactions on ice and use a pre-heated thermal cycler [75].
Increase Annealing Stringency:
- Temperature: Increase the annealing temperature in 2°C increments. The optimal temperature is usually 3–5°C below the primer Tm [48] [73]. Use a gradient thermal cycler for empirical determination.
- Time: Shorten the annealing time (e.g., to 5-15 seconds) to reduce opportunities for off-target primer binding [72].
Optimize Mg²⁺ and Primer Concentration: High Mg²⁺ concentration stabilizes DNA duplexes and can promote non-specific priming. Optimize the Mg²⁺ concentration in 0.2–1 mM increments, starting from the lower end of the recommended range [74] [75]. Reduce primer concentration if it exceeds 0.1–1 µM, as excess primer facilitates mispriming [48] [73].
Reduce Template and Cycle Number: Excessive template DNA can overwhelm the reaction's specificity. Reduce the amount by 2–5 fold [72]. Similarly, an excessive number of cycles (>35) can allow non-specific products to amplify to detectable levels; reduce the cycle number [48].
Utilize Additives and Advanced Protocols: Additives like DMSO (1-10%) or formamide (1.25-10%) can help denature GC-rich secondary structures and increase primer annealing stringency [73]. For persistently difficult assays, consider switching to a touchdown PCR protocol, which starts with a high annealing temperature and gradually decreases it, thereby favoring the accumulation of the specific product [48] [72].
Problem 3: Low Yield and Faint Bands
Insufficient product for downstream applications often results from suboptimal reaction efficiency or enzyme performance.
Systematic Troubleshooting Methodology
Increase Template and Cycle Number: Systematically increase the amount of input template. If the target is of low abundance, increase the number of cycles to 35-40 [72]. Ensure the template is of high quality and free of inhibitors.
Optimize Extension Time and Temperature: Ensure the extension time is sufficient for the polymerase to complete synthesis of the full amplicon. A general guideline is 1 minute per 1000 base pairs, but this can vary with polymerase processivity [73]. For long templates (>3 kb), a longer extension time or a slight reduction in extension temperature (to 68°C) can be beneficial [48].
Address Complex Templates (GC-Rich/Secondary Structures):
- Polymerase Choice: Use a polymerase with high processivity and thermostability, such as those engineered for GC-rich targets [48].
- Additives: Include PCR enhancers such as GC Enhancer, DMSO (1-10%), glycerol, or betaine. These co-solvents help denature stable secondary structures and lower the template's melting temperature, facilitating polymerase progression [74] [73].
- Denaturation: Increase the denaturation temperature (e.g., to 98°C) or duration during cycling [48].
Ensure Balanced Reaction Components:
- dNTPs: Use a fresh, balanced dNTP mix at 20-200 µM each. Unbalanced nucleotides increase error rates and can deplete polymerase activity [75] [73].
- Mg²⁺: This is a critical cofactor. If the concentration is too low, polymerase activity is severely impaired. Re-optimize the Mg²⁺ concentration to ensure it is sufficient, considering that dNTPs and EDTA chelate Mg²⁺ [74] [48].
The tables below consolidate key quantitative parameters for systematic optimization.
Table 1: Optimization of Critical PCR Component Concentrations
| Component | Recommended Range | Effect of Low Concentration | Effect of High Concentration |
|---|---|---|---|
| Mg²⁺ | 0.5 - 5.0 mM (optimize in 0.2-1 mM increments) [75] [73] | No or low yield [74] | Non-specific bands, increased error rate [48] |
| Primers | 0.1 - 1.0 µM each [48] [73] | Low yield [75] | Primer-dimer, non-specific bands [75] [73] |
| dNTPs | 20 - 200 µM each (balanced) [73] | Low yield [74] | Misincorporation, Mg²⁺ chelation [72] |
| Template DNA | Genomic: 1 ng - 1 µg; Plasmid: 1 pg - 10 ng per 50 µL reaction [75] | Low or no yield [48] | Non-specific bands, smearing [48] |
| Cycle Number | 25 - 40 cycles [48] | Low yield [72] | Non-specific products, smearing [72] |
Table 2: DNA Polymerase Selection Guide Based on Template Properties
| Template Challenge | Recommended Polymerase Properties | Example Enzymes | Supporting Additives |
|---|---|---|---|
| High Fidelity Required (Cloning) | Proofreading (3'→5' exonuclease), High Fidelity | Q5 (NEB), Pfu [75] [5] | Balanced dNTPs [48] |
| Long Amplicons (>5 kb) | High Processivity, Proofreading (often in a blend) | LongAmp Taq (NEB), blends of Taq and Pfu [70] [71] | Extended extension time [48] |
| GC-Rich Sequences | High Thermostability, High Processivity | Platinum II Taq (Thermo Fisher) [48] | DMSO (1-10%), GC Enhancer, Betaine [48] [73] |
| Routine, High-Specificity PCR | Hot-Start | Any commercial hot-start Taq [5] [71] | Optimized Mg²⁺ [74] |
Advanced Considerations: Multi-Template PCR and Amplification Bias
In modern applications like next-generation sequencing library preparation and metabarcoding, multi-template PCR is ubiquitous. A significant challenge in these reactions is non-homogeneous amplification efficiency, where different template sequences amplify at varying rates, drastically skewing the abundance data of the final product [76].
Recent research using deep learning models has revealed that sequence-specific factors, independent of traditional metrics like GC content, are a major cause. These models identified specific sequence motifs near priming sites that lead to poor amplification, often by facilitating adapter-mediated self-priming [76]. This finding challenges long-standing primer design assumptions and opens new avenues for designing amplicon libraries with inherently balanced amplification. For researchers performing quantitative multi-template PCR, this underscores the importance of sequence context beyond primary primer-design rules and suggests that computational tools may soon be integrated into experimental design to predict and correct for such biases [76].
Effective PCR troubleshooting transcends a simple checklist of actions; it requires a mechanistic understanding of the DNA polymerase at the heart of the reaction. The properties of the chosen enzyme—its fidelity, processivity, thermostability, and the specificity conferred by hot-start technology—interact directly with reaction components and cycling parameters to determine success. By systematically addressing problems of no product, non-specific bands, and low yield through the lens of enzyme function, researchers can not only rescue failed experiments but also proactively design robust and reliable PCR assays. This rigorous approach ensures the generation of high-quality data, forming a solid foundation for critical downstream applications in research and drug development.
The polymerase chain reaction (PCR) stands as a cornerstone technique in molecular biology, biomedical research, and clinical diagnostics. At the heart of this method lies DNA polymerase, a thermostable enzyme that catalyzes the synthesis of new DNA strands complementary to a target template. The efficiency and accuracy of this enzyme are not intrinsic properties alone; they are profoundly influenced by the reaction environment. The precise optimization of key reaction components—magnesium ions (Mg²⁺), deoxyribonucleotide triphosphates (dNTPs), and primer design—is therefore fundamental to successful PCR amplification. These components directly affect DNA polymerase kinetics, fidelity, and specificity, forming a complex interdependent system that determines experimental outcomes. This guide provides an in-depth examination of these critical parameters, offering evidence-based strategies to optimize their function in support of robust and reliable DNA amplification.
Magnesium Ion (Mg²⁺) Concentration: The Essential Cofactor
Function and Optimization of Mg²⁺
Magnesium ions serve as an essential cofactor for DNA polymerase activity. Mg²⁺ facilitates the formation of a functional complex between the enzyme and the DNA template and is directly involved in the catalytic reaction for phosphodiester bond formation [1]. Its concentration is arguably the most critical variable in PCR optimization.
A comprehensive 2025 meta-analysis of 61 studies established a clear logarithmic relationship between MgCl₂ concentration and DNA melting temperature, with an optimal concentration range between 1.5 and 3.0 mM [77]. Within this range, every 0.5 mM increase in MgCl₂ was associated with an average 1.2 °C increase in melting temperature [77]. This meta-analysis also revealed that template complexity significantly influences optimal Mg²⁺ requirements; genomic DNA templates generally require higher concentrations than simpler templates such as plasmid DNA [77].
Table 1: Effects and Optimization of Mg²⁺ Concentration in PCR
| Parameter | Optimal Range | Effect of Low Concentration | Effect of High Concentration |
|---|---|---|---|
| General MgCl₂ | 1.5 - 3.0 mM [77] | Reduced enzyme activity, low yield | Increased non-specific binding, primer-dimer formation |
| Standard Recommendation | ~1.5 mM [1] | Failed amplification | Band smearing on gels |
| Template Specific: Genomic DNA | Higher end of range | Inefficient amplification of complex targets | — |
| Impact on Tm | +1.2°C per 0.5 mM [77] | — | — |
Interaction with Other Reaction Components
Magnesium concentration is not isolated in its effect. It exists in a dynamic equilibrium with dNTPs, as Mg²⁺ binds to dNTPs to form the actual substrate recognized by DNA polymerase. Consequently, the Mg²⁺ concentration must exceed the total dNTP concentration to ensure enzyme activity. A common recommendation is to start with 1.5-2.0 mM Mg²⁺ for standard reactions, adjusting based on template and primer characteristics [78]. The presence of EDTA or other chelators in template preparations can sequester Mg²⁺, necessitating higher concentrations to compensate.
dNTP Concentration: The Building Blocks of Amplification
Balancing Quantity and Quality
Deoxyribonucleotide triphosphates (dNTPs) are the fundamental building blocks for new DNA synthesis. Their concentration in the reaction is a critical determinant of PCR yield, specificity, and fidelity. Optimal dNTP levels typically range between 0.2 to 0.4 mM (200-400 μM) for standard PCR applications [79]. This range provides sufficient substrates for DNA polymerase while minimizing misincorporation errors that can occur at higher concentrations.
The quality of dNTPs is equally crucial. Using ultra-pure dNTPs (e.g., ≥99% purity by HPLC assessment) ensures the integrity of the reaction and prevents degradation that can occur with repeated freeze-thaw cycles [79]. Proper storage at -20°C or -70°C is recommended to maintain stability.
Table 2: dNTP Optimization Guidelines for Various PCR Applications
| Application | Recommended dNTP Concentration | Key Considerations |
|---|---|---|
| Standard PCR | 0.2 - 0.4 mM (200-400 μM) [79] | Balanced with Mg²⁺ concentration; 50 μM of each dNTP is a common starting point [45]. |
| High-Fidelity PCR | Lower end of range (e.g., 0.2 mM) | Reduces misincorporation risk for high-fidelity polymerases [79]. |
| Long Amplicon PCR | Higher end of range (e.g., 0.4 mM) | Ensures sufficient substrates for extended synthesis [79]. |
| RT-PCR | 0.25 mM [79] | Used in visualized SARS-CoV-2 detection assays. |
Interrelationship with Mg²⁺ Concentration
The critical interaction between dNTPs and Mg²⁺ must be emphasized. Since Mg²⁺ binds stoichiometrically to dNTPs in the reaction, the total Mg²⁺ concentration must be adjusted relative to dNTP concentration. A general rule is that the Mg²⁺ concentration should be 0.5-1.0 mM higher than the total dNTP concentration [45]. For example, in a reaction with 0.2 mM dNTPs (50 μM of each dNTP), the Mg²⁺ concentration should be at least 1.0 mM before accounting for the enzyme's additional Mg²⁺ requirement.
Primer Design: The Foundation of Specificity
Core Principles for Effective Primers
Well-designed primers are the foundation of specific and efficient PCR amplification. They must accurately target the sequence of interest while avoiding secondary structures or non-specific binding. The following parameters represent the consensus for optimal primer design [80] [81] [45]:
Table 3: Comprehensive Primer Design Specifications
| Parameter | Optimal Range | Rationale |
|---|---|---|
| Length | 18-30 nucleotides (17-27 bp) [80] [82] | Balances specificity and binding efficiency. |
| Melting Temperature (Tm) | 60-65°C [80]; Ideally 52-58°C for both primers [45] | Optimal for polymerase function. |
| Tm Difference Between Primers | ≤2-5°C [80] [82] | Ensures simultaneous binding during annealing. |
| GC Content | 40-60% [45] [82] | Provides sequence complexity without excessive stability. |
| 3' End Specificity | Avoid complementary ends; end with G or C [45] | Prevents primer-dimer formation and increases priming efficiency. |
| Amplicon Length | 70-150 bp for qPCR; up to 500 bp for standard PCR [80] [81] | Compatible with standard cycling conditions. |
Advanced Considerations and Validation
Beyond these core parameters, several advanced considerations enhance primer success. Primers should be screened for secondary structures including hairpins, self-dimers, and heterodimers using tools like OligoAnalyzer, with a ΔG value weaker than -9.0 kcal/mol [80]. For gene expression analysis using RT-qPCR, designing primers to span an exon-exon junction reduces false positives from genomic DNA contamination [80] [81]. Furthermore, verifying primer specificity through NCBI BLAST analysis ensures uniqueness to the target sequence [80].
Annealing temperature (Ta) optimization is critical and should be set approximately 3-5°C below the primer Tm [80] [81]. Setting the Ta too low permits non-specific annealing, while too high a Ta reduces reaction efficiency. Modern tools like IDT SciTools or Geneious Prime can calculate precise Tm values based on actual reaction conditions, including salt concentrations [80] [82].
Integrated Optimization Strategies
Component Interdependence and Workflow
The optimization of Mg²⁺, dNTPs, and primers cannot be performed in isolation, as these components function within an interdependent system. A strategic, sequential approach yields the best results, beginning with primer design and validation, followed by Mg²⁺ titration, and finally fine-tuning of dNTP concentrations.
PCR Optimization Workflow
Troubleshooting Common PCR Problems
Even with careful optimization, challenges may persist. The following table addresses common PCR issues and their component-based solutions:
Table 4: Troubleshooting Guide for Common PCR Problems
| Problem | Potential Causes | Solution Approaches |
|---|---|---|
| No Product | Mg²⁺ too low; dNTPs too low; primers inefficient | Titrate Mg²⁺ upward; verify dNTP concentration; check primer design and Tm [78]. |
| Non-specific Bands/Smearing | Mg²⁺ too high; primer Tm too low; poor primer specificity | Reduce Mg²⁺; increase annealing temperature; redesign primers [78]. |
| Primer-Dimer Formation | Primer 3' end complementarity; excess primers; Mg²⁺ too high | Redesign primers; reduce primer concentration (0.2-1.0 μM); optimize Mg²⁺ [78]. |
| Low Yield | dNTPs too low; Mg²⁺ suboptimal; insufficient cycles | Titrate both dNTPs and Mg²⁺; increase cycle number for low-copy targets [79] [78]. |
The Scientist's Toolkit: Research Reagent Solutions
Successful PCR optimization relies on high-quality reagents and specialized enzyme formulations. The following table details essential solutions for overcoming common challenges:
Table 5: Research Reagent Solutions for PCR Optimization
| Reagent / Tool | Function / Application | Key Features |
|---|---|---|
| High-Purity dNTPs (e.g., SBS Genetech) [79] | Molecular biology grade nucleotides for reliable amplification | ≥99% HPLC purity; DNase/RNase-free; available as mix or individual components. |
| Inhibition-Resistant Polymerases (e.g., Taq C-66, Klentaq1 H101) [83] | Amplification from challenging samples (blood, soil, food) | Novel variants with superior resistance to PCR inhibitors like humic acid, hematin. |
| Hot-Start DNA Polymerases (e.g., ZymoTaq) [81] | Reduction of non-specific amplification and primer-dimers | Enzyme activated only at high temperatures, improving specificity. |
| Novel Taq Polymerase Variants [65] | Single-enzyme reverse transcription and DNA amplification | Engineered Taq pol variants with reverse transcriptase activity for simplified RT-PCR. |
| PCR Enhancers (e.g., DMSO, Betaine) [45] | Improvement of amplification efficiency for difficult templates | Aid in denaturation of GC-rich regions; reduce secondary structure. |
| Primer Design Tools (e.g., IDT SciTools, Geneious Prime, NCBI Primer-BLAST) [80] [82] | In silico primer design and validation | Calculate Tm under specific conditions; check for secondary structures; verify specificity. |
The precise optimization of Mg²⁺ concentration, dNTP levels, and primer design represents a critical triad that directly controls DNA polymerase function and PCR success. These components form an integrated system where each element influences the others, requiring a balanced, systematic approach to optimization. The strategies and data presented here provide a framework for researchers to develop robust, reliable PCR protocols tailored to their specific experimental needs, from basic research to advanced diagnostic applications. As PCR technology continues to evolve with novel polymerase variants and enhanced formulations [65] [83], these fundamental optimization principles will remain essential for harnessing the full potential of this indispensable technique.
The polymerase chain reaction (PCR) is a foundational technique in molecular biology, and its success is fundamentally governed by the function of DNA polymerase. This enzyme catalyzes the synthesis of new DNA strands, and its activity is directly influenced by the thermal cycler conditions set by the researcher [1]. Fine-tuning parameters such as annealing temperature and extension time is not merely a procedural step but a critical process to align the reaction conditions with the kinetic and thermodynamic properties of the specific DNA polymerase in use. Optimal conditions maximize the enzyme's efficiency, fidelity, and processivity, leading to high yields of specific products. Conversely, suboptimal conditions can lead to enzyme inefficiency, resulting in PCR failure, nonspecific amplification, or the generation of erroneous sequences [5]. This guide provides an in-depth framework for optimizing these crucial parameters within the context of modern DNA polymerase research.
The Critical Interplay Between DNA Polymerase and Thermal Parameters
DNA polymerases are not all created equal; their properties dictate the physical conditions of the reaction. Thermostability is a key characteristic, as the enzyme must withstand repeated denaturation temperatures, often above 90°C. Enzymes like Pfu DNA polymerase, derived from Pyrococcus furiosus, exhibit greater thermostability than Taq DNA polymerase, which can be beneficial for denaturing complex templates [5].
Furthermore, the processivity of an enzyme—the number of nucleotides it adds per binding event—directly influences the optimal extension time. Highly processive enzymes can synthesize long amplicons more rapidly [5]. Perhaps the most crucial advancement for specificity is the hot-start property. Antibody-mediated or chemically modified hot-start polymerases remain inactive until a high-temperature initial denaturation step, preventing nonspecific amplification and primer-dimer formation during reaction setup [5]. The optimization of annealing and extension parameters is, therefore, an exercise in creating the perfect environment for a specific DNA polymerase to function at its peak.
The diagram below illustrates how the core functional properties of DNA polymerase dictate the optimization of thermal cycling parameters.
Optimizing Annealing Temperature
Theoretical Foundation and Calculation
The annealing temperature (Ta) is the temperature at which primers bind to their complementary sequence on the template DNA. The primary goal is to find a temperature high enough to ensure perfect primer-template matching for specificity, but low enough to allow stable binding for the polymerase to initiate extension. The theoretical starting point is the primer's *melting temperature (Tm), which is the temperature at which 50% of the DNA duplex dissociates [84].
A common calculation for a preliminary Tm estimate is: Tm = 2°C × (A + T) + 4°C × (G + C) [84]
For routine PCR, the initial annealing temperature is typically set at 3–5°C below the calculated Tm of the lower-melting primer [85] [84]. However, this is only a starting point, and empirical optimization is often required.
Experimental Optimization Strategies
Gradient PCR: The most robust method for optimization is using a thermal cycler with a gradient function. This allows a single PCR reaction to be run with a range of annealing temperatures across different blocks simultaneously. The optimal Ta is identified as the highest temperature that yields the strongest, specific product band on an agarose gel [86].
Touchdown PCR: This strategy begins with an annealing temperature several degrees above the estimated Tm and gradually decreases it in subsequent cycles (e.g., by 1°C every cycle or every two cycles) until a lower, permissive temperature is reached. The initial high-temperature cycles promote the enrichment of specific targets, which are then amplified efficiently in later cycles, thereby enhancing specificity [84].
Universal Annealing Buffers: Recent innovations include specialized PCR buffers with isostabilizing components. These buffers, such as those used with Invitrogen Platinum DNA polymerases, allow for a universal annealing temperature of 60°C for a wide range of primers, drastically reducing optimization time without compromising yield or specificity [87].
Table 1: Summary of Annealing Temperature Optimization Methods
| Method | Principle | Procedure | Advantage |
|---|---|---|---|
| Gradient PCR [86] | Empirical testing of a temperature range in a single run. | Set a gradient (e.g., 50–65°C) across the thermoblock during the annealing step. | Fast, data-driven determination of optimal ( T_a ). |
| Touchdown PCR [84] | High-stringency initial cycles reduce nonspecific amplification. | Start 5–10°C above estimated ( T_m ), decrease 1°C every cycle/2 cycles to a lower limit. | Enhances specificity for difficult assays or complex templates. |
| Universal Annealing [87] | Buffer chemistry stabilizes primer binding across different ( T_m )s. | Use a proprietary buffer system with a fixed annealing temperature of 60°C. | Eliminates optimization; ideal for high-throughput screening. |
Optimizing Extension Time
Guidelines Based on Amplicon Length and Polymerase Type
Extension time is determined by the length of the target amplicon and the synthesis rate of the DNA polymerase. A general rule of thumb for standard polymerases like Taq is 1 minute per 1000 base pairs (kb) [85] [88]. However, this is a conservative estimate, and "fast" enzymes with high processivity can significantly reduce this requirement.
Table 2: Extension Time Guidelines for Different DNA Polymerases
| DNA Polymerase Type | Typical Extension Rate | Example: 2 kb Amplicon | Considerations |
|---|---|---|---|
| Standard Taq [85] [88] | 1 min/kb | 2 minutes | A conservative starting point. |
| Fast/Rapid Enzymes (e.g., SpeedSTAR HS, SapphireAmp) [88] | 10–15 sec/kb | 20–30 seconds | Reduces total cycling time significantly. |
| High-Fidelity/Proofreading (e.g., PrimeSTAR GXL) [88] | 15–30 sec/kb | 30–60 seconds | May be slower due to 3'→5' exonuclease activity. |
| Long-Range Enzymes (e.g., LA Taq) [88] | 1–2 min/kb (for >5 kb targets) | - | Designed for processivity over long distances. |
Co-amplification of Multiple Targets
When amplifying multiple targets of different lengths in a single tube (multiplex PCR), the extension time must be sufficient for the longest amplicon. Using a highly processive enzyme or a universal annealing buffer can help ensure that shorter amplicons are amplified efficiently without nonspecific products, even with a longer-than-ideal extension time [87].
An Integrated Workflow for Systematic Optimization
The following workflow provides a step-by-step protocol for a comprehensive optimization experiment, leveraging a thermal cycler with a gradient function.
Step 1: Calculate Primer Melting Temperature
- Use the formula provided in Section 3.1 or an online calculator from a reputable supplier (e.g., IDT's OligoAnalyzer) [84].
Step 2: Run a 2D Gradient PCR (Annealing and Denaturation)
- If available, a 2D-gradient function can optimize both annealing and denaturation temperatures in a single run. This is particularly useful for difficult templates (GC-rich, long) and can improve both specificity and yield [86].
- Protocol: Prepare a master mix containing your template (10 pg–100 ng, depending on source [85] [88]), primers (0.1–0.5 µM each [85]), dNTPs (200 µM each), appropriate MgCl₂ (start with 1.5–2.0 mM for Taq [85]), and a hot-start DNA polymerase. Load the mixture into the thermal cycler and set a 2D-gradient with an annealing temperature range (e.g., 50–65°C) and a denaturation temperature range (e.g., 92–98°C). Cycle for 25–35 cycles.
Step 3: Analyze Results and Refine
- Analyze PCR products by agarose gel electrophoresis.
- Select the temperature combination that yields the brightest, single band of the expected size.
- If nonspecific amplification persists, consider using a touchdown PCR protocol or slightly increasing the annealing temperature [84].
Step 4: Fine-tune with Additives
- For GC-rich templates (>65% GC), additives like DMSO (2.5–5%) can help denature secondary structures and improve yield [88].
- If the reaction fails, consider titrating MgCl₂ in 0.5 mM increments from 1.0 mM up to 4 mM, as Mg²⁺ is a critical cofactor for polymerase activity [85] [88].
The Scientist's Toolkit: Essential Reagents for PCR Optimization
Table 3: Key Research Reagent Solutions for PCR Optimization
| Reagent / Tool | Function in Optimization |
|---|---|
| Hot-Start DNA Polymerase (e.g., Platinum Taq, OneTaq HS) [87] [5] | Reduces nonspecific amplification and primer-dimer formation by inhibiting polymerase activity at room temperature. Essential for robust and reproducible results. |
| Gradient Thermal Cycler [86] | Enables empirical determination of optimal annealing temperature in a single experiment, saving time and precious samples. |
| MgCl₂ Solution (supplied separately with buffer) [85] [88] | Allows for fine-tuning of magnesium concentration, a critical variable that affects primer annealing, enzyme activity, fidelity, and specificity. |
| GC-Rich Enhancers / DMSO [88] | Additives that disrupt secondary structures in high-GC templates, facilitating primer binding and polymerase progression. |
| Universal Annealing Buffer (e.g., with Platinum SuperFi II) [87] | A proprietary buffer system that allows a fixed 60°C annealing temperature for most primers, simplifying protocol standardization and co-cycling of different assays. |
The precision of thermal cycler programming is a direct reflection of our understanding of DNA polymerase function. Methodical optimization of annealing temperature and extension time is not a one-time task but a critical investment in experimental reliability. By applying the systematic strategies outlined—from gradient and touchdown PCRs to leveraging modern enzyme and buffer systems—researchers can ensure their PCR assays achieve maximum specificity and yield. This rigorous approach to foundational techniques underpins advancements across biomedical research, from basic gene expression analysis to the development of next-generation molecular diagnostics.
The function of DNA polymerase extends far beyond simple DNA synthesis in the polymerase chain reaction (PCR). For researchers and drug development professionals targeting complex genomic regions, the intrinsic properties of the polymerase are paramount. Amplifying challenging templates, such as GC-rich sequences and long amplicons, often leads to reaction failure with standard polymerases due to incomplete amplification, nonspecific products, or truncated sequences. These challenges are frequently encountered in critical applications like sequencing tumor suppressor gene promoters, analyzing repetitive DNA expansions linked to diseases, and cloning large gene constructs. Success hinges on selecting a polymerase with a specific biochemical profile and optimizing the reaction environment to overcome the unique thermodynamic and structural barriers these templates present. This guide details the strategic optimization of PCR components and conditions to enable reliable amplification of these difficult targets, focusing on the core role of DNA polymerase functionality.
Understanding the Fundamental Challenges
The GC-Rich Template Problem
GC-rich sequences, typically defined as those with a guanine-cytosine (GC) content of 60% or greater, pose a significant challenge for several interconnected reasons.
- Thermodynamic Stability: A G-C base pair is stabilized by three hydrogen bonds, compared to only two for an A-T pair. This makes GC-rich duplexes more thermostable and resistant to denaturation, often requiring higher denaturation temperatures [89].
- Secondary Structure Formation: GC-rich regions are highly prone to forming stable intra-strand secondary structures, such as hairpin loops and stem-loops, during the cooler annealing and extension steps of PCR. These structures can physically block the progression of the DNA polymerase, leading to incomplete or truncated products [89] [53].
- Base Stacking Interactions: The increased stability of GC-rich regions is not solely due to hydrogen bonding. Strong base stacking interactions between adjacent G and C bases significantly contribute to the high melting temperature of these sequences [53].
- Primer-Related Issues: Primers designed for GC-rich targets are themselves GC-rich and therefore prone to forming self-dimers, cross-dimers, and secondary structures, which reduce the efficiency of specific target binding and can promote mispriming [53].
The Long Amplicon Amplification Hurdles
Amplifying long PCR products (generally considered to be >3-4 kilobases) introduces a different set of challenges that impact PCR efficiency and fidelity.
- Depurination: During the high-temperature denaturation steps, the DNA template can undergo hydrolytic depurination (loss of adenine or guanine bases). While this is negligible in short amplicons, the probability of a depurination event occurring within a long template is proportionally higher. These abasic sites can stall DNA polymerases, preventing the synthesis of full-length products [90].
- Fidelity and Error Accumulation: The longer the DNA sequence to be replicated, the greater the chance that the polymerase will incorporate a mismatched nucleotide. Standard non-proofreading polymerases like Taq cannot correct these errors. A single error early in the amplification process can be propagated through cycles, potentially compromising the sequence of the final product [90] [5].
- Reduced Processivity: Processivity refers to the number of nucleotides a polymerase can incorporate in a single template-binding event. Polymerases with low processivity are more likely to dissociate from long templates before synthesis is complete, leading to inefficient amplification [5].
- Time Constraints: Standard extension times (e.g., 1 minute per kb) may be insufficient for long amplicons, especially if the polymerase has low processivity or encounters secondary structures.
Table 1: Primary Challenges in Amplifying Complex Templates
| Challenge | Impact on PCR | Common Symptom on Gel |
|---|---|---|
| GC-Rich Secondary Structures | Polymerase stalling during extension; primer misannealing | Smear of truncated products; no product |
| High Melting Temperature (Tm) | Incomplete template denaturation; poor primer annealing | Weak or no amplification |
| Depurination (Long Amplicons) | Polymerase stalling at abasic sites | Failure to amplify full-length product; background smearing |
| Low Fidelity (Long Amplicons) | Incorporation of sequence errors during amplification | Mutated final product sequence |
Strategic Optimization: A Toolkit for Success
Overcoming these challenges requires a multi-faceted approach centered on polymerase choice and buffer composition.
DNA Polymerase Selection: The Core Engine
Choosing the right DNA polymerase is the most critical decision. The ideal enzyme for complex templates often combines high fidelity, strong processivity, and thermostability.
- High Fidelity for Long Amplicons: Proofreading polymerases (e.g., Q5, Pfu) possess 3'→5' exonuclease activity, which allows them to identify and excise misincorporated nucleotides. This results in error rates that can be >50–280 times lower than that of Taq polymerase, which is essential for accurately replicating long sequences [5] [91].
- Processivity for GC-Rich and Long Targets: Processivity is key for navigating through complex secondary structures and long stretches of DNA. Engineered polymerases that include a DNA-binding domain can exhibit a 2- to 5-fold enhancement in processivity, enabling them to synthesize long amplicons more efficiently [5].
- Thermostability: Hyperthermostable enzymes (e.g., those from Pyrococcus species) remain active longer during prolonged high-temperature denaturation steps needed to melt GC-rich structures. For instance, Pfu polymerase is about 20 times more stable at 95°C than Taq [5].
- Hot-Start Activation: To prevent non-specific amplification and primer-dimer formation at room temperature, use a hot-start polymerase. These enzymes are inactivated by antibodies or chemicals until the initial high-temperature denaturation step, vastly improving specificity and yield [5] [91].
Table 2: Key Characteristics of DNA Polymerases for Complex Templates
| Polymerase Characteristic | Importance for GC-Rich Templates | Importance for Long Amplicons | Example Enzymes |
|---|---|---|---|
| Proofreading (High Fidelity) | Secondary | Critical (reduces error accumulation) | Q5, Phusion, Pfu |
| High Processivity | Critical (unblocks secondary structures) | Critical (completes long synthesis) | Engineered polymerases (e.g., Platinum II) |
| Hyperthermostability | Critical (withstands high denaturation temps) | Beneficial (maintains activity over long runs) | Pfu, KOD, GBD |
| Hot-Start Activation | Beneficial (increases specificity) | Beneficial (increases specificity) | OneTaq Hot Start, Platinum Taq |
Buffer Composition and Additives
The reaction buffer creates the chemical environment for polymerization and can be modified to destabilize secondary structures.
- GC Enhancers: Many manufacturers offer proprietary GC Enhancer solutions. These often contain a combination of additives that help denature stable secondary structures and increase primer annealing stringency, enabling amplification of templates with up to 80% GC content [89].
- Betaine: Betaine (also known as N,N,N-trimethylglycine) is a common additive that reduces the melting temperature of GC-rich DNA by disrupting base stacking, thereby helping to resolve secondary structures like hairpins [89].
- Dimethyl Sulfoxide (DMSO): DMSO is a polar solvent that can interfere with the hydrogen bonding network of DNA, facilitating the denaturation of DNA strands and helping to unwind secondary structures that impede the polymerase [89] [53].
- Magnesium Ion (Mg²⁺) Concentration: Magnesium is an essential cofactor for polymerase activity. While standard concentrations are 1.5–2.0 mM, GC-rich PCRs often require optimization. Too little Mg²⁺ reduces polymerase activity, while too much can promote non-specific binding. A titration in 0.5 mM increments between 1.0 and 4.0 mM is recommended to find the optimal concentration [89] [91].
- dNTPs with Analogues: For extremely challenging GC-rich targets, incorporating a guanine analog like 7-deaza-2'-deoxyguanosine can improve yield. This analog pairs with cytosine but forms only two hydrogen bonds, reducing the overall stability of the duplex and minimizing secondary structure formation [89] [53].
Cycling Condition Modifications
Adjusting the thermal cycling protocol is a powerful and often necessary optimization step.
- Modified Denaturation: Using a higher denaturation temperature (e.g., 98°C) for the first few cycles can help fully denature exceptionally stable GC-rich templates. However, to minimize depurination of long templates, the duration at this temperature should be kept short [90] [53].
- Annealing Temperature Gradient: The primer annealing temperature (Ta) is critical. A temperature gradient PCR should be run to empirically determine the optimal Ta, which is often higher for GC-rich targets to increase specificity [89] [91].
- Modified Extension for Long Amplicons: For long products, using a slightly lower extension temperature (e.g., 68°C instead of 72°C) can dramatically improve the yield of full-length products by reducing depurination events during the longer extension time [90].
- Multiple Heat Pulses during Extension: A sophisticated strategy for GC-rich templates involves introducing brief, high-temperature heat pulses (e.g., to 98°C) during the extension step. This temporarily denatures stable secondary structures that have reformed, allowing the polymerase to continue synthesis. This method has successfully amplified 100% GC-rich sequences spanning nearly 3 kb [92].
- Slower Ramp Rates: Reducing the temperature transition speed between the denaturation and annealing steps (i.e., a slower ramp rate) can facilitate more specific primer binding to complex templates [53].
The following diagram illustrates a strategic workflow that integrates polymerase selection with buffer and cycling optimizations to tackle complex templates.
Detailed Experimental Protocols
Protocol for Amplifying GC-Rich Templates
This protocol uses a combination of a specialized polymerase, a GC enhancer, and optimized cycling conditions.
Research Reagent Solutions:
- DNA Polymerase: Q5 High-Fidelity DNA Polymerase (NEB #M0491) or OneTaq DNA Polymerase (NEB #M0480). Selected for high fidelity and supplied with a GC Enhancer.
- GC Enhancer: Proprietary solution containing additives like betaine to destabilize secondary structures.
- dNTP Mix: Balanced solution of dATP, dTTP, dCTP, dGTP. Freshly prepared or aliquoted to prevent freeze-thaw degradation.
- Primers: High-quality, HPLC-purified primers designed to avoid self-complementarity, especially at the 3' ends.
- Template DNA: High-purity template (e.g., column-purified) to minimize PCR inhibitors.
Methodology:
Reaction Setup (on ice):
- Prepare a 50 µL reaction mixture as follows. Note: Using a master mix for multiple reactions is recommended to minimize pipetting error.
- Template DNA: 1–100 ng (genomic DNA) or 1–10 ng (plasmid DNA).
- Q5 Reaction Buffer (5X): 10 µL.
- Q5 High GC Enhancer (5X): 10 µL.
- Forward Primer (10 µM): 2.5 µL.
- Reverse Primer (10 µM): 2.5 µL.
- dNTPs (10 mM each): 1 µL.
- Q5 DNA Polymerase: 0.5 µL (1 unit).
- Nuclease-Free Water: to 50 µL.
Thermal Cycling:
- Use the following program in a thermal cycler:
- Initial Denaturation: 98°C for 2 minutes.
- 35 Cycles of:
- Denaturation: 98°C for 10–20 seconds.
- Annealing: Use a gradient from 5°C below to 5°C above the calculated Tm of the primers. Determine optimal Ta empirically.
- Extension: 72°C for 30 seconds per kb.
- Final Extension: 72°C for 5 minutes.
- Hold: 4°C.
- Use the following program in a thermal cycler:
Analysis:
- Analyze 5–10 µL of the PCR product by agarose gel electrophoresis. A single, sharp band of the expected size indicates successful amplification.
Protocol for Amplifying Long Amplicons
This protocol emphasizes a proofreading polymerase, longer extension times, and a modified temperature profile to reduce depurination.
Research Reagent Solutions:
- DNA Polymerase: A high-fidelity, proofreading polymerase mix (e.g., Q5 Hot Start High-Fidelity or a Taq/Pfu blend). Essential for accuracy over long sequences.
- dNTP Mix: Balanced solution to prevent misincorporation due to limiting nucleotides.
- Mg²⁺ Solution: Separate from the buffer for precise titration.
- Template DNA: High-quality, intact DNA (e.g., minimally sheared genomic DNA).
Methodology:
Reaction Setup (on ice):
- Prepare a 50 µL reaction mixture.
- Template DNA: 10–200 ng (high complexity templates may require more).
- 5X Reaction Buffer: 10 µL.
- MgCl₂ (25 mM): Optimized concentration, start at 1.5–2.0 mM final.
- Forward Primer (10 µM): 2.5 µL.
- Reverse Primer (10 µM): 2.5 µL.
- dNTPs (10 mM each): 1 µL.
- DNA Polymerase Mix: 0.5–1.0 µL (as per mfr. recommendation).
- Nuclease-Free Water: to 50 µL.
Thermal Cycling:
- Use the following program, which features a short denaturation and lower extension temperature [90]:
- Initial Denaturation: 95°C for 2 minutes.
- 40 Cycles of:
- Denaturation: 94°C for 10 seconds. Short time to limit depurination.
- Annealing: 50–68°C for 1 minute.
- Extension: 68°C for 1 minute per kb. Lower temperature to limit depurination.
- Final Extension: 68°C for 10 minutes.
- Hold: 4°C.
- Use the following program, which features a short denaturation and lower extension temperature [90]:
Analysis:
- Analyze the product on a low-percentage agarose gel (e.g., 0.7–1.0%) to properly resolve the long amplicon. A single, sharp high-molecular-weight band is desired.
The Scientist's Toolkit: Essential Reagents
Table 3: Key Research Reagent Solutions for Complex Template PCR
| Reagent / Material | Function / Rationale | Example Products |
|---|---|---|
| High-Fidelity Proofreading Polymerase | Reduces error rate for accurate long amplicon synthesis. | Q5 Hot Start (NEB), Phusion (Thermo Fisher) |
| Polymerase with High Processivity | Navigates through secondary structures and long templates without dissociating. | Platinum II Taq (Thermo Fisher), LongAmp Taq (NEB) |
| Specialized GC Buffers & Enhancers | Proprietary additive mixes that help denature stable GC structures. | OneTaq GC Buffer & Enhancer (NEB), Q5 GC Enhancer (NEB) |
| Chemical Additives (DMSO, Betaine) | Destabilizes DNA secondary structures by altering melting behavior. | Molecular biology grade DMSO, Betaine solution |
| Hot-Start Polymerase Formulations | Inhibits activity at room temp to prevent non-specific priming and primer-dimer formation. | Antibody-mediated or chemically modified hot-start enzymes |
| Optimized dNTP / Analog Mixes | Provides balanced nucleotides; analogs like 7-deaza-dGTP can ease amplification. | dNTP sets, 7-deaza-2'-deoxyguanosine |
Leveraging High-Fidelity and High-Processivity Polymerases for Difficult Amplifications
The DNA polymerase enzyme serves as the fundamental engine of the polymerase chain reaction (PCR), directly determining the success, accuracy, and efficiency of nucleic acid amplification. Within the context of a broader thesis on DNA polymerase function in PCR research, this technical guide examines how engineered enzyme properties address the most persistent challenges in molecular biology. Early PCR methodologies relied on polymerases like Taq DNA polymerase, which, despite its thermostability, presented significant limitations including low fidelity (error-prone replication) and low processivity (limited nucleotides incorporated per binding event) [93]. These inherent constraints severely hampered the amplification of complex templates such as GC-rich sequences, long amplicons, and targets from suboptimal samples.
The evolution of PCR technology is, in essence, a history of DNA polymerase engineering. The development of hot-start polymerases through antibody-based or chemical inhibition marked a significant advancement by preventing non-specific amplification during reaction setup [94] [93]. A pivotal breakthrough emerged with the discovery and engineering of high-fidelity and high-processivity polymerases. These enzymes have expanded the possible applications of PCR by enabling accurate and efficient amplification of templates that were previously considered intractable, thus pushing the boundaries of genetic research, diagnostic assay development, and therapeutic discovery [95] [96].
Core Concepts: Fidelity and Processivity
DNA Polymerase Fidelity
Fidelity refers to the accuracy of a DNA polymerase in selecting and incorporating the correct nucleotide complementary to the template strand during DNA synthesis. It is quantitatively expressed as an error rate, typically defined as the number of misincorporated nucleotides per base pair per duplication event [97] [98]. Low-fidelity polymerases like Taq have error rates in the range of 1 × 10⁻⁵ to 2 × 10⁻⁵ [97] [96], meaning one error per 50,000 to 100,000 nucleotides incorporated. Such inaccuracies can lead to erroneous sequences in cloned DNA, incorrect experimental conclusions, and failed diagnostic tests.
High-fidelity polymerases achieve superior accuracy primarily through 3'→5' exonuclease proofreading activity [93]. This activity allows the enzyme to recognize and excise misincorporated nucleotides before continuing DNA synthesis. Engineered high-fidelity enzymes, such as Platinum SuperFi II DNA Polymerase, can reduce error rates to a level more than 300 times lower than that of Taq polymerase [96].
DNA Polymerase Processivity
Processivity is defined as the number of nucleotides a DNA polymerase incorporates into a growing DNA chain per single binding event with the template [95]. A highly processive enzyme can synthesize long stretches of DNA without dissociating, which is critical for efficiently amplifying long targets (>5 kb) or navigating through templates with complex secondary structures [94]. Polymerases with low processivity, in contrast, dissociate frequently, leading to incomplete synthesis and low product yield, especially for challenging amplicons.
Nature enhances the processivity of replicative polymerases through accessory proteins like sliding clamps. Inspired by this, a key protein engineering strategy involves fusing the polymerase domain to a sequence non-specific double-stranded DNA binding protein, such as Sso7d [95]. This fusion creates a molecular "tether" that increases the enzyme's affinity for the template, significantly boosting its processivity without compromising catalytic activity or stability [95]. This enhanced processivity is also crucial for direct PCR protocols, where the enzyme must withstand inhibitors present in crude samples without extensive nucleic acid purification [94] [96].
Enzyme Properties and Performance Data
The following tables summarize key performance metrics for various types of DNA polymerases, enabling informed selection for specific experimental needs.
Table 1: Comparative Error Rates of DNA Polymerases
| DNA Polymerase | Error Rate (errors/bp/duplication) | Fidelity Relative to Taq | Proofreading Activity |
|---|---|---|---|
| Taq | 1.0 × 10⁻⁵ to 2.0 × 10⁻⁵ [97] [96] | 1x [97] | No |
| AccuPrime-Taq HF | ~1.0 × 10⁻⁵ [97] | ~9x [97] | No (Blend) |
| Pfu | 1.0 × 10⁻⁶ to 2.0 × 10⁻⁶ [97] | 6-10x [97] | Yes (3'→5') |
| Pwo | ~1.0 × 10⁻⁶ [97] | ~10x [97] | Yes (3'→5') |
| Phusion Hot Start | 4.0 × 10⁻⁷ to 9.5 × 10⁻⁷ [97] | 24->50x [97] | Yes (3'→5') |
| Platinum SuperFi II | Not specified (vendor data) | >300x [96] | Yes (Engineered) |
Table 2: Polymerase Recommendations for Challenging PCR Applications
| Application Challenge | Recommended Polymerase Properties | Key Supporting Features |
|---|---|---|
| GC-Rich Templates (>65% GC) [94] | High Processivity, Hyperthermostable | DMSO or GC enhancer additives [94]; higher denaturation temperature (e.g., 98°C) [94] |
| Long Amplicons (>5 kb) [94] | High Processivity, High Fidelity | Polymerase blends (e.g., Taq + high-fidelity enzyme) [94]; engineered fusions (e.g., Sso7d) [95] |
| Direct PCR (from crude samples) [94] | High Processivity, Inhibitor Tolerant | Specially formulated lysis buffers; tolerance to inhibitors like heparin, humic acid, and heme [94] [96] |
| High-Throughput Cloning & Sequencing [97] | High Fidelity (>50x Taq) | Proofreading activity (3'→5' exonuclease) [97] [93]; optimized buffer systems |
| Multiplex PCR [94] | Hot-Start, Balanced Specificity | Specially formulated multiplex PCR buffer; primer sets with closely matched Tms [94] |
Engineering Strategies for Enhanced Polymerases
Protein Engineering to Boost Processivity
A seminal strategy for enhancing processivity involves the fusion of a standard polymerase domain to a heterologous, sequence non-specific double-stranded DNA-binding protein. Research has demonstrated that fusing the Sso7d protein from Sulfolobus solfataricus to the N-terminus of polymerases like Taq or Pfu significantly increases their processivity without negatively affecting catalytic activity or thermostability [95]. The engineered fusion enzyme exhibits a stronger grip on the DNA template, allowing it to synthesize longer fragments in a single binding event. This translates directly to improved PCR performance, particularly for long or difficult targets, as the enzyme is less likely to stall or fall off [95].
The Hot-Start Mechanism for Superior Specificity
Hot-start PCR is a widespread technique to improve amplification specificity. It employs an enzyme modifier—such as an antibody, affibody, aptamer, or chemical group—to inhibit the polymerase's activity at room temperature [94] [93]. This inactivation prevents the formation of non-specific products and primer-dimers during reaction setup. The modifier is released during the initial high-temperature denaturation step (typically >90°C), activating the enzyme. This controlled activation is particularly valuable for multiplex PCR and high-throughput workflows where reactions are assembled at ambient temperature, as it ensures high specificity and yield without requiring a cold setup [94].
Applications and Experimental Protocols
Amplifying GC-Rich Templates
Background: DNA templates with high GC content (>65%) form strong hydrogen bonds and stable secondary structures that can impede polymerase progression, leading to low yield or failed amplification [94].
Recommended Polymerase: A high-processivity polymerase with proofreading activity and enhanced stability, such as Platinum SuperFi II or Phusion Plus [94] [96] [93].
Detailed Protocol:
- Reaction Setup:
- Use the manufacturer's recommended buffer. Many modern high-fidelity buffers are pre-optimized for challenging templates.
- If problems persist, include a PCR enhancer such as DMSO (1-5%), formamide, or GC enhancer solutions. Note: These additives often lower the primer Tm, and the annealing temperature may need empirical optimization [94].
- Thermal Cycling Conditions:
- Initial Denaturation: 98°C for 30 seconds (for highly thermostable enzymes) [96].
- PCR Cycles (30-35 cycles):
- Final Extension: 72°C for 5-10 minutes.
Long-Range PCR
Background: Amplification of DNA targets longer than 5 kb requires a polymerase capable of synthesizing extensive stretches of DNA without dissociation [94].
Recommended Polymerase: A high-processivity, high-fidelity polymerase, often achieved through engineered fusions (e.g., Sso7d-based) or specialized blends [94] [95].
Detailed Protocol:
- Reaction Setup:
- Use a polymerase and master mix specifically designed for long-range PCR.
- Ensure template DNA is of high quality and integrity.
- Thermal Cycling Conditions:
- Initial Denaturation: 98°C for 30 seconds.
- PCR Cycles (30-35 cycles):
- Denaturation: 98°C for 10 seconds.
- Annealing & Extension: A two-step protocol is often effective. Combine annealing and extension at 68-72°C for a duration of 1-2 minutes per kb, depending on the enzyme's processivity [94]. The required time can be significantly shorter for highly processive enzymes (e.g., 1/2 to 1/3 the time needed for Taq) [94].
- Final Extension: 72°C for 10 minutes.
Sensitive Mutation Detection Using PNA Clamp PCR
Background: This technique uses Peptide Nucleic Acid (PNA) oligomers to suppress the amplification of wild-type DNA, allowing for the sensitive detection of rare mutant alleles in a background of excess wild-type sequences (e.g., in cancer research) [99].
Recommended Polymerase: A high-fidelity DNA polymerase is critical. The use of Taq polymerase can limit sensitivity because replication errors introduced by Taq in the PNA-binding site are amplified during PCR due to resulting mismatches between PNA and DNA. Switching to a high-fidelity enzyme can improve sensitivity by approximately 10-fold [99].
Detailed Protocol (Adapted from) [99]:
- Reaction Setup:
- 1x High-Fidelity Buffer (e.g., Phusion HF Buffer)
- 0.2 mmol/L dNTPs
- 0.15 μmol/L each of forward and reverse primer
- 0.25 μmol/L wild-type specific PNA oligomer
- 0.02 U/μL High-Fidelity DNA Polymerase (e.g., Phusion HS)
- Template DNA (e.g., 200 ng)
- Thermal Cycling Conditions:
- Initial Denaturation/Activation: 98°C for 30 seconds.
- PCR Cycles (45 cycles):
- Denaturation: 98°C for 10 seconds.
- PNA Annealing: 76°C for 10 seconds. This high-temperature step is key for specific PNA binding.
- Primer Annealing: 60°C for 20 seconds.
- Extension: 72°C for 20 seconds.
- Detection: Analyze products by real-time PCR with intercalating dyes like SYBR Green I, followed by melting curve analysis.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Advanced PCR Applications
| Reagent / Kit | Function / Application | Key Feature |
|---|---|---|
| Platinum SuperFi II DNA Polymerase [96] | High-fidelity PCR for cloning, sequencing, and mutagenesis. | >300x Taq fidelity; buffer allows universal 60°C annealing. |
| Phusion Plus DNA Polymerase [93] | High-fidelity amplification of challenging templates (GC-rich, long). | >100x Taq fidelity; improved performance with engineered domain. |
| Pfu DNA Polymerase [97] [93] | High-fidelity PCR requiring low error rates. | Native proofreading activity; error rate ~1x10⁻⁶. |
| Platinum Multiplex PCR Master Mix [94] | Simultaneous amplification of multiple targets in a single tube. | Specially formulated buffer for multiplexing; hot-start capability. |
| Direct PCR Kits (e.g., from cells or tissue) [94] | Amplification directly from crude samples without DNA purification. | Includes lysis buffers; uses highly processive, inhibitor-tolerant enzymes. |
| DMSO / GC Enhancer [94] | Additive to facilitate denaturation of GC-rich secondary structures. | Lowers DNA melting temperature; improves polymerase progression. |
Visualizing Experimental Workflows and Concepts
Engineering Strategy for High-Processivity Polymerases
Diagram Title: Polymerase Engineering via Sso7d Fusion
Comparative Workflow for Challenging Amplifications
Diagram Title: PCR Outcomes with Different Polymerases
The strategic application of high-fidelity and high-processivity DNA polymerases is indispensable for modern PCR research, directly addressing the functional limitations of earlier-generation enzymes. By understanding the mechanisms of polymerase fidelity and processivity, researchers can make informed decisions to overcome specific amplification challenges, from cloning GC-rich genes to detecting rare mutations in complex biological samples. The continued engineering of DNA polymerases—through proofreading domains, processivity-enhancing fusions, and sophisticated hot-start mechanisms—ensures that PCR will remain a cornerstone technology, driving discovery and innovation in genetics, medicine, and drug development.
Ensuring Reliability: Validation Protocols and Comparative Analysis of PCR Assays
The polymerase chain reaction (PCR) stands as a cornerstone of modern molecular biology and diagnostics, enabling the precise amplification and analysis of specific nucleic acid sequences. The technique's exponential power, capable of amplifying a single DNA molecule into billions of copies, necessitates rigorous validation to ensure data integrity and reliable conclusions [100]. The fidelity and performance of DNA polymerase, the core enzyme driving this process, are paramount to successful PCR assay development. This technical guide details the essential principles of PCR assay validation—sensitivity, specificity, and reproducibility—framed within the critical context of DNA polymerase function, providing researchers and drug development professionals with a comprehensive framework for developing robust, trustworthy assays.
The Central Role of DNA Polymerase in PCR
DNA polymerase is the fundamental engine of the PCR reaction. This thermostable enzyme synthesizes new DNA strands complementary to the target template by adding nucleotides to the 3' end of a primer in a 5' to 3' direction [1]. The properties of the DNA polymerase used directly influence every aspect of assay performance, including speed, fidelity, processivity, and robustness to inhibitors. Key advancements continue to be made through protein engineering; for instance, novel variants of Thermus aquaticus DNA polymerase I (Taq pol) have been engineered to possess reverse transcriptase activity, enabling single-enzyme, multiplex reverse transcription-PCR (RT-PCR), which simplifies workflows and reduces costs [65]. The choice of DNA polymerase and the optimization of its reaction conditions are, therefore, the foundational steps upon which a validated assay is built.
Core Principles of PCR Assay Validation
Assay validation is a fit-for-purpose process that confirms the reliability of an assay for its specific context of use (COU) [101]. For clinical research assays, which occupy the crucial space between Research Use Only (RUO) and fully certified In Vitro Diagnostics (IVD), rigorous validation is essential for generating credible, translatable data [101]. The three core principles, along with other key performance characteristics, are summarized in the table below.
Table 1: Key Performance Characteristics for PCR Assay Validation
| Validation Parameter | Definition | Typical Validation Experiment |
|---|---|---|
| Analytical Sensitivity | The lowest concentration of the analyte that can be reliably detected [101]. | Probit analysis of serial dilutions to determine the Limit of Detection (LOD) with ≥95% hit rate [56]. |
| Diagnostic Sensitivity | The true positive rate; ability to correctly identify subjects with the disease [101]. | Comparison against a reference method using well-characterized clinical samples [102]. |
| Analytical Specificity | The ability to distinguish the target from non-target analytes [101]. | Testing against a panel of non-target pathogens and interfering substances to check for cross-reactivity [56]. |
| Diagnostic Specificity | The true negative rate; ability to correctly identify subjects without the disease [101]. | Comparison against a reference method using samples known to be negative or containing other conditions [102]. |
| Precision/Reproducibility | The closeness of agreement between independent measurements under stipulated conditions [101]. | Repeated testing of the same samples within a run (intra-assay) and between different runs, operators, or days (inter-assay) [56]. |
| Inclusivity | The ability of the assay to detect all known strains or subtypes of the target organism [100]. | Testing the assay against a panel of well-defined, genetically diverse strains of the target [100]. |
| Linear Dynamic Range | The range of template concentrations over which the fluorescent signal is directly proportional to the input [100]. | Running a dilution series of a known standard and ensuring a linear fit with an R² value of ≥0.980 [100]. |
Sensitivity and the Limit of Detection (LOD)
Sensitivity defines the lowest quantity of an analyte that an assay can discern from zero. The Limit of Detection (LOD) is its quantitative measure, determined through statistical analysis (e.g., probit analysis) of dilution series, representing the concentration detectable in ≥95% of replicates [56]. For example, a novel multiplex respiratory assay demonstrated high sensitivity with LODs between 4.94 and 14.03 copies/µL [56]. Sensitivity is intrinsically linked to DNA polymerase efficiency. A highly efficient polymerase will generate more amplicon per cycle, leading to an earlier fluorescence threshold cycle (Cq) value, thereby improving the assay's ability to detect very low target concentrations [103]. Factors such as primer design, reagent purity, and reaction conditions all converge to influence the efficiency of the polymerase and, ultimately, the sensitivity of the assay.
Specificity, Inclusivity, and Exclusivity
Specificity ensures an assay detects only the intended target. This principle has two key components:
- Inclusivity: The assay must detect all genetic variants of the target pathogen or sequence. For an influenza A assay, this means reliable detection of subtypes like H1N1, H1N2, and H3N2 [100].
- Exclusivity (Cross-reactivity): The assay must not react with genetically similar non-targets or other organisms that may be present in the sample. An influenza A assay must not amplify influenza B [100].
Specificity is primarily governed by the primer and probe design and their interaction with the DNA polymerase. The annealing temperature and the buffer composition can influence the stringency of this binding. Assays often employ hydrolysis probes (e.g., TaqMan) to enhance specificity, as fluorescence is only generated upon specific probe hybridization and cleavage by the 5' to 3' exonuclease activity of the DNA polymerase [103]. One study validated specificity by testing against a panel of 47 reference strains and 14 non-target respiratory pathogens, observing no cross-reactivity [56].
Reproducibility and Precision
Reproducibility, or precision, measures the assay's consistency across repeated measurements. It is typically broken down into:
- Intra-assay precision: Repeatability within a single run.
- Inter-assay precision: Reproducibility between different runs, instruments, operators, or days [104].
This parameter directly tests the robustness of the entire PCR system, with the thermostability and lot-to-lot consistency of the DNA polymerase being critical factors. A well-formulated polymerase that maintains consistent activity over long-term storage and through numerous freeze-thaw cycles is essential for achieving high reproducibility. One validation study demonstrated exceptional precision with intra- and inter-assay coefficients of variation (CVs) for melting temperature (Tm) of ≤0.70% and ≤0.50%, respectively [56].
Experimental Protocols for Core Validation
Protocol 1: Determining the Limit of Detection (LOD)
- Prepare Standards: Create a serial dilution (e.g., 10-fold or 2-fold) of a standard material with a known concentration (e.g., copies/µL). The standard can be synthesized nucleic acid, or genomic DNA/RNA from a cultured organism [104].
- Amplify Replicates: Run each dilution in a minimum of 20 replicates using the optimized PCR conditions [56].
- Analyze Data: Use probit regression analysis to determine the concentration at which the target is detected in 95% of the replicates. This concentration is the LOD [56].
Protocol 2: Evaluating Specificity (Inclusivity and Exclusivity)
- In Silico Analysis: Perform BLAST analysis of all primer and probe sequences against genetic databases to check for potential cross-reactivity with non-target sequences and to ensure they align with all intended target variants [100].
- Wet-Lab Testing:
- Inclusivity Panel: Test the assay against a panel of at least 20-50 well-characterized strains or isolates representing the genetic diversity of the target organism [100].
- Exclusivity (Cross-reactivity) Panel: Test the assay against a panel of non-target organisms that are genetically related or likely to be found in the same sample matrix. This panel should include bacteria, viruses, and fungi [56] [105].
Protocol 3: Assessing Precision (Reproducibility)
- Sample Selection: Select at least two different samples with known concentrations, one near the LOD and one at a higher concentration [56].
- Intra-assay Precision: Test each sample multiple times (e.g., n=5) within the same PCR run.
- Inter-assay Precision: Test each sample across multiple separate runs, performed by different operators on different days [56].
- Statistical Analysis: Calculate the mean, standard deviation, and coefficient of variation (CV) for the quantitative results (Cq or Tm values). Lower CVs indicate higher precision [56].
Visualization of the PCR Assay Validation Workflow
The following diagram illustrates the comprehensive workflow for validating a PCR assay, integrating the core principles and their relationship to DNA polymerase performance.
The Scientist's Toolkit: Essential Reagents for PCR Validation
Table 2: Key Research Reagent Solutions for PCR Assay Validation
| Reagent / Material | Function in Validation | Key Considerations |
|---|---|---|
| DNA Polymerase | Catalyzes DNA synthesis; core enzyme determining efficiency, fidelity, and speed. | Thermostability, reverse transcriptase activity for RT-PCR, compatibility with probe chemistry (e.g., 5' nuclease activity), and tolerance to inhibitors [65]. |
| Quantified Standards | Calibrate the assay and determine LOD, linear range, and efficiency. | Should be traceable to an international standard. Used in serial dilution experiments [104]. |
| Primers & Probes | Confer sequence specificity for target detection. | Purity, stability, and specificity of sequence. Probe requires appropriate fluorophore and quencher [56]. |
| Inclusivity Panel | Validates the assay's ability to detect all target strains/subtypes. | A collection of well-characterized, genetically diverse target strains [100]. |
| Exclusivity Panel | Validates the assay does not cross-react with non-targets. | A collection of genomic DNA/RNA from non-target, closely related species [105]. |
| Internal PCR Control (IPC) | Monitors for PCR inhibition and verifies reaction integrity. | A non-target sequence (exogenous or endogenous) co-amplified with the target to identify reaction failure [104]. |
The validation of PCR assays based on the principles of sensitivity, specificity, and reproducibility is a non-negotiable standard for ensuring scientific rigor and reliability in both research and diagnostic applications. This process is not merely a series of independent tests but an integrated system where the performance of each component—especially the DNA polymerase—profoundly influences the whole. As enzyme engineering continues to advance, producing novel polymerases with enhanced capabilities such as intrinsic reverse transcriptase activity and greater multiplexing power [65], the fundamental need for thorough, methodical validation remains constant. By adhering to the structured framework and experimental protocols outlined in this guide, researchers can develop robust, fit-for-purpose PCR assays that generate trustworthy data, drive confident decision-making in drug development, and ultimately contribute to scientific progress.
Determining the Limit of Detection (LOD) and Reportable Range
This technical guide details the principles and practices for determining the Limit of Detection (LOD) and Reportable Range, framed within the context of PCR research. For researchers investigating DNA polymerase function, establishing a method's detection capabilities is paramount for interpreting amplification efficiency, sensitivity, and specificity. Robust LOD and range determination ensure data reliability when quantifying low-abundance targets, characterizing novel polymerase properties, or validating diagnostic assays [106] [107].
In PCR research, the Limit of Detection (LOD) represents the lowest number of target DNA copies per reaction that can be reliably distinguished from the absence of target, with a stated degree of confidence [106] [108]. The Reportable Range defines the concentration interval over which the method provides quantitative results with acceptable accuracy and precision, from the LOD to the Upper Limit of Quantification (ULOQ). Determining these parameters is critical when evaluating DNA polymerase performance, as factors like processivity, fidelity, and amplification efficiency directly influence the assay's sensitivity and dynamic range [107].
Theoretical Foundations of Detection Limits
Statistical Definitions: Error Types and Their Control
The statistical determination of LOD involves managing two types of potential errors. A Type I error (α), or false positive, occurs when the method indicates the target is present in a blank sample. A Type II error (β), or false negative, occurs when the method fails to detect the target in a sample that genuinely contains it at or above the LOD [106]. The relationship between these errors and detection capability is conceptualized through several key limits:
- Critical Level (LC): The signal threshold above which a response is considered indicative of target presence, typically set to control false positives to 5% (α=0.05) [106].
- Limit of Detection (LOD): The true concentration at which the method can reliably detect the analyte, set to ensure a low probability (typically β=0.05) of false negatives [106] [108].
- Limit of Quantification (LOQ): The lowest concentration at which the analyte can be quantified with acceptable precision and bias, representing the lower end of the Reportable Range [108] [109].
Conceptual Workflow for Limit Determination
The following diagram illustrates the logical relationship and workflow between key concepts and experimental steps in determining the Critical Level, LOD, and LOQ.
Methodologies for LOD and LOQ Determination
Multiple approaches exist for determining LOD and LOQ, each with specific applications and requirements. The choice of method depends on the analytical technique, the nature of the sample matrix, and regulatory considerations [109] [107].
Standard Deviation-Based Approaches
This method uses the statistical distribution of blank and low-concentration sample measurements.
- Limit of Blank (LOB): Determined from repeated measurements of a blank sample (no target). LOB = Mean
blank+ 1.645 × SDblank(assuming 95% one-sided confidence for normal distribution) [108]. - Limit of Detection (LOD): Requires measurements from both blank samples and samples with low analyte concentrations. LOD = LOB + 1.645 × SD
low concentration sample[108]. An alternative simplified calculation is LOD = Meanblank+ 3.3 × SDblankwhen α=β=0.05 and standard deviation is constant [106] [109]. - Limit of Quantitation (LOQ): The lowest concentration where quantification meets predefined bias and imprecision goals. Often calculated as LOQ = Mean
blank+ 10 × SDblankor as 5-10 times the LOD, depending on the required confidence level [109] [110].
Table 1: Standard Deviation-Based LOD/LOQ Calculations
| Parameter | Sample Type | Minimum Replicates (Guideline) | Typical Calculation Formula |
|---|---|---|---|
| LOB | Blank (no analyte) | 20-60 [108] | Meanblank + 1.645 × SDblank |
| LOD | Low concentration analyte | 20-60 [108] | LOB + 1.645 × SDlow conc or Meanblank + 3.3 × SDblank |
| LOQ | Low concentration analyte | 20-60 [108] | Meanblank + 10 × SDblank |
Signal-to-Noise Ratio
Commonly used in chromatographic methods and applicable to electrophoretic analysis of PCR products, this approach defines LOD as the concentration yielding a signal 2-3 times higher than the background noise, and LOQ as the concentration with a signal 10 times higher [106] [109]. For example, the European Pharmacopoeia defines LOD using a signal-to-noise ratio of 3:1 [106].
Calibration Curve-Based Approach
This method is appropriate when a linear calibration curve can be established. The LOD and LOQ are derived from the standard error of the regression and the slope, which represents the sensitivity of the method [109].
- LOD = 3.3 × σ / Slope
- LOQ = 10 × σ / Slope
Where σ is the standard deviation of the response (residual standard error of the regression) and the Slope is from the calibration curve [109]. This approach is particularly useful for quantitative PCR (qPCR) standard curves.
Table 2: Comparison of LOD/LOQ Determination Methods
| Method | Key Requirement | Advantages | Common Application in PCR Context |
|---|---|---|---|
| Standard Deviation of Blank/Low Sample | Multiple blank and low-concentration replicates | Directly accounts for matrix effects; statistically rigorous [108] | Validating sensitivity of diagnostic PCR assays |
| Signal-to-Noise Ratio | Measurable background noise | Simple, instrument-independent; practical for visual assessment [106] | Gel electrophoresis analysis of amplification products |
| Calibration Curve | Linear relationship between signal and concentration | Uses data from quantitative standards; incorporates method sensitivity [109] | qPCR standard curve analysis for copy number determination |
Experimental Protocol for LOD Determination in PCR
This protocol provides a step-by-step methodology for determining the LOD of a PCR assay, crucial for researchers characterizing DNA polymerase performance.
Sample Preparation
- Blank Sample: Prepare a reaction mixture containing all PCR components (buffer, primers, dNTPs, DNA polymerase) except the DNA template. Use nuclease-free water or the appropriate buffer as substitute [107].
- Low-Concentration Samples: Serially dilute the target DNA in the appropriate matrix to create concentrations expected to be near the detection limit. For absolute quantification, determine copy number using spectrophotometry or droplet digital PCR.
Data Acquisition
- Replication: Analyze a minimum of 10-20 replicates of the blank sample and each low-concentration sample. The CLSI EP17 guideline recommends up to 60 replicates for robust estimation, though 20 may suffice for verification [108].
- Randomization: Run samples in random order to avoid systematic bias.
- Inclusion of Controls: Include positive controls above the expected LOD and no-template controls (blanks) in each run.
Data Analysis and Calculation
- For qPCR Data: Record Cq (quantification cycle) values. Set a threshold for fluorescence detection consistent with the assay's linear amplification phase.
- Calculate LOB: From the blank replicates, calculate the mean and standard deviation (SD). LOB = Mean
blank+ 1.645 × SDblank(for 95% confidence) [108]. - Calculate LOD: Analyze results from the low-concentration samples. Identify the lowest concentration where ≥95% of replicates are detected (i.e., Cq value is significantly different from blank). Statistically, LOD = LOB + 1.645 × SD
low concentration sample[108]. - Verification: Confirm the calculated LOD by testing additional replicates at that concentration. No more than 5% of results should fall below the LOB [108].
The following workflow summarizes the key experimental steps for LOD determination:
The Scientist's Toolkit: Research Reagent Solutions
The following reagents and materials are essential for conducting robust LOD and reportable range studies in PCR research.
Table 3: Essential Research Reagents for LOD Determination in PCR
| Reagent/Material | Function in LOD Determination | Technical Considerations |
|---|---|---|
| DNA Polymerase | Catalyzes DNA amplification; its efficiency and processivity directly impact detection sensitivity. | Key determinant of amplification efficiency; fidelity affects error rate at low copy numbers [106]. |
| Synthetic DNA Target | Provides standardized template for calibration curves and LOD studies. | Allows precise copy number determination; essential for absolute quantification [107]. |
| Nuclease-Free Water | Serves as blank matrix and diluent for standards. | Must be verified as DNA-free to prevent false positives in blank samples [107]. |
| dNTPs | Building blocks for DNA synthesis. | Quality affects polymerase efficiency and amplification yield, particularly at low target concentrations. |
| Primers | Sequence-specific oligonucleotides that define the amplified region. | Specificity is critical to minimize background amplification and false positives in blanks [107]. |
| Probe (for qPCR) | Provides sequence-specific fluorescence detection in real-time PCR. | Signal-to-noise ratio of probe fluorescence directly influences detection capability [106]. |
| Buffer Components | Provides optimal chemical environment for polymerase activity. | Mg²⁺ concentration particularly critical for polymerase fidelity and amplification efficiency. |
Reportable Range Determination
The Reportable Range encompasses all concentrations between the LOQ and the Upper Limit of Quantification (ULOQ), where the method provides quantitative results with acceptable accuracy and precision [109].
- Lower End: Defined by the LOQ, where the analyte can be quantified with specified precision (e.g., ≤20% CV) and bias [108] [109].
- Upper End (ULOQ): The highest concentration where the method remains quantitatively reliable without signal saturation or significant deviation from linearity. In qPCR, this is often the concentration where amplification efficiency begins to decline significantly.
- Verification: Validate the reportable range by testing samples across the entire claimed range, ensuring accuracy and precision specifications are met at both the LOQ and ULOQ.
Accurate determination of LOD and Reportable Range is fundamental to method validation in PCR research. The selection of an appropriate determination strategy must align with the analytical method's underlying principles, whether based on statistical analysis of blank and low-concentration samples, signal-to-noise evaluation, or calibration curve characteristics. For scientists investigating DNA polymerase function, these parameters provide critical insights into enzyme performance and ensure the reliability of data used to draw conclusions about polymerase efficiency, sensitivity, and applicability to specific research or diagnostic applications. Properly established and validated detection limits and quantitative ranges form the foundation for scientifically defensible experimental results in molecular biology research.
Comparative Performance Analysis of Different DNA Polymerases
The function of DNA polymerase is fundamental to the polymerase chain reaction (PCR), serving as the core enzymatic engine that drives the amplification of specific DNA sequences. The selection of an appropriate DNA polymerase is a critical determinant of the success, accuracy, and efficiency of PCR research, influencing outcomes in fields ranging from basic molecular biology to clinical diagnostics and drug development. This whitepaper provides an in-depth technical analysis of the performance characteristics of various DNA polymerases, framing this analysis within the broader context of their mechanistic function in PCR. It synthesizes current data on error rates, fidelity, processivity, and application-specific suitability to equip researchers with the evidence needed to make informed reagent selections for their experimental pipelines.
Core Functions and Mechanism of DNA Polymerases in PCR
DNA polymerases are enzymes that catalyze the template-directed synthesis of DNA from deoxyribonucleoside triphosphates (dNTPs), a process essential for DNA replication and amplification [33]. In the context of PCR, their primary functions include:
- Template Binding: Recognizing and binding to single-stranded DNA templates at the primer-template junction.
- Nucleotide Incorporation: Catalyzing the step-wise addition of nucleotides to the 3'-end of a primer in a 5' to 3' direction, complementary to the template strand [33] [1].
- Processivity: The ability to incorporate multiple nucleotides per single binding event, significantly enhancing the rate of DNA synthesis [33].
High-fidelity DNA polymerases achieve remarkable accuracy through a multi-step mechanism involving both nucleotide selection and proofreading. The kinetic mechanism, as detailed in studies of bacteriophage T7 DNA polymerase, involves a substrate-induced conformational change from an open to a closed state after nucleotide binding [111]. This induced-fit mechanism is a key fidelity checkpoint; correct nucleotides promote a conformational change that optimally aligns catalytic residues for efficient phosphoryl transfer, whereas incorrect nucleotides disrupt this alignment, leading to their rapid release before incorporation [111] [112]. For polymerases with intrinsic 3'→5' exonuclease activity (proofreading), a second fidelity checkpoint exists. When a misincorporated nucleotide slows polymerization, the primer terminus can be transferred to the exonuclease site for excision before replication continues [112]. This proofreading activity can enhance overall fidelity by 10 to 100-fold [112].
Diagram 1: The kinetic pathway and fidelity checkpoints of high-fidelity DNA polymerases during PCR.
Quantitative Performance Comparison of DNA Polymerases
The performance of DNA polymerases varies significantly across commercial formulations, impacting key experimental outcomes such as accuracy, yield, and the presence of artifacts. The following tables summarize quantitative data from comparative studies.
Fidelity and Error Rate Analysis
Error rate, typically expressed as mutations per base pair per duplication, is a primary metric for assessing polymerase fidelity. Lower values indicate higher fidelity.
Table 1: DNA Polymerase Error Rates and Key Characteristics
| DNA Polymerase | Family | Proofreading | Error Rate (errors/bp/duplication) | Fidelity Relative to Taq |
|---|---|---|---|---|
| Taq | A | No | 1.0 × 10⁻⁵ to 2.0 × 10⁻⁵ [97] | 1x (Baseline) |
| AccuPrime-Taq HF | A | No | ~1.0 × 10⁻⁵ [97] | ~9x better [97] |
| KOD Hot Start | B | Yes | ~4x better than Taq [97] | >10x better [97] |
| Pfu | B | Yes | 1.0 × 10⁻⁶ to 2.0 × 10⁻⁶ [97] | 6-10x better [97] |
| Pwo | B | Yes | >10x lower than Taq [97] | >10x better [97] |
| Phusion Hot Start | B | Yes | 4.0 × 10⁻⁷ (HF buffer) [97] | >50x better [97] |
A comprehensive study employing direct sequencing of cloned PCR products from 94 unique DNA targets confirmed that proofreading polymerases (Pfu, Pwo, Phusion) exhibit significantly higher fidelity than non-proofreading enzymes like Taq. The study also reported that transition mutations were the predominant error type for high-fidelity enzymes [97].
Comprehensive Performance Metrics in Metabarcoding
Beyond raw error rate, other performance parameters are critical for specialized applications like DNA metabarcoding, which requires high accuracy to distinguish between species.
Table 2: Multi-Parameter Performance in Metabarcoding [113]
| Performance Parameter | KOD plus Neo (TOYOBO) | HotStart Taq (BiONEER) | Significance |
|---|---|---|---|
| Chimera Formation | Low | Low | p < 0.05 |
| Blast Top Hit Accuracy | High | High | p < 0.05 |
| Deletion Artifacts | Low | Low | p < 0.05 |
| Insertion Artifacts | Variable | Variable | p < 0.05 |
| Base Substitution | Variable | Variable | p < 0.05 |
| Amplification Bias | Statistically significant differences observed across all 14 tested kits | p < 0.05 |
A comparative analysis of 14 different PCR kits using a mock eukaryotic community DNA sample found statistically significant differences for all seven parameters measured [113]. The study highlighted that kits containing KOD plus Neo and HotStart Taq DNA polymerase performed particularly well in parameters critical for metabarcoding, such as low chimera formation, high BLAST hit accuracy, and few deletion artifacts, especially at an elevated annealing temperature of 65 °C [113].
Experimental Protocols for Performance Analysis
Standardized experimental workflows are essential for generating comparable data on polymerase performance. The protocols below outline key methods for assessing fidelity and general PCR performance.
Standard PCR Protocol for Baseline Performance
A foundational protocol for PCR using Taq polymerase involves the following steps and reagent concentrations [114]:
- Reagent Setup: In a PCR tube, combine the following components to form a standard reaction mixture.
- Thermal Cycling:
- Initial Denaturation: 95°C for 2 minutes.
- Amplification Cycles (25-35 cycles):
- Denaturation: 95°C for 30 seconds.
- Annealing: 55-72°C for 30 seconds (temperature is primer-specific).
- Extension: 72°C (or 75-80°C [1]) for 1 minute per kb of amplicon.
- Final Extension: 72°C for 5-10 minutes.
- Product Analysis: Analyze amplified DNA by agarose gel electrophoresis and ethidium bromide staining [114].
Direct Sequencing Protocol for Error Rate Determination
A robust method for determining error rates involves direct sequencing of cloned PCR products across a wide sequence space [97].
- PCR Amplification: Amplify a diverse set of plasmid templates (e.g., 94 unique targets) using the polymerase under test. Use a minimal amount of template DNA (e.g., 25 pg/reaction) to maximize the number of template doublings.
- Cloning: Purify PCR products and clone them into a suitable vector system (e.g., Gateway recombination cloning).
- Sequencing: Sequence a sufficient number of clones (e.g., 65-75 clones per experiment) to achieve statistical power.
- Data Analysis:
- Calculate Doublings: Determine the average number of template doublings per PCR reaction from the fold-amplification.
- Identify Mutations: Compare clone sequences to the known reference sequence to identify all mutations.
- Compute Error Rate: Apply the formula: Error Rate = (Number of mutations observed) / (Total bp sequenced × Number of doublings) [97].
Diagram 2: Experimental workflow for determining DNA polymerase error rates via direct sequencing of cloned PCR products.
The Scientist's Toolkit: Essential Research Reagents
Selecting the correct reagents is paramount for successful PCR. The following table details key solutions and their functions in polymerase performance experiments.
Table 3: Research Reagent Solutions for DNA Polymerase Studies
| Reagent Solution | Function in Performance Analysis | Key Considerations |
|---|---|---|
| High-Fidelity Polymerase Blends (e.g., Phusion, Pfu) | Provides high accuracy for cloning and sequencing applications; essential for low error rate experiments. | Often include proofreading activity; requires optimization of buffer conditions (e.g., HF vs GC buffer) [97]. |
| Standard Taq Polymerase | Serves as a baseline control for fidelity and yield comparisons; ideal for routine PCR. | Lacks proofreading activity; higher error rate makes it unsuitable for high-fidelity needs [97] [1]. |
| Mock Community DNA | A defined mix of DNA templates (e.g., 40 microalgal species) for standardized assessment of artifacts and bias in applications like metabarcoding [113]. | Allows quantitative comparison of chimeras, insertions, deletions, and amplification bias across polymerases. |
| dNTP Mix | The building blocks for DNA synthesis; quality and concentration directly impact fidelity and yield. | Imbalances can increase error rates; ultrapure dNTPs are recommended for high-fidelity applications. |
| Optimized Reaction Buffers | Provides optimal pH, ionic strength, and co-factors (especially Mg²⁺) for polymerase activity and fidelity. | Mg²⁺ concentration must be titrated for each polymerase and primer system to maximize performance [114]. |
The comparative performance analysis of DNA polymerases reveals a critical trade-off between speed, fidelity, and application-specific requirements. Proofreading enzymes from families B and A, such as Pfu, Phusion, and KOD, offer superior accuracy, making them indispensable for cloning, sequencing, and quantitative analyses where sequence integrity is paramount. In contrast, non-proofreading polymerases like Taq remain suitable for rapid diagnostic PCR and qualitative applications. The selection of a DNA polymerase must be guided by the experimental context within the broader thesis of PCR research. For gene expression analysis, a polymerase with reverse transcriptase activity might be engineered for a single-enzyme RT-PCR [65], while for sequencing ancient DNA samples, an enzyme with superior damage bypass fidelity might be chosen. Ultimately, understanding the kinetic mechanisms, error profiles, and biases of different DNA polymerases empowers researchers to strategically select the optimal enzyme, thereby ensuring the reliability and reproducibility of their scientific findings.
Verification of Commercial Kits vs. Laboratory-Developed Tests (LDTs)
The polymerase chain reaction (PCR) stands as a cornerstone of modern molecular biology, and its functionality is fundamentally dependent on the properties of DNA polymerase. The enzyme's characteristics—including thermostability, fidelity, specificity, and processivity—directly influence the accuracy, sensitivity, and reliability of any PCR-based assay [115]. These properties are paramount when evaluating two primary types of PCR tests used in diagnostic and research settings: commercially manufactured kits and laboratory-developed tests (LDTs).
The verification of these assays is a critical process, ensuring that test results are consistently accurate, precise, and reliable. For commercial kits, verification confirms that the manufacturer's stated performance specifications are met in the user's laboratory. For LDTs, the process is more extensive, involving a full analytical validation to establish these performance characteristics from the ground up [104]. Within the context of a broader thesis on DNA polymerase function, this whitepaper explores the rigorous methodologies required for the verification and validation of commercial kits and LDTs, providing a technical guide for researchers and drug development professionals.
The Role of DNA Polymerase in PCR Assay Performance
The choice of DNA polymerase is a critical variable that underpins the performance of both commercial kits and LDTs. Its properties dictate the efficiency and faithfulness of the amplification reaction.
- Specificity: Refers to the enzyme's ability to amplify only the intended target. Nonspecific amplification can drastically impact yield and sensitivity. Hot-start DNA polymerases, which are inactive at room temperature, are engineered to minimize the amplification of misprimed targets or primer-dimers during reaction setup, thereby vastly improving specificity [115].
- Fidelity: This is the accuracy of DNA sequence replication, defined by the enzyme's proofreading capability. DNA polymerases with 3′→5′ exonuclease activity can correct misincorporated nucleotides. High-fidelity enzymes are crucial for applications like cloning and sequencing, where error rates must be exceptionally low [115].
- Thermostability: Essential for withstanding repeated denaturation cycles. While Taq polymerase is thermostable, enzymes from hyperthermophilic organisms like Pyrococcus furiosus (Pfu) offer superior stability at high temperatures (≥95°C), which is beneficial for denaturing complex templates [115].
- Processivity: Defined as the number of nucleotides incorporated per enzyme binding event. Highly processive polymerases are more efficient at amplifying long templates, sequences with high GC content, secondary structures, and samples containing PCR inhibitors [115].
Comparative Analysis: Commercial Kits vs. LDTs
The decision to use a commercial kit or to develop an LDT is driven by factors such as clinical need, cost, turnaround time, and the availability of suitable commercial tests, particularly for novel or rare pathogens [104].
Table 1: Key Characteristics of Commercial Kits vs. Laboratory-Developed Tests (LDTs)
| Characteristic | Commercial Kits | Laboratory-Developed Tests (LDTs) |
|---|---|---|
| Development | Developed by manufacturer for broad application. | Designed and developed within an individual laboratory [104]. |
| Regulatory Status | Often CE-marked or FDA-approved [104]. | Traditionally subject to laboratory-specific validation; FDA oversight is evolving [116]. |
| Cost | Higher cost per test [56]. | Often more cost-effective; one study reported 86.5% cheaper than commercial kits [56]. |
| Flexibility & Speed | Limited flexibility; rapid introduction for common pathogens [104]. | Highly flexible; can be rapidly developed and adapted in response to new threats (e.g., SARS-CoV-2) [104] [117]. |
| Typical Applications | High-volume, routine testing of common pathogens. | Specialized, small-scale tests for rare pathogens, research applications, and outbreak response [104]. |
Performance Comparison Data
Studies have directly compared the performance of commercial kits and LDTs for specific applications, such as the detection of SARS-CoV-2. One such study found 100% positive and negative agreement between several commercial assays (Roche cobas, Cepheid Xpert) and LDT variants, despite differences in their limits of detection (LOD) [118].
Table 2: Analytical Performance Comparison of SARS-CoV-2 PCR Assays from a Clinical Study [118]
| Assay | Type | E gene LOD (copies/mL) | Secondary Target LOD (copies/mL) | Positive Percent Agreement |
|---|---|---|---|---|
| LDT-1 | Laboratory Developed | 455 | n.d. (N1) | Reference (100%) |
| cobas SARS-CoV-2 | Commercial | 24 | 31.4 (orf1a) | 100% |
| Xpert Xpress SARS-CoV-2 | Commercial | 100 | ≤7.5 (N2) | 100% |
Another study validating a novel FMCA-based multiplex PCR LDT for respiratory pathogens reported high sensitivity with limits of detection between 4.94 and 14.03 copies/µL, and a clinical evaluation demonstrating 98.81% agreement with reference RT-qPCR methods [56].
Experimental Verification and Validation Protocols
A robust verification or validation plan is essential to ensure any PCR assay meets its intended purpose. The process is continuous, involving ongoing monitoring even after the assay is implemented [104].
The Verification and Validation Workflow
The following diagram outlines the key stages in the process of verifying a commercial kit or validating an LDT, from initial planning to ongoing quality control.
Detailed Methodologies for Key Experiments
Determining Limit of Detection (LOD)
The LOD is the lowest concentration of the analyte that can be reliably detected by the assay.
- Procedure:
- Sample Preparation: Create a dilution series of the target analyte (e.g., from cultured virus, recombinant RNA, or plasmid DNA) in a relevant matrix like viral transport medium (VTM) [56] [118]. For SARS-CoV-2 LOD determination, one study used serial dilutions of AccuPlex Reference Material from 1000 to 10 genome copies/mL [118].
- Testing Replicates: Test each dilution with a sufficient number of replicates (e.g., 20 replicates for a novel FMCA assay [56] or at least 3 technical replicates in another study [118]).
- Data Analysis: The LOD is determined by probit analysis as the concentration detectable with ≥95% probability [56]. It can be reported as copies per reaction or normalized to copies per volume of original sample (e.g., copies/mL) [118].
Assessing Analytical Specificity
Specificity testing ensures the assay detects only the intended target and does not cross-react with related organisms or substances.
- Procedure:
- Panel Creation: Assemble a panel of non-target organisms that are genetically similar or could be present in the same sample type. For a respiratory panel LDT, this included 10 respiratory viruses and 4 bacteria [56]. Another SARS-CoV-2 assay specificity panel included endemic coronaviruses (OC43, NL63), influenza, RSV, and others [118].
- In silico Analysis: Use tools like BLAST to check primer and probe sequences for potential cross-reactivity during the design phase [56].
- Experimental Testing: Run the assay against the specificity panel. A lack of amplification confirms the assay's specificity [56] [118].
Evaluating Precision
Precision measures the assay's reproducibility under defined conditions, including both repeatability (intra-assay) and reproducibility (inter-assay).
- Procedure:
- Sample Selection: Use at least two concentrations of the target (e.g., 2x LOD and 5x LOD) in a suitable matrix [56].
- Intra-assay Precision: Analyze each concentration multiple times (e.g., 5 replicates) in a single run by the same operator.
- Inter-assay Precision: Analyze each concentration in separate runs, conducted on different days and/or by different operators.
- Data Analysis: Calculate the coefficient of variation (CV%) for the quantitative results (e.g., Ct values or Tm values). One LDT validation reported exceptional intra- and inter-assay CVs of ≤0.70% and ≤0.50%, respectively [56].
The Scientist's Toolkit: Essential Research Reagent Solutions
The development and verification of PCR assays rely on a suite of essential reagents and components, each playing a critical role in the reaction's success.
Table 3: Key Reagents and Materials for PCR Assay Development and Verification
| Reagent/Material | Function | Technical Considerations |
|---|---|---|
| DNA Polymerase | Enzymatically synthesizes new DNA strands. | Selection is critical; consider hot-start for specificity, proofreading for fidelity, and processivity for complex templates [115]. |
| Primers & Probes | Provide sequence-specificity for target amplification and detection. | Designed against conserved genomic regions; checked for specificity via BLAST. Probes can be labeled with different fluorescent dyes for multiplex detection [56] [117]. |
| Reference Materials | Used for calibration, LOD determination, and quality control. | Include cultured pathogen, recombinant RNA, plasmid DNA, or synthetic oligonucleotides [104] [118]. |
| Internal Control | Co-amplified control to monitor extraction efficiency and PCR inhibition. | Typically a human housekeeping gene (e.g., RNase P) or an exogenous control spiked into the sample [56] [118]. |
| Nucleic Acid Extraction Kits | Isolate and purify DNA/RNA from clinical samples. | Extraction efficiency significantly impacts overall assay sensitivity; must be verified as part of the assay system [104] [118]. |
The verification of commercial kits and validation of LDTs are foundational to generating trustworthy data in both clinical and research environments. This process is intrinsically linked to the core function of DNA polymerase, whose biochemical properties—specificity, fidelity, thermostability, and processivity—directly dictate the performance boundaries of any PCR assay. A rigorous, methodical approach is required, encompassing the determination of analytical sensitivity (LOD), specificity, precision, and clinical agreement.
While commercial kits offer the advantage of convenience and regulatory clearance, LDTs provide unparalleled flexibility and cost-effectiveness, proving indispensable for rapid outbreak response and specialized research applications. As the PCR kits market continues to grow and evolve, driven by technological innovation and emerging diagnostic needs, the principles of thorough assay verification and validation will remain paramount. By adhering to detailed experimental protocols and international guidelines, researchers and scientists can ensure their molecular tools are fit for purpose, ultimately advancing drug development and scientific discovery.
Quantitative PCR (qPCR) is a cornerstone technique in molecular biology, diagnostics, and drug development. Its function relies fundamentally on the activity of DNA polymerase, the enzyme that catalyzes the replication of DNA sequences during thermal cycling. The precision of this enzyme directly impacts the accuracy, sensitivity, and reproducibility of the entire assay. However, the extreme sensitivity of qPCR also makes it vulnerable to subtle variations in reagents, sample quality, and protocol execution, which can lead to irreproducible and unreliable results.
To address these challenges, the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines were established. Originally published in 2009 [119] and recently updated to MIQE 2.0 [120], these guidelines provide a standardized framework for the design, execution, and reporting of qPCR experiments. For scientists and drug development professionals, adherence to MIQE and diagnostic validation standards is not merely a publication formality but a critical practice that ensures data integrity, facilitates peer review, and bolsters the credibility of scientific findings. This guide details the core principles of these standards within the context of modern PCR research.
The MIQE Guidelines: Ensuring Reproducible qPCR Research
The Evolution and Purpose of MIQE
The MIQE guidelines were created to combat a lack of consensus and insufficient experimental detail in qPCR publications. The primary aim is to ensure the reliability of results, promote consistency between laboratories, and increase experimental transparency [119]. The recently released MIQE 2.0 guidelines reflect advances in qPCR technology and offer updated recommendations tailored to the evolving complexities of contemporary applications [120].
Transparent, clear, and comprehensive description of all experimental details is necessary to ensure the repeatability and reproducibility of qPCR results. The guidelines encourage researchers to provide all necessary information without undue burden, thereby promoting more rigorous and reproducible qPCR research [120].
Core MIQE Requirements and Their Connection to DNA Polymerase
The MIQE checklist covers all aspects of a qPCR experiment. Key items directly related to the function of DNA polymerase include:
- Assay Validation: Detailed information on primer and probe sequences, including a unique identifier like a TaqMan Assay ID or the amplicon context sequence, must be provided [121]. This allows others to verify the specificity of the assay.
- Sample Handling: The guidelines outline best practices for sample collection, storage, and nucleic acid extraction to prevent degradation and the introduction of inhibitors that could affect DNA polymerase activity [120].
- qPCR Protocol and Analysis: The guidelines emphasize that quantification cycle (Cq) values must be converted into efficiency-corrected target quantities [120]. Assumptions of 100% PCR efficiency are often incorrect; applying an efficiency correction is essential for accurate interpretation [1]. Furthermore, the dynamic range and limits of detection for each assay must be established.
Adhering to these requirements ensures that the performance of the DNA polymerase—and by extension, the entire qPCR assay—can be properly evaluated and replicated.
Validation Standards for Diagnostic qPCR Assays
While MIQE provides a broad framework for research, diagnostic assays, whether laboratory-developed tests (LDTs) or commercial kits, require even more rigorous validation to ensure patient safety and accurate clinical decision-making.
The Need for Diagnostic Validation
The validation process begins with defining the clinical purpose of the assay, as this guides all subsequent steps [104]. For in-house LDTs, comprehensive validation is essential. However, even the use of a CE-marked or FDA-approved commercial kit does not eliminate the need for local verification. Factors such as staff competency, equipment maintenance, and workflow can affect performance, making it necessary for laboratories to verify the manufacturer's claims [104].
Key Analytical Validation Parameters
The table below summarizes the core parameters that must be established for a robust diagnostic qPCR assay.
Table 1: Key Analytical Validation Parameters for Diagnostic qPCR Assays
| Validation Parameter | Description | Experimental Methodology |
|---|---|---|
| Inclusivity | Measures the assay's ability to detect all intended target strains or variants [100]. | Test against a panel of well-defined, certified strains (e.g., up to 50) reflecting the genetic diversity of the target [100]. |
| Exclusivity (Cross-reactivity) | Assesses the assay's ability to avoid detection of genetically related non-targets [100]. | Test against a panel of near-neighbor organisms and common background flora that are not targets of the assay [100]. |
| Linear Dynamic Range | The range of template concentrations over which the fluorescent signal is directly proportional to the input [100]. | Run a 7-10 fold dilution series of a known standard in triplicate. Plot Cq values against the log of the concentration; the linear portion defines the range. Acceptable linearity (R²) is typically ≥0.980 [100]. |
| Limit of Detection (LOD) | The lowest concentration of the target that can be reliably detected [104]. | Probit analysis on multiple replicates of low-concentration samples. The LOD is often defined as the concentration at which 95% of replicates test positive [104]. |
| Limit of Quantification (LOQ) | The lowest concentration of the target that can be reliably quantified with acceptable precision and accuracy [100]. | Determined by assessing precision (CV) and accuracy across a dilution series. The LOQ is the lowest level where the CV remains below an acceptable threshold (e.g., 25-35%) [100]. |
| Precision | The random variation of repeated measurements [122]. | Measured by running multiple technical and biological replicates. Precision is reported as the Coefficient of Variation (CV), calculated as (Standard Deviation / Mean) × 100% [122]. |
| Accuracy | How closely measurements match the true quantity [122]. | Established by testing samples with known concentrations (e.g., international standards) and comparing the measured value to the expected value [122]. |
Advanced Data Analysis and Precision in qPCR
Moving Beyond the Cq Value
Robust qPCR data analysis requires more than just reporting raw Cq values. MIQE 2.0 emphasizes that Cq values should be converted into efficiency-corrected target quantities [120]. This is crucial because low PCR efficiency, potentially caused by suboptimal DNA polymerase performance or reaction inhibitors, requires more cycles to reach the detection threshold, leading to a higher Cq value and an underestimation of the true target quantity [1].
Advanced data processing methods can further improve accuracy. A "taking-the-difference" approach, which subtracts the fluorescence in one cycle from the next, has been shown to reduce background estimation error compared to traditional background subtraction methods [123]. Furthermore, statistical models such as weighted linear regression and weighted linear mixed models provide more accurate and precise estimates of the initial DNA amount and amplification efficiency [123].
The Critical Role of Replicates and Precision
Precision is paramount in qPCR, as high variation can obscure true biological differences or lead to false positive/negative results in diagnostics [122]. There are two primary types of replicates:
- Technical Replicates: Repetitions of the same sample in multiple wells. They measure system variation (inherent to the pipetting, instruments, and reagents) and help detect outliers [122].
- Biological Replicates: Different samples from the same experimental group. They account for the true biological variation within a population [122].
A balance must be struck between the number of replicates and cost. While triplicate technical replicates are common in research, the optimal number depends on the required precision and the inherent variability of the assay [122].
Table 2: Essential Reagent Solutions for a Validated qPCR Workflow
| Research Reagent Solution | Function in qPCR Experiment |
|---|---|
| Thermostable DNA Polymerase (e.g., Taq) | Enzymatically synthesizes new DNA strands by extending primers; its fidelity and processivity are fundamental to amplification efficiency and accuracy [1]. |
| Reverse Transcriptase (for RT-qPCR) | Converts RNA templates into complementary DNA (cDNA) for subsequent amplification; thermostability is a key performance factor [65]. |
| Sequence-Specific Primers & Probes | Provide the specificity of the assay by binding to the target sequence. Primers initiate synthesis, while probes (e.g., TaqMan) enable real-time detection [121]. |
| Passive Reference Dye | An inert fluorescent dye used to normalize the reporter dye signal, correcting for variations in well volume and optical anomalies, thereby improving precision [122]. |
| Internal & Extraction Controls | Non-target nucleic acids co-processed with the sample to monitor the efficiency of nucleic acid extraction and check for the presence of PCR inhibitors [104]. |
Emerging Trends: Engineering DNA Polymerase for Advanced Applications
The field of qPCR continues to evolve, driven in part by the engineering of novel DNA polymerases. Recent research focuses on creating multifunctional enzymes to simplify and improve assays. For instance, novel Taq polymerase variants have been engineered that possess intrinsic reverse transcriptase (RT) activity [65]. These single-enzyme variants can catalyze both cDNA synthesis and DNA amplification in one tube, eliminating the need for a separate viral reverse transcriptase. This innovation:
- Simplifies Workflows: Enables single-enzyme, single-tube RT-qPCR.
- Reduces Costs: Removes the need for a second enzyme.
- Enables Multiplexing: Some variants are capable of performing quantitative multiplex detection of various RNA targets in a single reaction, a significant advancement for molecular diagnostics [65].
Such engineering highlights the central role of DNA polymerase in expanding the capabilities of PCR technology and underscores the importance of detailed enzyme characterization as mandated by validation guidelines.
Experimental Protocol: A Template for qPCR Assay Validation
The following protocol provides a generalized methodology for validating a qPCR assay, incorporating MIQE and diagnostic standards.
Protocol: Validation of a qPCR Assay for a Novel RNA Virus Target
1. Assay Design and In Silico Analysis
- Primer/Probe Design: Design oligonucleotides targeting a conserved region of the viral genome using appropriate software.
- In Silico Specificity Check: Use databases (e.g., BLAST) to verify that the primers/probe bind exclusively to the target virus and not to human DNA/RNA or other common flora (exclusivity) [100].
- In Silico Inclusivity Check: Analyze sequence diversity of the target virus to ensure primers/probe will bind to all relevant strains/clades [100].
2. Determination of Linear Dynamic Range and PCR Efficiency
- Standard Curve Preparation: Serially dilute (e.g., 10-fold dilutions) a synthetic standard of the target RNA or a plasmid containing the target sequence with a known concentration. Use at least 5 dilution points, run in triplicate.
- qPCR Run: Amplify the dilution series using the optimized protocol.
- Data Analysis: Plot the Cq values against the logarithm of the starting concentration. Perform linear regression. The correlation coefficient (R²) indicates linearity (aim for ≥0.980), and the slope is used to calculate PCR efficiency: Efficiency % = (10^(-1/slope) - 1) × 100. An efficiency between 90-110% is generally acceptable [100].
3. Determination of Limit of Detection (LOD) and Limit of Quantification (LOQ)
- Sample Preparation: Prepare multiple replicates (e.g., n=12 or more) of samples at very low concentrations near the expected detection limit.
- qPCR Run and Analysis: Test all replicates. The LOD is the concentration at which 95% of the replicates are positive, typically determined by probit analysis. The LOQ is the lowest concentration where the Coefficient of Variation (CV) is below a predefined acceptable limit (e.g., 35%) [104] [100].
4. Assessment of Precision (Repeatability and Reproducibility)
- Intra-assay Precision: Run multiple technical replicates (e.g., n=8) of at least two samples (low and high concentration) within the same run. Calculate the CV for each sample.
- Inter-assay Precision: Run the same samples across three different runs, on different days, by different operators. Calculate the overall CV to assess reproducibility.
5. Experimental Assessment of Specificity (Inclusivity/Exclusivity)
- Wet-Lab Testing: Test the assay against a panel of genomic nucleic acids from the target strains (for inclusivity) and from near-neighbor non-target organisms (for exclusivity) to confirm in silico predictions [100].
The following diagram illustrates the logical workflow and key decision points in this validation process.
For researchers, scientists, and drug development professionals, rigorous adherence to the MIQE guidelines and diagnostic validation standards is non-negotiable. These frameworks ensure that the powerful technique of qPCR, underpinned by the fundamental function of DNA polymerase, produces data that is reliable, reproducible, and fit for purpose. As PCR technology advances with novel engineered enzymes and applications, a commitment to transparent and thorough reporting will remain the bedrock of scientific integrity and clinical utility.
Conclusion
The function of DNA polymerase is the cornerstone of a successful PCR, directly determining the assay's specificity, yield, accuracy, and reliability. A deep understanding of its characteristics—from thermostability and fidelity to processivity—enables informed enzyme selection for diverse applications, from basic research to critical diagnostics. Strategic optimization and rigorous troubleshooting are essential for overcoming challenges associated with complex templates and reaction setup. Furthermore, adherence to stringent validation protocols and comparative analysis ensures data integrity, which is paramount for drug development and clinical decision-making. Future directions will likely involve the continued engineering of novel polymerases with enhanced properties, further integrating PCR into point-of-care diagnostics and personalized medicine, solidifying its indispensable role in advancing biomedical science.
References
- 1. Polymerase Chain Reaction (PCR) - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK589663/]
- 2. Polymerase Chain Reaction: A Toolbox for Molecular ... [https://pubmed.ncbi.nlm.nih.gov/39955471/]
- 3. What is Polymerase Chain Reaction (PCR) [https://www.addgene.org/protocols/pcr/]
- 4. A comprehensive review of methodological and ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002058]
- 5. DNA Polymerase—Four Key Characteristics for PCR [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/dna-polymerase-characteristics.html]
- 6. PCR for Sanger Sequencing [https://www.thermofisher.com/us/en/home/life-science/sequencing/sanger-sequencing/sanger-dna-sequencing/pcr-sanger-sequencing.html]
- 7. Working with specific applications and conditions [https://www.takarabio.com/learning-centers/pcr/faq/applications-and-conditions?srsltid=AfmBOooaxiF-s3PD-ywmf3sbo5O6PXo2gYWF97NydfkgpY6k8t2BIIin]
- 8. Quantitative PCR Basics [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/quantitative-pcr?srsltid=AfmBOoqXD4w6j3gJFdpAW1zMkWN9UlDlp9iqZ79OzdPrvxdmAL0AhR4T]
- 9. Real-time polymerase chain reaction [https://en.wikipedia.org/wiki/Real-time_polymerase_chain_reaction]
- 10. Advancing quantitative PCR with color cycle multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417387/]
- 11. Taq polymerase [https://en.wikipedia.org/wiki/Taq_polymerase]
- 12. Thermostable DNA polymerase [https://en.wikipedia.org/wiki/Thermostable_DNA_polymerase]
- 13. Modified DNA for polymerases troubleshooting - PMC PCR [https://pmc.ncbi.nlm.nih.gov/articles/PMC5243913/]
- 14. DNA Polymerase Thermostability [https://www.neb.com/en-us/products/pcr-qpcr-and-amplification-technologies/pcr-qpcr-and-amplification-technologies/dna-polymerase-thermostability?srsltid=AfmBOopxY8fse4CB9i_gZuNAa4L3JQm_8MVAzgUjD-bfBTUH8wAu1n5K]
- 15. DNA Polymerase Selection Chart [https://www.neb.com/en-us/tools-and-resources/selection-charts/dna-polymerase-selection-chart?srsltid=AfmBOoqhOzs8SPuxj6o7Q0sMh6YzEg8nbf6jF5ItA0qkFbd8G3iTJ-hY]
- 16. PCR Fidelity Calculator | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/pcr-fidelity-calculator.html]
- 17. How is Fidelity Measured? [https://www.neb.com/en-us/applications/dna-amplification-pcr-and-qpcr/pcr/high-fidelity-pcr/how-is-fidelity-measured?srsltid=AfmBOooUJ4eXtU1Y3KB6lL_YL06eUOkuS2eLfDihPufAydCgYOPhoCPh]
- 18. amplification by DNA ( Polymerase ) Chain Reaction PCR [http://vlabs.iitkgp.ac.in/biochem/Exp2/theory.html]
- 19. Polymerase chain reaction [https://en.wikipedia.org/wiki/Polymerase_chain_reaction]
- 20. Polymerase Chain Reaction (PCR) – Variations of DNA ... [https://info.abmgood.com/polymerase-chain-reaction-pcr-dna-polymerase]
- 21. Polymerases [https://www.genomics-online.com/resources/16/5013/polymerases/]
- 22. DNA polymerases engineered by directed evolution to ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2014.00565/full]
- 23. Engineering polymerases for new functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6745271/]
- 24. Building better polymerases: Engineering the replication of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7863901/]
- 25. Polymerase Chain Reaction (PCR) - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4102308/]
- 26. How does PCR work and what are its 3 steps? | INTEGRA [https://www.integra-biosciences.com/united-states/en/blog/article/how-does-pcr-work]
- 27. Three Steps of PCR Cycle – Denaturation, Annealing, and ... [https://www.letstalkacademy.com/three-steps-of-pcr-cycle-denaturation-annealing-extension/]
- 28. PCR Cycling Parameters—Six Key Considerations for ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html]
- 29. Standard PCR Protocol [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/pcr/standard-pcr?srsltid=AfmBOoot652tj0yaHyHpjBVInSVivqAkpSvH8sjLptdGPpKt24yDDxiE]
- 30. Introduction to Quantitative PCR (qPCR) Gene Expression ... [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/gene-expression-analysis-real-time-pcr-information/introduction-gene-expression.html]
- 31. Comparative Performance of Digital PCR and Real-Time RT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12474457/]
- 32. PCR and Thermal Cycler Technology: Fundamentals, ... [https://www.labx.com/resources/pcr-and-thermal-cycler-technology-fundamentals-applications-and-emerging-trends-in-dna/5577]
- 33. DNA polymerase [https://en.wikipedia.org/wiki/DNA_polymerase]
- 34. Techniques used to study the DNA polymerase reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2846202/]
- 35. Kinetics of the DNA polymerase pyrococcus kodakaraensis [https://www.sciencedirect.com/science/article/abs/pii/S000925090600011X]
- 36. FAQ: What does exonuclease activity mean for a DNA ... [https://www.neb.com/en-us/faqs/2023/07/25/what-does-exonuclease-activity-mean-for-a-dna-polymerase?srsltid=AfmBOorsQPfDO53aXLUrSI6XdqMr5huo7BjPS-KKgPD1UR02agMJ2EfA]
- 37. rtpcr: a package for statistical analysis and graphical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530193/]
- 38. Polymerase Chain Reaction (PCR) Fact Sheet [https://www.genome.gov/about-genomics/fact-sheets/Polymerase-Chain-Reaction-Fact-Sheet]
- 39. PCR Basics | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-basics.html]
- 40. DNA Polymerase Selection Chart [https://www.neb.com/en-us/tools-and-resources/selection-charts/dna-polymerase-selection-chart?srsltid=AfmBOooawk1crGltr2XiEmX9120SbE5x-VE7-Gywhzcw5gZfAT9O5XFI]
- 41. The importance of selecting the right DNA polymerase for ... [https://www.vhbio.com/selecting-dna-polymerase-pcr-application/]
- 42. Common Cloning Applications and Strategies [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/molecular-cloning/cloning/common-applications-strategies.html]
- 43. A qPCR method for genome editing efficiency ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6906436/]
- 44. PCR Setup—Six Critical Components to Consider [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-component-considerations.html]
- 45. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 46. Standard PCR Protocol [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/pcr/standard-pcr?srsltid=AfmBOoprPyu1yy5B6A2UWhi8yPDv4VPoy1WZVt2hB1ruFdQ8KYrmVAtP]
- 47. Polymerase/DNA interactions and enzymatic activity [https://www.nature.com/articles/srep12066]
- 48. PCR Troubleshooting Guide | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-troubleshooting.html]
- 49. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOorD3-_DJMbWudUbXV49kFwgYU0QqrJJLhNj73xck1s5WFUzjNvf]
- 50. Types, mechanisms, and applications in long-range PCR [https://www.sciencedirect.com/science/article/abs/pii/S0300908422000499]
- 51. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOop1Dy7Xz_4MZnY9qH5wakWnMCK-fDeYB8J9RbD8lsexOuMsFvMs]
- 52. Optimizing PCR amplification of GC-rich nicotinic ... [https://pubmed.ncbi.nlm.nih.gov/40486497/]
- 53. 5 easy tips to address problems amplifying GC-rich regions [https://bitesizebio.com/24002/problems-amplifying-gc-rich-regions-problem-solved/]
- 54. Evolution of Multiplex PCR: Benefits, Challenges & Key ... [https://www.eurogentec.com/en/news/111_evolution-of-multiplex-pcr-benefits-challenges-key-applications]
- 55. Impact of Multiplex PCR on Diagnosis of Bacterial and Fungal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12026191/]
- 56. Development and clinical validation of a novel multiplex ... [https://www.nature.com/articles/s41598-025-16434-2]
- 57. How is Hot-Start Technology Beneficial For Your PCR [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/hot-start-technology-benefits-PCR.html]
- 58. Hot start PCR [https://en.wikipedia.org/wiki/Hot_start_PCR]
- 59. Hot Start PCR with heat-activatable primers [https://pmc.ncbi.nlm.nih.gov/articles/PMC2582603/]
- 60. Hot Start PCR vs Standard PCR – What You Need to Know [https://www.icnbiomed.com/pcr-methodologies-compared-hot-start-pcr-vs-standard-pcr-what-you-need-to-know/]
- 61. Development of a Simple Direct and Hot-Start PCR Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10379934/]
- 62. dNTP Hot Start PCR Protocol [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/pcr/dntp-mediated-hot-start-pcr?srsltid=AfmBOoofXVh-cm3y99pst1Q6h_18-BPn3ggzPaDvGAXX7nhTqcqCw9p5]
- 63. Basic Principles of RT-qPCR [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/basic-principles-rt-qpcr.html]
- 64. Reverse transcription polymerase chain reaction [https://en.wikipedia.org/wiki/Reverse_transcription_polymerase_chain_reaction]
- 65. Engineering of novel DNA polymerase variants for single ... [https://www.nature.com/articles/s41598-025-10211-x]
- 66. The Impact of the Variability of RT-qPCR Standard Curves ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12029521/]
- 67. Comparative Performance of Digital PCR and Real-Time ... [https://www.mdpi.com/1999-4915/17/9/1259]
- 68. Reverse transcription: Process and applications [https://www.abcam.com/en-us/knowledge-center/dna-and-rna/rtpcr]
- 69. Reverse transcription-quantitative PCR assays for ... [https://www.sciencedirect.com/science/article/pii/S2405844025008837]
- 70. Anatomy of a Polymerase - How Function and Structure are ... [https://www.neb.com/en-us/tools-and-resources/feature-articles/anatomy-of-a-polymerase-how-structure-effects-function?srsltid=AfmBOoq8LjFuPwx-_UqmwoaEvnE95tUZ8S7HL9sTaL1Ie7HNO5ZUbpqL]
- 71. PCR Amplification | An Introduction to PCR Methods [https://www.promega.com/resources/guides/nucleic-acid-analysis/pcr-amplification/]
- 72. Troubleshooting your PCR [https://www.takarabio.com/learning-centers/pcr/faq/troubleshooting?srsltid=AfmBOopLGxrKDgaeI4FunpsvxNSf2ovqtdrAErqegXu7-88hpdApzyg9]
- 73. Optimizing PCR: Proven Tips and Troubleshooting Tricks [https://www.the-scientist.com/optimizing-pcr-proven-tips-and-troubleshooting-tricks-71660]
- 74. PCR Troubleshooting: Common Problems and Solutions [https://www.mybiosource.com/learn/pcr-troubleshooting-common-problems-and-solutions/]
- 75. PCR Troubleshooting Guide [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/pcr-troubleshooting-guide?srsltid=AfmBOooHUSIBkEmjfD-eP09NfUlXJ1x6mTeVQ0CgPu8hCy1kx6UIMjnm]
- 76. Predicting sequence-specific amplification efficiency in ... [https://www.nature.com/articles/s41467-025-64221-4]
- 77. Comprehensive review and meta-analysis of magnesium ... [https://pubmed.ncbi.nlm.nih.gov/40436087/]
- 78. PCR Optimization: Factors Affecting Reaction Specificity ... [https://www.mybiosource.com/learn/pcr-optimization-factors-affecting-reaction-specificity-and-efficien/]
- 79. Optimizing dNTP Concentration in PCR Reactions for Effi... [https://www.sbsgenetech.com/blog/dntp-concentration-in-pcr-reaction?srsltid=AfmBOopYON-hRRokrZb2N8N--1eZkFt5038AQfkkPdE0fYsYIORgNZ0S]
- 80. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 81. How to Design Primers for PCR Experiments [https://www.zymoresearch.com/blogs/blog/how-to-design-primers-for-pcr-experiments?srsltid=AfmBOooGsDkvtXlzxPOzki8L-CWClIdbbtBPSDSAPT8aF527zOAlYRQc]
- 82. How to Design PCR Primers in Geneious Prime [https://www.geneious.com/tutorials/primer-design]
- 83. Live culture-based qPCR screening of Taq DNA ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1624735/full]
- 84. Faster PCR Optimization [https://bitesizebio.com/31937/optimizing-your-pcr-faster/]
- 85. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOooAHwz8CsNdhezfXBELxGuZTzv3eIReMwA5FyR1-9CAMg3wWP23]
- 86. Improve Yield and Specificity of Your PCR Using the 2D- ... [https://www.eppendorf.com/it-en/lab-academy/life-science/molecular-biology/improve-yield-and-specificity-of-your-pcr-using-the-2d-gradient-function/]
- 87. How to Simplify PCR Optimization Steps for Primer Annealing [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/pcr-annealing-optimization-universal-annealing.html]
- 88. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOoqavQYE6t734Jz-F95XmxIfr6VI4NIQYCH7N6xNlLNWyDJOHNgi]
- 89. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOoq7CKIaulPp7WnoHKzesF7EDEcEirGlOPYeGcGIE0Dg3nTleFyn]
- 90. PCR amplification: Long PCR products [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/introduction/amplification-of-long-pcr-products]
- 91. PCR Troubleshooting Guide [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/pcr-troubleshooting-guide?srsltid=AfmBOoq6-ffvCbfYN8NXBmD64yPCUEaiVJh2ZsU8E2-6wgYGFxLjkV4n]
- 92. Multiple Heat Pulses During PCR Extension Enabling ... [https://pubmed.ncbi.nlm.nih.gov/22220596/]
- 93. The History of PCR [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/history-pcr.html]
- 94. PCR Methods - Top Ten Strategies [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-methods.html]
- 95. A novel strategy to engineer DNA polymerases for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC373405/]
- 96. Platinum SuperFi II DNA Polymerase-High Fidelity PCR ... [https://www.thermofisher.com/us/en/home/life-science/pcr/pcr-enzymes-master-mixes/platinum-high-fidelity-pcr-enzyme.html]
- 97. Error Rate Comparison during Polymerase Chain Reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4150459/]
- 98. High-Fidelity PCR [https://www.neb.com/en-us/applications/dna-amplification-pcr-and-qpcr/pcr/high-fidelity-pcr?srsltid=AfmBOoprIpFeNxQam7bQhbzI7DZkK_RE4Mj7O-fPRum3hxBdgcCMnvEH]
- 99. High-Fidelity DNA Polymerase Enhances the Sensitivity of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2438201/]
- 100. Quantitative PCR validation for research scientists: Part 1 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 101. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 102. Validation of the Clinical Performance and Reproducibility ... [https://pubmed.ncbi.nlm.nih.gov/40280407/]
- 103. Quantitative PCR Basics [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/quantitative-pcr?srsltid=AfmBOorheQCc3_xj_eKXxxC6Ppq_GAERiwHUFO9TV5Sf21XuzDoixbU6]
- 104. Validating Real-Time Polymerase Chain Reaction (PCR) Assays [https://pmc.ncbi.nlm.nih.gov/articles/PMC7834634/]
- 105. Developmental validation of a real-time PCR assay for the ... [https://www.sciencedirect.com/science/article/abs/pii/S1872497308000975]
- 106. The Limit of Detection | LCGC International [https://www.chromatographyonline.com/view/limit-detection]
- 107. A tutorial for computing limits of detection and ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267021011685]
- 108. Limit of Blank, Limit of Detection and Limit of Quantitation [https://pmc.ncbi.nlm.nih.gov/articles/PMC2556583/]
- 109. Method Validation Essentials, Limit of Blank ... [https://www.biopharminternational.com/view/method-validation-essentials-limit-blank-limit-detection-and-limit-quantitation]
- 110. Laboratory Detection Limits Explained [https://mytapscore.com/blogs/tips-for-taps/how-low-can-you-go-lab-detection-limits-explained?srsltid=AfmBOorZgvAkWLrfk-wcE4y0rxmTrPV8IQAByrrnrpcuoo1fmMIeqhY0]
- 111. The kinetic and chemical mechanism of high-fidelity DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3047511/]
- 112. Kinetic Approaches to Understanding the Mechanisms of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3010682/]
- 113. Comparing PCR-generated artifacts of different ... [https://www.sciencedirect.com/science/article/pii/S2534970822000163]
- 114. Standard PCR Protocol [https://www.sigmaaldrich.com/US/en/technical-documents/protocol/genomics/pcr/standard-pcr?srsltid=AfmBOoqP6JMjJf3ENFATXHB6UIbu2fuda9wOnDkW5MJkY6k6k8Zccee4]
- 115. DNA Polymerase—Four Key Characteristics for PCR [https://www.thermofisher.com/ru/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/dna-polymerase-characteristics.html]
- 116. Laboratory Developed Tests [https://www.fda.gov/medical-devices/in-vitro-diagnostics/laboratory-developed-tests]
- 117. Development and validation of novel RT-PCR assay for ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202325000933]
- 118. Comparison of commercial assays and laboratory developed tests for detection of SARS-CoV-2 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7482591/]
- 119. The MIQE guidelines: minimum information for publication ... [https://pubmed.ncbi.nlm.nih.gov/19246619/]
- 120. MIQE 2.0: Revision of the Minimum Information for ... [https://pubmed.ncbi.nlm.nih.gov/40272429/]
- 121. MIQE Guidelines | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/why-choose-taqman-assays/publish-real-time-pcr-results.html]
- 122. Quantitative PCR (qPCR) Data Analysis [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/gene-expression-analysis-real-time-pcr-information/precision-qpcr.html]
- 123. Comparison of Analytic Methods for Quantitative Real-Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4642823/]