Taq Polymerase: The Yellowstone Discovery That Revolutionized Biomedicine
This article details the discovery of Taq polymerase, from its origins in the hot springs of Yellowstone National Park to its pivotal role in perfecting the Polymerase Chain Reaction (PCR).
Taq Polymerase: The Yellowstone Discovery That Revolutionized Biomedicine
Abstract
This article details the discovery of Taq polymerase, from its origins in the hot springs of Yellowstone National Park to its pivotal role in perfecting the Polymerase Chain Reaction (PCR). Aimed at researchers and drug development professionals, it provides a comprehensive examination of Taq's enzymatic properties, its vast applications in molecular diagnostics and drug development, and a critical comparison with high-fidelity polymerases. The scope extends to practical guidance on optimizing PCR protocols, troubleshooting common issues, and validating results, particularly through methods like quantitative PCR. By synthesizing foundational knowledge with advanced methodological insights, this resource supports the effective application of Taq polymerase in cutting-edge biomedical research.
From Yellowstone Hot Springs to Lab Benches: The Origin Story of a Revolutionary Enzyme
The discovery of Thermus aquaticus by Thomas Brock in the hot springs of Yellowstone National Park represents a cornerstone discovery in microbiology that fundamentally reshaped our understanding of life's limits. Prior to Brock's work, scientific consensus held that life could not exist at temperatures much above 73°C [1]. However, Brock's pioneering field research from 1965 to 1975, funded by the National Science Foundation, directly challenged this dogma [2]. His initial observations of brightly colored bacterial filaments thriving in the Octopus Hot Spring at temperatures exceeding 80°C revealed the existence of a previously unknown world of extremophiles—organisms that thrive in conditions once considered inhospitable to life [1] [3]. This discovery of Thermus aquaticus not only opened an entirely new field of scientific inquiry but also serendipitously provided the essential biological tool that would later revolutionize molecular biology and biotechnology: the heat-stable Taq DNA polymerase [2] [4].
The broader thesis of this research underscores the profound importance of basic, curiosity-driven scientific exploration. Brock's investigation into the pink bacterial filaments was motivated by fundamental questions about the limits of life rather than immediate commercial application [3]. Yet, this basic research ultimately laid the foundation for the polymerase chain reaction (PCR) technology, which has since become indispensable in fields ranging from medical diagnostics to forensic science [5] [6]. This article traces the complete trajectory of Brock's discovery, from the initial observation in Yellowstone's extreme environments to the isolation and characterization of T. aquaticus, and finally to the subsequent identification and application of its thermostable polymerase, illustrating how fundamental ecological research can yield tools of transformative power.
The Discovery:Thermus aquaticusand the Upper Temperature Limit of Life
Thomas Brock and the Yellowstone Research Initiative
Thomas Dale Brock, then a professor of bacteriology at Indiana University, was a microbiologist whose interests were shifting toward microbial ecology when he began studying microorganisms in diverse habitats, including intertidal pools, freshwater lakes, and cold springs [1] [2]. His passion for field ecology and a fortuitous travel bug led him to establish a research station in Yellowstone National Park [1]. In a 1964 visit, Brock's scientific curiosity was sparked by a park ranger's talk near a thermal pool, where he observed vivid colors that the ranger attributed to "blue-green algae" [3]. This chance encounter ignited a decade-long systematic research program into the microbial life of Yellowstone's geothermal features.
Brock's approach combined rigorous field sampling with meticulous laboratory analysis. From 1965 to 1975, he and his team collected samples from various extreme environments throughout Yellowstone, including hot pots, geyser pools, fumaroles (steam vents), and thermal basins [3]. This sustained fieldwork was critical, as the protected status of Yellowstone as a national park preserved these unique habitats from development or destruction, making prolonged international research possible [1]. Brock's methodology exemplifies the importance of interdisciplinary field research in ecological and environmental sciences, particularly for understanding complex ecosystems found in many national park locations [3].
Isolation and Characterization of a Novel Extremophile
In 1969, Brock and his undergraduate student Hudson Freeze published their landmark paper introducing Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile [1] [2] [7]. They isolated this novel organism from Mushroom Spring, where it was thriving at temperatures of 70°C (160°F) [2] [3]. This discovery was monumental because it provided the first definitive evidence of an organism not merely surviving but reproducing at such high temperatures, effectively disproving the established upper temperature limit for life [1].
Table 1: Characterization of Thermus aquaticus and Its Habitat
| Characteristic | Description |
|---|---|
| Discovery Location | Mushroom Spring, Yellowstone National Park [2] [3] |
| Discovery Year | 1969 [2] |
| Discoverers | Thomas D. Brock and Hudson Freeze [2] |
| Growth Temperature Range | 45°C to 80°C [8] |
| Optimal Growth Temperature | ~70°C [2] [3] |
| Classification | Thermophilic, gram-negative bacterium [7] |
| Cell Morphology | Rod-shaped; forms "rotund bodies" from fused cell associations [7] |
| Global Distribution | Found in hot springs worldwide and even man-made hot water systems [1] |
Subsequent research revealed that T. aquaticus was not merely a Yellowstone curiosity but a ubiquitous organism in high-temperature environments worldwide, found in hot springs across Japan, New Zealand, and Iceland, as well as in more mundane settings like the hot water supply at Indiana University and soil in tropical-temperature greenhouses [1]. Electron microscopy studies of its cellular structure showed that T. aquaticus has a typical gram-negative tripartite cell envelope, consisting of a plasma membrane, a thin middle layer, and a thicker irregular outer layer [7]. The organism's unique adaptation to thermal extremes extends to its macromolecules; research confirmed that its ribosomes and RNA possess exceptional thermal stability, a prerequisite for functionality at high temperatures [7].
From Bacterium to Biotechnology: The Taq Polymerase Revolution
Enzymatic Properties of Taq DNA Polymerase
The true significance of Thermus aquaticus emerged with the isolation and characterization of its DNA polymerase, now famously known as Taq polymerase. This enzyme, first isolated by Alice Chien et al. in 1976, possesses remarkable thermostability that makes it ideally suited for the high-temperature processes required in DNA amplification [5] [9]. Taq polymerase is an 832-amino acid protein with a molecular weight of approximately 94 kDa, classified within the Family A DNA polymerases alongside E. coli DNA polymerase I [9].
Table 2: Biochemical Properties of Taq DNA Polymerase
| Property | Specification |
|---|---|
| Molecular Weight | 93,920 Da [9] |
| Specific Activity | 292,000 units/mg [9] |
| Optimal Polymerization Temperature | 75-80°C [5] [9] |
| Polymerization Rate at 70°C | 60-100 nucleotides/second [5] [9] |
| Thermal Half-life | >2 hours at 92.5°C; 40 minutes at 95°C; 9 minutes at 97.5°C [5] [9] |
| Processivity | Extends a primer 50-60 nucleotides on average before dissociating [9] |
| Exonuclease Activity | 5'→3' polymerase activity; lacks 3'→5' proofreading activity [5] [9] |
| Error Rate | Approximately 1 in 9,000-10,000 nucleotides [5] [6] |
| Cofactor Requirement | Mg²⁺ (1.5-4.0 mM optimal) [9] |
The exceptional heat resistance of Taq polymerase stems from its origin in a thermophilic organism whose entire cellular machinery has evolved to function at high temperatures. Unlike polymerases from mesophilic organisms, Taq can withstand repeated exposure to the near-boiling temperatures (95°C) required to denature double-stranded DNA without significant loss of activity [5]. This property proved to be the key innovation that enabled the automation and widespread adoption of the polymerase chain reaction. However, a significant limitation of Taq polymerase is its lack of 3' to 5' exonuclease proofreading activity, which results in a relatively high error rate compared to proofreading enzymes [5]. This has led to the development of other thermostable polymerases with higher fidelity for applications requiring extreme accuracy.
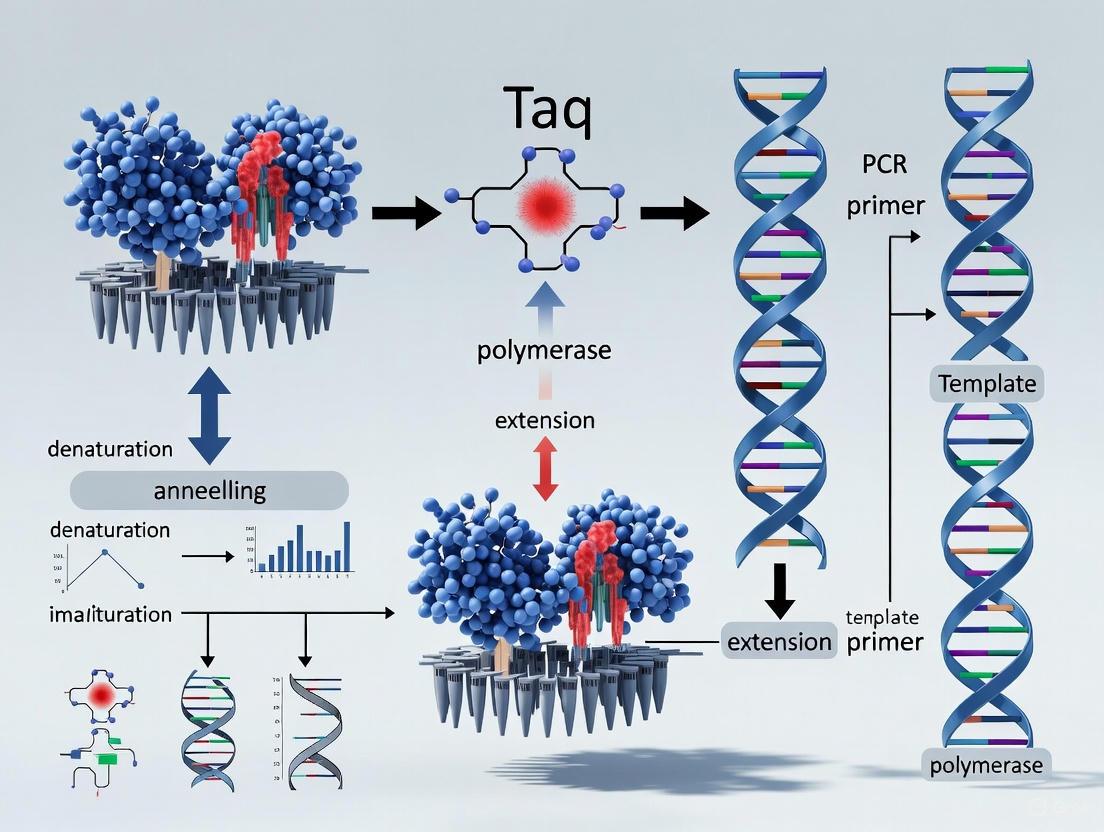
The PCR Breakthrough: Taq Polymerase Enables Molecular Biology Revolution
The invention of the polymerase chain reaction by Kary Mullis at Cetus Corporation in 1983 created an urgent need for a heat-stable DNA polymerase [5]. Early PCR protocols used the Klenow fragment of E. coli DNA polymerase, which was inactivated by the high denaturation temperatures, requiring fresh enzyme to be added after each cycle—a tedious and inefficient process [5] [10]. The incorporation of Taq polymerase into the PCR process in the late 1980s solved this critical limitation, allowing the entire reaction to be automated in a single tube within a thermal cycler [5] [10].
The PCR process leverages the unique properties of Taq polymerase in a three-step cycling process:
- Denaturation: Double-stranded DNA is heated to 94-95°C to separate the complementary strands.
- Annealing: The temperature is lowered to 50-65°C to allow specific primers to hybridize to their complementary sequences on the target DNA.
- Extension: Taq polymerase synthesizes new DNA strands at 72°C, its optimal polymerization temperature.
Each cycle theoretically doubles the amount of target DNA, enabling exponential amplification of specific sequences from just a few copies to millions in a matter of hours [6]. The thermostability of Taq allows this process to be repeated 25-40 times without adding fresh enzyme, making PCR both practical and efficient [5]. This automation, coupled with the enzyme's activity at high temperatures which increases primer specificity and reduces nonspecific amplification, transformed PCR from a cumbersome technique into the powerful, ubiquitous tool it is today [5] [10]. For this breakthrough, Kary Mullis was awarded the 1993 Nobel Prize in Chemistry [5].
Diagram 1: The PCR process leveraging Taq polymerase's thermostability for exponential DNA amplification.
Experimental Protocols: From Isolation to Application
Original Isolation and Culturing Methodology forThermus aquaticus
The initial isolation of Thermus aquaticus by Brock and Freeze followed a systematic approach to sampling, culturing, and characterization that can be replicated for other extremophiles:
Sample Collection: Environmental samples were collected from the outflow channels of Mushroom Spring and other thermal features in Yellowstone, where temperatures ranged from 45°C to 100°C. Samples included water, sediment, and bacterial mat material [3].
Enrichment and Isolation: Samples were inoculated into a dilute nutrient broth (tryptone-yeast extract) and incubated at 70°C for 24-48 hours. This selective temperature inhibited mesophilic contaminants while promoting the growth of thermophilic organisms [2] [7].
Pure Culture Techniques: Following enrichment, pure cultures were obtained through streak-plating on nutrient agar plates containing castione (0.1%), yeast extract (0.1%), and a salts solution, incubated at 70°C in a humidified chamber to prevent desiccation [7].
Morphological Characterization: Initial characterization included Gram staining (revealing gram-negative rods) and examination of unique morphological features such as the formation of "rotund bodies"—spherical structures resulting from the association of multiple cells with fused outer envelope layers [7].
Temperature Range Determination: The optimal growth temperature and thermal limits were established by incubating pure cultures across a temperature gradient from 40°C to 85°C, with growth monitored by turbidity measurements [2].
Electron Microscopy: For ultrastructural analysis, cells were fixed in glutaraldehyde and osmium tetroxide, embedded in epoxy resin, thin-sectioned, and stained with lead citrate and uranyl acetate before examination with transmission electron microscopy [7].
Standard PCR Protocol Utilizing Taq Polymerase
The following protocol represents the standard methodology for DNA amplification using native Taq DNA polymerase:
Reaction Setup:
- 1X PCR Buffer (typically 10 mM Tris-HCl, pH 8.3, 50 mM KCl)
- 1.5-2.5 mM MgCl₂ (concentration must be optimized for each primer-template system)
- 200 μM of each dNTP (dATP, dCTP, dGTP, dTTP)
- 0.2-1.0 μM of each forward and reverse primer
- 0.5-2.5 units of Taq DNA polymerase per 50 μL reaction
- 10-1000 ng of template DNA
- Nuclease-free water to volume
Thermal Cycling Parameters:
- Initial Denaturation: 94-95°C for 2-5 minutes (activates hot-start versions and ensures complete denaturation)
- Amplification Cycles (25-35 cycles):
- Denaturation: 94-95°C for 20-60 seconds
- Annealing: 50-65°C for 20-60 seconds (temperature primer-specific)
- Extension: 72°C for 1 minute per kilobase of amplicon
- Final Extension: 72°C for 5-10 minutes to ensure complete extension of all products
- Hold: 4-10°C indefinitely
Product Analysis:
Research Reagent Solutions for Taq Polymerase Applications
Table 3: Essential Research Reagents for PCR-Based Experiments
| Reagent Solution | Function and Application |
|---|---|
| Native Taq DNA Polymerase | Thermostable enzyme for standard PCR amplification; lacks proofreading activity [5] [9] |
| Hot-Start Taq Variants | Antibody- or chemically-modified Taq; reduces non-specific amplification by inhibiting activity until high temperatures [10] |
| Stoffel Fragment | N-terminal truncated version (61 kDa); lacks 5'→3' exonuclease activity; more thermostable and tolerates broader Mg²⁺ range [9] |
| dNTP Mix | Deoxynucleoside triphosphates (dATP, dCTP, dGTP, dTTP); building blocks for DNA synthesis [6] |
| PCR Buffer with MgCl₂ | Provides optimal ionic environment and pH (typically Tris-HCl); Mg²⁺ is essential cofactor for polymerase activity [9] |
| Olignucleotide Primers | Short, single-stranded DNA sequences (18-25 nucleotides) that define the start points for DNA synthesis [6] |
Impact and Applications: From Basic Research to Global Biotechnology
Transformative Applications Across Scientific Disciplines
The integration of Taq polymerase into PCR catalyzed advancements across diverse fields:
Molecular Biology and Genomics: PCR with Taq polymerase enabled direct cloning of DNA or cDNA, genetic fingerprinting, analysis of allelic sequence variations, and direct nucleotide sequencing [6]. Most significantly, it made possible the sequencing of the entire human genome by providing sufficient amplified material for analysis [4].
Medical Diagnostics and Infectious Disease Detection: PCR revolutionized clinical testing by enabling extremely sensitive detection of pathogenic organisms. It has been successfully applied to detect HIV, hepatitis viruses, human papillomaviruses, Mycobacterium tuberculosis, Chlamydia trachomatis, and many other pathogens with superior sensitivity and specificity compared to traditional culture methods [5] [6]. During the COVID-19 pandemic, PCR tests relying on Taq polymerase became the global standard for SARS-CoV-2 detection [4].
Forensic Science: The ability to amplify minute amounts of DNA from crime scene evidence has transformed forensic investigation, enabling DNA profiling from hair follicles, saliva, skin cells, and other biological materials previously insufficient for analysis [5] [3].
Genetic Disease Diagnosis: PCR facilitates the diagnosis of hereditary conditions including hemophilia, cystic fibrosis, sickle cell anemia, muscular dystrophy, Huntington's disease, and numerous other genetic disorders through detection of characteristic mutations [6].
Environmental Microbiology and Biotechnology: PCR allows monitoring of microbial populations in environmental samples without cultivation, tracking pollution, assessing ecosystem health, and conserving species [3]. It has been used to detect indicator bacteria like E. coli and Legionella in water supplies and to identify novel extremophiles in diverse habitats [6].
Technical Limitations and Enzyme Engineering Advancements
Despite its revolutionary impact, native Taq polymerase has several limitations that have driven the development of improved enzymes:
Error Rate and Lack of Proofreading: The absence of 3'→5' exonuclease activity results in an error rate of approximately 1 in 9,000-10,000 bases, making Taq unsuitable for applications requiring high fidelity such as cloning and long-range sequencing [5] [6].
Inhibitor Sensitivity: Taq polymerase can be inhibited by various compounds commonly found in clinical and environmental samples, including heparin, hemoglobin, humic acids, and tannins [9].
Limited Amplicon Size: The moderate processivity of Taq (averaging 50-60 nucleotides per binding event) restricts efficient amplification of very long DNA fragments (>5 kb) [9].
These limitations have spurred the development of engineered polymerases and alternatives:
- Pfu Polymerase: Isolated from Pyrococcus furiosus in 1991, this enzyme possesses 3'→5' proofreading capability, reducing error rates 10-fold compared to Taq [10].
- Hot-Start Modifications: Antibody-based or chemical inhibition of Taq prevents activity at room temperature, dramatically reducing non-specific amplification and primer-dimer formation [10].
- Chimeric Enzymes: Fusion proteins like Phusion DNA Polymerase combine a Pyrococcus-like enzyme with a processivity-enhancing domain, offering superior speed, fidelity, and tolerance to inhibitors [10].
- Taq Mutants: Site-directed mutagenesis and domain-swapping experiments have created Taq variants with improved characteristics, including those with restored proofreading activity from E. coli and "domain-tagged" versions with enhanced DNA-binding capability [5].
Diagram 2: Technical limitations of native Taq polymerase and corresponding biotechnology solutions.
The discovery of Thermus aquaticus in Yellowstone's extreme environments and the subsequent characterization of its thermostable polymerase represents one of the most impactful examples of how basic, curiosity-driven research can yield unexpected and transformative applications. Thomas Brock's initial investigation into the pink bacterial filaments of Mushroom Spring was motivated by fundamental questions about the limits of life, not commercial potential [3]. Yet this basic research ultimately provided the essential tool that made PCR practical, launching a revolution in molecular biology that continues to accelerate scientific discovery across disciplines.
The legacy of this discovery extends beyond the laboratory, highlighting the critical importance of preserving natural environments like Yellowstone National Park as reservoirs of biological diversity and sources of scientific insight. The unique thermal features of Yellowstone, protected from development, served as the exclusive source of the original T. aquaticus strain that spawned a multi-billion dollar biotechnology industry [1] [3]. This case has prompted ongoing discussions about benefit-sharing arrangements for biological resources from protected areas, with the National Park Service conducting environmental impact studies to determine appropriate frameworks for managing such resources [1].
Future research directions continue to build upon this foundation. The study of extremophiles has expanded dramatically, with scientists discovering organisms thriving in increasingly extreme conditions and exploiting their unique enzymes for industrial and biomedical applications. Protein engineering efforts continue to develop enhanced versions of Taq polymerase with improved characteristics, while synthetic biology approaches explore the creation of entirely novel enzymes. The ongoing exploration of Earth's extreme environments, guided by Brock's pioneering work, promises to yield new biological tools and insights that will continue to drive innovation in biotechnology and medicine for decades to come.
The 1976 isolation and characterization of Taq DNA polymerase by Alice Chien and colleagues represents a landmark achievement in enzymology that ultimately revolutionized molecular biology. This in-depth technical analysis examines Chien's pioneering methodology for purifying the heat-stable DNA polymerase from Thermus aquaticus, detailing the experimental protocols that enabled the critical discovery. The characterization of this thermostable enzyme laid the essential groundwork for the polymerase chain reaction (PCR) technology that would emerge nearly a decade later, transforming genetic research, clinical diagnostics, and therapeutic development. Within the broader context of Taq polymerase research, Chien's work exemplifies how fundamental biochemical characterization of extremophilic organisms can yield tools of extraordinary practical significance, enabling breakthroughs across biomedical sciences and drug development.
The discovery of Thermus aquaticus by Thomas Brock and Hudson Freeze in 1969 revealed a bacterium thriving in the near-boiling thermal springs of Yellowstone National Park (80-85°C), challenging fundamental assumptions about the temperature limits of life [11] [1]. This extremophilic organism represented a rich source of thermostable enzymes, but remained primarily of ecological interest until Alice Chien, then a Master's student at the University of Cincinnati, undertook the systematic characterization of its DNA polymerase [11] [12].
The broader significance of Taq polymerase research lies in its resolution of a critical bottleneck in molecular biology: the need for a heat-stable DNA-synthesizing enzyme that could withstand the denaturing temperatures required for DNA amplification. Prior to Chien's work, available DNA polymerases from mesophilic organisms like E. coli were heat-labile, requiring fresh enzyme addition after each thermal denaturation cycle in early PCR attempts [5] [13]. Chien's isolation and biochemical characterization of Taq polymerase provided the essential reagent that would transform PCR from a cumbersome manual process to an automated technique capable of exponential DNA amplification [11] [12].
Table 1: Key Milestones in Early Taq Polymerase Research
| Year | Breakthrough | Key Researchers | Significance |
|---|---|---|---|
| 1969 | Discovery of Thermus aquaticus | Brock and Freeze | First identification of extreme thermophile bacterium [1] |
| 1976 | Isolation and characterization of Taq polymerase | Chien, Edgar, and Trela | First purification and biochemical analysis of heat-stable DNA polymerase [12] |
| 1985 | Polymerase Chain Reaction concept | Mullis et al. | Development of DNA amplification method using heat-labile polymerase [11] |
| 1988 | PCR with Taq polymerase | Mullis et al. | Adaptation of PCR using thermostable Taq polymerase [11] |
| 1989 | Science "Molecule of the Year" | - | Recognition of Taq polymerase's significance [11] |
| 1993 | Nobel Prize in Chemistry | Kary Mullis | Award for invention of PCR method [11] |
Experimental Characterization: Methodology and Protocols
Source Organism and Growth Conditions
Chien's experimental protocol began with cultivation of the source organism, Thermus aquaticus strain YT-1, originally isolated from Yellowstone National Park's Mushroom Spring [1] [14]. The bacterium was grown in a complex medium containing tryptone and yeast extract, with incubation at 75°C for approximately 15 hours to reach late-log phase growth [12]. This high-temperature cultivation was essential for inducing the heat-stable enzymes that enable the organism's survival in thermal environments.
Enzyme Purification Protocol
The purification methodology developed by Chien et al. employed multiple chromatographic techniques to isolate active DNA polymerase from cellular lysates:
Cell Lysis and Initial Processing: Harvested cells were resuspended in Tris-HCl buffer (pH 7.3) containing 2-mercaptoethanol and disrupted using sonication. The crude lysate was initially clarified by centrifugation at 30,000 × g for 20 minutes [12].
Nucleic Acid Precipitation: Streptomycin sulfate was added to a final concentration of 1.5% to precipitate nucleic acids, which were removed by centrifugation. This critical step eliminated contaminating DNA and RNA that could interfere with subsequent purification [12].
DEAE-Cellulose Chromatography: The supernatant was applied to a DEAE-cellulose column equilibrated with Tris-HCl buffer (pH 7.3). The column was washed with the same buffer, and bound proteins were eluted using a linear KCl gradient (0-0.3 M). DNA polymerase activity typically eluted at approximately 0.2 M KCl [12].
Hydroxyapatite Chromatography: Active fractions from the DEAE-cellulose column were pooled and applied to a hydroxyapatite column. Proteins were eluted with a linear potassium phosphate gradient (0.05-0.30 M, pH 7.3). This step effectively separated the DNA polymerase from the bulk of contaminating proteins [12].
Phosphocellulose Chromatography: The most active fractions from hydroxyapatite chromatography were dialyzed and applied to a phosphocellulose column. After washing, bound proteins were eluted with a linear KCl gradient (0.05-0.50 M). This final purification step yielded enzyme of sufficient purity for biochemical characterization [12].
The entire purification procedure was conducted at room temperature, demonstrating the enzyme's stability under standard laboratory conditions despite its thermophilic origin.
Figure 1: Taq Polymerase Purification Workflow - Multi-step chromatographic process developed by Chien et al. for isolating Taq polymerase from T. aquaticus cultures.
Activity Assay Methodology
Chien employed a standardized DNA synthesis assay to track polymerase activity throughout purification:
Reaction Conditions: The assay mixture contained Tris-HCl (pH 7.4), MgCl₂, 2-mercaptoethanol, dATP, dGTP, dCTP, and ³H-labeled dTTP as the radioactive tracer [12].
Template-Primer System: Activated calf thymus DNA served as the template-primer complex, providing initiation sites for DNA synthesis [12].
Incubation and Quantification: Reactions were incubated at 74°C for 30 minutes, then terminated by cooling and adding trichloroacetic acid. Acid-insoluble radioactivity was collected on filters and measured by liquid scintillation counting to quantify DNA synthesis [12].
One unit of enzyme activity was defined as the amount catalyzing the incorporation of 10 nmoles of deoxyribonucleotide into acid-insoluble material in 30 minutes at 74°C [12].
Biochemical Properties and Key Findings
Chien's characterization revealed exceptional thermal stability that distinguished Taq polymerase from previously known DNA polymerases. The enzyme demonstrated optimal activity at 75-80°C, with a specific activity of 6,180 units/mg of protein [12] [5]. This thermostability proved to be the defining characteristic that would later enable automated PCR.
Table 2: Biochemical Properties of Taq Polymerase Characterized by Chien et al.
| Property | Characteristic | Experimental Conditions | Significance |
|---|---|---|---|
| Optimal Temperature | 75-80°C | DNA synthesis assay in Tris-HCl buffer | Ideal for high-temperature DNA synthesis [12] |
| Thermal Stability | Half-life >2h at 92.5°C, 40min at 95°C | Incubation at elevated temperatures | Withstands DNA denaturation temperatures [5] |
| Molecular Weight | ~63,000 Da | Sedimentation analysis | Smaller than E. coli DNA polymerase I [12] |
| Divalent Cation Requirement | Mg²⁺ optimal | Metal ion dependence assay | Essential for catalytic activity [12] |
| pH Optimum | 7.4-7.8 | pH profile in buffered systems | Compatible with standard reaction conditions [12] |
| Processivity | ~150 nucleotides/sec at 75°C | DNA synthesis rate measurement | High extension rate at elevated temperatures [5] |
The enzyme demonstrated an absolute requirement for Mg²⁺, with optimal activity at 2-4 mM concentration. Interestingly, Chien noted that the polymerase was strongly inhibited by KCl concentrations above 50 mM, with complete inhibition occurring at 100 mM [12]. The molecular weight was estimated at approximately 63,000 Da based on sedimentation analysis, notably smaller than E. coli DNA polymerase I (109,000 Da) [12].
Perhaps most significantly, Chien's thermal stability experiments demonstrated that Taq polymerase retained nearly full activity after prolonged incubation at high temperatures, including 30 minutes at 95°C [12]. This exceptional thermostability would prove to be the enzyme's most valuable property for PCR applications.
Research Reagents and Experimental Tools
The characterization of Taq polymerase relied on specific reagents and methodologies that defined both the initial studies and subsequent applications in molecular biology.
Table 3: Essential Research Reagents for Taq Polymerase Studies
| Reagent/Material | Function in Research | Technical Specification | Application Context |
|---|---|---|---|
| Thermus aquaticus YT-1 | Source organism for native Taq polymerase | Extreme thermophile; optimal growth at 70-75°C [1] | Initial enzyme purification; natural source studies |
| Recombinant E. coli expression system | Production of recombinant Taq polymerase | E. coli with cloned Taq gene; high GC content (70%) [12] | Large-scale enzyme production; commercial applications |
| DEAE-Cellulose | Anion exchange chromatography | Weak anion exchanger; separation by charge characteristics [12] | Initial purification step; nucleic acid removal |
| Phosphocellulose | Cation exchange chromatography | Strong cation exchanger; binds DNA polymerases [12] | High-resolution purification; final polishing step |
| Activated calf thymus DNA | Template-primer for activity assays | DNase I-treated DNA; provides primer sites [12] | Enzyme activity measurement; kinetic characterization |
| dNTP substrates | DNA synthesis substrates | dATP, dGTP, dCTP, dTTP; ³H-dTTP for radiolabeling [12] | Polymerase activity assays; fidelity studies |
Impact and Applications in Research and Development
Transformation of PCR Technology
The incorporation of Taq polymerase into PCR protocols addressed the fundamental limitation of earlier amplification methods that used the heat-labile Klenow fragment of E. coli DNA polymerase I [11] [13]. This innovation eliminated the need for manual enzyme addition after each denaturation cycle, enabling automation and making PCR accessible to diverse research and clinical applications [5].
The exceptional thermostability of Taq polymerase allowed PCR to be performed at higher temperatures, increasing reaction specificity by reducing nonspecific primer binding [5]. Furthermore, the enzyme's temperature optimum of 72°C for extension facilitated more efficient and complete DNA synthesis during each cycle [11].
Commercial and Therapeutic Implications
The commercialization of Taq polymerase created a multi-billion dollar industry, with Hoffmann-La Roche purchasing the PCR and Taq patents from Cetus Corporation for $330 million [5]. The enzyme's critical role in genetic research positioned it as an essential tool across multiple sectors:
Drug Discovery and Development: Taq polymerase enabled rapid gene identification, cloning, and expression analysis central to target validation and mechanistic studies [12] [15].
Clinical Diagnostics: PCR-based tests for infectious diseases (HIV, tuberculosis, hepatitis), genetic disorders, and cancer mutations became routine clinical tools [5] [16].
Forensic Science: DNA fingerprinting using PCR revolutionized criminal investigations and paternity testing [11] [17].
Biotechnology Research: Site-directed mutagenesis, genetic engineering, and gene expression analysis all leveraged Taq polymerase-based PCR [12].
Figure 2: Research Impact Pathway - The trajectory from basic enzyme characterization to diverse scientific and commercial applications.
Limitations and Enzyme Engineering
Despite its transformative impact, Taq polymerase has recognized limitations that have driven subsequent enzyme engineering efforts:
Fidelity Considerations: Taq polymerase lacks 3'→5' proofreading exonuclease activity, resulting in an error rate of approximately 1 in 9,000 nucleotides [5]. This relatively low fidelity can limit applications requiring high sequence accuracy.
Thermostability Constraints: While exceptionally heat-stable compared to mesophilic enzymes, Taq polymerase does show progressive inactivation at temperatures above 90°C, with a half-life of 9 minutes at 97.5°C [5].
These limitations have spurred the development of engineered variants and novel thermostable polymerases with improved properties:
Proofreading Enzymes: DNA polymerases from hyperthermophilic archaea like Pyrococcus furiosus (Pfu) offer 3'→5' exonuclease activity and higher replication fidelity [12].
Recombinant Variants: Engineered forms including Klentaq (lacking 5'→3' exonuclease domain) and hot-start mutants provide enhanced specificity for particular applications [5].
Chimeric Enzymes: Domain-swapping experiments have created hybrid polymerases combining the thermostability of Taq with proofreading domains from other organisms [5].
Alice Chien's systematic isolation and characterization of Taq polymerase exemplifies how fundamental biochemical research on seemingly obscure biological systems can yield tools of transformative power. Her detailed methodological approach provided the essential foundation for understanding this exceptional enzyme's properties, enabling the PCR revolution that would emerge years later. The ongoing optimization and engineering of DNA polymerases for specific research and diagnostic applications continues to build upon this foundational work, demonstrating the enduring impact of rigorous enzyme characterization in advancing biomedical science and therapeutic development.
The invention of the Polymerase Chain Reaction (PCR) by Kary Mullis in 1983 represented a paradigm shift in molecular biology, virtually dividing biology into "the two epochs of before PCR and after PCR" [18]. This revolutionary technique allowed for the exponential amplification of specific DNA sequences, creating millions of copies from a single fragment in a matter of hours. The core principle of PCR involves repeated cycles of DNA denaturation, primer annealing, and DNA synthesis. However, the initial PCR methodology faced a critical limitation: the DNA polymerase employed from E. coli was heat-labile and became irreversibly denatured during the high-temperature DNA denaturation step (approximately 95°C) required at the beginning of each cycle [5] [17]. This necessitated the tedious and costly addition of fresh enzyme after each denaturation step, severely hampering the technique's efficiency, potential for automation, and broad application [5] [19]. The quest for a heat-stable DNA polymerase was therefore not merely an optimization but a fundamental requirement to unlock PCR's full potential, leading researchers to explore extremophilic microorganisms thriving in high-temperature environments.
The Discovery of Thermus aquaticus and Taq Polymerase
The solution to PCR's central problem emerged from the hot springs of Yellowstone National Park. In the 1960s, biologist Thomas Brock challenged the long-accepted notion that life could not survive at extreme temperatures [1] [17]. His research led to the discovery of a novel bacterium, Thermus aquaticus (Taq), in the Octopus Hot Spring, where it was found thriving at temperatures above 80°C [1]. This was the first organism known to exist at such high temperatures, fundamentally changing scientific understanding of the limits of life [17].
The heat-stable DNA polymerase from T. aquaticus was first isolated by Alice Chien and colleagues in 1976 [5] [20]. This enzyme, later named Taq polymerase, was identified as a key candidate for PCR due to its inherent ability to withstand the protein-denaturing conditions of the reaction [5]. The connection between Mullis's PCR problem and this extant biological resource was serendipitous; while searching for a solution, Mullis and his colleagues at Cetus Corporation discovered the sample of T. aquaticus that Brock had deposited in the American Type Culture Collection [17]. This discovery marked the beginning of a new era for PCR, replacing the E. coli DNA polymerase and transforming the technique into the powerful tool it is today.
Technical Characterization of Taq Polymerase
Biochemical and Enzymatic Properties
Taq polymerase is a 94 kDa thermostable DNA polymerase that functions as a DNA-dependent DNA polymerase [20]. Its enzymatic activity is localized to the C-terminus, while its 5' to 3' exonuclease activity resides in the N-terminus [20]. A significant characteristic is its lack of 3' to 5' exonuclease proofreading activity, which contributes to its relatively low replication fidelity compared to other polymerases like Pfu DNA polymerase [5] [20].
Table 1: Key Enzymatic Properties of Taq Polymerase
| Property | Specification | Significance |
|---|---|---|
| Optimal Temperature Range | 75-80°C [5] [20] | Ideal for primer extension at high temperatures |
| Polymerization Rate | ~150 nucleotides/second at 75-80°C [5] [20] | Enables rapid amplification |
| Thermal Stability | Half-life: >2 hours at 92.5°C; 40 minutes at 95°C; 9 minutes at 97.5°C [5] [20] | Survives multiple PCR denaturation cycles |
| Error Rate | Approximately 10⁻⁵ mutations per base per duplication [20] | Lacks proofreading capability |
| Optimal pH | 8.0-9.4 [20] | Compatible with standard PCR buffers |
Table 2: Reaction Optimization Parameters for Taq Polymerase
| Parameter | Optimal Condition | Effect of Deviation |
|---|---|---|
| Mg²⁺ Concentration | ~2 mM (must be optimized) [20] | Critical cofactor; affects yield, specificity, and fidelity |
| KCl Concentration | ~50 mM [20] | Reduces electrostatic repulsion; higher concentrations increase specificity for short products |
| dNTPs | Required for catalytic activity [20] | Essential DNA building blocks |
| Hot-Start Activation | Chemical, antibody-based, or aptamer-mediated inhibition [20] | Reduces non-specific amplification and primer-dimer formation |
PCR Workflow with Taq Polymerase
The following diagram illustrates the standard PCR workflow utilizing Taq polymerase, highlighting its role in the cyclical amplification process.
The standard PCR protocol begins with an initial denaturation step (95°C for 2-10 minutes) to fully separate the DNA strands [21]. This is followed by repeated cycles (typically 25-40) of three core steps executed at specific temperatures optimized for Taq polymerase:
- Denaturation: The reaction is heated to 95°C for 10-30 seconds, melting the double-stranded DNA into single strands [21].
- Annealing: The temperature is lowered to 50-65°C for approximately 30 seconds, allowing the primers to hybridize to their complementary sequences on the single-stranded DNA templates [21].
- Extension: The temperature is raised to 72°C (Taq's optimal extension temperature) for 1 minute per kilobase of target DNA, during which Taq polymerase synthesizes new DNA strands by adding nucleotides to the 3' ends of the primers [5] [21].
Key Research Reagents and Their Functions
Table 3: Essential Research Reagents for PCR with Taq Polymerase
| Reagent | Function | Technical Notes |
|---|---|---|
| Taq DNA Polymerase | Enzyme that catalyzes DNA-dependent DNA synthesis | Thermostable; requires Mg²⁺ as cofactor; lacks 3'-5' proofreading activity [5] [20] |
| Primers | Short, single-stranded DNA oligonucleotides that define the start and end of the target sequence | Typically 18-25 nucleotides long; designed for specific annealing temperature [5] |
| dNTPs (deoxynucleoside triphosphates) | The four building blocks (dATP, dCTP, dGTP, dTTP) for new DNA strands | Added in equimolar concentrations to the reaction mixture [20] |
| MgCl₂ (Magnesium Chloride) | Essential cofactor for Taq polymerase activity | Concentration must be optimized (typically 1.5-2.5 mM); critical for reaction efficiency [20] |
| Reaction Buffer | Provides optimal ionic environment and pH for Taq activity | Typically contains Tris-HCl (pH 8.0-9.0) and KCl (~50 mM) [20] |
| Template DNA | The DNA sample containing the target sequence to be amplified | Can be genomic DNA, cDNA, plasmid DNA, etc.; purity and quantity affect amplification [21] |
Experimental Advancements Enabled by Taq Polymerase
Quantitative PCR (qPCR) and Gene Expression Analysis
The integration of Taq polymerase was pivotal in the development of quantitative PCR (qPCR), which allows for the real-time quantification of DNA amplification [21]. This is achieved by monitoring fluorescence at each cycle, with the quantification cycle (Cq) indicating when the fluorescence signal exceeds a background threshold [21]. The heat stability of Taq is crucial for the TaqMan probe assay, a widely used qPCR method. In this assay, a probe with a 5' fluorescent reporter and a 3' quencher hybridizes to the target sequence. During the extension phase, the inherent 5' to 3' exonuclease activity of Taq polymerase cleaves the probe, separating the reporter from the quencher and generating a fluorescent signal proportional to the amount of amplified product [5] [21].
The following diagram illustrates the molecular mechanism of the TaqMan probe assay, showcasing the critical role of Taq's exonuclease activity.
Critical Experimental Protocols and Methodologies
Hot-Start PCR Protocol: This technique is essential for improving PCR specificity when using Taq polymerase. It involves inhibiting the polymerase's activity during reaction setup at room temperature to prevent non-specific priming and primer-dimer formation [20]. Methods include:
- Antibody-Based Inhibition: A neutralizing antibody binds to the active site of Taq polymerase, which is denatured at the initial high-temperature step, releasing active enzyme [20].
- Chemical Modification: The polymerase is chemically modified to be inactive until a prolonged high-temperature incubation reverses the modification [20].
- Physical Separation: Essential reaction components are physically separated (e.g., by a wax barrier) from the polymerase until the first denaturation step [20].
Two-Step RT-qPCR for Gene Expression: This common protocol for mRNA quantification leverages Taq polymerase's stability [21].
- cDNA Synthesis (Reverse Transcription): Total RNA or mRNA is used as a template with reverse transcriptase to generate complementary DNA (cDNA). This can be primed using oligo-dT primers, random hexamers, or gene-specific primers [21].
- qPCR Amplification: The synthesized cDNA is used as a template in a standard qPCR reaction containing Taq polymerase, sequence-specific primers, and a detection method (e.g., SYBR Green or TaqMan probes). The amplification is monitored in real-time to quantify the initial amount of the target transcript [21].
Impact and Applications in Research and Drug Development
The incorporation of Taq polymerase into PCR protocols fundamentally transformed biomedical research and drug development. Its thermostability enabled the automation of PCR in a single closed tube, dramatically increasing throughput, reliability, and accessibility [5]. This automation was a critical step in making PCR a ubiquitous technique in laboratories worldwide.
In the field of diagnostics, Taq polymerase-based PCR became the gold standard for detecting a wide array of pathogens, including HIV, tuberculosis, and hepatitis, due to its high sensitivity and specificity [5]. The COVID-19 pandemic highlighted its enduring significance, as shortages of Taq polymerase directly impacted the global production capacity for SARS-CoV-2 test kits [5]. In drug development, PCR is indispensable for gene cloning, site-directed mutagenesis (for which Michael Smith shared the 1993 Nobel Prize with Mullis) [22], and the quantification of gene expression to understand drug mechanisms and effects [21]. Furthermore, forensic science and molecular paleontology were revolutionized by the ability to analyze minute or degraded DNA samples [19].
Kary Mullis's quest for a heat-stable DNA polymerase was not merely a technical improvement but the pivotal solution that unlocked the full potential of PCR. The discovery and characterization of Taq polymerase from the extremophile Thermus aquaticus provided the robust, thermostable engine required to automate and scale the polymerase chain reaction. This breakthrough transformed PCR from a cumbersome manual process into a highly efficient, automated, and ubiquitous technology. The synergy between Mullis's conceptual framework and the unique biochemical properties of Taq polymerase created a powerful tool that has since become fundamental to molecular biology, medical diagnostics, and drug development. Its role in enabling real-time quantitative PCR and a multitude of other applications underscores its profound and lasting impact on science and medicine, cementing its place as one of the most significant biological discoveries of the 20th century.
The integration of the thermostable Taq DNA polymerase into the polymerase chain reaction (PCR) workflow represents a paradigm-shifting synergy that transformed molecular biology from a specialized discipline into a ubiquitous technological foundation. This integration, framed within the broader thesis of Taq polymerase research, was not merely an incremental improvement but a fundamental reconfiguration of biochemical processes that enabled unprecedented scalability and automation [23]. The discovery of Thermus aquaticus by Thomas D. Brock in the thermal springs of Yellowstone National Park in 1969 unlocked a biological resource that would ultimately catalyze a methodological revolution [4] [5] [24]. The subsequent isolation of its thermostable DNA polymerase by Chien et al. in 1976 provided the critical component that would address a fundamental constraint in molecular amplification—the thermal lability of enzymatic function at DNA denaturation temperatures [5] [20].
When Kary Mullis conceptualized PCR in 1983, the initial process relied on the Klenow fragment of E. coli DNA polymerase I, which necessitated manual enzyme replenishment after each denaturation cycle due to thermal inactivation [5] [20]. This cumbersome process severely limited throughput, scale, and practical application. The strategic incorporation of Taq polymerase created a seamless, automated workflow by leveraging the enzyme's remarkable ability to withstand repeated exposure to temperatures exceeding 90°C [25] [26]. This integration represents a quintessential example of architectural innovation in science, where existing concepts were reconfigured into a transformative new framework that fundamentally changed how researchers approach DNA manipulation, analysis, and application [23]. The resulting synergy between enzyme properties and technological process has propelled advances across diverse fields including clinical diagnostics, forensic science, biomedical research, and environmental DNA analysis [25] [26] [24].
Historical Foundation: From Extreme Environments to Laboratory Workhorses
The discovery of Thermus aquaticus emerged from basic curiosity-driven research into the limits of biological existence. Thomas Brock's investigation of the microbial communities in Yellowstone's hot springs, where temperatures often exceed 80°C, led to the identification and characterization of this extreme thermophile in 1969 [4] [24]. This foundational discovery, with no immediate applied purpose, exemplified the value of basic scientific exploration and would ultimately provide the key to one of molecular biology's most significant methodological challenges.
The chronological path from discovery to innovation reveals how separate research trajectories converged to create a transformative technology:
The critical turning point came in 1988 when Saiki and colleagues demonstrated that Taq polymerase could replace the E. coli enzyme in PCR, eliminating the need for manual intervention and enabling automation through thermal cycling [5] [23]. This integration constituted a disruptive innovation that fundamentally altered molecular biology methodologies, creating a seamless workflow where previous implementations had been fragmented and labor-intensive [23]. The deletion of the enzyme replenishment step exemplifies the innovation principle of "deleting the part or process step"—a simplification that yielded exponential improvements in efficiency and accessibility [23]. The recognition of this breakthrough with the 1993 Nobel Prize in Chemistry for Kary Mullis underscored its transformative impact, while subsequent applications during the COVID-19 pandemic highlighted its enduring significance in global public health [25] [27].
Technical Characteristics: Quantitative Profiling of Taq Polymerase
Biochemical and Kinetic Properties
Taq polymerase functions as a 94 kDa molecular machine with DNA synthesis activity localized to its C-terminus and 5'→3' exonuclease activity at the N-terminus [20]. Unlike many bacterial DNA polymerases, it lacks 3'→5' exonuclease proofreading activity, which has profound implications for its fidelity and appropriate application contexts [5] [28] [20]. The enzyme demonstrates exceptional thermal tolerance, with a half-life of approximately 40 minutes at 95°C and optimal polymerization activity between 75-80°C, where it can incorporate 150 nucleotides per second [5] [26] [20]. This thermostability is the cornerstone of its utility in PCR, allowing it to remain active through repeated denaturation cycles that would irreversibly denature mesophilic polymerases.
Table 1: Enzymatic Properties of Taq DNA Polymerase
| Parameter | Specification | Significance in PCR Workflow |
|---|---|---|
| Optimal Temperature | 75-80°C | Compatible with standard extension steps at 72°C |
| Thermal Stability | Half-life: >2 hr at 92.5°C, 40 min at 95°C, 9 min at 97.5°C | Withstands repeated denaturation cycles |
| Polymerization Rate | 150 nucleotides/second at 75-80°C | Enables rapid amplification (~1kb in <10 seconds) |
| Processivity | ~50 nucleotides/binding event | Efficient for amplicons <3-4kb |
| Fidelity (Error Rate) | ~1 error per 6,000-9,000 nucleotides [5] [28] | Suitable for many applications but limited for cloning |
| Size | 94 kDa | Standard molecular weight for reagent formulation |
Biochemical Optimization Parameters
The enzymatic activity of Taq polymerase is critically dependent on specific buffer components that stabilize its structure and facilitate catalysis. Divalent cations, particularly Mg²⁺, serve as essential cofactors with optimal concentrations typically between 1.5-2.0 mM, though this must be optimized based on specific reaction conditions [29] [20]. Monovalent cations such as K⁺ also play crucial roles, with approximately 50 mM KCl generally providing optimal activity, though adjustments can enhance specificity for shorter amplicons or improve efficiency for longer products [29] [20]. The enzyme functions within a pH optimum of 8.0-9.4, typically maintained by Tris-HCl buffers in commercial formulations [20]. Deoxynucleoside triphosphates (dNTPs) are typically used at 200 µM each, though lower concentrations (50-100 µM) can enhance fidelity at the cost of reduced yield [29].
Table 2: Optimization Parameters for Taq Polymerase in PCR
| Component | Optimal Concentration | Effect of Deviation |
|---|---|---|
| Mg²⁺ | 1.5-2.0 mM | Too low: no product; Too high: nonspecific amplification |
| KCl | ~50 mM | Higher concentrations increase specificity for short products |
| dNTPs | 200 µM each | Lower concentrations (50-100 µM) increase fidelity |
| Primers | 0.1-0.5 µM each | Higher concentrations may cause spurious amplification |
| Template DNA | 1pg-10ng (plasmid), 1ng-1µg (genomic) | Higher concentrations can decrease specificity |
| Enzyme | 0.5-2.0 units/50µL reaction | Excessive enzyme increases nonspecific products |
Integrated PCR Workflow: Methodological Framework
Procedural Framework and Thermal Cycling
The integration of Taq polymerase establishes a streamlined three-step PCR workflow that can be automated through programmable thermal cycling. This process leverages the enzyme's thermostability to create a seamless transition between the essential stages of DNA amplification:
The initial denaturation at 95°C for 2 minutes ensures complete separation of DNA strands before cycling commences [29] [26]. During the denaturation phase of each cycle (typically 15-30 seconds at 95°C), the double-stranded DNA melts into single strands while Taq polymerase retains activity despite brief exposure to these denaturing temperatures [26]. The annealing phase then cools the reaction to a temperature 5°C below the primer melting temperature (typically 50-60°C), allowing specific hybridization of oligonucleotide primers to their complementary sequences [25] [29]. The extension phase at 68-72°C represents the optimal temperature for Taq polymerase activity, during which the enzyme synthesizes new DNA strands at approximately 60-150 nucleotides per second depending on the exact temperature [5] [29] [26]. For a standard 500bp amplicon, a 45-second extension is typically sufficient, while longer products require proportionally longer extension times (approximately 1 minute per kilobase) [29].
Research Reagent Solutions: Essential Materials
Table 3: Research Reagent Solutions for Taq-Based PCR
| Reagent | Function | Optimization Considerations |
|---|---|---|
| Taq DNA Polymerase | Catalyzes DNA synthesis | Thermostable; lacks proofreading; 5'→3' exonuclease activity |
| Primers | Target sequence recognition | 20-30 nucleotides; 40-60% GC content; Tm within 5°C of each other |
| dNTPs | DNA synthesis building blocks | 200 µM each; quality affects fidelity and yield |
| MgCl₂ | Essential enzyme cofactor | Concentration critical (1.5-2.0 mM typical); chelated by dNTPs |
| Reaction Buffer | Maintains optimal pH and ionic strength | Typically Tris-based, pH 8.0-8.8; may include KCl and (NH₄)₂SO₄ |
| Template DNA | Amplification target | 1pg-10ng plasmid; 1ng-1µg genomic; quality affects specificity |
| Hot Start Modifiers | Reduce nonspecific amplification | Antibodies, chemical modifications, or aptamers that inhibit Taq until initial denaturation |
Advanced Applications: Expanding the Diagnostic and Research Toolkit
Real-Time PCR and Reverse Transcription Applications
The integration of Taq polymerase has enabled sophisticated molecular detection platforms that extend beyond basic DNA amplification. In real-time PCR (qPCR), the inherent 5'→3' exonuclease activity of Taq is leveraged for probe hydrolysis in TaqMan assays, allowing simultaneous amplification and detection without post-processing [25] [20]. This enables precise quantification of initial template concentrations through monitoring of fluorescence accumulation during exponential amplification phases [25]. The quantification cycle (Cq) provides a reliable metric for target abundance, with efficiency corrections essential for accurate interpretation across clinical and biological contexts [25].
Remarkably, recent research has revealed that under optimized buffer conditions, Taq polymerase can exhibit reverse transcriptase activity, enabling its use as a single-enzyme solution for RT-qPCR [27]. This discovery, particularly relevant during the COVID-19 pandemic when reagent availability became constrained, demonstrates that Taq alone can execute CDC SARS-CoV-2 TaqMan RT-qPCR assays with sensitivity to as few as 2 copies/μL of input viral genomic RNA [27]. The "Gen 6 A" buffer system, characterized by specific compositions of Tris-HCl, (NH₄)₂SO₄, KCl, and MgCl₂, promotes this relaxed substrate specificity, allowing Taq to utilize RNA templates for cDNA synthesis before proceeding with DNA amplification [27].
Clinical and Research Applications
The implementation of Taq polymerase in PCR workflows has established the gold standard for numerous clinical and research applications. In infectious disease diagnostics, PCR enables rapid detection of viral pathogens including HIV, herpes simplex virus, SARS-CoV-2, hepatitis viruses, and human papillomavirus, as well as bacterial species such as Mycobacterium tuberculosis, Chlamydia trachomatis, and Neisseria meningitidis [25]. The technique's extreme sensitivity and specificity facilitate early detection of fulminant diseases including meningitis and sepsis, allowing timely therapeutic intervention [25]. In genetic testing, PCR screens for specific alleles and disease-associated mutations both in utero and in adult samples, enabling carrier status determination and prenatal diagnosis [25] [5]. Additional applications span forensic analysis, DNA sequencing, in vitro mutagenesis, and environmental DNA monitoring, demonstrating exceptional methodological versatility [25] [24].
Quality Considerations: Optimization and Troubleshooting
Contamination Control and Fidelity Considerations
A significant challenge in Taq-based PCR arises from the enzyme's exceptional sensitivity, which can detect minimal nucleic acid contamination [25]. This issue is compounded by findings that commercial Taq preparations may contain contaminating bacterial DNA, including 16S rRNA and beta-lactamase antibiotic resistance genes, potentially originating from the expression systems used in enzyme production [20]. Such contamination poses particular challenges for highly sensitive applications including pathogen detection and digital droplet PCR. Decontamination strategies include ultraviolet irradiation, DNase treatment (with subsequent heat inactivation), serial dilution of enzyme preparations, and adsorption using nylon membrane disks [20].
The fidelity limitations of Taq polymerase, with an error rate of approximately 1 per 6,000-9,000 nucleotides [5] [28], stem from its lack of 3'→5' proofreading activity [28]. While sufficient for many applications including routine genotyping and qualitative detection, this error rate necessitates careful consideration for applications requiring high sequence accuracy such as cloning and sequencing. For these applications, high-fidelity polymerases with proofreading capability such as Q5 DNA Polymerase (with 280× higher fidelity than Taq) or polymerase blends may be preferable [28]. The intrinsic processivity of Taq (approximately 50 nucleotides per binding event) also limits its effectiveness for amplifying fragments beyond 3-4 kb, though this can be addressed through specialized polymerase formulations or enzyme blends [28].
Technical Optimization Strategies
Several methodological enhancements can address common challenges in Taq-based PCR workflows. Hot-start activation techniques—employing antibody-based inhibition, chemical modifications, or physical separation—reduce nonspecific amplification and primer-dimer formation by preventing enzymatic activity during reaction setup at lower temperatures [29] [20]. Additive incorporation of DMSO, BSA, or betaine can improve amplification efficiency for templates with strong secondary structure or high GC content [26]. Magnesium optimization through titration in 0.5 mM increments represents one of the most critical adjustments for challenging amplification targets, as Mg²⁺ concentration directly affects enzyme processivity, fidelity, and primer annealing [29]. For quantitative applications, efficiency correction using standard curves or amplification curve analysis is essential for accurate interpretation of Cq values, as assumptions of 100% efficiency can introduce substantial quantification errors [25].
The integration of Taq polymerase into the PCR workflow exemplifies how strategic synergy between fundamental biological discovery and methodological innovation can catalyze transformative scientific advancement. This integration, emerging from basic research on extremophile microorganisms, created a streamlined, automated DNA amplification process that has become foundational to modern molecular biology, clinical diagnostics, and biotechnology [23] [24]. The deletion of the enzyme replenishment step through Taq's thermostability represents an architectural innovation that fundamentally reconfigured the PCR process, enabling exponential improvements in efficiency, scalability, and accessibility [23].
Future directions in polymerase engineering continue to build upon this foundation, with developments including high-fidelity variants, chimeric enzymes with enhanced processivity through DNA-binding domain fusions, and specialized formulations for challenging applications such as long-range PCR and multiplex assays [28]. The recent discovery of Taq's reverse transcriptase activity under optimized buffer conditions further demonstrates the potential for methodological innovation even with well-characterized enzymes [27]. As molecular diagnostics continues to evolve, the fundamental synergy between Taq polymerase and PCR workflows established a paradigm for biotechnological innovation that continues to inspire new generations of methodological advancement across diverse scientific disciplines.
The story of Taq polymerase is a testament to how fundamental, curiosity-driven research can catalyze a technological revolution. The enzyme's journey began not in a corporate laboratory, but in the hot springs of Yellowstone National Park. In the 1960s, microbiologist Thomas Brock was studying microbial life in extreme environments [1]. His research led to the identification of a new bacterium, Thermus aquaticus, which thrived in the near-boosting waters of the Octopus Hot Spring at temperatures above 80°C [1] [5]. This discovery challenged the prevailing scientific belief that nothing could live above 73°C [1]. The heat-stable properties of this bacterium were later identified by master's student Alice Chien et al. in 1976, who isolated its DNA polymerase, now famously known as Taq polymerase [5] [12].
For years, this discovery remained a biological curiosity. The pivotal moment arrived in 1983 when Kary Mullis, a chemist working at Cetus Corporation, invented the Polymerase Chain Reaction (PCR) method [30] [18]. The initial PCR process used a DNA polymerase from E. coli that was heat-labile and had to be replenished after every heating cycle, making the procedure inefficient and laborious [5]. The integration of Taq polymerase, with its inherent thermostability, was the key innovation that transformed PCR from a conceptual technique into a robust, automated, and highly efficient tool [5] [12]. For this breakthrough, Kary Mullis was awarded the Nobel Prize in Chemistry in 1993 [30] [18]. The Nobel committee recognized that his invention had "been of major importance in both medical research and forensic science" [30]. This synergy between a basic ecological discovery and an applied technical problem unlocked a multi-billion dollar industry, demonstrating the profound commercial potential of fundamental scientific research.
The Scientific Breakthrough and Its Recognition
The PCR Revolution Enabled by Taq Polymerase
The Polymerase Chain Reaction is a technique for amplifying a specific segment of DNA across several orders of magnitude, generating thousands to millions of copies. The core principle involves repeated cycles of heating and cooling to facilitate DNA melting and enzymatic replication. The critical challenge was the high heat (over 90°C) required to separate the double-stranded DNA molecules in each cycle; this heat would denature and inactivate the DNA polymerases used in the initial protocols [5].
Taq polymerase, isolated from Thermus aquaticus, provided the perfect solution. As a thermostable enzyme, it could withstand the denaturing temperatures without losing activity. Its optimal temperature for activity is 75–80°C, and it has a half-life of greater than 2 hours at 92.5°C, allowing it to remain active throughout the PCR process [5]. This eliminated the need to add fresh enzyme after each cycle, enabling the entire reaction to be automated in a single tube within a thermal cycler machine [5]. This specific property turned PCR into a simple, specific, and powerful technique, "virtually dividing biology into the two epochs of before PCR and after PCR" [18].
Nobel Prize Accolade
The immense significance of PCR was formally recognized in 1993 when the Royal Swedish Academy of Sciences awarded the Nobel Prize in Chemistry solely to Kary B. Mullis [30]. The prize motivation was explicitly "for his invention of the polymerase chain reaction (PCR) method" [30]. The Nobel Foundation's facts page highlights that "analyzing genetic information requires quite a large amount of DNA" and that PCR allows a small amount of DNA to be "copied in large quantities over a short period of time" [30]. This recognition underscored the transformative nature of the technique, which became a cornerstone of modern molecular biology, medical diagnostics, and forensic science.
Table: Key Properties of Taq Polymerase that Enabled the PCR Revolution
| Property | Description | Impact on PCR |
|---|---|---|
| Thermostability | Half-life >2 hours at 92.5°C; remains intact at DNA denaturation temperatures (~95°C) [5]. | Eliminated need to add enzyme each cycle, enabling full automation in a thermal cycler. |
| Temperature Optimum | Optimal polymerization rate at 75–80°C [5]. | Well-suited for the primer annealing and extension steps of PCR, ensuring efficient DNA synthesis. |
| Lack of 3' to 5' Proofreading | No exonuclease proofreading activity [5]. | Results in relatively low replication fidelity, which is a drawback for some applications but sufficient for many. |
| Ion Dependence | Activity promoted by small amounts of KCl and Mg²⁺ ions [5]. | Requires optimized buffer conditions for maximal performance in reactions. |
Experimental Protocol: Demonstrating PCR with Taq Polymerase
The following is a standard methodology for a basic PCR amplification, as enabled by Taq polymerase.
Objective: To amplify a specific target DNA sequence from a complex template (e.g., genomic DNA).
Principles: The reaction relies on thermal cycling between three temperatures: a high temperature to denature double-stranded DNA, a lower temperature for specific primer annealing, and an intermediate temperature for DNA synthesis by Taq polymerase.
Materials and Reagents:
- Template DNA: Contains the target sequence to be amplified.
- Thermostable DNA Polymerase: Recombinant Taq polymerase (e.g., 1.25–2.5 units per reaction).
- Oligonucleotide Primers: Two single-stranded DNA primers (typically 18–25 nucleotides long) that flank the target sequence.
- Deoxynucleoside Triphosphates (dNTPs): A mixture of dATP, dCTP, dGTP, and dTTP, providing the building blocks for new DNA strands.
- PCR Buffer: A Tris-based buffer containing MgCl₂ (a cofactor for the polymerase), KCl, and stabilizers.
- Nuclease-Free Water: To bring the reaction to the final volume.
- Thermal Cycler: An instrument programmed to rapidly heat and cool the reaction tubes.
Procedure:
- Reaction Setup: On ice, prepare a 25–50 µL reaction mixture containing:
- 1X PCR Buffer
- 200 µM of each dNTP
- 0.2–1.0 µM of each primer
- 10–100 ng of template DNA
- 1.25 U of Taq DNA Polymerase
- Nuclease-free water to volume.
- Initial Denaturation: Place the reaction tube in the thermal cycler and program an initial denaturation step at 95°C for 2–5 minutes to fully denature the template DNA and activate the hot-start Taq polymerase (if used).
- Amplification Cycles (Repeat 25–35 times):
- Denaturation: 95°C for 30 seconds. This separates the double-stranded DNA into single strands.
- Annealing: 50–65°C for 30 seconds. The temperature is set based on the melting temperature (Tm) of the primers, allowing them to bind (anneal) to their complementary sequences on the single-stranded DNA templates.
- Extension: 72°C for 1 minute per kilobase of target DNA. At this optimal temperature for Taq, the polymerase synthesizes a new DNA strand by extending from the primers, copying the template.
- Final Extension: A single step at 72°C for 5–10 minutes to ensure any remaining single-stranded DNA is fully extended.
- Hold: 4–10°C indefinitely.
- Analysis: Analyze the PCR product by agarose gel electrophoresis to confirm the size and quantity of the amplified DNA.
The Multi-Billion Dollar DNA Polymerase Market
The commercialization of PCR and Taq polymerase created an entire industry. The DNA polymerase market, a direct beneficiary of this technology, is a multi-million dollar sector with robust growth, projected to reach nearly three-quarters of a billion dollars within a decade [31].
Market Size and Growth Projections
The global DNA polymerase market is experiencing significant growth, driven by its critical role in molecular diagnostics, genetic research, and biotechnology. Market forecasts, while varying slightly between sources, consistently show a strong upward trajectory.
Table: DNA Polymerase Market Size and Growth Forecasts (2024–2035)
| Metric | Source 1: Biospace/Towards Healthcare | Source 5: Research Nester | Source 9: Future Market Insights |
|---|---|---|---|
| Base Year (2024) | USD 395.21 million [31] | - | USD 374.8 million [32] |
| 2025 Market Size | USD 420 million [31] | USD 145.68 million [33] | USD 397.7 million [32] |
| Projected Year | 2034 [31] | 2035 [33] | 2035 [32] |
| Projected Market Size | USD 721.42 million [31] | USD 179.33 million [33] | USD 725.8 million [32] |
| Forecast Period CAGR | 6.24% (2025–2034) [31] | 2.1% (2026–2035) [33] | 6.2% (2025–2035) [32] |
Note: Discrepancies in market size values are likely due to different segmentation and valuation methodologies used by each research firm. However, all sources affirm a positive and substantial growth trend.
Key Market Drivers and Segments
The expansion of the DNA polymerase market is fueled by several key factors:
- Prevalence of Genetic and Infectious Diseases: The escalating burden of genetic disorders (e.g., cystic fibrosis, Huntington's disease) and infectious diseases drives the demand for PCR-based diagnostic tests, which rely on DNA polymerases [31] [32].
- Growth in Precision Medicine and Genomics: The rise of personalized medicine, next-generation sequencing (NGS), and gene editing technologies like CRISPR-Cas9 creates a sustained need for high-quality, high-fidelity DNA polymerases for research and development [31] [33].
- Technological Advancements: Continuous improvements in PCR diagnostics, sequencing platforms, and the development of specialized polymerases (e.g., for fast or long-range PCR) further stimulate market growth [31].
The market is segmented to cater to diverse application needs:
- By Type: The Taq DNA polymerase segment dominated the market in 2024 due to its low cost and essential role in standard PCR [31]. However, the high-fidelity DNA polymerase segment is expected to grow rapidly, driven by demand for accurate DNA replication in sequencing and diagnostic applications [31].
- By Application: The PCR segment held the largest revenue share in 2024, while the DNA sequencing segment is anticipated to be the fastest-growing, propelled by government-funded genomic projects [31].
- By End User: Academic & research institutes were the dominant end user in 2024, but pharmaceutical & biotechnology companies are projected to be the fastest-growing segment due to their role in drug discovery and development [31].
The Scientist's Toolkit: Key Research Reagent Solutions
Modern laboratories have a suite of specialized DNA polymerases at their disposal, each engineered for specific applications.
Table: Essential DNA Polymerases and Their Research Applications
| Research Reagent | Function and Key Characteristics | Primary Research Applications |
|---|---|---|
| Standard Taq Polymerase | Thermostable, family A polymerase. Lacks 3'→5' proofreading activity, leading to relatively low fidelity but high processivity [5] [12]. | Routine PCR for genotyping, cloning, and diagnostic assays. Ideal when cost-effectiveness is prioritized over ultimate accuracy. |
| High-Fidelity Polymerases (e.g., Pfu) | Thermostable, family B polymerases. Possess 3'→5' exonuclease (proofreading) activity, resulting in significantly lower error rates [5] [12]. | Gene cloning, mutagenesis studies, NGS library prep, and any application where sequence accuracy is critical (e.g., synthetic biology). |
| Reverse Transcriptase-PCR Enzymes (e.g., Tth) | DNA polymerases with inherent reverse transcriptase activity in the presence of Mn²⁺ ions [12]. | Single-tube RT-PCR for amplifying RNA targets. Used in gene expression analysis and viral RNA detection. |
| Ready-to-Use Master Mixes | Pre-mixed solutions containing DNA polymerase, dNTPs, MgCl₂, and optimized reaction buffers [31]. | Standardizes and simplifies PCR setup, reduces contamination risk, and increases workflow efficiency in high-throughput settings. |
Visualization of Workflows and Relationships
The PCR Cycle Enabled by Taq Polymerase
The following diagram illustrates the repetitive temperature cycles of the Polymerase Chain Reaction, a process made simple and automated by the thermostability of Taq polymerase.
From Discovery to Commercialization
This diagram outlines the logical pathway from the initial discovery of Thermus aquaticus to the development of a global multi-billion dollar industry, highlighting key milestones and driving factors.
The journey of Taq polymerase from a curious enzyme in a Yellowstone hot spring to the core of a Nobel Prize-winning technology and a global market underscores an essential paradigm in science. It demonstrates that fundamental, exploratory research, even when its applications are not immediately apparent, is an invaluable investment. The synergy between Brock's discovery of an extremophile, Mullis's inventive genius in creating PCR, and the subsequent commercial development by the biotechnology industry created a positive feedback loop that has propelled decades of innovation. Today, the DNA polymerase market continues to evolve, driven by the relentless demand for better diagnostics, deeper genomic understanding, and novel therapeutic approaches. The story of Taq is far from over; it serves as a powerful reminder that the next transformative tool in life sciences may be hiding in plain sight, waiting for a curious mind to reveal its potential.
Powering Modern Biomedicine: Key Applications of Taq Polymerase in Research and Diagnostics
The Polymerase Chain Reaction (PCR) stands as a cornerstone technique in molecular biology, enabling the exponential amplification of specific DNA sequences from minimal starting material. The discovery of thermostable DNA polymerases, particularly Taq polymerase from Thermus aquaticus, revolutionized this process by allowing reaction automation and significantly improving reliability. This technical guide examines the core PCR mechanism framed within the broader significance of Taq polymerase research, providing researchers and drug development professionals with detailed experimental protocols and optimization strategies essential for successful nucleic acid amplification.
The Core PCR Mechanism
The fundamental PCR process consists of three temperature-dependent steps repeated for 25-40 cycles: denaturation, annealing, and extension. These steps facilitate the targeted replication of DNA sequences through precise thermal cycling [25] [34].
Denaturation
The first step involves heating the reaction mixture to 94-98°C for 15-30 seconds, causing the separation of double-stranded DNA into single strands by breaking the hydrogen bonds between complementary bases [26] [25]. This process provides the necessary single-stranded templates for primer binding. For the initial cycle, a longer denaturation period of 2 minutes is often recommended to ensure complete separation of all DNA strands [35].
Annealing
Following denaturation, the temperature is lowered to 50-65°C for 15-30 seconds to allow short, synthetic DNA primers to bind flanking regions of the target sequence [25] [35]. The optimal annealing temperature is primer-specific and typically set 5°C below the calculated melting temperature (Tm) of the primers [35] [36]. Proper annealing temperature is critical for specific amplification, as higher temperatures enhance specificity while lower temperatures may promote nonspecific binding.
Extension
During this final step, the temperature is raised to 68-72°C, enabling the DNA polymerase to synthesize new DNA strands by adding nucleotides to the 3' ends of the annealed primers [26] [34]. Taq polymerase incorporates nucleotides at a rate of approximately 60-150 bases per second [26] [36]. Extension time is determined by the length of the target amplicon, with a general guideline of 1 minute per 1000 base pairs [35].
Table 1: Core PCR Steps and Parameters
| Step | Temperature Range | Duration | Key Function |
|---|---|---|---|
| Denaturation | 94-98°C | 15-30 seconds | Separates double-stranded DNA into single strands |
| Annealing | 50-65°C | 15-30 seconds | Allows primers to bind to complementary target sequences |
| Extension | 68-72°C | 1 min/kb | Synthesizes new DNA strands from primer templates |
Taq Polymerase: A Revolutionary Enzyme
The isolation of Taq DNA polymerase from the thermophilic bacterium Thermus aquaticus in 1976 marked a pivotal advancement in PCR technology [34]. Unlike the previously used Klenow fragment of E. coli DNA polymerase, which denatured at high temperatures, Taq polymerase exhibits remarkable thermostability, retaining enzymatic activity after repeated exposure to temperatures above 90°C [26] [34]. This property eliminated the need to add fresh enzyme after each denaturation cycle, enabling PCR automation and dramatically improving amplification efficiency, specificity, and yield [34].
Taq polymerase functions optimally at 70-75°C and can remain active at temperatures as high as 92°C, with a half-life of approximately 40 minutes at 95°C [26] [36]. The enzyme demonstrates 5′→3′ polymerase activity but lacks 3′→5′ proofreading exonuclease activity, resulting in a relatively high error rate of approximately 1×10⁻⁴ to 2×10⁻⁵ errors per base per duplication [26] [34]. This limitation makes Taq polymerase less suitable for applications requiring high fidelity, though it remains ideal for routine amplification where maximum accuracy is not critical.
Table 2: Taq Polymerase Properties and Performance
| Property | Specification | Performance Impact |
|---|---|---|
| Optimal Temperature Range | 70-75°C | Compatible with PCR cycling parameters |
| Thermostability | Half-life of ~40 min at 95°C | Survives repeated denaturation cycles |
| Processivity | 60-150 nucleotides/second | Rapid amplification of target sequences |
| Fidelity | Error rate: 1×10⁻⁴ to 2×10⁻⁵ | Suitable for routine, not high-fidelity, applications |
| Amplicon Size Range | Up to 5 kb | Appropriate for most standard amplification targets |
Advanced Taq Engineering and Novel Variants
Recent research has focused on engineering enhanced Taq polymerase variants with improved properties. A notable advancement is the Taq D732N mutant, which contains a single amino acid change (aspartic acid to asparagine at position 732) that confers unexpected reverse transcriptase activity and strand-displacement capability [37]. This gain-of-function mutation enables the enzyme to catalyze RT-PCR and RT-LAMP assays without additional enzymes, expanding its application scope [37]. The D732N variant also demonstrates faster PCR amplification, reducing required extension times by 2-3 times compared to wild-type Taq polymerase [37].
Quantitative Analysis in Real-Time PCR
Quantitative PCR (qPCR) builds upon the core PCR mechanism by enabling real-time monitoring of amplification progress through fluorescent detection systems. The quantification cycle (Cq), defined as the cycle number at which fluorescence exceeds a predetermined threshold, serves as the primary quantitative measurement [25]. PCR efficiency, calculated from standard curves, typically ranges between 90-105% (equivalent to an efficiency value of 1.9-2.05) for optimal reactions [25] [38]. Advanced analysis methods, including weighted linear regression and mixed models, have demonstrated improved accuracy in quantifying initial template concentrations, particularly when combined with the "taking-the-difference" data preprocessing approach that subtracts fluorescence in consecutive cycles [38].
Experimental Protocols and Optimization
Standard PCR Protocol for a 500 bp Amplicon
Source: [35]
Critical Reaction Components and Optimization
DNA Template
- Plasmid or viral DNA: 1 pg–10 ng
- Genomic DNA: 1 ng–1 µg
- Higher DNA concentrations may reduce specificity, particularly with high cycle numbers [35] [36]
Primers
- Length: 20-30 nucleotides
- GC content: 40-60%
- Tm: 55-70°C (within 5°C for primer pairs)
- Final concentration: 0.1-1 µM (typically 0.1-0.5 µM) [35] [36]
Magnesium Concentration
- Optimal range: 1.5-2.0 mM for Taq DNA polymerase
- Magnesium serves as a essential cofactor for polymerase activity [35] [36]
- Concentration must be optimized in 0.5 mM increments, as excessive magnesium promotes nonspecific amplification while insufficient magnesium reduces yield [35]
Deoxynucleotides (dNTPs)
- Standard concentration: 200 µM of each dNTP
- Lower concentrations (50-100 µM) can enhance fidelity but reduce yields [35] [36]
Taq DNA Polymerase
- Recommended amount: 0.5–2.0 units per 50 µl reaction (ideally 1.25 units) [35]
Troubleshooting Common Issues
- Nonspecific amplification: Increase annealing temperature, reduce primer/enzyme concentration, optimize Mg²⁺ concentration [35] [36]
- Low yield: Increase template concentration, extend extension time, check primer design and reaction components [35]
- No product: Verify enzyme activity, check primer specificity, ensure adequate Mg²⁺ concentration, confirm thermal cycler calibration [35]
Research Reagent Solutions
Table 3: Essential PCR Reagents and Their Functions
| Reagent | Function | Optimal Concentration |
|---|---|---|
| Taq DNA Polymerase | Catalyzes DNA synthesis by adding nucleotides to growing DNA strands | 0.5–2.0 units/50 µl reaction [35] |
| Primers | Provide starting points for DNA synthesis by binding flanking regions of target sequence | 0.1–1 µM each primer [36] |
| dNTPs | Building blocks for new DNA strands (dATP, dCTP, dGTP, dTTP) | 200 µM each [35] |
| MgCl₂ | Essential cofactor for polymerase activity; stabilizes primer-template complexes | 1.5–2.0 mM (requires optimization) [35] [36] |
| Reaction Buffer | Maintains optimal pH and ionic strength for enzymatic activity | 1X concentration [35] |
Visualization of PCR Workflow
Diagram 1: PCR Cycle Workflow
Diagram 2: PCR Component Relationships
Applications in Research and Diagnostics
The core PCR mechanism, enabled by Taq polymerase, serves as the foundation for numerous applications across biomedical research and clinical diagnostics. These include genetic disorder screening, infectious disease detection (including COVID-19 diagnosis), forensic analysis, cancer research, and personalized medicine [26] [25]. Real-time PCR platforms incorporating high-resolution melting (HRM) analysis further extend these capabilities, enabling precise species identification in pathogens such as Plasmodium falciparum and Plasmodium vivax in malaria diagnostics [39]. The continued evolution of Taq polymerase variants with enhanced capabilities promises to further expand PCR applications in drug development and molecular diagnostics.
The core PCR mechanism of denaturation, annealing, and extension represents a elegantly simple yet powerful process that has revolutionized molecular biology. The discovery and continued optimization of Taq polymerase have been instrumental in transforming PCR into an automated, robust, and indispensable technique. Ongoing research continues to enhance our understanding of this fundamental process and develop improved enzyme variants with expanded capabilities, ensuring PCR remains at the forefront of biomedical research and diagnostic applications for the foreseeable future.
The discovery of Taq DNA polymerase, a thermostable enzyme isolated from the thermophilic bacterium Thermus aquaticus found in Yellowstone National Park's thermal springs, revolutionized molecular biology by enabling the automation of polymerase chain reaction (PCR) [20]. This breakthrough eliminated the need to replenish enzymes after each denaturation cycle, transforming PCR from a laborious technique into a efficient, automated process that has become fundamental to modern molecular diagnostics [20]. The exceptional thermostability of Taq polymerase, with a half-life of 40 minutes at 95°C, allows it to withstand the repeated high-temperature cycles required for DNA denaturation, making it ideally suited for PCR applications [20]. Its catalytic optimum at 75-80°C, where it can incorporate 150 nucleotides per second, further enhances its utility in rapid thermal cycling protocols [20].
The impact of this discovery extends profoundly into pathogen detection, where Taq polymerase serves as the foundational enzyme driving PCR-based diagnostic platforms worldwide. Molecular diagnostics heavily relies on Taq polymerase for detecting pathogenic nucleic acids with exceptional sensitivity and specificity [33]. The global market for DNA polymerase, dominated by Taq polymerase, is projected to grow from USD 145.68 million in 2025 to USD 179.33 million by 2035, reflecting its critical role in healthcare and research applications [33]. This growth is fueled by increasing demands for molecular diagnostics, with the Taq polymerase segment alone expected to capture over 50.3% of the market share by 2035 [33]. The COVID-19 pandemic particularly highlighted the indispensable value of this enzyme, as it became the workhorse for millions of diagnostic tests that enabled pandemic monitoring and control efforts globally [33].
Principles of PCR-Based Pathogen Detection
Fundamental Mechanisms
PCR operates through a cyclic three-step process that exponentially amplifies target DNA sequences. The process begins with denaturation, where double-stranded DNA is separated into single strands at high temperatures (typically 94-95°C). Next, annealing occurs at lower temperatures (50-65°C), allowing specific primers to bind complementary sequences flanking the target region. Finally, extension at 72°C enables Taq polymerase to synthesize new DNA strands by adding nucleotides to the 3' ends of the primers [40]. These cycles are repeated 30-40 times, theoretically generating billions of copies of the target sequence from a single template molecule [40].
The exceptional utility of Taq polymerase in PCR stems from its fundamental biochemical properties. Unlike the Klenow fragment of E. coli DNA polymerase originally used in PCR, Taq polymerase remains active after repeated exposure to high temperatures, eliminating the need for enzyme replenishment between cycles [20]. The enzyme demonstrates optimal activity at neutral to slightly alkaline pH (8.0-9.4) and requires magnesium ions as essential cofactors at approximately 2 mM concentration for maximum efficiency [20]. A significant functional characteristic is its possession of 5' to 3' exonuclease activity while lacking 3' to 5' proofreading capability, resulting in an error rate of approximately 10⁻⁵ mutations per base per duplication [20]. This balance of thermostability and functionality makes Taq polymerase particularly suitable for diagnostic applications where reliability and efficiency are paramount.
Advanced Detection Methodologies
Real-time PCR (qPCR) represents a significant advancement over conventional PCR, enabling both amplification and simultaneous quantification of target DNA through fluorescence detection. In probe-based qPCR, Taq polymerase's 5' to 3' exonuclease activity cleaves fluorescently-labeled probes during amplification, releasing fluorophores that generate measurable signals proportional to the amount of amplified DNA [20]. This methodology allows for precise quantification of pathogen load through determination of cycle threshold (Ct) values, which represent the number of amplification cycles required for the fluorescence signal to cross a detection threshold [41]. Lower Ct values indicate higher initial target concentrations, enabling not just detection but also quantification of pathogen levels in clinical samples.
Reverse Transcription PCR (RT-PCR) expands detection capabilities to RNA viruses by incorporating an initial reverse transcription step to convert RNA to complementary DNA (cDNA) before amplification. This approach has proven indispensable for detecting RNA viruses such as SARS-CoV-2 and HIV [41] [42]. The TaqPath COVID-19 PCR Kit, for instance, utilizes this methodology, specifically targeting the ORF1ab, N, and S genes of SARS-CoV-2 for comprehensive detection [42]. Recent innovations like the reverse transcription-hairpin occlusion system (RT-HOS) further enhance this technology, enabling one-pot, one-step multiplex detection of short RNA molecules like microRNAs, with potential applications in cancer diagnostics and other fields [43].
Detection of Specific Pathogens
SARS-CoV-2 Detection
Molecular detection of SARS-CoV-2 primarily focuses on conserved genomic regions to ensure diagnostic reliability. The TaqPath COVID-19 Diagnostic PCR Kit exemplifies standard methodology, simultaneously targeting three viral genes: ORF1ab, which remains relatively stable across viral variants; the nucleocapsid (N) gene, essential for viral structure and replication; and the spike (S) protein gene, which exhibits specificity for SARS-CoV-2 but also accumulates mutations indicative of emerging variants [42]. This multi-target approach provides robust detection while monitoring for genetic changes that might affect diagnostic accuracy or indicate variant emergence.
SARS-CoV-2 detection employs diverse specimen types, with nasopharyngeal swabs representing the gold standard during acute infection phases [44]. The virus demonstrates differential distribution across body compartments, with respiratory samples showing peak viral loads during the second week of illness, while fecal samples may remain positive for up to four weeks post-infection [44]. Proper sample handling critically impacts test reliability, with optimal preservation achieved by storing nasopharyngeal swabs at +4°C in RNA extraction buffer [44]. When stored in viral transport media, samples maintain stability at room temperature for up to two days, though refrigeration is recommended for longer storage intervals [44].
Table 1: SARS-CoV-2 Detection Targets and Characteristics
| Target Gene | Function | Detection Significance | Stability |
|---|---|---|---|
| ORF1ab | Encodes non-structural proteins involved in viral replication | Relatively stable across variants; accurate detection | High |
| N gene | Encodes nucleocapsid protein for viral RNA packaging | Highly expressed; sensitive detection target | High |
| S gene | Encodes spike protein for host cell entry | Specific to SARS-CoV-2; hotspot for mutations | Lower due to variant emergence |
The clinical interpretation of SARS-CoV-2 PCR results requires understanding viral persistence patterns. While most individuals clear detectable virus within weeks, exceptional cases demonstrate prolonged RNA detection, particularly among immunocompromised patients. A notable case report documented SARS-CoV-2 RNA persistence for 147 days in an HIV-infected patient with severe immunosuppression (CD4 count: 25 cells/mL) [41]. This persistence reflected ongoing viral replication rather than residual RNA detection, as evidenced by remarkably low Ct values (7.17) indicating high viral load [41]. Such cases highlight the importance of considering patient immune status when interpreting PCR results and making clinical decisions.
HIV Detection and Monitoring
HIV diagnostics employ PCR technology for both direct detection and treatment monitoring. Viral load testing utilizes qPCR to quantify HIV RNA in plasma, providing critical information for assessing disease progression and monitoring antiretroviral therapy efficacy [41]. Additionally, PCR applications in HIV care include drug resistance genotyping through amplification and sequencing of viral genes, and CD4 cell counting through quantitative analysis of specific DNA sequences, though flow cytometry remains the standard for CD4 enumeration [41].
The complex relationship between HIV and SARS-CoV-2 infection underscores the importance of robust PCR diagnostics in immunocompromised populations. The noted case of prolonged SARS-CoV-2 infection in a treatment-naïve HIV patient illustrates how severe immunosuppression (CD4 count <50 cells/μL) can compromise viral clearance, necessitating extended isolation and specialized treatment approaches [41]. In such cases, PCR monitoring guides clinical management decisions, with viral clearance only achieved after implementation of antiretroviral therapy restored immune function [41].
Advanced Technologies and Experimental Protocols
Innovative PCR Methodologies
Recent advancements in PCR technology significantly enhance multiplexing capabilities for comprehensive pathogen detection. Color Cycle Multiplex Amplification (CCMA) represents a groundbreaking approach that dramatically expands detection capacity by utilizing fluorescence permutations rather than combinations [45]. In CCMA, each DNA target produces a pre-programmed sequence of fluorescence increases across multiple channels, with rationally designed blockers creating deliberate delays in Ct values between different fluorescence signals [45]. This innovative methodology theoretically enables detection of up to 136 distinct DNA targets using just four fluorescence channels, vastly expanding diagnostic capabilities without requiring instrumentation modifications [45].
The reverse transcription-hairpin occlusion system (RT-HOS) enables one-pot, one-step multiplex miRNA detection compatible with both standard Taq polymerase and high-fidelity DNA polymerases [43]. This system integrates three critical functions—reverse transcription primer, fluorescent probe, and reverse primer—into a unified mechanism that operates at higher temperatures than conventional methods, enhancing specificity and reducing contamination risk [43]. The methodology demonstrates exceptional performance characteristics, with a wide linear dynamic range from 7.5 × 10⁸ to 7.5 × 10¹ copies per reaction and amplification efficiencies consistently exceeding 90% [43]. When applied to gastrointestinal cancer detection, this approach achieved AUC values of 0.917-0.989, significantly outperforming the conventional CEA marker (AUC=0.611) [43].
Table 2: Comparison of Nucleic Acid Detection Technologies
| Technology | Targets/Identification | Quantitative Capability | Turnaround Time | Cost Efficiency |
|---|---|---|---|---|
| Standard qPCR | Limited (4-6 targets) | Excellent | Fast (1-2 hours) | High |
| CCMA | High (up to 136 targets theoretically) | Excellent | Fast (1-2 hours) | High |
| NGS | Comprehensive | Moderate | Slow (days) | Lower |
| Microarray | Moderate | Limited | Moderate | Moderate |
Essential Research Reagent Solutions
Table 3: Key Research Reagents for PCR-Based Pathogen Detection
| Reagent Solution | Function | Application Examples |
|---|---|---|
| Taq DNA Polymerase | Thermostable enzyme for DNA amplification | All PCR-based pathogen detection systems |
| Reverse Transcriptase | Converts RNA to cDNA for RNA virus detection | SARS-CoV-2, HIV viral load testing |
| Fluorogenic Probes | Sequence-specific detection with 5' reporter and 3' quencher | Real-time PCR detection (TaqMan) |
| Primers | Target-specific oligonucleotides for amplification | Target gene selection (ORF1ab, N, S for SARS-CoV-2) |
| dNTPs | Nucleotide substrates for DNA synthesis | Essential component for all PCR reactions |
| Magnesium Chloride | Cofactor for polymerase activity | Optimization of reaction efficiency |
| Buffer Systems | Maintain optimal pH and ionic conditions | Tris-HCl buffers at pH 8.0-9.4 |
Experimental Protocol: SARS-CoV-2 Detection via RT-qPCR
Sample Collection and Processing: Collect nasopharyngeal swabs using appropriate synthetic fiber swabs with plastic or wire shafts. Place swabs immediately in sterile transport media, maintaining cold chain (2-8°C) if processing within 48 hours, or freeze at -80°C for longer storage [44]. For RNA extraction, employ automated systems such as the MagMAX Viral/Pathogen II Nucleic Acid Isolation Kit, following manufacturer specifications [42].
Reverse Transcription and qPCR Setup: Prepare reaction mix containing TaqPath 1-Step Multiplex Master Mix, SARS-CoV-2 specific primers and probes targeting ORF1ab, N, and S genes, and template RNA [42]. Include appropriate controls: positive extraction control, negative extraction control, and positive amplification control. Perform reverse transcription at 50°C for 10-15 minutes, followed by polymerase activation at 95°C for 2-5 minutes [42].
Amplification and Analysis: Conduct 40-45 amplification cycles of denaturation (95°C for 10-30 seconds) and annealing/extension (60°C for 30-60 seconds) [42]. Monitor fluorescence accumulation in real-time across all channels. Analyze amplification curves to determine Ct values, with positive results typically indicated by Ct values <40 [41]. Specimens are considered positive if 2 or more targets demonstrate exponential amplification, while single-target positives may suggest emerging variants and require confirmation [42].
Visualization of Diagnostic Workflows
The visualization above outlines the comprehensive workflow for SARS-CoV-2 detection, from sample collection through result interpretation. This standardized protocol ensures reliable identification of positive cases while flagging potential variant strains that might exhibit dropout in a single target channel.
Challenges and Quality Considerations
Pre-analytical and Analytical Factors
PCR-based pathogen detection faces several technical challenges that require careful management. Sample collection quality profoundly impacts test sensitivity, with improper nasopharyngeal sampling potentially contributing to false-negative rates as high as 30% despite the inherent sensitivity of RT-qPCR methodology [44]. The anatomical location of sampling proves critical, as ACE2 receptor expression—the primary binding target for SARS-CoV-2—is higher in the distal compared to proximal nasal regions [44]. Additionally, viral persistence patterns vary significantly between specimen types, with fecal samples potentially remaining positive weeks after respiratory samples convert to negative, complicating clearance determinations [44].
Reagent contamination represents another significant challenge in molecular diagnostics. Taq polymerase preparations frequently contain contaminating bacterial DNA, possibly originating from expression vector systems used during manufacture [20]. Studies have detected beta-lactamase antibiotic resistance genes in 11 of 16 commercial Taq polymerase preparations and 16S rRNA in 15 of 16 products tested [20]. Such contamination poses particular problems for highly sensitive applications like digital droplet PCR and when detecting low-abundance bacterial targets. Effective decontamination strategies include ultraviolet irradiation (though this may reduce enzyme activity), DNase treatment (requiring subsequent heat inactivation), serial dilution of polymerase preparations, and adsorption using nylon membrane disks [20].
Interpretation Complexities in Clinical Context
Result interpretation requires understanding of cycle threshold (Ct) values and their clinical correlations. Lower Ct values indicate higher viral loads, with values below 30-35 generally suggesting presence of replicating virus [41]. However, definitive Ct cutoffs for infectivity remain challenging to establish, as evidenced by cases where patients with persistently low Ct values (<10) nonetheless demonstrated clinical recovery [41]. The prolonged RNA detection in immunocompromised patients—documented up to 147 days in severe HIV immunosuppression—further complicates result interpretation and infection control decisions [41].
Variant emergence presents additional interpretive challenges, as mutations in target regions can potentially lead to detection failures. The multi-target design of modern SARS-CoV-2 tests provides a safeguard against this phenomenon, with single-target amplification patterns potentially signaling variant emergence rather than test failure [42]. This approach balances diagnostic reliability with surveillance capability, enabling simultaneous patient management and public health monitoring.
The trajectory of PCR-based diagnostics points toward increasingly multiplexed platforms capable of simultaneous pathogen detection and characterization. Technologies like CCMA demonstrate the potential to expand dramatically the number of targets detectable in single reactions, enabling comprehensive syndromic testing for patients presenting with non-specific symptoms [45]. Such advancements align with growing recognition that syndromic testing panels providing rapid identification of multiple potential pathogens significantly impact clinical decision-making and antimicrobial stewardship [45]. The integration of high-fidelity polymerases with proofreading capability into novel detection systems like RT-HOS further enhances application range, particularly for quantitative analyses requiring maximal accuracy [43].
The economic landscape of PCR diagnostics continues evolving, with the DNA polymerase market projected to sustain steady growth driven by expanding molecular diagnostic applications [33]. The established instrumentation base for qPCR systems in clinical laboratories worldwide provides a foundation for implementing advanced methodologies without requiring capital-intensive new equipment [45]. This existing infrastructure, combined with ongoing methodological innovations, positions PCR technology to maintain its central role in pathogen detection despite emerging competition from alternative platforms like CRISPR-based systems and next-generation sequencing.
In conclusion, Taq polymerase remains the cornerstone of modern molecular pathogen detection, its enduring utility evidenced by its indispensable role during the COVID-19 pandemic and its ongoing evolution through methodological advancements. From its origins in thermal springs to its current status as a diagnostic workhorse, this remarkable enzyme has fundamentally transformed disease detection and management. The continuing innovation in PCR technologies—from enhanced multiplexing capabilities to streamlined reaction systems—ensures this powerful diagnostic platform will address future infectious disease challenges with increasing sophistication, sensitivity, and efficiency.
The discovery of Taq polymerase, a heat-resistant enzyme derived from the extremophile Thermus aquaticus, revolutionized molecular biology by enabling the polymerase chain reaction (PCR) technique. This breakthrough transformed genomic research, making rapid DNA amplification and analysis a routine laboratory practice. Quantitative PCR (qPCR) and its derivative, reverse transcription qPCR (RT-qPCR), have since become cornerstone technologies in drug development. These methods provide precise, quantitative insights into gene expression patterns, enabling the identification and validation of genetic biomarkers critical for diagnosing diseases and developing targeted therapies. This whitepaper explores the integral role of qPCR and RT-qPCR in modern biomarker discovery and gene expression analysis, framing their impact within the broader context of the revolutionary discovery of Taq polymerase.
The development of PCR represents a landmark achievement in scientific innovation, fundamentally altering the landscape of biological research. The critical breakthrough came with the isolation of Taq polymerase from Thermus aquaticus, a thermophilic bacterium discovered in the hot springs of Yellowstone National Park [4]. This enzyme's remarkable heat stability enabled the automation of PCR through thermal cycling, eliminating the need to add fresh enzyme during each cycle and dramatically increasing the method's practicality and efficiency [23].
The innovation trajectory began with the discovery of DNA structure, followed by the invention of PCR, which became a radical innovation when combined with Taq polymerase and automated thermocyclers [23]. This combination ultimately served as a disruptive innovation that transformed bioscience, paving the way for sequencing the human genome and creating new fields of research [23]. The foundational role of Taq polymerase is now extended through qPCR technologies, which provide the quantitative precision necessary for modern biomarker discovery and validation in pharmaceutical development.
qPCR and RT-qPCR in Biomarker Discovery: Methodological Framework
Core Principles and Data Interpretation
RT-qPCR enables accurate quantification of gene expression by measuring the accumulation of PCR products in real-time through fluorescent detection systems. The fundamental measurement in qPCR analysis is the Cycle threshold (Ct), also known as quantification cycle (Cq), which represents the intersection between an amplification curve and a threshold line, providing a relative measure of target concentration in the reaction [46]. Accurate data interpretation requires proper establishment of two key parameters:
- Baseline: The background fluorescence level during initial cycles (typically 3-15), established using the fluorescence intensity when little change occurs [46].
- Threshold: A point sufficiently above the baseline where a significant increase in fluorescence is detected, typically set within the exponential phase of amplification where all amplification curves are parallel [46].
Proper calculation of PCR efficiency is crucial for reliable results. Efficiency is calculated using serial dilutions of DNA template and the formula: Efficiency (%) = (10−1/Slope−1) × 100, with acceptable ranges between 85-110% [46].
Quantitative Approaches in Gene Expression Analysis
Two primary methods are employed for quantifying qPCR data:
- Absolute Quantification: Determines the exact copy number of targets using a standard curve, essential for applications like viral load measurement and gene copy number determination [46].
- Relative Quantification: Compares gene expression between samples relative to a reference gene (control), commonly used for comparative expression studies across different experimental conditions [46].
The relative quantification approach typically employs one of two calculation methods:
- Livak Method: Used when PCR efficiencies of target and reference genes are between 90-100% [46].
- Pfaffl Method: Applies an efficiency correction factor when reaction efficiencies differ [46].
Integrative Approaches: Machine Learning-Enhanced Biomarker Discovery
Advanced biomarker discovery now combines qPCR validation with machine learning algorithms to analyze complex transcriptomic data. A recent study on Thermus thermophilus HB8 demonstrated this integrative approach, analyzing transcriptomic data from 65 samples under various abiotic stresses to identify key stress-responsive genes [47].
Machine Learning Classification and Gene Selection
The research applied multiple supervised machine learning algorithms to classify samples and prioritize informative genetic features. Performance across models demonstrated exceptional classification accuracy [47]:
Table 1: Machine Learning Model Performance in Biomarker Classification
| Machine Learning Model | Classification Performance (AUC) |
|---|---|
| Extreme Gradient Boosting (XGBoost) | 1.00 |
| Random Forest (RF) | 0.99 |
Feature importance analysis consistently identified three candidate genes—TTHA0029, TTHA1720, and TTHA1359—as central to stress adaptation mechanisms [47]. Subsequent RT-qPCR validation confirmed significant upregulation of TTHA0029 and TTHA1720 under salt and hydrogen peroxide stress, suggesting their roles in redox regulation and ionic homeostasis [47].
Similar methodologies have been successfully applied in oncology research. A study on breast cancer transcriptomic data utilized five gene selection approaches—LASSO, Membrane LASSO, Surfaceome LASSO, Network Analysis, and Feature Importance Score—to identify diagnostic biomarkers [48]. Through Recursive Feature Elimination and Genetic Algorithms, researchers developed eight-gene panels that achieved F1 Macro ≥80% across cell line and patient datasets [48].
Table 2: Significant Prognostic Biomarkers Identified via Machine Learning
| Biomarker | Predictive Capability |
|---|---|
| MFSD2A, TMEM74, SFRP1, UBXN10, CACNA1H, ERBB2, SIDT1, TMEM129, MME, FLRT2, CA12, ESR1, TBC1D9 | Significant predictive capabilities for up to five years of survival |
| TBC1D9, UBXN10, SFRP1, MME | Significant for relapse-free survival after five years |
Experimental Protocols and Workflows
RT-qPCR Experimental Workflow
The following diagram illustrates the comprehensive workflow for RT-qPCR analysis in biomarker validation:
Data Analysis Procedure
Proper analysis of RT-qPCR data requires meticulous attention to technical details:
- Baseline Correction: Establish baseline using fluorescence from early cycles (typically 5-15) to account for background fluorescence [49].
- Threshold Setting: Set threshold within the exponential phase where amplification curves are parallel, ensuring consistent ∆Ct measurements across samples [49].
- Ct Determination: Record Ct values for all samples and reference genes.
- Efficiency Calculation: Calculate PCR efficiency using serial dilutions: Efficiency (%) = (10−1/Slope−1) × 100 [46].
- Normalization: Normalize target gene expression to reference genes using either:
- ∆Ct method (for similar efficiencies between target and reference genes)
- ∆∆Ct method (for comparing relative expression between samples) [46]
- Statistical Analysis: Perform appropriate statistical tests to determine significance of expression changes.
For machine learning integration, the validated gene expression data serves as input for feature selection algorithms, creating a virtuous cycle of discovery and validation [47] [48].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Research Reagents for qPCR-based Biomarker Discovery
| Reagent/Equipment | Function | Technical Considerations |
|---|---|---|
| Taq Polymerase | Enzyme for DNA amplification | Heat-stable; optimal activity at 70-80°C [4] |
| Reverse Transcriptase | Synthesizes cDNA from RNA | Essential for RT-qPCR; requires RNA template |
| Fluorescent Dyes (e.g., SYBR Green) | Binds to double-stranded DNA | Fluorescence increases with product accumulation [46] |
| Primers | Sequence-specific amplification | Must be validated for specificity and efficiency |
| Reference Genes (e.g., ACTB, GAPDH) | Endogenous controls for normalization | Must have stable expression across all samples [46] |
| Standard Curve Templates | For absolute quantification | Enables copy number determination [46] |
The discovery of Taq polymerase from extremophile bacteria represents more than a historical footnote—it exemplifies how fundamental biological discoveries can catalyze transformative technological innovations. The development of qPCR and RT-qPCR methodologies, built upon this foundation, continues to drive advances in drug development by enabling precise gene expression analysis and biomarker validation. The integration of these established laboratory techniques with emerging machine learning approaches creates a powerful paradigm for identifying and validating genetic targets with unprecedented efficiency and accuracy. As these methodologies continue to evolve, they promise to accelerate the translation of basic biological discoveries into clinically relevant therapeutic interventions, extending the legacy of Taq polymerase discovery into new frontiers of pharmaceutical innovation.
The field of forensic science and genetic identity testing has been fundamentally transformed by two pivotal discoveries: the polymerase chain reaction (PCR) and the thermostable Taq polymerase. This whitepaper details the technical foundations of DNA fingerprinting and paternity testing, framing these methodologies within the broader thesis that the discovery and application of Taq polymerase represented a revolutionary innovation in bioscience. For researchers and drug development professionals, understanding this evolution is critical, as it underscores how a basic science discovery—the isolation of a heat-resistant enzyme from a thermophilic bacterium—became the cornerstone of modern genetic analysis [23] [15]. The journey from the initial discovery of DNA's structure to the completion of the Human Genome Project was paved with such incremental and radical innovations, with Taq polymerase serving as a prime example of a discovery that fundamentally changed operational paradigms across molecular biology, medicine, and forensic science [23].
This document provides an in-depth technical guide to the core protocols and applications of genetic identity testing. It summarizes critical quantitative data for key enzymes and genetic markers, details experimental methodologies, and visualizes core workflows and signaling pathways, with a specific focus on the role of Taq polymerase in enabling these technologies.
The Discovery and Significance of Taq Polymerase
A Journey from Basic Discovery to Applied Innovation
The story of Taq polymerase is a testament to the profound impact of basic, curiosity-driven research. The sequence of discovery and innovation unfolded over several decades:
- 1969: Biologist Thomas Brock and undergraduate student Hudson Freeze, funded by the National Science Foundation, first identified and described the thermophilic bacterium Thermus aquaticus from the hot springs of Yellowstone National Park. Their research was motivated by fundamental scientific curiosity about life in extreme environments, without foresight of its eventual application [15].
- 1976: Alice Chien, David Edgar, and John Trela at the University of Cincinnati successfully purified a heat-stable DNA polymerase from T. aquaticus [5] [50] [15]. This initial characterization revealed the enzyme's remarkable ability to withstand high temperatures, a property inherent to its bacterial source.
- 1983: Kary Mullis conceived the polymerase chain reaction (PCR) technique at Cetus Corporation [23] [5] [9]. The initial PCR process was cumbersome, requiring the manual addition of fresh DNA polymerase after each denaturation cycle because the heat destroyed the enzyme [10].
- Late 1980s: The integration of Taq polymerase into the PCR protocol marked the transformative innovation. Its thermostability eliminated the need for repeated enzyme addition, allowing the entire process to be automated in a thermocycler [23] [5] [10]. This turned PCR from a specialized, laborious technique into a rapid, robust, and ubiquitous tool.
This trajectory exemplifies the "innovation algorithm" wherein a foundational discovery (T. aquaticus), a radical idea (PCR), and incremental improvements (commercial thermocyclers) combined to create a disruptive technology [23]. The use of Taq polymerase in PCR substantially increased the specificity of the reaction and the yield of the desired product, thereby enabling large-scale analyses like the Human Genome Project and revolutionizing diagnostic and forensic applications [23] [5] [50].
Key Biophysical and Biochemical Properties
Taq polymerase is a 94 kDa DNA-dependent DNA polymerase with optimal activity at 75–80°C [5] [9] [50]. Its critical feature is thermostability, with a half-life of greater than 2 hours at 92.5°C and approximately 40 minutes at 95°C, allowing it to endure the repeated high-temperature denaturation steps required for PCR [5] [9].
The enzyme exhibits a 5'→3' polymerase activity and a 5'→3' exonuclease activity, but it lacks 3'→5' exonuclease proofreading capability [5] [9]. This absence of proofreading activity results in a relatively low replication fidelity, with an error rate estimated between 1 in 9,000 and 3 x 10⁻⁵ errors per nucleotide polymerized [5] [9]. The enzyme is moderately processive, extending a primer by an average of 50–60 nucleotides before dissociating, and can incorporate nucleotides at a rate of approximately 150 nucleotides per second at its optimal temperature [9].
Table 1: Key Biophysical and Functional Properties of Taq Polymerase
| Property | Specification | Significance in PCR |
|---|---|---|
| Optimal Temperature | 75–80°C [9] | Matches the primer extension step in PCR. |
| Thermal Stability | Half-life >2 hrs at 92.5°C; ~40 min at 95°C [5] | Survives repeated DNA denaturation cycles. |
| Molecular Weight | 94 kDa [50] | - |
| Polymerase Activity | 5'→3' direction [50] | Essential for DNA strand synthesis. |
| Exonuclease Activity | 5'→3' present; 3'→5' proofreading absent [5] [9] | Lack of proofreading contributes to error rate. |
| Fidelity (Error Rate) | ~1x10⁻⁴ to ~3x10⁻⁶ [9] | Higher error rate than proofreading enzymes. |
| Processivity | ~50-60 nucleotides [9] | Number of nucleotides added per binding event. |
| Metal Ion Cofactor | Requires Mg²⁺ [5] [9] | Essential for catalytic activity; concentration is optimized. |
Recent single-molecule studies using single-walled carbon nanotube transistors have provided unprecedented insight into Taq polymerase's dynamics at PCR temperatures. These studies have directly observed two distinct types of conformational closures: rapid, ~20-microsecond "transient closures" used to test nucleotide complementarity, and longer "catalytic closures" for nucleotide incorporation. On average, even complementary substrate pairs undergo five transient testing closures for every catalytic incorporation event at 72°C, highlighting a dynamic fidelity-checking mechanism [51].
Genetic Identity Testing: Core Methodologies
Historical Evolution of Genetic Markers
The foundation of genetic identity testing was laid by Sir Alec Jeffreys in 1984 with the development of DNA fingerprinting [52] [53]. His method targeted minisatellites, also known as variable number of tandem repeats (VNTRs), which are regions of DNA with sequences 6-100 base pairs in length repeated multiple times [53]. The technique relied on restriction enzymes to cut the DNA, Southern blotting for separation, and radioactive probes for detection, producing a complex bar-code-like pattern unique to each individual [53].
This method was later refined with the introduction of PCR and the analysis of microsatellites, or short tandem repeats (STRs) [52] [53]. STRs are shorter repetitive sequences of 1-7 base pairs that are abundant and randomly scattered throughout the human genome [53]. The shift to PCR-based STR analysis provided greater sensitivity, allowing analysis of minute or degraded DNA samples, higher throughput, and easier standardization and data sharing between laboratories [52] [53].
The Modern Workflow: From Sample to Profile
The standard workflow for forensic DNA analysis involves a series of meticulously controlled steps to ensure reliability and reproducibility [54].
- DNA Extraction: The process of releasing DNA from cellular material. Common methods include silica-based columns, Chelex-100, or organic extraction using phenol-chloroform [52].
- Quantitation: Determining the quantity of human DNA present in the extract to ensure optimal amplification in subsequent steps [54].
- Amplification (PCR): The targeted amplification of specific STR loci using fluorescently labeled primers in a reaction mix containing Taq polymerase, buffers, dNTPs, and magnesium [54].
- Separation: The amplified DNA fragments are separated by size using capillary electrophoresis [54].
- Analysis & Interpretation: The data is analyzed by software to generate an electropherogram, which displays the alleles (peaks) present at each locus. The results are interpreted by comparing evidence samples to reference samples [54].
The following diagram illustrates this core forensic DNA analysis workflow.
Essential Genetic Markers for Identity Testing
Ideal DNA loci for forensic genetics are highly polymorphic, easy to characterize, simple to interpret, and have a low mutation rate [52]. The current gold standard in the United States and many other countries is the analysis of autosomal short tandem repeats (STRs). The FBI's Combined DNA Index System (CODIS) database, for instance, originally used 13 core STR loci and has been updated to require data from 20 autosomal STR markers for upload to the national database [54].
Table 2: Core Genetic Marker Types Used in Identity Testing
| Marker Type | Unit Length (bp) | Key Features | Primary Applications |
|---|---|---|---|
| Short Tandem Repeats (STRs) | 1 - 7 [53] | Highly polymorphic, PCR-friendly, easily standardized. | Modern forensic casework, paternity testing, CODIS database [52] [54]. |
| Variable Number of Tandem Repeats (VNTRs) | 6 - 100 [53] | Highly polymorphic, requires larger DNA amounts, no PCR. | Early DNA fingerprinting (historical) [53]. |
| Single Nucleotide Polymorphisms (SNPs) | 1 [52] | Abundant, useful for degraded DNA, lower discrimination per locus. | Ancestry inference, phenotyping, analyzing highly degraded samples [52]. |
The following diagram illustrates the process of STR analysis, from the collection of a biological sample to the generation of a DNA profile for database comparison.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents and materials essential for conducting PCR-based genetic identity tests, such as DNA fingerprinting and paternity testing.
Table 3: Essential Reagents and Materials for PCR-Based Genetic Identity Testing
| Item | Function | Key Considerations |
|---|---|---|
| Taq DNA Polymerase | Enzyme that catalyzes the template-directed synthesis of DNA during PCR [5] [9]. | Thermostability is critical. Lack of proofreading activity limits fidelity. Hot-start versions reduce nonspecific amplification [10]. |
| Primers | Short, single-stranded DNA sequences that define the start points for DNA synthesis and target specific STR loci [5]. | Must be precisely designed for the target loci. Often fluorescently labeled for detection in capillary electrophoresis. |
| Deoxynucleotide Triphosphates (dNTPs) | The building blocks (dATP, dCTP, dGTP, dTTP) used by the polymerase to synthesize new DNA strands [50]. | Required in the reaction mix at optimal concentrations to ensure efficient and accurate amplification. |
| Magnesium Chloride (MgCl₂) | A necessary cofactor for Taq polymerase activity; Mg²⁺ ions facilitate the polymerase reaction [5] [9]. | Concentration must be optimized, as it significantly impacts reaction specificity and yield. |
| Thermal Cycler | Instrument that automatically cycles through the precise temperatures required for DNA denaturation, primer annealing, and extension [23]. | Enabled the automation of PCR, making large-scale analysis feasible. |
| Capillary Electrophoresis Instrument | Separates fluorescently labeled PCR amplicons by size and detects them using a laser, generating an electropherogram [54]. | Essential for resolving and analyzing the lengths of amplified STR fragments. |
Detailed Experimental Protocol: STR Analysis for Forensic Identification
This protocol provides a detailed methodology for generating a DNA profile from a biological sample using multiplex PCR of STR loci, reflecting standard procedures used in accredited forensic laboratories [54].
Sample Collection and DNA Extraction
- Sample Collection: Collect biological evidence using sterile swabs or tools. For reference samples, collect buccal (cheek) swabs or liquid blood preserved in EDTA [52] [54]. Dry stains should be stored in paper envelopes at room temperature in a controlled environment; liquid samples should be refrigerated or frozen [54].
- DNA Extraction: Use a validated method such as silica-based column extraction or Chelex-100 protocol.
- Silica-based method: Lyse cells, bind DNA to a silica membrane in the presence of a chaotropic salt, wash away impurities, and elute pure DNA [52].
- Chelex-100 method: Boil the sample in a Chelex-100 slurry to lyse cells and denature proteins, then centrifuge to pellet contaminants, leaving DNA in the supernatant [52].
DNA Quantitation
- Procedure: Use a quantitative PCR (qPCR) method or a fluorescent DNA-binding dye assay to determine the concentration of human DNA in the extract.
- Purpose: Accurate quantitation is critical to standardize the amount of DNA input into the PCR, typically between 0.5-1.0 ng for forensic STR kits, to ensure optimal and balanced amplification of all loci [54].
PCR Amplification of STR Loci
- Reaction Setup: Prepare a master mix on ice to include the following components:
- Taq DNA Polymerase: Use a hot-start formulation to prevent non-specific amplification during setup [10].
- PCR Buffer: Provides optimal pH and salt conditions (e.g., 10 mM Tris-HCl, pH ~8.3, 50 mM KCl) [9].
- MgCl₂: Typically at a final concentration of 1.5 - 2.5 mM (requires optimization) [9].
- dNTPs: Usually 200 µM of each dNTP.
- Multiplex Primer Set: A mixture of primers targeting multiple CODIS core STR loci. Primers are fluorescently labeled with different dyes for multiplex detection.
- Template DNA: 0.5 - 1.0 ng of quantified human genomic DNA.
- Thermal Cycling Conditions: Program the thermal cycler as follows:
- Initial Denaturation: 95°C for 5-10 minutes (activates hot-start Taq).
- Cycling (28-32 cycles):
- Denaturation: 95°C for 30 seconds.
- Annealing: 58-60°C for 30 seconds (primer-specific).
- Extension: 72°C for 45-60 seconds.
- Final Extension: 72°C for 10-30 minutes.
- Hold: 4°C or 15°C ∞.
Analysis of Amplified Products
- Capillary Electrophoresis: Dilute the PCR product appropriately and combine with a size standard and formamide. Denature the mixture and inject it into the capillary array. Apply a voltage to separate the DNA fragments by size [54].
- Data Interpretation: Software generates an electropherogram. Analysts review the peaks, assigning allele calls based on their size relative to the internal standard. The profile is a string of numbers representing the allele pairs at each locus (e.g., D8S1179: 12,15) [54].
Comparison and Statistical Analysis
- Evidence Comparison: Compare the DNA profile from the crime scene evidence to reference profiles from a suspect and/or victim. A full match across all loci provides strong evidence linking the suspect to the evidence [54].
- Database Search: Upload the unknown forensic profile to CODIS to search for matches against known offenders, arrestees, and other crime scenes [54].
- Statistical Analysis: Calculate the statistical significance of a match using population genetics databases. The random match probability (RMP) estimates the probability that an unrelated individual selected at random from a population would have the same DNA profile [54].
The integration of Taq polymerase into the PCR workflow stands as a defining innovation in genetic analysis, enabling the robust, automated, and highly sensitive technologies that underpin modern forensic science and paternity testing. What began as basic research into extremophilic microorganisms culminated in a tool that has fundamentally altered the landscape of molecular biology, justice, and personal identification. The continued evolution of DNA polymerases with higher fidelity, greater processivity, and enhanced resistance to inhibitors promises to further refine these techniques. For the research and drug development community, the story of Taq polymerase serves as a powerful reminder that fundamental, curiosity-driven science is not an end in itself, but a vital and indispensable ingredient in the recipe for transformative innovation.
TA cloning represents one of the most straightforward and efficient methods for the direct cloning of polymerase chain reaction (PCR) products. This technique exploits the terminal transferase activity of Taq DNA polymerase, which preferentially adds a single adenosine to the 3'-ends of amplified DNA fragments. These 3'A overhangs can then be ligated directly with vectors featuring complementary 3'T overhangs, eliminating the need for restriction enzymes and simplifying the cloning process. This technical guide explores the fundamental principles of TA cloning, details experimental protocols, and examines its significance within the broader context of Taq polymerase research, which has revolutionized molecular biology and continues to enable advancements in genetic research and drug development.
The discovery and characterization of Taq DNA polymerase from the thermophilic bacterium Thermus aquaticus marked a pivotal advancement in molecular biology, primarily for its transformative role in enabling the polymerase chain reaction (PCR). Beyond its thermostability, a secondary enzymatic property—terminal transferase activity—has been equally instrumental in simplifying subsequent cloning steps. This activity facilitates TA cloning, a method that leverages the enzyme's tendency to add a single, non-template-directed deoxyadenosine (A) to the 3' ends of PCR products [55] [56].
TA cloning eliminates the need for complex procedures such as restriction enzyme digestion of inserts or the use of adapters, providing a rapid and highly efficient subcloning strategy [57]. Its simplicity and robustness have made it a cornerstone technique in molecular biology laboratories worldwide, supporting a wide array of applications from basic genetic research to the development of sophisticated therapeutic agents. The continued dominance of Taq polymerase in the market, with its segment forecasted to capture over 50.3% share by 2035 [33], is a testament to its enduring utility, driven by high processivity and tolerance to elevated temperatures.
The Core Principle of TA Cloning
Biochemical Basis
The fundamental principle of TA cloning hinges on the enzymatic property of Taq DNA polymerase to catalyze the non-template-dependent addition of a single nucleotide—almost exclusively an adenosine (A)—to the 3'-termini of double-stranded PCR products [55] [56]. This activity is attributed to the enzyme's lack of 3' to 5' proofreading exonuclease activity [57]. The resulting PCR fragments thus possess single 3'A overhangs on both ends.
For cloning, a specialized plasmid vector, known as a "T-vector," is employed. This vector is linearized and engineered to present single 3'thymidine (T) overhangs on both ends [56]. The complementarity between the insert's 3'A overhangs and the vector's 3'T overhangs allows for stable hybridization. In the presence of DNA ligase, these fragments are covalently joined, creating a circular recombinant plasmid ready for transformation into a bacterial host [58].
Key Differentiator from Other DNA Polymerases
It is crucial to note that not all DNA polymerases exhibit this terminal transferase activity. Proofreading DNA polymerases (e.g., Pfu polymerase, Vent polymerase), which possess 3' to 5' exonuclease activity for error correction, typically generate blunt-ended PCR products [57]. Consequently, PCR products amplified with these high-fidelity enzymes are not directly compatible with standard TA cloning vectors unless an additional enzymatic "A-tailing" step is performed using Taq polymerase [58].
TA Cloning Workflow: A Step-by-Step Guide
Insert Preparation
The insert is typically generated by PCR amplification using Taq DNA polymerase. To maximize the efficiency of the 3'A addition, PCR primers should be designed with guanines (G) at their 5' ends [58]. A final extension step of 5-10 minutes at 72°C is recommended to ensure complete A-tailing of all PCR products [58].
Following amplification, the PCR product must be analyzed and purified. This involves:
- Gel Electrophoresis: Running 5-10% of the PCR reaction on an agarose gel to confirm the presence and size of a single, specific amplification product [58].
- Purification: Removing primers, enzymes, and non-specific fragments. For clean reactions, spin columns can be used. If non-specific products are present, gel extraction of the correct band is necessary to prevent cloning of unwanted fragments [58].
Table 1: Troubleshooting Insert Preparation
| Issue | Potential Cause | Solution |
|---|---|---|
| No 3'A overhangs | Use of a proofreading polymerase | Add Taq polymerase in a final polishing step or use an enzyme mix |
| Low cloning efficiency | Impure PCR product (primers, non-specific bands) | Gel-purify the target band before ligation |
| Multiple bands | Non-specific primer binding | Optimize PCR conditions or re-design primers |
Ligation Reaction
The purified A-tailed insert is ligated into the T-vector using T4 DNA ligase. The molar ratio of insert to vector is critical for success. A ratio of 3:1 (insert:vector) is often optimal [58]. Using too much insert can lead to ligation of multiple copies, while too little will result in mostly empty vectors.
A standard ligation reaction is set up as follows:
- pGEM-T Vector: 50 ng (≈ 1 µl) [58]
- PCR Insert: Amount calculated for a 3:1 molar ratio (e.g., 50 ng for a 1 kb fragment) [58]
- 2X Rapid Ligation Buffer: 5 µl (or 10X Buffer at 1.2 µl)
- T4 DNA Ligase: 1 µl (3 Weiss units)
- Nuclease-Free Water: to a final volume of 10 µl
The reaction is mixed gently and incubated at room temperature (for 15-60 minutes with rapid buffer) or overnight at 4°C (with standard buffer) [58].
Transformation and Screening
The ligation mixture is used to transform competent E. coli cells (e.g., JM109, DH5α) via chemical or electrical transformation [58]. After a brief outgrowth in SOC medium, cells are plated on LB agar containing ampicillin (or the appropriate antibiotic for the T-vector), IPTG, and X-Gal.
The pGEM-T and many common T-vectors utilize blue-white screening. The vector contains a β-galactosidase gene (lacZα) interrupted by the cloning site. Successful insertion of a PCR fragment disrupts the lacZα gene, resulting in white colonies. In contrast, cells containing empty, re-ligated vectors produce functional β-galactosidase, resulting in blue colonies [58]. Putative positive (white) colonies are then screened by colony PCR or restriction digestion to verify the presence and correct size of the insert.
Research Reagent Solutions
A successful TA cloning experiment relies on a suite of specialized reagents. The following table details the essential components and their functions.
Table 2: Essential Reagents for TA Cloning
| Reagent / Kit | Function | Key Characteristics |
|---|---|---|
| Taq DNA Polymerase | Amplifies the DNA fragment of interest and adds 3'A overhangs. | Lacks 3'→5' proofreading activity; thermostable. |
| T-Vector (e.g., pGEM-T) | Linearized cloning vector with 3'T overhangs. | Allows direct ligation of A-tailed inserts; contains antibiotic resistance and LacZα for screening. |
| T4 DNA Ligase | Catalyzes the formation of a phosphodiester bond between the insert and vector. | Joins cohesive ends; requires ATP and Mg²⁺. |
| Competent E. coli Cells | Host for propagating the recombinant plasmid after ligation. | High transformation efficiency (e.g., JM109, DH5α strains). |
| Selection Plates (LB/Amp, IPTG, X-Gal) | For blue-white screening of transformants. | Antibiotic selects for transformants; IPTG induces LacZα; X-Gal turns blue in its presence. |
Advancements and Market Context
The development of TA cloning was a direct consequence of in-depth Taq polymerase research. Its discovery solved a major bottleneck in PCR cloning, eliminating the reliance on restriction sites and significantly accelerating laboratory workflows [55] [57]. The impact of this is reflected in the substantial market for DNA polymerases, which is poised to grow from approximately USD 138.46 million in 2025 to USD 156.91 million by 2034, with Taq polymerase consistently maintaining a dominant revenue share [59].
The technique has been further refined into "Universal TA Cloning," where any double-stranded DNA fragment—including those generated with proofreading polymerases or those with blunt or sticky ends—can be adapted for high-efficiency cloning into a single T-vector [55]. This is often achieved by hemi-phosphorylation of the DNA fragments to enable directional cloning. The technique remains especially valuable when compatible restriction sites are unavailable for subcloning, solidifying its role as a versatile tool in the molecular biologist's toolkit [55] [57].
Table 3: DNA Polymerase Market Overview and Projections
| Metric | Value | Source / Timeframe |
|---|---|---|
| Global DNA Polymerase Market Size (2025) | USD 138.46 million | Precedence Research [59] |
| Projected Market Size (2034) | USD 156.91 million | Precedence Research [59] |
| CAGR (2025-2034) | 1.40% | Precedence Research [59] |
| Taq Polymerase Segment Share (Projected 2035) | >50% | Research Nester [33] |
| Key Growth Driver | Surge in demand for molecular diagnostics (e.g., PCR-based pathogen detection) | [33] [59] |
TA cloning stands as a powerful testament to how a deep understanding of a single enzyme's properties can yield a methodology that simplifies and accelerates entire domains of scientific inquiry. By exploiting the inherent terminal transferase activity of Taq DNA polymerase, this technique provides a direct, efficient, and robust pathway for cloning PCR products. Its continued relevance in modern laboratories, amidst a landscape of advanced cloning techniques, is a function of its simplicity and reliability. As research in genomics and personalized medicine continues to expand, driving the demand for Taq polymerase and related products [33] [59], the principles of TA cloning will remain a fundamental component of genetic engineering, supporting ongoing discoveries in basic research and therapeutic development.
The discovery of Thermus aquaticus (Taq) in the hot springs of Yellowstone National Park by Thomas Brock in the 1960s was a fundamental scientific breakthrough that challenged the long-accepted notion that life could not survive in extreme environments [17]. This thermophilic bacterium, thriving at near-boiling temperatures, became the source of Taq polymerase, a thermostable DNA polymerase whose isolation was first reported by Alice Chien et al. in 1976 [5]. The significance of this enzyme was fully realized years later by Kary Mullis at Cetus Corporation, who incorporated it into his revolutionary method for amplifying DNA, the Polymerase Chain Reaction (PCR) [17] [5]. Unlike the DNA polymerase from E. coli originally used in PCR, which was destroyed by the high temperatures required to denature DNA, Taq polymerase could withstand repeated heating to 95°C, eliminating the need to add fresh enzyme after each cycle and paving the way for the automation of PCR [17] [5]. This key advantage transformed PCR from a cumbersome technique into a robust, high-throughput method, a contribution for which Mullis received the Nobel Prize in Chemistry in 1993 [5].
The next pivotal innovation in this narrative was the development of the 5' nuclease assay, first reported in 1991 by researcher David Gelfand at Cetus Corporation [60]. This assay ingeniously harnessed a previously known but underutilized property of Taq polymerase: its 5'→3' exonuclease activity [61]. This activity allows the enzyme to cleave DNA that is hybridized to its template strand during replication. The assay, christened "TaqMan" due to its mechanistic resemblance to the Pac-Man video game where the polymerase "chews" its way along the DNA strand, introduced a fluorogenic probe to enable the direct detection of specific PCR products in real-time [62] [60]. By coupling the principles of PCR with fluorescence-based detection, TaqMan technology provided a method for precise quantification of nucleic acids, moving beyond the simple qualitative detection offered by traditional PCR with gel electrophoresis [63] [64]. This review will provide an in-depth technical examination of the TaqMan assay, detailing its core principles, components, and methodologies, framed within the transformative impact of Taq polymerase research on molecular biology.
The Core Principle: Harnessing 5' Nuclease Activity for Real-Time Detection
The fundamental innovation of the TaqMan assay is its use of the 5'→3' exonuclease activity of Taq polymerase to generate a fluorescent signal directly proportional to the amount of amplified DNA [61] [60]. This process integrates probe hydrolysis directly into the PCR amplification process, allowing for real-time monitoring of the reaction.
The TaqMan Probe Chemistry
A TaqMan probe is a short, target-specific oligonucleotide that is dual-labeled with two key molecules [65] [60]:
- A Reporter Dye: A fluorophore (e.g., FAM or VIC) attached to the 5' end of the probe.
- A Quencher Molecule: A non-fluorescent quencher (NFQ) attached to the 3' end.
When the probe is intact, the proximity of the quencher to the reporter dye suppresses the reporter's fluorescence through a mechanism called Förster Resonance Energy Transfer (FRET) [60]. The quencher absorbs the energy from the excited reporter and dissipates it as heat, resulting in no detectable fluorescent signal [66]. Modern TaqMan probes often include an additional component known as a Minor Groove Binder (MGB). The MGB is a small molecule attached to the quencher that fits into the minor groove of the DNA double helix, significantly increasing the probe's melting temperature (Tm) and stabilizing its binding to the target [62] [66]. This allows for the use of shorter probes, which provides better sequence discrimination, particularly for targets that are difficult to design, such as those with high AT content [62].
The Hydrolysis Process During PCR
The orchestrated sequence of events during a TaqMan real-time PCR cycle is as follows [65] [66] [60]:
- Denaturation: The reaction temperature is raised (typically to 95°C) to melt the double-stranded DNA template into single strands.
- Annealing: The temperature is lowered to allow both the PCR primers and the TaqMan probe to anneal to their complementary sequences on the target DNA. The probe binds to an internal region of the amplicon, between the forward and reverse primer sites.
- Extension: Taq polymerase extends the primer and synthesizes a new DNA strand. When the polymerase encounters the bound TaqMan probe, its intrinsic 5'→3' exonuclease activity cleaves the probe.
- Signal Detection: The cleavage of the probe separates the reporter dye from the quencher. Once physically apart, the quencher can no longer suppress the reporter's fluorescence. The reporter dye is now free to fluoresce when excited by the instrument's light source.
With each subsequent PCR cycle, more probes are hydrolyzed, and the cumulative fluorescence intensity increases in direct proportion to the amount of amplified product [62]. This process is visualized in the diagram below.
TaqMan Assay Components and Workflow
A successful TaqMan experiment relies on a optimized set of reagents and a streamlined workflow designed for specificity and precision.
Key Research Reagent Solutions
The table below details the essential components of a TaqMan reaction mix and their critical functions [65] [67] [62].
Table 1: Essential Components of a TaqMan Real-Time PCR Reaction
| Component | Function | Key Features |
|---|---|---|
| TaqMan Assay | Contains target-specific primers & probe(s) | Pre-optimized for high efficiency; includes FAM/VIC dye-labeled MGB-NFQ probe. |
| DNA Template | The target nucleic acid to be amplified & quantified | Can be genomic DNA or cDNA. Must be free of inhibitors. |
| TaqMan Master Mix | Provides reaction buffer, salts, dNTPs, & enzyme | Contains thermostable Taq DNA polymerase with 5' nuclease activity, MgCl₂, and optimized salt concentrations. |
| Passive Reference Dye | Normalizes for well-to-well variations | An inert dye (e.g., ROX) included in the master mix that does not participate in the PCR. |
Optimized Experimental Workflow
The standard procedure for a TaqMan gene expression assay is detailed below, highlighting steps critical for data reproducibility [65] [66].
- Assay Design and Selection: For common targets, over 20 million predesigned and performance-guaranteed TaqMan assays are available, which eliminates the need for laborious design and optimization [67] [62]. For novel targets, custom assays can be designed using specialized bioinformatics tools. These assays are designed to work at or near 100% efficiency, maximizing sensitivity and accuracy [62].
- Reaction Setup: The reaction is assembled by combining the TaqMan assay (primers and probe), TaqMan Master Mix, and the template DNA or cDNA into a single well of a PCR plate. This closed-tube system significantly reduces the risk of amplicon contamination compared to methods requiring post-PCR handling [64].
- Real-Time PCR Amplification: The plate is loaded into a quantitative PCR instrument, which executes a thermocycling protocol typically consisting of:
- Initial Denaturation: One cycle at 95°C for 10-20 minutes to activate the hot-start enzyme and fully denature the template.
- Amplification (40-50 cycles):
- Denature: 95°C for 15 seconds.
- Anneal/Extend: 60°C for 1 minute (during which fluorescence data is collected).
- Data Analysis: The instrument's software records the fluorescence intensity of the reporter dye at each cycle. The cycle threshold (Ct), the cycle number at which the fluorescence crosses a predetermined threshold, is determined. The Ct value is inversely proportional to the starting quantity of the target nucleic acid, enabling precise quantification through comparison to standard curves or reference genes [66].
Applications and Experimental Data
The specificity, sensitivity, and quantitative nature of TaqMan assays have made them the gold standard for a diverse range of applications in research and molecular diagnostics.
Key Application Areas
- Gene Expression Analysis: TaqMan Gene Expression Assays are widely used to precisely quantify messenger RNA (mRNA) transcript levels, normalized to endogenous control genes [67] [60].
- SNP Genotyping and Pharmacogenomics: TaqMan Genotyping Assays use two allele-specific probes, each labeled with a different reporter dye (e.g., FAM and VIC), to distinguish between single nucleotide polymorphisms (SNPs) [65] [64]. This is crucial for studies linking genetic variation to drug response.
- Pathogen Detection and Quantification: The assay is ideal for determining viral load (e.g., HIV, Hepatitis) in clinical specimens or for the specific identification of bacterial pathogens in food safety and clinical microbiology [63] [60] [64].
- Copy Number Variation (CNV) Analysis: TaqMan Copy Number Assays are run in duplex with a reference assay to determine the relative copy number of a specific gene or genomic sequence [65] [67].
Performance Data from a Representative Study
A study comparing a TaqMan assay to conventional PCR for detecting the mecA gene in staphylococci provides concrete performance metrics, summarized in the table below [63].
Table 2: Performance Metrics of a TaqMan Assay for mecA Gene Detection [63]
| Parameter | Result | Context / Implication |
|---|---|---|
| Total Isolates Tested | 222 | Included S. aureus and coagulase-negative staphylococci. |
| DNA Extraction Methods | High-salt & Qiagen kit | Qiagen kit eliminated PCR inhibition (0% vs. 7.2% with high-salt). |
| Assay Agreement | 96% (197/206) | High concordance between TaqMan and conventional PCR methods. |
| Time to Result | ~2 hours | Compared to 2 days for conventional PCR with gel electrophoresis. |
| Sensitivity | Target detected <30 cycles | Positive samples consistently showed early amplification. |
Detailed Experimental Protocol: mecA Gene Detection [63]
- Bacterial Isolates: 222 staphylococcal isolates from patients in four hospitals, including controls (S. aureus ATCC 43300, mecA-positive; ATCC 25923, mecA-negative).
- DNA Extraction:
- Method 1 (High-Salt): Cells were incubated with lysostaphin and proteinase K, followed by protein precipitation with NaCl and ethanol. This method resulted in a 7.2% rate of PCR inhibition for the TaqMan assay, which could be overcome by a 1:5 sample dilution.
- Method 2 (Qiagen Kit): Cells were treated with lysozyme and lysostaphin, followed by processing using the QIAamp Tissue Kit. This method resulted in no instances of PCR inhibition.
- TaqMan PCR Conditions:
- A different set of primers was designed for optimal performance with the TaqMan chemistry.
- The reaction utilized the TaqMan 5' nuclease PCR kit and was run on an ABI Prism 7700 Sequence Detector.
- The process eliminated the need for agarose gels, staining, and UV visualization.
- Data Interpretation: Results were considered positive if the target DNA was detected before 30 PCR cycles. Discrepant results (9 samples) were largely attributed to potential low-level DNA cross-contamination during frequent sample handling, as they were negative upon repeat testing with freshly extracted DNA.
The trajectory from the discovery of Thermus aquaticus in the extreme environment of Yellowstone to the development of the sophisticated TaqMan assay epitomizes the profound impact of basic scientific research. The initial characterization of Taq polymerase, driven by fundamental curiosity about extremophiles, provided the essential enzyme that made PCR a practical and revolutionary tool. The subsequent ingenuity of harnessing this enzyme's 5' nuclease activity gave rise to TaqMan technology, which transformed PCR from a qualitative technique into a precise, quantitative, and efficient method for nucleic acid analysis.
The significance of this journey is reflected in the technology's pervasive adoption. TaqMan assays have been cited in over 200,000 scientific publications and are integral to fields as diverse as gene discovery, pharmacogenomics, infectious disease diagnosis, and genetically modified organism (GMO) detection [67] [62]. The ability to obtain reliable, quantitative data in a high-throughput format has accelerated drug development and basic research alike. Furthermore, the ongoing relevance of this technology is evidenced by its role in modern applications such as digital PCR [67]. The story of Taq polymerase and the TaqMan assay underscores how investigating fundamental biological mechanisms—from the ecology of hot springs to the enzymatic properties of a polymerase—can yield tools that permanently alter the landscape of scientific inquiry and clinical application.
Maximizing Fidelity and Specificity: A Practical Guide to Taq Polymerase Protocols
Thermus aquaticus DNA polymerase, or Taq polymerase, revolutionized molecular biology by enabling the polymerase chain reaction (PCR). However, its utility is tempered by a characteristically high error rate compared to many other DNA polymerases. This whitepaper analyzes the structural and functional basis for Taq's lack of proofreading activity, quantifying its replication fidelity against high-fidelity polymerases. We detail the experimental methodologies used to determine polymerase error rates and discuss the critical implications of fidelity in research and diagnostic applications. This analysis is framed within the broader context of Taq polymerase research, underscoring the enduring significance of fundamental enzymological discovery.
The discovery of Thermus aquaticus by Thomas Brock and Hudson Freeze in the hot springs of Yellowstone National Park was a foundational moment in microbiology, revealing that life could thrive at near-boiling temperatures [1] [68]. This basic research proved to be of immense practical value when the DNA polymerase isolated from this bacterium, Taq polymerase, became the cornerstone of the Polymerase Chain Reaction (PCR) [17]. Its thermostability—a necessary adaptation to its natural environment—allowed it to withstand the high-temperature denaturation steps of PCR, eliminating the need to add fresh enzyme after every cycle and thus automating the process [5]. This transformed PCR from a cumbersome technique into a powerful, ubiquitous tool that underpins modern molecular biology, clinical diagnostics, and drug development.
However, as PCR was adopted for increasingly sensitive applications, from cloning to sequencing, a key limitation of Taq polymerase emerged: its relatively low replication fidelity. This whitepaper provides an in-depth analysis of the structural basis for Taq's error rate, its quantification relative to other enzymes, and the experimental methods used to measure it, framing this discussion within the ongoing research efforts to understand and engineer better polymerases.
The Molecular Basis of Fidelity and Taq's Structural Deficit
DNA polymerase accuracy, or fidelity, is maintained through a multi-step process. The primary checkpoint is geometric selection at the polymerase active site, where the correct incoming nucleotide is positioned for efficient incorporation due to proper Watson-Crick base pairing. Incorrect nucleotides create a suboptimal architecture, slowing incorporation and increasing the chance they will dissociate [69] [70]. Many DNA polymerases possess a secondary checkpoint known as proofreading: a 3'→5' exonuclease activity that provides an additional layer of protection against replication errors.
The Missing Proofreading Domain in Taq
A defining characteristic of Taq polymerase is its lack of a functional `3'→5' exonuclease (proofreading) activity [5] [71]. While the Taq polymerase protein retains a vestigial structural domain homologous to the proofreading domain in other polymerases like E. coli DNA Polymerase I, this domain is dramatically altered and is not functional [5]. Consequently, when Taq misincorporates a nucleotide, it cannot excise the error and correct it. The mismatched base remains, creating a permanent mutation in the newly synthesized DNA strand. This fundamental structural deficit is the primary cause of its high error rate.
The following diagram illustrates the fidelity mechanisms in DNA polymerases, highlighting the pathway Taq polymerase lacks.
In contrast to Taq, proofreading polymerases like Pfu (from Pyrococcus furiosus) and Q5 (an engineered enzyme) possess a functional 3'→5' exonuclease domain. When a mismatch is detected, the growing DNA chain is transferred from the polymerase active site to the exonuclease domain, the incorrect nucleotide is excised, and the chain is returned for continued synthesis [69] [70]. This process reduces error rates by orders of magnitude.
Quantitative Error Rate Comparison of DNA Polymerases
The error rate of a DNA polymerase is typically expressed as the number of errors incorporated per base pair per duplication event (errors/bp/duplication). Measurements using various sequencing methods have consistently shown that Taq polymerase has a significantly higher error rate than proofreading enzymes.
Table 1: DNA Polymerase Fidelity Comparison
| DNA Polymerase | Proofreading Activity | Reported Error Rate (errors/bp/duplication) | Fidelity Relative to Taq | Primary Source/Reference |
|---|---|---|---|---|
| Taq | No | 1.0 × 10⁻⁵ to 2.0 × 10⁻⁵ [72] | 1X | [73] [72] |
| Taq | No | 1.5 × 10⁻⁴ (PacBio SMRT Sequencing) [70] | 1X | [70] |
| AccuPrime Taq | No | ~1.0 × 10⁻⁵ [73] | ~5-10X better | [73] |
| KOD | Yes | ~1.2 × 10⁻⁵ [70] | ~12X better | [70] |
| Pfu | Yes | 1.0 × 10⁻⁶ to 2.0 × 10⁻⁶ [73] [72] | ~6-30X better | [73] [70] |
| Deep Vent | Yes | 4.0 × 10⁻⁶ [70] | ~44X better | [70] |
| Phusion | Yes | 3.9 × 10⁻⁶ [70] | ~39X better | [70] |
| Q5 | Yes | 5.3 × 10⁻⁷ [70] | 280X better | [70] |
The data in Table 1 demonstrates that proofreading polymerases (Pfu, Deep Vent, Q5) consistently exhibit error rates that are more than an order of magnitude lower than that of Taq. Engineered enzymes like Q5 High-Fidelity DNA Polymerase represent the pinnacle of fidelity, with an error rate 280 times lower than Taq [70].
Experimental Methodologies for Measuring Fidelity
Determining polymerase error rates requires sophisticated assays that can detect rare mutations. The evolution of these protocols mirrors advancements in sequencing technology.
The lacZ-Based Forward Mutation Assay
This classic method, pioneered by Kunkel and refined by Barnes, involves amplifying a reporter gene (often the lacZα gene) via PCR [73] [70]. The PCR products are cloned into a vector and transformed into bacteria. The functional lacZ gene produces blue colonies on X-Gal plates, while mutations that disrupt the gene result in white colonies. The ratio of white to blue colonies provides an indirect measure of the error frequency. A significant limitation is that only mutations within a small, functionally critical region of the gene are detected, and the specific types of mutations are not identified without further sequencing [73] [70] [74].
Direct Sequencing of Cloned PCR Products
With the decreasing cost of Sanger sequencing, it became feasible to directly sequence cloned PCR products to identify all mutations within an amplicon. This method provides a more direct and comprehensive view of the mutation spectrum (types and locations of errors) [73]. However, to achieve statistical significance for high-fidelity polymerases, a very large number of clones must be sequenced, making it a low-throughput option for accurate quantification of modern enzymes [70].
Next-Generation Sequencing (NGS) Approaches
NGS platforms (e.g., Illumina) overcome the throughput limitations of Sanger sequencing by generating millions of reads, allowing for the detection of extremely low error rates. However, these systems themselves have an error rate that can interfere with accurate fidelity measurement, often requiring complex molecular barcoding strategies to distinguish polymerase errors from sequencing errors [70].
Single-Molecule Real-Time (SMRT) Sequencing
PacBio SMRT sequencing is considered a gold standard for modern fidelity measurement. It sequences individual DNA molecules repeatedly to generate a highly accurate consensus sequence, with a demonstrated background error rate of about 9.6 × 10⁻⁸ [70]. This is low enough to accurately quantify the fidelity of proofreading polymerases without the need for an intermediate cloning or amplification step, capturing the true spectrum of polymerase errors with high confidence [70].
The workflow for these key experimental methods is summarized below.
The Scientist's Toolkit: Essential Reagents for Fidelity Research
The following table details key reagents and materials used in polymerase fidelity research, as derived from the cited experimental protocols.
Table 2: Key Research Reagent Solutions for Fidelity Experiments
| Reagent/Material | Function in Experiment | Specific Example |
|---|---|---|
| Target Plasmid DNA | Provides a defined, "error-free" template for PCR amplification. | Plasmid containing the lacZ gene or other suitable target [73] [70]. |
| Test DNA Polymerase | The enzyme whose fidelity is being evaluated under optimized buffer conditions. | Taq, Pfu, Q5, etc. [73] [70]. |
| Cloning Vector & Host | Allows for the separation and propagation of individual PCR products for analysis. | M13 bacteriophage or other vectors; Competent E. coli cells [70]. |
| Selection Medium | Enables phenotypic screening for mutations. | Agar plates with X-Gal for blue/white screening [70] [74]. |
| Sequencing Platform | Determines the nucleic acid sequence of PCR products to identify mutations. | Sanger sequencer, Illumina, or PacBio SMRT sequencer [73] [70]. |
Implications for Research and Drug Development
The choice of DNA polymerase has profound consequences. In cloning and protein expression, mutations introduced by a low-fidelity polymerase can alter or abolish the function of the encoded protein, leading to failed experiments and incorrect conclusions [73]. In diagnostics, Taq's error rate can be a source of false positives or negatives, particularly in assays designed to detect single-nucleotide changes [5]. For next-generation sequencing library preparation, using a high-fidelity polymerase is paramount to ensure that observed variants are biological and not artifacts of the amplification process.
Furthermore, Taq's lack of proofreading limits its effectiveness in amplifying long DNA fragments (>3-4 kb). Misincorporated bases cause the enzyme to stall and dissociate, as it cannot remove the inhibitory mismatch. This limitation can be overcome by blending Taq with a small amount of a proofreading enzyme, which cleans up the errors and allows for the amplification of fragments ≥20 kb [69] [71].
The discovery of Thermus aquaticus and the subsequent isolation of Taq polymerase were seminal events that catalyzed a revolution in biotechnology. The enzyme's thermostability made PCR practical, but its lack of proofreading activity and consequent high error rate have driven decades of further research. This analysis has detailed the structural basis for this fidelity deficit, quantitatively compared Taq to modern proofreading enzymes, and outlined the sophisticated experimental protocols used to measure error rates. The enduring legacy of Taq polymerase is not only the technique it enabled but also the clear need it established for continuous enzyme engineering, pushing the field toward ever-higher standards of accuracy to meet the demands of advanced research and precision medicine.
The discovery of Thermus aquaticus (Taq) in the hot springs of Yellowstone National Park by Thomas Brock in the 1960s unlocked a biological marvel: a thermostable DNA polymerase [23] [17]. This enzyme, Taq polymerase, became the cornerstone of the Polymerase Chain Reaction (PCR) revolution, transforming molecular biology by providing a reliable method for DNA amplification [23] [9]. The significance of this discovery was cemented when Kary Mullis and colleagues integrated Taq polymerase into PCR, eliminating the need to add fresh enzyme after each denaturation cycle and thereby enabling automation of the process [9] [17]. This integration was not merely an incremental improvement but a radical innovation that fundamentally altered bioscience research, allowing for large-scale analyses that culminated in projects like the Human Genome Project [23]. The story of Taq polymerase exemplifies how a discovery, coupled with innovative application, can reshape scientific paradigms. Within this context, a deep understanding and meticulous optimization of the reaction environment—specifically the critical roles of Mg2+, KCl, and pH—is essential for harnessing the full potential of this transformative enzyme. These components are not passive bystanders; they are active regulators of enzyme fidelity, specificity, and efficiency [75] [76] [77].
The Biochemical Foundations of PCR Optimization
The performance of Taq DNA polymerase is governed by its interaction with the reaction buffer's chemical components. Taq polymerase is an 832-amino acid protein with optimal polymerization activity at 75–80°C [9]. Similar to the Klenow fragment of E. coli DNA polymerase I, its structure can be conceptualized as a "partly open right hand" with palm, fingers, and thumb domains responsible for catalysis and DNA binding [9] [78]. A key feature of Taq polymerase is the presence of a 5'→3' exonuclease activity and the notable absence of a 3'→5' proofreading activity, resulting in an error rate estimated between 3 × 10^–4 and 3 × 10^–6 errors per nucleotide polymerized [9]. The binding of this enzyme to the primed-template DNA junction is a structure-specific interaction characterized by significant thermodynamic changes, including a large negative heat capacity change (ΔCp), which dictates that the driving forces for binding shift from entropy-driven at lower temperatures to enthalpy-driven at physiological temperatures [78]. This intricate interaction is highly sensitive to the ionic environment, making the optimization of Mg2+, KCl, and pH not just beneficial but imperative for successful amplification.
Core Components: Functions and Optimization Strategies
Magnesium Ions (Mg2+): The Essential Cofactor
Magnesium ions (Mg2+) serve as an essential cofactor for Taq DNA polymerase, directly activating the enzyme for catalysis [77]. The Mg2+ ion facilitates the formation of the phosphodiester bond by binding to a dNTP's alpha phosphate group, enabling the removal of beta and gamma phosphates and the subsequent attachment of the nucleotide to the growing DNA chain [77].
- Optimal Concentration Range: The typical optimal final concentration for MgCl2 is 1.5–2.0 mM, though this must be optimized for each primer-template system [75]. Some polymerases, such as the Stoffel fragment, perform optimally in a broader range of 3.5–4 mM MgCl2 [9].
- Consequence of Deviation:
- Too Low (<1.5 mM): Results in dramatically reduced enzyme activity or complete PCR failure due to insufficient cofactor availability [75] [77].
- Too High (>2.0 mM): Promotes non-specific amplification, increases error rates (reduces fidelity), and can lead to the formation of primer-dimers [75] [79] [77].
- Optimization Protocol: A magnesium titration should be performed by supplementing the reaction buffer with MgCl2 in 0.5 mM increments up to 4 mM, followed by analysis of amplification specificity and yield on an agarose gel [75]. It is critical to note that dNTPs and any chelating agents (like EDTA) in the sample can bind Mg2+, reducing the free concentration available for the polymerase [75] [76].
Table 1: Optimization of Magnesium Chloride in PCR
| Parameter | Optimal Range | Effect of Low Concentration | Effect of High Concentration | Optimization Method |
|---|---|---|---|---|
| MgCl₂ Concentration | 1.5 - 2.0 mM (standard Taq) [75]; 3.5 - 4.0 mM (Stoffel fragment) [9] | No PCR product; enzyme inactivity [75] [77] | Non-specific products; reduced fidelity; primer-dimer formation [75] [79] | Titrate in 0.5 mM increments up to 4 mM [75] |
Potassium Chloride (KCl): Regulating Nucleic Acid Stability
Potassium chloride (KCl) acts as a neutralizer of the negative charges on the phosphate backbone of DNA. By offsetting these repulsive charges, KCl stabilizes the DNA duplex, influencing the efficiency of both denaturation and primer annealing [76].
- Optimal Concentration Range: A final concentration of 50 mM KCl is standard in most PCR buffers [75] [76]. However, this concentration can be adjusted to fine-tune specificity based on amplicon length.
- Consequence of Deviation:
- High KCl (70-100 mM): Preferentially permits denaturation of short DNA molecules, making it more effective for the amplification of shorter products (<1 kb) [76].
- Low KCl (<50 mM): More effective for the amplification of longer products, as it facilitates the denaturation of larger DNA molecules [76]. It is important to note that concentrations significantly above 50 mM can be inhibitory to Taq polymerase [76].
Table 2: Optimization of Potassium Chloride in PCR
| Parameter | Optimal Range | Effect of Low Concentration | Effect of High Concentration | Application Guidance |
|---|---|---|---|---|
| KCl Concentration | ~50 mM [75] [76] | Favors denaturation of long templates; better for long-range PCR [76] | Favors denaturation of short templates; better for amplicons <1 kb [76]. >50 mM can inhibit Taq [76]. | Adjust based on product length: Lower for long PCR, higher for short PCR [76] |
pH and Buffering System: Maintaining Enzymatic Integrity
The pH of the reaction buffer is critical for maintaining the structural integrity and catalytic function of Taq DNA polymerase. The enzyme exhibits maximal activity in a slightly alkaline environment [9].
- Optimal pH Range: Maximal enzymatic activity for Taq polymerase is achieved in a buffer at pH 8.3 (composed of 10 mM Tris-HCl) for standard PCR, though one study reported optimal activity in a Taps-KOH buffer at pH 9.4 [75] [9].
- Critical Consideration - Temperature Dependence: The pKa of the standard Tris-HCl buffer has a strong temperature dependence, approximately -0.031 ΔpKa/°C [78]. This means a buffer adjusted to pH 8.3 at 25°C will have a significantly higher pH (more basic) at the typical denaturation temperature of 95°C. This drift can affect reaction components. Therefore, for highly sensitive applications, it is crucial to ensure the buffer is pH-adjusted at the temperature for which the specified pH is intended, or to use buffers with lower temperature dependence [78].
- Consequence of Deviation: A pH that is too low (acidic) can inactivate the enzyme and increase the rate of DNA depurination, particularly detrimental for long-range PCR. A pH that is too high (basic) can also reduce enzyme efficiency [9] [76].
Diagram 1: A sequential workflow for optimizing key PCR buffer components. The process is iterative; evaluation results guide which parameter to re-adjust.
Advanced Optimization: Addressing Challenging Templates
GC-Rich and AT-Rich Templates
Challenging templates require deviations from standard conditions. GC-rich templates (>65% GC content) form strong secondary structures that impede polymerase progression, while AT-rich templates can have stability issues [76].
- GC-Rich Strategies:
- Denaturation: Use a higher denaturation temperature (98°C) to ensure complete strand separation [76].
- Additives: Incorporate DMSO at 2–10% or betaine at 1–2 M to destabilize secondary structures and homogenize DNA thermal stability [76] [79].
- Polymerase Selection: Use polymerases specifically engineered for GC-rich templates [76].
- AT-Rich Strategies: For templates with >80–85% AT content, the extension temperature can be lowered to 60–65°C to improve replication reliability [76].
Long-Range PCR and Fidelity Considerations
Amplifying products greater than 5 kb demands special attention to template quality and reaction conditions to prevent truncation [75] [76].
- Template Quality: DNA integrity is paramount; avoid depurination by keeping denaturation times to a minimum and ensuring DNA is resuspended in a neutral buffered solution (pH 7–8), not water [76].
- Extension Time and Temperature: Use longer extension times and a lower extension temperature of 68°C to reduce the depurination rate [76].
- High-Fidelity Polymerases: For applications like cloning where accuracy is critical, replace standard Taq with a high-fidelity polymerase (e.g., Pfu, KOD) that possesses 3'→5' proofreading activity, reducing error rates by up to 10-fold [9] [79].
The Scientist's Toolkit: Essential Reagents for PCR Optimization
Table 3: Key Research Reagent Solutions for PCR Optimization
| Reagent | Critical Function | Considerations for Optimization |
|---|---|---|
| Taq DNA Polymerase | Thermally stable enzyme that synthesizes new DNA strands. | Lacks proofreading activity. Standard concentration is 1.25 units per 50 µl reaction [75]. |
| dNTP Mix | The building blocks (dATP, dTTP, dCTP, dGTP) for DNA synthesis. | Typical concentration is 200 µM of each dNTP. Higher concentrations can increase yield but reduce fidelity [75]. |
| Primers | Short oligonucleotides that define the start and end of the amplicon. | Final concentration 0.1–0.5 µM each. Should have matched Tm within 5°C, 40–60% GC content, and be free of secondary structure [75]. |
| MgCl₂ Solution | Essential cofactor for DNA polymerase activity. | Concentration is the most critical variable to titrate. Start with 1.5 mM and optimize from 1.0 to 4.0 mM [75] [77]. |
| PCR Buffer (with KCl) | Provides the ionic environment and pH for the reaction. | Typically contains ~50 mM KCl. Concentration can be adjusted to influence duplex stability based on amplicon length [75] [76]. |
| Template DNA | The target DNA to be amplified. | Use high-quality, purified DNA. For genomic DNA, use 1 ng–1 µg; for plasmid DNA, use 1 pg–10 ng [75]. |
| Additives (DMSO/Betaine) | Assist in denaturing complex secondary structures in GC-rich templates. | DMSO is used at 2.5–10%; Betaine at 1–2 M. Required for many difficult templates [76] [79]. |
The discovery of Taq polymerase was a pivotal moment in science, but its true power is unlocked only through precise biochemical optimization. As this guide has detailed, the triumvirate of Mg2+, KCl, and pH forms the foundation of a robust PCR reaction, each component exerting a profound influence on specificity, yield, and fidelity. Mastering these parameters empowers researchers to push the boundaries of their work, from routine genotyping to the amplification of the most challenging templates. In the spirit of the innovation that brought us Taq polymerase itself, continuous refinement and understanding of these fundamental reaction conditions will continue to drive discovery and innovation across the biological sciences.
The discovery of Thermus aquaticus (Taq) DNA polymerase in the hot springs of Yellowstone National Park marked a revolutionary turning point in molecular biology, enabling the automation of the polymerase chain reaction (PCR) and transforming genetic analysis across diverse fields from medical diagnostics to forensic science [17] [5]. This thermostable enzyme, with an optimal temperature of 75–80°C and the ability to withstand the DNA denaturation temperatures of 95°C required for PCR, replaced the E. coli DNA polymerase that necessitated replenishment after each cycle [17] [9]. However, a significant challenge emerged alongside its widespread adoption: Taq polymerase exhibits residual enzymatic activity at lower temperatures encountered during reaction setup and initial thermal cycling. This activity facilitates the nonspecific amplification of untargeted sequences, including primer dimers and mis-primed products, which occurs when primers anneal to partially complementary sites or to each other under the less stringent conditions present before the reaction mixture is fully heated [80] [81] [82]. These nonspecific products compete for reaction resources, reducing the yield and sensitivity of the desired amplification and compromising the reliability of downstream applications [81]. The development of Hot-Start PCR strategies represents a critical advancement built upon the foundation of Taq polymerase, specifically designed to inhibit this premature enzymatic activity and thereby suppress nonspecific amplification, enhancing the specificity, sensitivity, and overall robustness of one of molecular biology's most essential techniques [80].
Core Mechanisms of Non-Specific Amplification
Understanding the sources of non-specific amplification is fundamental to appreciating the solutions offered by Hot-Start technologies. The primary errors encountered in standard PCR can be categorized into two main types.
- Mis-priming: This occurs during the lower temperature conditions of reaction setup and the initial thermal cycler ramp. At these sub-optimal temperatures, primers can bind to regions of the template DNA with partial complementarity. When Taq polymerase possesses activity at these temperatures, it can extend these mis-matched primers, synthesizing off-target products that do not correspond to the intended amplicon [81] [82].
- Primer-Dimer Formation: Under the same low-stringency conditions, the primers themselves can anneal to each other via complementary bases, particularly at their 3' ends. The polymerase can then extend these hybridized primers, creating short, artifactual products known as "primer-dimers." These artifacts can be efficiently amplified in subsequent PCR cycles, consuming dNTPs, primers, and enzyme, thereby drastically reducing the efficiency of target amplification, especially for low-copy-number templates [81] [83].
The following diagram illustrates the logical workflow of how these non-specific products are generated and how Hot-Start strategies intervene to prevent them.
Hot-Start PCR: Fundamental Principles and Commercialized Strategies
The core principle of Hot-Start PCR is to reversibly inhibit the DNA polymerase's activity during the reaction setup and initial denaturation phases, only activating it after the reaction mixture has reached a high temperature (typically >90°C). This ensures that the enzyme becomes functional only when the stringency is high enough to promote specific primer-template hybridization, effectively preventing the extension of nonspecific complexes formed at lower temperatures [80]. Several sophisticated strategies have been developed to implement this principle, each with distinct mechanisms and advantages, as summarized in the table below.
Table 1: Commercial Hot-Start PCR Strategies and Their Characteristics
| Strategy | Mechanism of Inhibition | Activation Requirement | Key Characteristics |
|---|---|---|---|
| Antibody-Based | A neutralizing antibody binds the polymerase's active site [9]. | High-temperature incubation (e.g., 95°C for 2–5 min) denatures the antibody, releasing active polymerase [9]. | Easy to use; rapid activation; one of the most common methods. |
| Chemical Modification | Polymerase is covalently modified with inert chemical groups, blocking its activity [81]. | Extended high-temperature incubation (e.g., 95°C for 10–15 min) cleaves the inactivating groups [80] [81]. | Robust inhibition; requires longer initial denaturation. |
| Physical Separation | A critical component (e.g., Mg²⁺ or polymerase) is physically separated by a wax or paraffin barrier [9]. | Initial denaturation melt step melts the barrier, mixing components at high temperature [9]. | Manual and tedious; less common in modern kits. |
| Primer-Based (OXP) | Primers are synthesized with thermolabile phosphotriester (OXP) modifications at the 3'-end [81]. | High temperature cleaves OXP groups, converting primers to a natural, extendable form [81]. | Targets the primer instead of the enzyme; highly specific. |
| Protein-Based (MutS) | A thermostable mismatch-recognizing protein (e.g., MutS) is added to the reaction [82]. | MutS binds to mispaired primer-template complexes at the extension step, blocking polymerase access [82]. | Suppresses both mis-priming and polymerase-generated mutations. |
Experimental Protocols for Key Hot-Start Methodologies
Protocol: Standard Hot-Start PCR with a Commercial Enzyme
This protocol is adapted for use with a typical antibody-inactivated Hot-Start Taq polymerase [80] [83].
Reaction Setup (on ice):
- Prepare a master mix in a thin-walled 0.2 mL PCR tube on ice.
- Reagents for a 50 μL reaction:
- Sterile distilled water: Q.S. to 50 μL
- 10X PCR Buffer (with MgCl₂): 5 μL
- dNTP Mix (10 mM total): 1 μL
- Forward Primer (20 μM): 1 μL
- Reverse Primer (20 μM): 1 μL
- DNA Template (1–1000 ng): variable
- Hot-Start Taq DNA Polymerase (e.g., 5 U/μL): 0.5–1.0 μL
- Gently mix the reaction by pipetting up and down 20 times. Do not vortex.
Thermal Cycling:
- Initial Denaturation/Activation: 95°C for 2–5 minutes. This critical step fully activates the Hot-Start enzyme. [80]
- Amplification (25–35 cycles):
- Denature: 95°C for 20–30 seconds.
- Anneal: 52–65°C (primer-specific) for 20–30 seconds.
- Extend: 72°C for 1 minute per kilobase.
- Final Extension: 72°C for 5–10 minutes.
- Hold: 4°C.
Protocol: Hot-Start PCR with Thermolabile Modified Primers (OXP)
This protocol utilizes primers modified with 4-oxo-1-pentyl (OXP) groups, which requires a slightly modified thermal profile to ensure complete deprotection [81].
Reaction Setup:
- The reaction mixture is assembled similarly to the standard protocol, but using OXP-modified primers instead of standard primers. All other components, including a standard Taq polymerase, remain the same.
Thermal Cycling:
- Extended Initial Denaturation/Deprotection: 95°C for 10–12 minutes. This extended step is necessary for the complete thermal conversion of the OXP-modified primers to their natural, extendable phosphodiester form. [81]
- Amplification (25–35 cycles): Standard cycling as in Section 4.1.
- Final Extension and Hold: Standard steps as in Section 4.1.
Protocol: Hot-Start PCR Using a Thermostable MutS Protein
This innovative approach adds a recombinant thermostable MutS protein to the reaction to physically block extension from mismatched primers [82].
Reaction Setup (on ice):
- Prepare the standard master mix as in Section 4.1, using a standard (non-Hot-Start) Taq polymerase.
- Add Thermostable MutS Protein (e.g., from T. thermophilus) to a final concentration of 0.5–2.0 μM.
- Mix gently and proceed directly to thermal cycling.
Thermal Cycling:
- Initial Denaturation: 95°C for 2 minutes.
- Amplification (25–35 cycles): Standard cycling. During the extension step, MutS binds to any mispaired primer-template complexes, sterically hindering the DNA polymerase and suppressing the amplification of non-specific products. [82]
- Final Extension and Hold: Standard steps.
The Scientist's Toolkit: Essential Reagents for Hot-Start PCR
Successful implementation of Hot-Start PCR relies on a suite of specialized reagents. The selection below covers core components and advanced tools for optimization.
Table 2: Essential Research Reagent Solutions for Hot-Start PCR
| Reagent / Kit | Function & Application | Key Features |
|---|---|---|
| Hot-Start Taq DNA Polymerase | Core enzyme for amplification; inhibited at low temps. | Available in antibody-based or chemically modified formats; essential for all standard Hot-Start protocols [80]. |
| OXP-Modified Primers | Gene-specific primers with thermolabile 3' blocks. | Enable primer-based Hot-Start; synthesized with phosphotriester modifications; require extended initial denaturation [81]. |
| Thermostable MutS Protein | Mismatch-binding protein for error suppression. | Suppresses both non-specific amplification and polymerase-generated mutations; added directly to PCR mix [82]. |
| dNTP Mix | Building blocks (dATP, dCTP, dGTP, dTTP) for DNA synthesis. | High-purity, neutral pH solutions are critical for efficient amplification and high yield [83]. |
| Optimized PCR Buffer | Provides optimal ionic and pH conditions for Taq activity. | Typically contains Tris-HCl, KCl, and sometimes MgCl₂; Mg²⁺ concentration is a key optimization parameter [9] [83]. |
| PCR Additives (DMSO, BSA, Betaine) | Enhancers to improve specificity and yield of difficult amplicons. | DMSO reduces secondary structure; BSA counters inhibitors; Betaine stabilizes DNA melting—all used empirically [83]. |
Troubleshooting and Optimization of Hot-Start PCR
Despite the advantages of Hot-Start PCR, optimization is often required for challenging templates or primer sets. The flowchart below outlines a systematic approach to diagnosing and resolving common issues.
The development of Hot-Start PCR stands as a pivotal innovation built upon the foundational discovery of Taq polymerase, directly addressing its inherent limitation of low-temperature activity to achieve unparalleled amplification specificity. From simple physical separation to sophisticated molecular strategies involving antibody neutralization, chemical modification, and novel primer or protein-based mechanisms, Hot-Start technologies have become the standard for reliable PCR [80] [81] [82]. The strategic inhibition of DNA polymerase until a critical high-temperature threshold is crossed ensures that primer extension occurs only under stringent conditions, effectively suppressing the nonspecific amplification that once plagued conventional PCR assays.
The implications of this technological refinement extend far beyond basic research. In clinical diagnostics, the enhanced specificity of Hot-Start PCR is critical for the accurate detection of low-abundance pathogens and genetic mutations, forming the basis for numerous FDA-approved tests [5] [84]. In next-generation sequencing and biodefense, it ensures the fidelity of library preparation and the reliable identification of biohazardous agents [81] [82]. The ongoing evolution of Hot-Start methods, including the integration of mutant and high-fidelity enzymes, continues to push the boundaries of PCR applications. As the Taq polymerase market grows—projected to reach significant value—its continued integration with advanced Hot-Start technologies will undoubtedly underpin future breakthroughs in genomics, personalized medicine, and molecular diagnostics, securing its legacy as an indispensable tool in the life sciences for years to come [31] [85] [84].
The discovery of Taq DNA polymerase from Thermus aquaticus revolutionized molecular biology by enabling the polymerase chain reaction (PCR) to become a simple, automated process [12]. However, this breakthrough introduced a persistent challenge: bacterial DNA contamination in the enzyme preparations themselves. This contamination presents a significant obstacle for diagnostic applications aiming to detect low-abundance bacterial pathogens, as it can lead to false-positive results [86] [87] [88]. This technical guide explores the sources and nature of this contamination, summarizes quantitative data on its prevalence, and details established experimental protocols for its mitigation, thereby ensuring the reliability of sensitive molecular assays in both research and clinical diagnostics.
The inherent heat stability of Taq polymerase, isolated from the thermophilic bacterium Thermus aquaticus, was the key innovation that transformed PCR from a cumbersome technique into a robust and widely adopted technology [12] [20]. Despite its profound impact, a critical caveat soon emerged. Taq polymerase preparations, particularly those produced recombinantly in E. coli, are frequently contaminated with exogenous bacterial DNA [20]. This DNA originates from the expression host's genome or the plasmid vectors used for recombinant production, which often carry antibiotic resistance genes like blaTEM (encoding for beta-lactamase) as selectable markers [87].
This contaminating DNA becomes a substantial problem in highly sensitive applications, such as:
- Diagnosing bacterial infections with low pathogen loads (e.g., sepsis) [86] [89].
- Detecting bacterial DNA in environmental samples [88].
- Broad-range bacterial detection using primers targeting conserved genes like 16S rRNA [86] [88].
In these contexts, the contaminating DNA acts as an amplifiable template, leading to false-positive outcomes that can compromise diagnostic accuracy and research integrity [86] [87].
Identifying and Quantifying Contamination
The primary source of contaminating DNA is the manufacturing process of the polymerase itself. Investigations have revealed that the contamination often consists of fragmented DNA rather than complete genes, though amplifiable sequences are common [87]. The most frequently encountered contaminants include:
- 16S ribosomal RNA (rRNA) gene sequences: A common target for universal bacterial detection [86] [20].
- Antibiotic resistance genes: Notably, the blaTEM gene fragment is a prevalent contaminant due to its use in plasmid vectors [87].
Notably, one study found that 11 out of 16 commercial Taq polymerase batches were contaminated with beta-lactamase genes, and 15 out of 16 contained 16S rRNA sequences [20]. Contamination has also been traced to other reagents, such as monoclonal antibodies used in hot-start formulations [20].
Quantitative Data on Contamination Levels
The table below summarizes findings from various studies on the amount and impact of contaminating DNA in Taq polymerase preparations.
Table 1: Quantitative Profile of Contaminating DNA in Taq Polymerase
| Study / Context | Contaminant Identified | Estimated Level / Impact | Detection Method |
|---|---|---|---|
| Carroll et al. (1999) [86] | Bacterial 16S rRNA gene sequences | Sufficient to cause false-positives in nested PCR | Gel electrophoresis, Southern hybridization |
| Spangler et al. (2009) [88] | General bacterial DNA | Estimates of 10–1000 genome equivalents per Unit of enzyme | Quantitative PCR (qPCR) |
| Kulakov et al. (2019) [87] | blaTEM gene fragments | 10²–10⁴ copies of contaminating fragments per unit of enzyme | PCR, hybridization with oligonucleotide probes |
| Ultrapure Commercial Preps (e.g., Amplitaq LD) [86] | Bacterial 16S rRNA gene sequences | <10 copies of bacterial 16S rRNA gene per 2.5-μl aliquot | Nested PCR |
These quantitative data highlight that even "ultrapure" commercial preparations can contain sufficient DNA to interfere with exceptionally sensitive assays.
Experimental Protocols for Mitigation
Several methods have been developed to remove or neutralize contaminating DNA in Taq polymerase. The choice of method involves a trade-off between effectiveness, practicality, and potential impact on enzyme activity.
Restriction Endonuclease Pretreatment
This method uses a restriction enzyme to cleave contaminating DNA into fragments that are no longer viable PCR templates.
- Principle: The restriction enzyme Sau3AI (or other frequently cutting enzymes) is used to digest double-stranded contaminating DNA present in the PCR master mix prior to the addition of the sample template [86].
- Detailed Protocol:
- Prepare Master Mix: Combine water, PCR buffer, MgCl₂, and Taq DNA polymerase.
- Digestion Step: Add 1.0 Unit of Sau3AI per Unit of Taq DNA polymerase. Incubate the mixture at 37°C for 30 minutes [86].
- Enzyme Inactivation: Heat the mixture to 95°C for 2 minutes to inactivate Sau3AI. This step does not significantly harm Taq polymerase due to its thermostability.
- Complete PCR Setup: After the mixture cools, add deoxynucleoside triphosphates (dNTPs), primers, and the template DNA. Commence standard PCR amplification [86].
- Considerations: This method is effective, simple to incorporate into existing protocols, and the enzyme is active in standard PCR buffers. However, the choice of restriction enzyme should be considered carefully to avoid cleaving the target sequence of interest [86].
Optimized Dilution of Taq Polymerase
A simple yet effective method to reduce background signal is to systematically dilute the Taq polymerase.
- Principle: Contaminating DNA decreases linearly with enzyme dilution. However, the amplification efficiency for a specific, low-abundance target remains constant until the polymerase becomes limiting [88].
- Detailed Protocol:
- Dilution Series: Perform a cross-titration experiment. Prepare a series of dilutions of the Taq polymerase (e.g., 2-fold serial dilutions).
- Spiked Control: For each polymerase dilution, run a parallel dilution series of a known, low-quantity target DNA or bacteria relevant to the assay.
- qPCR Analysis: Perform qPCR and analyze the Cycle Threshold (Cq) values. The optimal polymerase concentration is the most dilute point that does not alter the Cq for the spiked target compared to higher concentrations, while minimizing the signal from the negative (water) control [88].
- Considerations: This method can yield a greater than 10-fold reduction in background signal without compromising sensitivity for the true target. It is a practical and cost-effective first step for assay optimization [88].
DNase I Treatment with Careful Inactivation
DNase I can be used to digest contaminating DNA, but its subsequent inactivation is critical.
- Principle: DNase I enzymatically degrades DNA in the reagent mix before PCR.
- Reported Protocol and Challenges:
- DNase I is added to the master mix and incubated.
- Inactivation requires heating to 80-95°C, sometimes with the addition of a reducing agent like dithiothreitol (DTT) [88] [20].
- A significant drawback is that incomplete inactivation of DNase I can lead to degradation of the sample template DNA upon its addition, reducing PCR sensitivity. The high-temperature inactivation step can also partially denature and reduce the activity of Taq polymerase [88] [20].
Other Reported Methods
- Ultraviolet (UV) Irradiation: UV light induces pyrimidine dimers in DNA, making it unamplifiable. However, UV also damages the Taq polymerase itself, significantly reducing PCR sensitivity and is not recommended for most applications [88] [20].
- Psoralen/UVA Treatment: Psoralen intercalates into DNA and, upon UVA exposure, cross-links the strands. This method is effective but requires optimization of multiple variables (psoralen concentration, UVA dose), making it complex for routine use [88].
- Nylon Membrane Adsorption: A 2021 study found that incubating Taq polymerase with nylon membrane disks for 24 hours adsorbed contaminating DNA without decreasing enzyme activity, offering a potential physical removal method [20].
The following workflow diagram summarizes the logical relationship between the contamination problem and the primary mitigation strategies discussed:
The Scientist's Toolkit: Essential Reagents for Contamination Mitigation
Successful implementation of the above protocols requires specific reagents and materials. The following table lists key solutions and their functions.
Table 2: Key Research Reagent Solutions for Decontamination Protocols
| Reagent / Material | Function / Principle | Example Application / Note |
|---|---|---|
| Sau3AI Restriction Enzyme | Cleaves contaminating bacterial DNA into non-amplifiable fragments. | Core reagent for the restriction enzyme pretreatment protocol [86]. |
| DNase I (RNase-free) | Enzymatically degrades all DNA in a solution. | Requires careful heat inactivation to avoid degrading sample template [88] [20]. |
| Silica-coated Magnetic Beads | Bind and physically remove nucleic acids from solution. | Can be used to purify Taq polymerase post-DNase treatment or as a standalone decontamination step [90]. |
| Nylon Membrane Disks | Adsorb contaminating DNA from the Taq polymerase solution. | Physical method reported to not decrease Taq activity [20]. |
| Hot-Start Taq Polymerase | Reduces non-specific amplification and primer-dimer formation. | While not a decontamination method, it improves overall assay specificity, helping to manage background [20]. |
| Broad-Host-Range 16S Primers | Amplify conserved bacterial 16S rRNA gene sequences. | Essential for testing and quantifying the level of bacterial DNA contamination [86] [88]. |
The contamination of Taq polymerase preparations with bacterial DNA is a well-documented and persistent challenge that stems directly from the enzyme's biological origin and production methods. As molecular diagnostics continues to push towards lower limits of detection, addressing this contamination becomes not merely an optimization step, but a fundamental requirement for assay validity. The methods detailed here—restriction enzyme pretreatment, systematic Taq dilution, and others—provide researchers and clinicians with a practical toolkit to mitigate this issue. The choice of method depends on the specific application, required sensitivity, and available resources. By critically assessing and implementing these strategies, the scientific community can continue to leverage the full power of PCR, ensuring that the revolutionary legacy of Taq polymerase is not undermined by the "uninvited guests" within its own preparations.
The discovery of thermostable DNA polymerase from Thermus aquaticus (Taq) revolutionized molecular biology by enabling the polymerase chain reaction (PCR) as we know it today. While Taq polymerase serves as a workhorse for routine amplification, its limitations become apparent when confronting challenging templates such as GC-rich sequences and long amplicons. This technical guide explores the mechanistic basis of these challenges and presents optimized strategies based on current research, enabling researchers to successfully amplify even the most recalcitrant targets. Through understanding Taq's conformational dynamics, fidelity mechanisms, and biochemical requirements, scientists can implement specialized protocols that push the boundaries of conventional PCR applications.
The isolation of Taq DNA polymerase from the thermophilic bacterium Thermus aquaticus, discovered in the thermal springs of Yellowstone National Park in 1969, marked a pivotal advancement in molecular biology [20]. This extreme thermophile provided a source of heat-stable enzymes that could withstand the repeated heating cycles required for PCR, eliminating the need to replenish enzymes after each denaturation step [20]. The inherent thermostability of Taq polymerase, with a temperature optimum of 72°C and half-life of 40 minutes at 95°C, made it ideally suited for automated thermal cycling [20] [91].
Despite its transformative impact, Taq polymerase presents specific limitations when dealing with challenging templates. The enzyme lacks 3'→5' exonuclease "proofreading" activity, resulting in an error rate of approximately 10⁻⁵ mutations per base per duplication [73] [20]. Furthermore, its performance can be compromised by structural features of GC-rich templates and requires optimization for efficient amplification of long fragments. Understanding these limitations in the context of Taq's molecular mechanisms provides the foundation for developing effective strategies to overcome amplification challenges.
Table 1: Key Characteristics of Taq DNA Polymerase
| Property | Specification | Significance for PCR |
|---|---|---|
| Size | ~94 kDa [20] | Standard molecular weight for DNA polymerases |
| Catalytic Activity | 150 nucleotides/second at 75-80°C [20] | Fast extension rates under optimal conditions |
| 5'→3' Exonuclease | Present [20] [91] | Enables probe hydrolysis in qPCR applications |
| 3'→5' Proofreading | Absent [73] [20] | Higher error rate compared to proofreading enzymes |
| Fidelity | ~10⁻⁵ error rate [73] | Suitable for routine applications requiring precision |
| Thermostability | 40 min at 95°C [20] | Withstands multiple denaturation cycles |
| Terminal Transferase | Adds single A-overhang [91] | Facilitates TA cloning |
Mechanistic Challenges: Template Structures and Polymerase Dynamics
The GC-Rich Template Challenge
GC-rich DNA sequences (defined as ≥60% GC content) present two fundamental challenges for amplification. First, the presence of three hydrogen bonds in G-C base pairs compared to two in A-T pairs creates greater thermal stability, requiring higher denaturation temperatures [92] [93]. This increased stability is primarily due to base stacking interactions rather than hydrogen bonding alone [94]. Second, GC-rich regions readily form stable secondary structures such as hairpin loops that can block polymerase progression [92] [93] [94]. These structures persist at standard PCR denaturation temperatures and can lead to truncated products, blank gels, or DNA smears [92].
Recent single-molecule studies have revealed unprecedented details of Taq polymerase dynamics. Using single-walled carbon nanotube transistors, researchers recorded Taq molecules processing matched or mismatched template–dNTP pairs from 22° to 85°C [51]. The technique distinguished whole-enzyme closures of nucleotide incorporations from rapid 20-μs closures of Taq's fingers domain that test complementarity and orientation [51]. Surprisingly, even complementary substrate pairs averaged five transient closures between each catalytic incorporation at 72°C, revealing a multi-step fidelity checking mechanism [51].
Diagram 1: GC-rich amplification challenges
Long Amplicon Amplification Difficulties
Amplifying long DNA fragments (>5 kb) presents distinct challenges related to polymerase processivity and reaction conditions. Taq polymerase performs best when amplifying DNA fragments <2 kb but can amplify longer fragments efficiently under defined reaction conditions including dNTP concentration, pH, and MgCl₂ concentration relative to total dNTP concentration [91]. Processivity—the number of nucleotides added per binding event—becomes increasingly important for long amplicons, as frequent dissociation and reassociation can lead to incomplete products.
The terminal structure of PCR products also varies among polymerases. While Taq and related enzymes typically yield products containing 3'-dA overhangs suitable for TA cloning, high-fidelity polymerases with proofreading activity primarily generate amplification products with blunt ends [95]. This distinction has important implications for downstream applications including cloning strategies.
Quantitative Comparison of Polymerase Performance
Fidelity Measurements and Error Rates
Polymerase fidelity is a critical consideration for applications requiring high accuracy, such as gene cloning, protein expression, and next-generation sequencing. Error rates are typically measured using either blue-white screening or direct sequencing approaches, with the latter being more accurate as it detects all mutation types [95].
Table 2: Error Rate Comparison of DNA Polymerases
| Polymerase | Published Error Rate (errors/bp/duplication) | Fidelity Relative to Taq | Key Features |
|---|---|---|---|
| Taq | 1–20 × 10⁻⁵ [73] | 1x | Standard for routine PCR |
| AccuPrime-Taq HF | N/A | 9x better [73] | Optimized for high fidelity |
| KOD Hot Start | N/A | 4-50x better [73] | High thermostability |
| Pfu | 1-2 × 10⁻⁶ [73] | 6-10x better | Proofreading activity |
| Phusion Hot Start | 4 × 10⁻⁷ (HF buffer) [73] | >50x better | Highest fidelity |
| Pwo | Comparable to Pfu [73] | >10x better | Proofreading activity |
Performance Under Suboptimal Conditions
Commercial Taq polymerase formulations demonstrate varying tolerance to suboptimal PCR conditions. One study comparing different enzyme systems found that QIAGEN Taq DNA Polymerase with PCR Buffer showed greater tolerance to a wide range of annealing temperatures (50-60°C) and magnesium concentrations (1.5-4.0 mM) without optimization compared to rival enzymes [91]. This robustness is particularly valuable when amplifying challenging templates where optimal conditions may not be easily predicted.
For long amplicon amplification, performance disparities become more pronounced with increasing fragment length. While product concentrations from different Taq systems were similar for a short amplicon (0.5 kb), one study observed an approximately 10-fold difference between enzyme systems when amplifying a 7.3 kb target [91].
Experimental Protocols and Optimization Strategies
Optimizing Magnesium Concentration
Magnesium ion (Mg²⁺) serves as an essential cofactor for Taq polymerase, binding to dNTPs at the α-phosphate group to facilitate removal of β and gamma phosphates and catalyze phosphodiester bond formation [92]. Standard PCR reactions typically use 1.5 to 2 mM MgCl₂, but GC-rich templates may require optimization.
Protocol: Magnesium Titration for GC-Rich Templates
- Prepare a master mix containing all reaction components except MgCl₂
- Aliquot the master mix into separate tubes
- Add MgCl₂ to create a concentration gradient from 1.0 to 4.0 mM in 0.5 mM increments
- Run PCR using otherwise identical conditions
- Analyze results by agarose gel electrophoresis to identify the optimal concentration that maximizes specific product yield while minimizing non-specific amplification [92] [93]
Touchdown PCR for Enhanced Specificity
Touchdown PCR increases specificity by gradually reducing the annealing temperature during initial cycles, favoring accumulation of the most specific amplicons early in the reaction [95].
Protocol: Standard Touchdown PCR
- Set initial annealing temperature 5-10°C above the calculated Tm of primers
- Perform 5 cycles at this elevated temperature
- Reduce annealing temperature by 2°C and perform another 5 cycles
- Continue stepwise reduction until reaching the calculated Tm of primers
- Complete amplification with 25+ cycles at the final annealing temperature
Example: For primers with Tm of 68°C, use:
- 5 cycles at 72°C
- 5 cycles at 70°C
- >25 cycles at 68°C [95]
A-Tailing for Blunt-End Cloning
When using high-fidelity polymerases that generate blunt ends, Taq polymerase can be used to add 3' A-overhangs for TA cloning.
Protocol: A-Tailing Reaction
- Purify the PCR product to remove all residual polymerase (critical step)
- Prepare reaction mix:
- Purified PCR product (0.15-1.5 pmol)
- dATP (0.2 mM final concentration)
- PCR buffer with Mg²⁺ (1X final concentration, 1.5 mM MgCl₂)
- Taq DNA polymerase (1 U)
- ddH₂O to 50 µl
- Incubate for 20 minutes at 72°C
- Proceed immediately to TA cloning for optimal efficiency [95]
Diagram 2: GC-rich PCR troubleshooting workflow
Advanced Solutions for Intractable Templates
Specialized Polymerases and Additives
For particularly challenging GC-rich templates, specialized polymerase formulations can overcome limitations of standard Taq. Enzymes such as OneTaq DNA Polymerase (NEB #M0480) and Q5 High-Fidelity DNA Polymerase (NEB #M0491) are specifically engineered for difficult amplicons, with fidelity 2x and 280x that of Taq, respectively [92] [93]. These polymerases are often supplemented with GC Enhancers containing additives that inhibit secondary structure formation and increase primer stringency [92].
Effective additives for GC-rich PCR include:
- DMSO, Glycerol, Betaine: Reduce secondary structures that inhibit polymerase [92] [93]
- Formamide, Tetramethyl ammonium chloride: Increase primer annealing stringency [92]
- 7-deaza-2'-deoxyguanosine: dGTP analog that improves yield of GC-rich regions [92] [94]
Slow-Down PCR Methodology
Slow-down PCR represents an alternative methodology specifically designed for GC-rich templates. This approach incorporates 7-deaza-2'-deoxyguanosine (a dGTP analog) into the PCR mixture and uses a standardized cycling protocol with lowered ramp rates and additional cycles compared to standard PCR [94]. The method improves amplification by reducing the stability of GC-rich secondary structures while maintaining polymerase processivity.
Commercial Kits for Challenging Templates
Several manufacturers offer complete systems optimized for challenging amplifications:
- OneTaq Hot Start 2X Master Mix with GC Buffer: Specifically tailored for GC-rich sequences [92]
- Q5 High-Fidelity DNA Polymerase with GC Enhancer: Provides high fidelity for GC-rich targets up to 80% GC content [92]
- Q5 Blood Direct 2X Master Mix: Works with amplicons up to 75% GC content and offers increased resistance to inhibitors in blood samples [93]
Table 3: Research Reagent Solutions for Challenging PCR
| Reagent | Function | Application Examples |
|---|---|---|
| GC Enhancer | Contains additives to inhibit secondary structure formation | Amplification of GC-rich templates up to 80% GC content [92] |
| Q-Solution | Modifies DNA melting behavior, facilitates denaturation | GC-rich templates or those with high secondary structure [91] |
| DMSO | Reduces DNA secondary structure stability | General use for difficult templates [92] [93] |
| Betaine | Equalizes Tm differences between AT and GC base pairs | GC-rich template amplification [93] |
| 7-deaza-2'-deoxyguanosine | dGTP analog that incorporates into DNA | Slow-down PCR for GC-rich regions [92] [94] |
| CoralLoad PCR Buffer | Contains gel-tracking dyes for direct loading | Time-saving immediate gel analysis [91] |
The strategies outlined in this technical guide demonstrate that successful amplification of challenging templates requires both understanding the mechanistic basis of amplification failures and implementing systematic optimization approaches. From its discovery in thermal springs to sophisticated single-molecule analyses of its conformational dynamics, Taq polymerase continues to be a fundamental tool in molecular biology. By applying specialized protocols, reagent systems, and troubleshooting methodologies, researchers can overcome even the most formidable amplification challenges presented by GC-rich sequences and long amplicons. The continued refinement of these approaches ensures that PCR remains a robust and versatile technique across diverse research applications.
The discovery of Thermus aquaticus in the hot springs of Yellowstone National Park and the subsequent isolation of its DNA polymerase, Taq polymerase, marked a pivotal breakthrough in molecular biology [5] [96]. This thermostable enzyme became the cornerstone of the polymerase chain reaction (PCR), a technology that would revolutionize genetic research, clinical diagnostics, and drug development [97]. The core attribute that enabled this revolution was the enzyme's remarkable ability to retain activity at the high temperatures required for DNA denaturation, a property known as thermostability [9]. For researchers and drug development professionals, a precise understanding of Taq polymerase's thermostability—specifically the critical balance between its operational temperature and its functional half-life—is not merely academic but a fundamental aspect of experimental design and efficiency. This guide provides an in-depth technical examination of these parameters, supported by quantitative data and practical methodologies.
The Fundamentals of Taq Polymerase Thermostability
Taq polymerase is an 832-amino acid protein with a molecular weight of approximately 94 kDa [9] [20]. Its thermostability is an intrinsic property, evolved to function in a thermophilic bacterium that thrives at high temperatures [5]. This stability is quantified by its half-life—the time required for the enzyme to lose 50% of its activity at a given temperature.
The relationship between temperature and half-life is inverse and non-linear; as temperature increases, the half-life decreases precipitously. This occurs because high temperatures disrupt the weak interactions (hydrogen bonds, hydrophobic effects, etc.) that stabilize the enzyme's tertiary structure, leading to irreversible denaturation. Understanding this relationship is paramount for selecting appropriate cycling conditions in PCR, especially in protocols with extended run times or high denaturation temperatures.
Table 1: Taq Polymerase Half-Life at Critical Temperatures
| Temperature (°C) | Half-Life | Experimental Context |
|---|---|---|
| 97.5 °C | ~9 minutes | Standard PCR denaturation temperature [9] |
| 95 °C | ~40 minutes | Common PCR denaturation temperature [9] [20] |
| 92.5 °C | >2 hours | Lower denaturation temperature offering high stability [5] |
Beyond mere survival, thermostability enables practical efficiency. Before Taq, PCR required the manual addition of fresh DNA polymerase after each denaturation cycle, a laborious and impractical process [97]. A thermostable enzyme eliminated this need, allowing for the automation of PCR in thermal cyclers and making it a high-throughput technique central to modern labs [97] [96].
Quantitative Analysis of Temperature and Enzymatic Activity
The activity of Taq polymerase is not static across temperatures; it exhibits a distinct temperature optimum for catalytic function. While the enzyme remains stable at high temperatures, its activity is maximized within a specific range. This section breaks down the key quantitative relationships between temperature and enzyme performance.
Temperature Optimum and Polymerization Rate
Taq polymerase demonstrates maximal polymerization activity at 75–80 °C [9] [5]. Within this range, the enzyme can incorporate nucleotides at a rate of 150-250 nucleotides per second [9] [5]. This high rate of synthesis is ideal for the primer extension step of PCR, ensuring rapid and complete amplification.
Activity drops significantly as the temperature deviates from this optimum. For example, at 70 °C, the extension rate falls to about 60 nucleotides/second, and at 55 °C, it plummets to just 24 nucleotides/second [5]. This underscores the importance of maintaining an appropriate extension temperature during PCR protocol design.
Divalent Cation Dependence
Taq polymerase is absolutely dependent on divalent cations, primarily Mg²⁺, which acts as a cofactor during catalysis [9] [20]. The optimal concentration of MgCl₂ is typically around 1.5-2.0 mM, but this can vary and often needs empirical optimization for specific primer-template systems [9] [20]. It is crucial to note that Mg²⁺ concentration influences both enzyme activity and product specificity; excess Mg²⁺ can reduce fidelity and increase non-specific amplification [20].
Thermostability vs. Thermoactivity: A Critical Distinction
A key concept is the difference between an enzyme's thermostability (how long it can withstand heat) and its thermoactivity (how well it performs at a given temperature). Some engineered fragments of Taq, like the Stoffel fragment, exhibit greater thermostability (e.g., a half-life of 80 minutes at 95°C) but lower thermoactivity at temperatures above 80°C compared to the full-length enzyme [9]. This trade-off highlights that the "most stable" enzyme variant is not always the most efficient for a given application.
Table 2: Comparative Properties of Full-Length Taq and the Stoffel Fragment
| Property | Full-Length Taq Polymerase | Stoffel Fragment |
|---|---|---|
| Molecular Weight | ~94 kDa | ~61 kDa [9] |
| Temperature Optimum | 75–80 °C | 75–80 °C [9] |
| Half-life at 95°C | ~40 minutes | ~80 minutes [9] |
| Processivity | 50-60 nucleotides | 5-10 nucleotides [9] |
| Optimal [Mg²⁺] | 1.5-2.0 mM | 3.5-4.0 mM [9] |
Experimental Protocols for Assessing Thermostability and Activity
This section outlines core methodologies used to characterize Taq polymerase thermostability and activity, providing a framework for researchers to validate enzyme performance.
Protocol 1: Determining Half-Life at Elevated Temperatures
Objective: To quantitatively determine the functional half-life of a Taq polymerase preparation at a specific temperature (e.g., 95°C).
Materials:
- Purified Taq polymerase [98]
- Standard PCR buffer (e.g., 10 mM Tris-HCl, pH 8.3, 50 mM KCl) [9]
- Water bath or thermal cycler set to target temperature
Method:
- Sample Preparation: Dilute the Taq polymerase in its standard storage buffer to a consistent concentration in multiple tubes.
- Heat Challenge: Place all tubes in a heating block pre-set to the target temperature (e.g., 95°C). At defined time intervals (e.g., 0, 10, 20, 40, 60 minutes), remove one tube and immediately place it on ice.
- Activity Assay: Using a standardized PCR amplification or a primer extension assay (see Protocol 2), measure the remaining enzymatic activity in each heat-treated sample [9] [99].
- Data Analysis: Plot the percentage of remaining activity (with the 0-minute sample as 100%) against the heating time. The time point at which activity drops to 50% is the half-life.
Protocol 2: Primer Extension Assay for Polymerization Activity
Objective: To measure the polymerization rate and processivity of Taq polymerase at various temperatures.
Materials:
- DNA Template: A single-stranded DNA molecule of known sequence.
- Primer: A complementary oligonucleotide, optionally labeled with a 5'-fluorescent dye or radioisotope for detection [99].
- dNTP Mix: Solution containing all four deoxyribonucleoside triphosphates.
- Stop Solution: EDTA to chelate Mg²⁺ and halt the reaction.
Method:
- Reaction Setup: Combine template, primer, dNTPs, and reaction buffer in separate tubes. Equilibrate each tube at a different temperature (e.g., 22°C, 37°C, 55°C, 70°C, 75°C).
- Initiation: Start the reaction by adding Taq polymerase to each tube.
- Termination: After a short, fixed incubation period (e.g., 1-2 minutes), add stop solution.
- Analysis: Denature the products and separate them by high-resolution polyacrylamide gel electrophoresis (PAGE). The length of the extended primer products, visualized by fluorescence or autoradiography, indicates the processivity and relative activity at each temperature [99].
Protocol 3: Purification of Recombinant Taq Polymerase
The following workflow, adapted from modern purification studies, leverages thermostability to achieve high-purity enzyme preparations, a key concern for sensitive applications like diagnostics [98].
The Scientist's Toolkit: Essential Reagents for Taq Polymerase Research
The following table details key reagents and their functions for working with Taq polymerase, based on established protocols and commercial practices.
Table 3: Key Research Reagent Solutions for Taq Polymerase Experiments
| Reagent / Material | Function / Explanation | Typical Concentration / Notes |
|---|---|---|
| Tris-HCl Buffer | Maintains optimal pH for enzyme activity (pH 8.0-9.4) [9] [20]. | 10-50 mM; pKa is temperature-dependent. |
| Potassium Chloride (KCl) | Monovalent salt that neutralizes DNA backbone charge, promoting primer-template annealing and polymerase binding [9] [20]. | ~50 mM; higher concentrations can inhibit enzyme activity [5]. |
| Magnesium Chloride (MgCl₂) | Essential divalent cation cofactor for polymerase activity [9] [20]. | 1.5-2.0 mM (must be optimized); absolutely required. |
| dNTP Mix | The four deoxyribonucleoside triphosphates (dATP, dCTP, dGTP, dTTP) are the building blocks for DNA synthesis. | Balanced equimolar solution; high-quality dNTPs are critical for fidelity. |
| Nickel-NTA Agarose | Affinity resin for purifying recombinant His-tagged Taq polymerase [98]. | Used in protocol 3.2; imidazole is used for elution. |
| DNase I (RNase-free) | Enzyme used during purification to digest residual E. coli genomic DNA contaminants, preventing false positives in PCR [20] [98]. | Requires subsequent heat-inactivation. |
Advanced Considerations and Future Directions
Fidelity and the Proofreading Limitation
A significant shortcoming of Taq polymerase is its lack of a 3′→5′ exonuclease ("proofreading") activity [9] [5]. This results in a relatively high error rate, estimated at approximately 1 error per 10,000-100,000 nucleotides incorporated [5] [20]. For applications requiring high accuracy, such as cloning or sequencing, this is a critical limitation. This drove the development of alternative thermostable polymerases from other thermophiles and archaea, such as Pfu DNA polymerase, which possesses proofreading capability and higher fidelity [5] [97].
Hot-Start Techniques for Enhanced Specificity
Even at room temperature, Taq polymerase possesses residual activity, which can lead to non-specific priming and the formation of primer-dimers during reaction setup [20] [97]. Hot-Start PCR techniques mitigate this by chemically, antibody-based, or aptamer-based inhibition of the enzyme until the first high-temperature denaturation step is reached [20] [97]. This controlled activation significantly improves assay specificity, sensitivity, and yield, making it a standard practice in diagnostic and quantitative PCR.
Engineering the Next Generation
The limitations of wild-type Taq have spurred extensive protein engineering efforts to create superior enzymes. Strategies include:
- Ancestral Sequence Reconstruction (ASR): Leveraging phylogenetic analysis to infer and resurrect ancient, often more stable, versions of proteins [100].
- Directed Evolution: Using iterative rounds of random mutagenesis and screening to select for variants with enhanced properties like thermostability or fidelity [100] [101].
- Rational Design: Using structural information to introduce specific stabilizing mutations, such as adding disulfide bonds or improving hydrophobic core packing [100] [101].
These approaches have yielded enzymes like Phusion DNA Polymerase, a chimeric enzyme that combines high fidelity, processivity, and robust thermostability, demonstrating the power of modern protein engineering to push the boundaries of PCR technology [97].
Taq vs. High-Fidelity Polymerases: Selecting the Right Tool for Your Research
The discovery of thermostable DNA polymerases, a foundational achievement in molecular biology, transformed the polymerase chain reaction (PCR) from a cumbersome process into an automated, high-fidelity technique central to modern bioscience. The fidelity of a DNA polymerase—its accuracy in copying a DNA template—is a critical parameter influencing the success of diverse applications, from cloning and sequencing to drug development. This whitepaper provides a comparative analysis of the error rates of three prominent DNA polymerases: Taq, Pfu, and KOD. We summarize quantitative fidelity data obtained from multiple methodological approaches, detail the experimental protocols for fidelity measurement, and contextualize these findings within the broader narrative of Taq polymerase research. The data underscores that while Taq polymerase enabled the PCR revolution, the superior fidelity of proofreading enzymes like Pfu and KOD is indispensable for applications where sequence integrity is paramount.
The history of PCR is inextricably linked to the discovery and application of Thermus aquaticus (Taq), a thermophilic bacterium discovered by Thomas Brock in the hot springs of Yellowstone National Park [17] [3]. This organism thrived in near-boiling water, a fact that challenged established notions about the limits of life. The enzyme ultimately isolated from this bacterium, Taq polymerase, proved to be remarkably thermostable [5]. This single property was the key innovation that allowed for the automation of PCR. Prior to its use, scientists were required to manually replenish the heat-labile E. coli DNA polymerase after every denaturation cycle, a process described as laborious and a "great burden in terms of labour and cost" [17] [10].
The introduction of Taq polymerase meant that a single aliquot of enzyme could withstand dozens of high-temperature cycles, enabling the entire PCR process to be carried out in a closed tube within a thermal cycler [5] [10]. This breakthrough propelled PCR from a specialized technique to a ubiquitous tool in laboratories worldwide, fundamentally accelerating progress in genetics, medicine, and biotechnology. Kary Mullis was awarded the 1993 Nobel Prize in Chemistry for the invention of PCR, a feat made practical by the properties of Taq polymerase [5]. However, a significant drawback of this pioneering enzyme soon became apparent: its relatively low replication fidelity [5] [74]. This limitation spurred the search for and development of more accurate, high-fidelity enzymes like Pfu and KOD, which are the focus of this comparative analysis.
Defining and Measuring DNA Polymerase Fidelity
What is DNA Polymerase Fidelity?
DNA polymerase fidelity refers to the accuracy with which an enzyme synthesizes a new DNA strand complementary to its template. This accuracy is crucial for maintaining sequence integrity during DNA replication. Fidelity is commonly expressed as an error rate, defined as the number of misincorporated nucleotides per base synthesized per duplication event (errors/bp/duplication) [73] [102]. The inverse of the error rate yields the accuracy, representing the number of bases synthesized per single error [102]. For example, an enzyme with an error rate of 1 x 10⁻⁶ possesses an accuracy of 1,000,000, meaning it incorporates one error for every million nucleotides synthesized [102].
The intrinsic fidelity of a DNA polymerase is largely determined by its proofreading activity. Many thermostable polymerases, including Pfu and KOD, possess an associated 3'→5' exonuclease activity that serves as a proofreading mechanism. When a mismatched nucleotide is incorporated, the polymerase stalls, and the nascent DNA strand is translocated to the exonuclease domain where the incorrect nucleotide is excised. The strand is then returned to the polymerase active site for continued synthesis with the correct nucleotide [102] [74]. In contrast, Taq polymerase lacks this 3'→5' proofreading activity, which is a primary reason for its higher error rate [5] [74].
Methodologies for Determining Fidelity
Several experimental assays have been developed to quantify polymerase error rates, each with distinct advantages and limitations.
Blue/White Colony Screening (Barnes Assay): This method involves using PCR to amplify a reporter gene, such as lacZ, which is then cloned and transformed into bacteria. Colonies containing error-free PCR products metabolize a substrate to produce a blue color, while clones with inactivating mutations in the reporter gene remain white. The ratio of white to blue colonies provides an indirect measure of the mutation frequency [102]. A significant limitation is that only mutations within a small, critical region of the gene that disrupt function are detected, obscuring the full spectrum of errors [73] [102].
Sanger Sequencing of Cloned PCR Products: This approach involves sequencing individual cloned PCR products to directly identify all mutations within the sequenced region. This method provides a more direct and comprehensive readout of the types and frequencies of errors than colony screening [73] [102]. However, its throughput has traditionally been limited by cost and labor, making it challenging to accumulate the vast number of sequenced bases required for highly accurate measurements of ultra-high-fidelity enzymes [73].
Next-Generation Sequencing (NGS): NGS platforms overcome the throughput limitations of Sanger sequencing by enabling the sequencing of millions of PCR products in a single run. This generates a statistically powerful dataset ideal for quantifying low error rates [102]. Single-Molecule, Real-Time (SMRT) Sequencing (e.g., PacBio) is a particularly powerful NGS method for fidelity studies because it sequences PCR products directly without an intermediary amplification step and derives a highly accurate consensus sequence for each read, minimizing sequencing-based background noise. The background error rate for one such SMRT sequencing fidelity assay was reported to be 9.6 x 10⁻⁸ errors/base, making it suitable for quantifying the fidelity of proofreading polymerases [102].
The following diagram illustrates a generalized workflow for a DNA polymerase fidelity experiment, incorporating elements from these different methodologies.
Quantitative Error Rate Comparison
Direct comparison of polymerase error rates can be challenging due to methodological differences between studies. However, controlled experiments and data from standardized assays provide a clear hierarchy of fidelity among Taq, Pfu, and KOD polymerases.
The table below summarizes key error rate data from multiple sources, including a study that used direct sequencing of 94 unique DNA targets to ensure a broad analysis [73] and data from New England Biolabs obtained via SMRT sequencing [102].
Table 1: DNA Polymerase Fidelity Comparison
| DNA Polymerase | Proofreading Activity | Error Rate (errors/bp/duplication) | Accuracy (1/error rate) | Fidelity Relative to Taq | Key Characteristics |
|---|---|---|---|---|---|
| Taq | No | ~4.3 x 10⁻⁵ [73] | ~23,256 | 1x | Standard for basic PCR; lowest fidelity |
| 1.5 x 10⁻⁴ [102] | 6,456 | 1x | |||
| KOD | Yes | ~1.2 x 10⁻⁵ [102] | 82,303 | 12x [102] | High fidelity and high processivity |
| Pfu | Yes | ~1-2 x 10⁻⁶ [73] | ~500,000-1,000,000 | 6-10x [73] | Archaeal family B enzyme; benchmark for high fidelity |
| 5.1 x 10⁻⁶ [102] | 195,275 | 30x [102] | |||
| Phusion | Yes | ~4 x 10⁻⁷ (HF buffer) [73] | 2,500,000 | >50x [73] | Engineered enzyme; often cited as highest fidelity |
Note on Discrepancies: The absolute error rates and relative fidelities for a given polymerase can vary between sources, as seen for Pfu. This highlights the impact of different measurement methods (e.g., Sanger vs. SMRT sequencing), template sequences, and reaction conditions. Despite these variations, the consistent trend across all studies is unambiguous: Pfu and KOD polymerases exhibit a significantly lower error rate (by a factor of 10 or more) than Taq polymerase [73] [102].
Mutation Spectra
Beyond the sheer number of errors, the type of mutations generated, or the "mutation spectrum," can also vary between enzymes. The study by McInerney et al. (2014) that sequenced 94 unique targets reported that for high-fidelity enzymes like Pfu, Phusion, and Pwo, transition mutations (purine to purine or pyrimidine to pyrimidine substitutions) predominated, with little bias for the type of transition [73]. The mutation spectra were broadly similar among these high-fidelity enzymes.
Experimental Protocols for Key Fidelity Studies
To provide context for the data in Table 1, this section outlines the detailed methodologies from two pivotal studies.
Protocol: Direct Sequencing Using Multiple DNA Targets (McInerney et al.)
This protocol is derived from the study that utilized 94 unique plasmid templates to interrogate a large DNA sequence space [73].
- Template DNA: 94 different plasmid constructs, each containing a unique glycosyltransferase gene insert from Arabidopsis thaliana cDNA. Insert sizes ranged from 360 bp to 3.1 kb.
- PCR Amplification: Each polymerase was used to amplify all 94 targets.
- Polymerases Tested: Taq, AccuPrime-Taq High Fidelity, KOD Hot Start, cloned Pfu, Phusion Hot Start, Pwo.
- Reaction Conditions: Vendor-recommended buffers were used. A small amount of plasmid template (25 pg/reaction) was used to maximize the number of template doublings. PCR was performed for 30 cycles.
- Cloning and Sequencing: The PCR products were purified and cloned into a plasmid vector using the Gateway recombination system. Multiple clones for each PCR product were Sanger sequenced.
- Data Analysis:
- The sequenced fragments were aligned to the known, original template sequence to identify mutations.
- The number of template doublings in each PCR reaction was calculated based on the measured fold-amplification.
- The error rate was calculated using the formula: (Total number of mutations observed) / (Total number of base pairs sequenced × Number of doublings).
Protocol: High-Throughput Fidelity Assay via SMRT Sequencing (NEB)
This protocol describes the modern approach used by New England Biolabs to achieve highly accurate fidelity measurements with low background noise [102].
- Template DNA: A plasmid containing a lacZ amplicon, virtually devoid of nucleotide errors to minimize background.
- PCR Amplification: The target amplicon is amplified with the polymerase of interest.
- Library Preparation and Sequencing: The PCR products are prepared as a library for PacBio SMRT sequencing without an intermediary cloning or amplification step.
- Data Analysis:
- The instrument sequences the same molecule multiple times to generate a highly accurate consensus sequence for each individual read.
- These consensus sequences are compared to the known template sequence to identify true replication errors.
- The error rate is calculated from the number of substitutions, insertions, and deletions identified across tens to hundreds of millions of sequenced bases, normalized per base per doubling.
The Scientist's Toolkit: Essential Research Reagents
Selecting the appropriate polymerase and associated reagents is critical for experimental success. The following table details key reagents and their functions in PCR-based research.
Table 2: Essential Reagents for PCR-Based Research
| Reagent | Function & Importance | Example Use-Cases |
|---|---|---|
| Standard Taq Polymerase | General-purpose enzyme for routine PCR where ultimate fidelity is not critical. | Colony PCR, genotyping, educational demonstrations. |
| High-Fidelity Polymerase (e.g., Pfu, KOD) | Essential for applications requiring accurate DNA sequence. Proofreading activity drastically reduces mutation frequency. | Cloning for protein expression, site-directed mutagenesis, NGS library prep. |
| Hot-Start Polymerases | Engineered to be inactive at room temperature, preventing nonspecific amplification and primer-dimer formation during reaction setup. Activated by initial denaturation step. | High-throughput setups, multiplex PCR, amplification from complex templates (e.g., genomic DNA). |
| dNTP Mix | The building blocks (dATP, dCTP, dGTP, dTTP) for DNA synthesis. Quality and concentration affect yield, fidelity, and specificity. | All PCR applications. |
| MgCl₂ / Reaction Buffer | Provides optimal ionic environment and co-factors (Mg²⁺ is essential) for polymerase activity. Concentration can dramatically impact specificity and fidelity. | All PCR applications; often optimized for specific template-primer systems. |
| PCR Enhancers / Additives | Chemicals (e.g., DMSO, betaine, glycerol) that can help denature templates with high GC-content or stable secondary structures. | Amplifying difficult templates, GC-rich regions. |
Implications for Research and Drug Development
The choice of DNA polymerase has profound implications in research and pharmaceutical development. The high error rate of Taq polymerase (~10⁻⁵) means that in a standard 1 kb PCR, approximately 1 in 10 molecules will contain a mutation after 25 cycles. For cloning applications, this necessitates the sequencing of multiple clones to identify a correct one, increasing time and cost [73] [74].
In contrast, using a high-fidelity enzyme like Pfu (error rate ~10⁻⁶) reduces the frequency of mutated 1 kb molecules to roughly 1 in 100. This dramatically increases the likelihood of obtaining a correct clone on the first attempt, streamlining workflows in structural genomics and the production of recombinant proteins for structural studies or therapeutic candidates [73]. In the development of gene therapies and vaccines, where the correct DNA sequence is non-negotiable, the use of ultra-high-fidelity polymerases is indispensable to ensure product safety and efficacy.
The following diagram summarizes the logical relationship between polymerase structure, function, and its ultimate application, rooted in the foundational discovery of Taq.
The journey from the discovery of Thermus aquaticus in the hot springs of Yellowstone to the routine use of high-fidelity PCR in laboratories worldwide is a powerful example of how basic research can drive transformative innovation. The initial adoption of Taq polymerase was a radical innovation that automated PCR. However, the recognition of its fidelity limitations spurred a subsequent wave of incremental and architectural innovations, leading to the isolation and engineering of superior enzymes like Pfu and KOD.
The quantitative data presented in this analysis unequivocally demonstrates the superior fidelity of Pfu and KOD polymerases over the foundational Taq enzyme. This fidelity, primarily conferred by 3'→5' proofreading activity, makes these enzymes the indispensable choice for any application where DNA sequence integrity is critical. As research continues to push into larger-scale cloning projects, synthetic biology, and precision medicine, the demand for even more accurate and efficient DNA synthesizing enzymes will undoubtedly continue, writing the next chapter in the story that began with a curious bacterium in a hot spring.
The invention of the Polymerase Chain Reaction (PCR) by Kary Mullis in 1983, for which he was awarded the Nobel Prize in Chemistry a decade later, revolutionized molecular biology [9]. However, this revolution was contingent upon a critical discovery made years earlier: the isolation of a thermostable DNA polymerase from the thermophilic bacterium Thermus aquaticus [9]. This enzyme, Taq polymerase, was discovered in 1976 in bacteria endemic to the hot springs of Yellowstone National Park [9]. Its inherent ability to withstand the high temperatures required for PCR's repetitive denaturation steps—without being irreversibly inactivated—eliminated the need to add fresh enzyme after each cycle, thus automating the procedure and making it efficient and widely accessible [9] [103].
The deployment of Taq polymerase in PCR not only streamlined a powerful technique but also catalyzed a new field of enzyme phylogeny. As more DNA polymerases were discovered and characterized, they were classified into families based on sequence homology and structural similarities [9] [104]. Taq polymerase, homologous to Escherichia coli DNA polymerase I, became the archetype for Family A polymerases [9] [104]. In parallel, a distinct group, Family B, was identified, comprising enzymes from archaea like Pyrococcus furiosus (Pfu polymerase) and Thermococcus kodakarensis (KOD polymerase) [103] [105]. This classification framework helps scientists understand the evolutionary relationships between these enzymes and provides a critical roadmap for selecting the appropriate polymerase for specific biotechnological and diagnostic applications, a choice that balances factors such as fidelity, processivity, and thermostability [104] [105].
Structural and Mechanistic Divergence Between Families
The functional differences between Family A and Family B polymerases are rooted in distinct structural features. All DNA polymerases resemble a right hand, with "fingers," "palm," and "thumb" subdomains that are responsible for nucleotide binding, catalysis, and template binding, respectively [104]. Despite this common architecture, key structural variations define each family and dictate their catalytic behavior.
Family A Architecture
Family A polymerases, like Taq polymerase, are generally simpler in structure. They possess a 5′→3′ polymerase activity and an associated 5′→3′ exonuclease or "nick-translation" activity, which is important for DNA repair in vivo [9]. A defining structural characteristic of Family A enzymes is the absence of a 3′→5′ exonuclease (proofreading) domain [9] [103]. This lack of proofreading capability is a primary contributor to their relatively higher error rate. The structure of the Klenow fragment of E. coli DNA polymerase I was the first high-resolution structure obtained for any DNA polymerase and served as a model for understanding Family A enzymes [104].
Family B Architecture
Family B polymerases are characterized by a more complex structure that includes an integral 3′→5′ exonuclease domain [103] [105]. This domain provides proofreading activity, allowing the enzyme to detect and excise misincorporated nucleotides during DNA synthesis, thereby significantly increasing replication fidelity [74] [104]. While they possess this 3′→5′ exonuclease activity, Family B polymerases lack 5′→3′ exonuclease activity [105]. Some archaeal Family B polymerases also contain a uracil-binding pocket, part of a DNA repair mechanism that prevents them from amplifying uracil-containing templates, which can be a limitation in certain applications like bisulfite sequencing [74].
The catalytic mechanism, however, is conserved. Both families require a DNA template, a primer with a free 3′ hydroxyl group, and the four deoxyribonucleoside triphosphates (dNTPs) [104]. The nucleotidyl-transfer reaction is dependent on two divalent metal ions (typically Mg²⁺) that coordinate the incoming dNTP and facilitate the nucleophilic attack by the primer's 3′-OH group on the α-phosphate of the dNTP [104].
Diagram 1: Core catalytic requirements and structural domains of Family A and Family B DNA polymerases.
Comparative Functional Analysis and Quantitative Performance
The structural differences between Family A and B polymerases translate directly into distinct functional profiles, which are quantifiable through key performance metrics. These metrics guide the selection of the optimal enzyme for a given application.
Fidelity and Error Rate
Fidelity is arguably the most significant differentiator. It refers to the enzyme's ability to incorporate the correct nucleotide and is inversely related to the error rate [74].
- Family A (Taq): Lacks proofreading activity, leading to an error rate estimated between 1 x 10⁻⁴ to 1 x 10⁻⁵ errors per base per duplication [73] [105]. This translates to roughly 1 error per 1,000–10,000 bases synthesized [103] [105].
- Family B (Pfu, KOD): Possesses proofreading activity, reducing the error rate to a range of 1 x 10⁻⁶ to 1 x 10⁻⁷, or 1 error per 1,000,000 to 10,000,000 bases [103]. Direct sequencing studies show that proofreading enzymes like Pfu and Pwo have error rates more than 10 times lower than Taq polymerase [73].
Processivity and Extension Rate
Processivity is the number of nucleotides incorporated per single enzyme-binding event, and the extension rate is the speed of synthesis [74].
- Family A (Taq): Is moderately processive, incorporating 50–60 nucleotides per binding event and extending primers at a high rate of ~150 nucleotides per second [9] [105]. This makes it fast and efficient for standard PCR.
- Family B (Pfu): Tends to be less processive and slower, with an extension rate of only ~25 nucleotides per second [105]. The proofreading activity inherently slows the polymerization process as the enzyme checks for errors [74]. However, some Family B enzymes like KOD polymerase are exceptions, offering high processivity and fast extension rates (100–130 nucleotides/second) [103].
Thermostability
While all enzymes used in PCR are thermostable, their stability at extreme temperatures varies.
- Family A (Taq): Has a half-life of 45–96 minutes at 95°C and 9 minutes at 97.5°C [9]. This is sufficient for most standard PCR protocols.
- Family B (Pfu): Is significantly more thermostable, with a half-life at 95°C about 20 times longer than that of Taq [74]. This hyperthermostability is advantageous for challenging templates or long extension times.
Table 1: Quantitative Functional Comparison of Representative Family A and Family B DNA Polymerases
| Functional Characteristic | Taq Polymerase (Family A) | Pfu Polymerase (Family B) | KOD Polymerase (Family B) |
|---|---|---|---|
| Fidelity (Error Rate) | 1–20 x 10⁻⁵ [73] | ~1.3 x 10⁻⁶ [73] | ~1 in 10⁶ [103] |
| Fidelity Relative to Taq | 1x | ~10x higher [74] [105] | ~50x higher [73] |
| Processivity (nucleotides/binding event) | 50–60 [9] | <20 [103] | 10–15x greater than Pfu [103] |
| Extension Rate (nucleotides/second) | ~150 [105] | ~25 [105] | 100–130 [103] |
| Thermostability (Half-life at 95°C) | 45–96 minutes [9] | ~20x Taq [74] | Similar to Pfu [103] |
| 5′→3′ Exonuclease | Present [9] | Absent [105] | Absent [105] |
| 3′→5′ Exonuclease (Proofreading) | Absent [9] | Present [105] | Present [103] |
Experimental Methodologies for Fidelity and Novel Enzyme Screening
The quantitative comparison of DNA polymerases relies on robust experimental assays. Furthermore, the drive to improve enzymatic properties has led to the development of high-throughput screening methods for engineered variants.
Measuring Fidelity via Direct Sequencing
A direct method for determining error rates involves sequencing cloned PCR products [73].
- PCR Amplification: A target gene (e.g., a 1.9 kb fragment of the lacZ gene or a diverse set of 94 unique DNA targets) is amplified using the test polymerase [73].
- Cloning: The resulting PCR products are cloned into a plasmid vector.
- Sequencing and Analysis: Individual clones are Sanger sequenced, and the sequences are compared to the known, original template sequence. Mutations are identified and counted. The error rate is calculated based on the total number of mutations, the total number of bases sequenced, and the number of template doublings that occurred during PCR [73]. Next-generation sequencing can also be used to sequence amplicons directly, providing a deep analysis of errors [74].
Screening for Novel Polymerase Variants Using Live Culture PCR (LC-PCR)
To evolve polymerases with new properties, such as resistance to PCR inhibitors, researchers use directed evolution and efficient screening protocols. The Live Culture PCR (LC-PCR) workflow is a streamlined method for this purpose [106].
- Library Creation: A gene for a polymerase (e.g., Taq or Klentaq1) is randomly mutagenized using error-prone PCR to create a vast library of variants [106].
- Cell Culture & Expression: The mutant genes are cloned into an expression vector and transformed into a bacterial host (e.g., E. coli). Single colonies are picked and grown in 96-well plates with an inducer to express the polymerase variants [106].
- Direct PCR Screening: Instead of purifying the enzymes, a small aliquot of the intact, growing bacterial culture is added directly to a PCR master mix. The cells serve as the source of both the DNA template (e.g., bacterial 16S rDNA) and the thermostable polymerase enzyme. The PCR is run in the presence of a challenging inhibitor (e.g., chocolate or blood extract) and a fluorescent dye like SYBR Green [106].
- Variant Selection: Clones that successfully produce an amplification curve under inhibitory conditions are identified by real-time PCR. These hits, which express inhibitor-resistant polymerase variants, are selected from the stored master plate for further sequencing and large-scale purification [106].
Diagram 2: Live Culture PCR (LC-PCR) workflow for screening Taq polymerase variants with enhanced properties like inhibitor resistance.
Application-Oriented Selection and the Scientist's Toolkit
The choice between Family A and Family B polymerases is not a matter of superiority but of suitability for the specific experimental goal. The distinct properties of each family make them ideal for different applications.
Application-Based Enzyme Selection
- Standard PCR and Real-Time qPCR: Family A (Taq) is the gold standard. Its high extension speed and inherent 5′→3′ exonuclease activity (which enables hydrolysis probe/TaqMan assays) make it perfect for routine amplification and quantitative detection [105]. Hot-start modifications, achieved via antibody inhibition or chemical modification, are critical to prevent non-specific amplification during reaction setup [74] [105].
- Cloning, Mutagenesis, and Protein Expression: Family B (Pfu, KOD) is strongly preferred. Their high fidelity ensured by proofreading activity is essential to avoid introducing mutations into the cloned sequence, which could alter the function of the expressed protein [74] [105].
- Amplification of Damaged or Challenging Templates: Specialized Y-Family polymerases (e.g., Dpo4 from archaea) show promise. Unlike Family A or B, these enzymes can bypass DNA lesions that normally block replication. Blending a Y-family polymerase with Taq can facilitate the amplification of damaged DNA from forensic or ancient samples [107].
- Reverse Transcription PCR (RT-PCR): Engineered Family A variants are emerging as all-in-one solutions. Recent protein engineering has created novel Taq variants with enhanced reverse transcriptase activity, enabling single-enzyme, single-tube RT-PCR without the need for viral reverse transcriptases, simplifying multiplexed RNA detection [108].
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and materials used in foundational polymerase experiments, particularly fidelity assays and novel enzyme screening.
Table 2: Key Research Reagents for DNA Polymerase Fidelity and Screening Experiments
| Reagent / Material | Function / Purpose | Example in Context |
|---|---|---|
| pGEM-T Vector | A TA-cloning vector for easy insertion of PCR products. | Used for cloning PCR products (e.g., lacZ amplicons) for subsequent sequencing in fidelity assays [73] [107]. |
| lacZ Gene Template | A reporter gene where mutations can be easily detected via colorimetric screening (blue/white colonies). | Serves as a defined DNA target for polymerase fidelity measurements in the classic forward mutation assay [73] [74]. |
| SYBR Green I Dye | A fluorescent dsDNA-binding dye for real-time detection of PCR amplification. | Used in Live Culture PCR (LC-PCR) to monitor amplification in real-time and identify positive clones expressing functional polymerases [106]. |
| PCR Inhibitors (Blood, Humic Acid) | Complex biological substances used as challenging reaction conditions. | Employed in screening assays to select for engineered polymerase variants with enhanced resistance to common inhibitors found in clinical or environmental samples [106]. |
| TaqMan Hydrolysis Probes | Fluorescently-labeled probes that are cleaved by the 5′→3′ exonuclease activity of Taq polymerase. | Enable specific, real-time detection of target sequences in qPCR and are a key application of Family A enzymes [108]. |
| Hot-Start Antibodies/Chemicals | Inhibitors that block polymerase activity at room temperature. | Essential for preventing non-specific priming and primer-dimer formation during reaction setup, improving PCR specificity [74] [105]. |
The discovery of Taq polymerase provided more than just a practical solution for PCR; it unlocked a systematic, phylogenetic understanding of DNA polymerases through the Family A and B classification. The ongoing structural and functional comparisons between these families provide a fundamental framework that directs experimental design in molecular biology. Driven by the demands of modern biotechnology and diagnostics, protein engineering is pushing the boundaries of these natural enzymes. Researchers are now creating novel variants—such as Taq polymerases with intrinsic reverse transcriptase activity for streamlined RNA detection or enhanced resistance to PCR inhibitors from complex samples [108] [106]. This continuous evolution of enzymes ensures that the powerful technique of PCR will remain adaptable and relevant for addressing future challenges in life science research and medicine.
The fidelity of DNA replication is a cornerstone of genetic inheritance and cellular viability. Central to this process is the 3' to 5' exonuclease activity, an intrinsic proofreading function of many replicative DNA polymerases. This activity serves as a critical frontline defense by recognizing and excising misincorporated nucleotides, thereby enhancing replication accuracy by 100 to 1000-fold [109]. While this mechanism is a universal feature of high-fidelity polymerases, its absence in ubiquitous tools like Taq polymerase underscores its biological significance and has profound implications for the accuracy of techniques such as PCR. This whitepaper delves into the molecular mechanics of proofreading, presents quantitative fidelity comparisons, and explores the experimental evidence that illuminates its role as a guardian of genomic integrity, all within the context of the revolutionary yet imperfect discovery of Taq polymerase.
The accurate duplication of genomic DNA is fundamental to life. It is estimated that for both prokaryotic and eukaryotic cells, DNA is replicated with an exceptionally high fidelity, with one error occurring only once per 10^8 to 10^10 nucleotides polymerized [109]. This astonishing accuracy is not the product of a single mechanism but is achieved through a multi-tiered system involving:
- Nucleotide Selectivity: The inherent ability of the DNA polymerase to choose the correct nucleoside triphosphate based on Watson-Crick base pairing.
- Exonucleolytic Proofreading: The immediate correction of misincorporation events by the 3'→5' exonuclease activity.
- Post-replicative DNA Mismatch Repair (MMR): A subsequent system that corrects errors missed by the first two processes [109].
The discovery and subsequent ubiquitous adoption of Taq DNA polymerase from the thermophilic bacterium Thermus aquaticus revolutionized molecular biology by enabling the polymerase chain reaction (PCR) [17]. However, a critical shortcoming was identified: Taq polymerase lacks 3'→5' proofreading activity [9] [5]. This deficiency results in a relatively high error rate, which has driven extensive research into the mechanisms of high-fidelity DNA replication and the development of novel, high-fidelity enzymes for applications where sequence accuracy is paramount, such as cloning and next-generation sequencing [110].
The Molecular Mechanism of 3'→5' Proofreading
The proofreading mechanism is an elegant example of intramolecular quality control. DNA polymerases with proofreading capability contain distinct polymerase (pol) and exonuclease (exo) active sites within a single polypeptide (or, in the case of E. coli Pol III, within a single complex) [109].
The proofreading process can be broken down into a series of coordinated steps, as illustrated below:
Diagram 1: The stepwise mechanism of 3'→5' exonucleolytic proofreading.
- Nucleotide Misincorporation: During replication, the polymerase occasionally incorporates an incorrect nucleotide that does not form a proper Watson-Crick base pair with the template strand [109].
- Kinetic Delay and Partitioning: The resulting mispair causes a distortion in the geometry of the primer terminus. This suboptimal architecture slows the catalytic reaction, introducing a lag time known as kinetic delay [110] [111]. This delay provides a critical window for the polymerase to halt further extension and initiate the proofreading process.
- Transfer to Exonuclease Site: The distorted primer terminus is recognized by the polymerase, which triggers the physical translocation of the 3' end of the growing DNA strand from the polymerase active site to the distant exonuclease active site [109] [111]. The mechanism of this transfer, while not fully understood, is a key step in ensuring fidelity.
- Excision of Incorrect Nucleotide: Once in the exonuclease active site, the 3'→5' exonuclease activity hydrolytically cleaves the phosphodiester bond, releasing the misincorporated nucleotide as a deoxynucleoside monophosphate [112].
- Transfer Back to Polymerase Site: Following excision, the now-corrected primer terminus is transferred back to the polymerase active site.
- Incorporation of Correct Nucleotide: With the proper geometry restored, the polymerase can then resume DNA synthesis by inserting the correct nucleotide [111].
This proofreading activity is highly discriminatory. A mismatched basepair at the primer terminus is the preferred substrate for the exonuclease activity over a correct basepair, ensuring that the system is activated primarily when an error occurs [113].
Quantitative Analysis of Proofreading Fidelity
The contribution of proofreading to overall replication fidelity is not merely qualitative; it has been rigorously quantified using advanced sequencing technologies. The following table compiles error rates for various DNA polymerases, demonstrating the profound impact of proofreading activity.
Table 1: Fidelity Measurements of DNA Polymerases by SMRT Sequencing [110]
| DNA Polymerase | Proofreading Activity | Substitution Rate (per base per doubling) | Accuracy (1 / Substitution Rate) | Fidelity Relative to Taq |
|---|---|---|---|---|
| Q5 High-Fidelity | Yes | 5.3 × 10^−7 | 1,870,763 | 280X |
| Phusion | Yes | 3.9 × 10^−6 | 255,118 | 39X |
| Deep Vent | Yes | 4.0 × 10^−6 | 251,129 | 44X |
| Pfu | Yes | 5.1 × 10^−6 | 195,275 | 30X |
| Taq | No | 1.5 × 10^−4 | 6,456 | 1X |
| Deep Vent (exo-) | No | 5.0 × 10^−4 | 2,020 | 0.3X |
The data unequivocally shows that polymerases possessing 3'→5' exonuclease activity (e.g., Q5, Phusion, Deep Vent) have error rates that are one to three orders of magnitude lower than Taq polymerase. The dramatic, 125-fold increase in the error rate observed for the exonuclease-deficient Deep Vent (exo-) mutant compared to its wild-type counterpart directly quantifies the contribution of proofreading to the fidelity of a single enzyme [110]. It is estimated that proofreading alone enhances the overall fidelity of DNA synthesis by a factor of 10^2 to 10^3 [109].
Experimental Paradigms for Measuring Fidelity
Understanding how polymerase fidelity is measured is crucial for interpreting the data in Table 1. Over time, methodologies have evolved to become more precise and high-throughput.
Table 2: Evolution of DNA Polymerase Fidelity Assays
| Assay Method | Key Feature | Principle | Limitations |
|---|---|---|---|
| Blue/White Screening (lacZα) [110] | Phenotypic selection | Errors in the lacZα gene disrupt function, leading to white instead of blue colonies in E. coli. | Indirect measurement; cannot resolve all single-base errors; relies on bacterial transformation efficiency. |
| Sanger Sequencing [110] | Direct sequencing of cloned products | PCR products are cloned, and individual clones are sequenced to identify mutations. | Lower throughput limits the total number of nucleotides sequenced, reducing statistical power for high-fidelity enzymes. |
| Barcoded Illumina Sequencing [110] | High-throughput | PCR products are fragmented, barcoded, and sequenced on Illumina platforms, generating millions of reads. | Lower threshold for error detection (~1 × 10^−6) is near the intrinsic error rate of high-fidelity polymerases. |
| PacBio SMRT Sequencing [110] | Single-molecule, real-time sequencing | Long reads allow direct sequencing of PCR products without an intermediary cloning step. Derives a highly accurate consensus sequence from multiple passes on the same molecule. | Very low background error rate (9.6 × 10^−8), making it the gold standard for quantifying ultra-high-fidelity polymerases. |
The following diagram outlines a generalized modern workflow for assessing polymerase fidelity using next-generation sequencing:
Diagram 2: A high-level workflow for determining DNA polymerase error rates using sequencing-based fidelity assays.
Beyond Intrinsic Proofreading: Extrinsic and Trans Proofreading
The classic model of proofreading is an intrinsic, cis activity—a polymerase correcting its own errors. However, recent research has revealed a more dynamic and collaborative system at the replication fork, particularly in eukaryotes.
In the established model of eukaryotic replication, DNA polymerase ε (Polε) primarily synthesizes the leading strand, while DNA polymerase δ (Polδ) synthesizes the lagging strand. Both are high-fidelity B-family polymerases with proofreading activity. A long-standing paradox, however, was that defects in Polδ proofreading had a much stronger mutator effect than analogous defects in Polε [114].
Elegant in vivo experiments in yeast, combining mutations in the polymerase (nucleotide selectivity) domain of one polymerase with proofreading defects in the other, provided a resolution. These studies demonstrated that Polδ can proofread errors made by Polε (a form of extrinsic or trans proofreading), but Polε cannot proofread errors made by Polδ [114]. This one-sided, extrinsic proofreading explains why Polδ contributes more significantly to overall mutation avoidance. The following diagram illustrates this sophisticated interplay:
Diagram 3: Model of extrinsic proofreading at the eukaryotic replication fork, where Polδ proofreads errors for Polε.
This concept extends even to polymerases entirely lacking an exonuclease domain. For example, DNA polymerase α (Polα), which initiates each Okazaki fragment on the lagging strand, is exonuclease-deficient. Evidence suggests that a separate, standalone nuclear 3'–5' exonuclease (exoN) can proofread errors made by Polα, thereby ensuring the fidelity of the initial steps of lagging strand synthesis [115].
The Scientist's Toolkit: Key Reagents for Fidelity Research
| Research Reagent / Material | Function in Proofreading Research |
|---|---|
| High-Fidelity Polymerases (e.g., Q5, Pfu) | Engineered or native polymerases with robust 3'→5' exonuclease activity; used as positive controls and for high-fidelity applications. |
| Exonuclease-Deficient Mutants (e.g., Deep Vent exo-) | Polymerases with inactivating mutations in the exonuclease active site; critical for isolating the contribution of proofreading to overall fidelity. |
| Defined Template DNA (e.g., lacZ plasmid) | A well-characterized DNA sequence used as a template in fidelity assays; any mutations in the amplified product are easily identified. |
| Phosphorothioate-Modified Primers | Primers with sulfur substituted for oxygen in the phosphate backbone; these bonds are resistant to exonuclease cleavage, allowing researchers to block proofreading activity for specific experiments [111]. |
| SMRT Sequencing (PacBio) | A next-generation sequencing technology capable of directly sequencing long PCR amplicons with a very low background error rate, enabling accurate quantification of polymerase error spectra [110]. |
The 3' to 5' exonuclease proofreading activity is a sophisticated and indispensable mechanism for maintaining the low mutation rates essential for life. It operates as a precise molecular editor, working in concert with the polymerase active site to ensure that the genetic code is replicated with extraordinary accuracy. The study of this activity has been profoundly informed by the limitations of one of molecular biology's most important tools, Taq polymerase. The drive to overcome the inherent error-proneness of Taq has not only led to the development of superior enzymes for biotechnology but has also deepened our fundamental understanding of DNA replication fidelity. From the intrinsic correction of mismatches to the recently elucidated complexities of trans proofreading between replicative polymerases, the study of exonucleolytic proofreading continues to reveal the elegant and layered strategies cells employ to safeguard their genetic information.
The discovery of Taq DNA polymerase from the thermophilic bacterium Thermus aquaticus, found in the hot springs of Yellowstone National Park, marked a revolutionary turning point for molecular biology [4] [24] [5]. Its thermostability enabled the automation of the Polymerase Chain Reaction (PCR), transforming DNA amplification from a laborious process into a routine laboratory technique [23] [17]. However, as PCR and other DNA amplification technologies have evolved to meet more demanding applications, the limitations of native Taq polymerase have become apparent. Its lack of 3'→5' exonuclease (proofreading) activity results in a relatively high error rate, making it unsuitable for applications requiring high fidelity, such as cloning and sequencing [74] [5]. Furthermore, its moderate processivity can hinder the amplification of long templates or sequences with complex secondary structures [74].
This landscape has driven the discovery and engineering of a diverse array of DNA polymerases, each designed to excel in specific parameters. From the hyperthermophilic archaea, enzymes like Pfu (Pyrococcus furiosus) and KOD (Thermococcus kodakarensis) were isolated, offering superior fidelity and thermostability [74] [116]. Through directed evolution and protein engineering, even more advanced polymerases have been developed, pushing the boundaries of speed, accuracy, and robustness [117]. This technical guide provides an in-depth comparison of these enzymes, benchmarking their performance across the critical metrics of processivity, speed, and accuracy to inform researchers and drug development professionals in their selection of the optimal polymerase for advanced applications.
Core Performance Metrics: Definitions and Experimental Determinations
A rigorous comparison of DNA polymerases requires a clear understanding of the key performance metrics and the experimental methods used to quantify them.
Fidelity: Quantifying Replication Accuracy
Fidelity refers to the accuracy of a DNA polymerase in replicating a DNA sequence, defined as the inverse of the error rate (number of misincorporated nucleotides per total nucleotides polymerized) [74]. The primary mechanism for high fidelity is proofreading activity, a 3'→5' exonuclease function that recognizes and excises misincorporated nucleotides [74] [116].
- Experimental Protocols for Fidelity Measurement:
- Colony Screening (lacZ Assay): A PCR-amplified fragment of the lacZ gene is cloned. Colonies with error-free PCR products remain blue, while those with mutations turn white, allowing for a calculated error rate [74].
- Sanger Sequencing: Cloned PCR fragments are sequenced to identify mutations introduced during amplification [74].
- Next-Generation Sequencing (NGS): PCR amplicons are directly sequenced, providing a comprehensive and direct measurement of the error rate across the entire product [74]. This is considered the gold standard.
Fidelity is often expressed relative to Taq DNA polymerase. While proofreading enzymes like Pfu naturally exhibit ~10x the fidelity of Taq, engineered "next-generation" high-fidelity polymerases can achieve fidelity >50–300x that of Taq [74].
Processivity and Speed: Efficiency of DNA Synthesis
Processivity is defined as the number of nucleotides incorporated by a DNA polymerase per single binding event [74]. A highly processive enzyme remains bound to the DNA template for longer, synthesizing more product without dissociating. This is crucial for amplifying long targets, GC-rich sequences, and in the presence of PCR inhibitors [74] [116].
Speed, or elongation rate, is the number of nucleotides synthesized per second per enzyme molecule. This directly impacts PCR cycling times [5] [116].
- Experimental Determination: Processivity and speed can be assessed by measuring the successful amplification of DNA templates of varying lengths and complexities. A polymerase with high processivity will efficiently generate long amplicons (>10 kb) that low-processivity enzymes cannot. Elongation rate is measured by quantifying the time required to synthesize a defined length of DNA under optimal conditions [116].
Thermostability: Withstanding Denaturing Conditions
Thermostability is a measure of how long a polymerase retains its activity and structure at high temperatures. This is vital for PCR, where repeated exposure to temperatures >90°C is required for DNA denaturation [74]. The half-life of an enzyme at a given temperature (e.g., 97.5°C) is a key quantitative measure of its thermostability [5].
Comparative Performance Analysis of Major Polymerases
The table below provides a quantitative and qualitative comparison of key DNA polymerases, highlighting their performance across the critical metrics.
Table 1: Benchmarking DNA Polymerase Performance Characteristics
| Polymerase | Source Organism | Fidelity (Relative to Taq) | Proofreading Activity | Processivity (nucleotides/binding event) | Elongation Rate (nt/s) | Optimal Extension Temperature | Key Characteristics and Applications |
|---|---|---|---|---|---|---|---|
| Taq | Thermus aquaticus | 1x | No | Moderate | ~60-150 [5] [116] | 72°C [5] | Standard PCR, genotyping; low cost; has A-overhang for TA cloning [5]. |
| Pfu | Pyrococcus furiosus | ~10x | Yes (3'→5' exonuclease) | Low (<20) [116] | Slow [74] | 75°C [74] | High-fidelity PCR, cloning; slower and less processive than KOD [74] [116]. |
| KOD | Thermococcus kodakarensis | ~10x [116] | Yes (3'→5' exonuclease) | Very High (10-15x Pfu) [116] | 100-130 [116] | 75°C [116] | Fast, high-fidelity, long-range PCR; amplifies GC-rich targets; resistant to inhibitors [116]. |
| Engineered High-Fidelity | Directed Evolution | >50-300x [74] | Yes (Strong) | Variable (often engineered for high processivity) | Variable (often optimized) | 72-75°C | Cloning, sequencing, mutagenesis; engineered for exceptional accuracy [74]. |
| KOD exo(-) | Engineered variant of KOD | Lower than KOD | No (3'→5' exonuclease deficient) | Very High | Very High | 75°C | Real-time PCR, fast PCR; retains high processivity without proofreading [116]. |
Analysis of Performance Trade-offs and Synergies
The data in Table 1 reveals inherent trade-offs and synergies between enzyme properties:
- The Fidelity-Speed Trade-off: Native proofreading enzymes like Pfu traditionally sacrificed speed for accuracy. The discovery of KOD polymerase demonstrated that this trade-off is not absolute, as KOD combines high fidelity with a rapid elongation rate [116].
- Engineering Synergies: Protein engineering has successfully broken these trade-offs. For example, fusion proteins incorporating a DNA-binding domain can enhance processivity 2- to 5-fold without compromising other activities [74]. Furthermore, the KOD exo(-) variant was created to remove proofreading activity for applications where extreme speed is more critical than ultimate fidelity [116].
- Uracil Tolerance: A significant limitation of many archaeal proofreading polymerases (including Pfu and native KOD) is their inability to amplify uracil-containing templates, as they stall at these sites. This is a problem for techniques like dUTP/UDG carryover prevention and bisulfite sequencing. Engineered, uracil-tolerant KOD variants overcome this limitation, expanding their application range [116].
Advanced Experimental Workflows
The following diagrams illustrate core experimental workflows for assessing polymerase performance and applying engineered enzymes in advanced research.
Workflow for Determining Polymerase Fidelity
Diagram 1: Polymerase Fidelity Assay Workflow. The process begins with PCR amplification of a target gene using the polymerase under evaluation. The resulting amplicons are purified, cloned into a vector, and transformed into bacteria. Individual colonies are picked, and the plasmid DNA is sequenced (ideally via NGS). The sequence data is compared to the known template sequence to calculate the error rate and, consequently, the fidelity of the polymerase [74].
Application: One-Pot miRNA Multiplex RT-qPCR
Advanced polymerase engineering enables novel assay configurations. The RT-HOS (Reverse Transcription-Hairpin Occlusion System) method allows for one-pot, one-step multiplex miRNA detection, driven by either Taq or high-fidelity polymerases [43].
Diagram 2: RT-HOS miRNA Detection Principle. The system uses an RT fluorescent primer and a complementary hairpin quencher. Upon hybridization to the miRNA target, reverse transcription occurs. The polymerase's strand-displacement activity then releases the hairpin quencher, separating the fluorophore from the quencher and generating a fluorescent signal that can be monitored in real-time during qPCR [43]. This method demonstrates high specificity and a wide linear dynamic range, showcasing the practical application of engineered polymerase properties in diagnostics.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of advanced PCR-based experiments relies on a suite of specialized reagents and materials.
Table 2: Key Research Reagent Solutions for Polymerase Applications
| Reagent/Material | Function and Importance in Polymerase Applications |
|---|---|
| Hot-Start Taq DNA Polymerase | Antibody- or chemically-modified enzyme inhibited at room temperature. Critical for preventing nonspecific amplification and primer-dimer formation during reaction setup, greatly improving specificity and yield in PCR and high-throughput applications [74]. |
| High-Fidelity Master Mix | Optimized buffer solutions containing a proofreading DNA polymerase, dNTPs, and Mg²⁺. Critical for applications requiring high accuracy like cloning and site-directed mutagenesis, ensuring low error rates in the amplified product [74]. |
| dUTP/UDG Decontamination System | Incorporation of dUTP into PCR products followed by Uracil-DNA Glycosylase (UDG) treatment. Critical for preventing carryover contamination between PCR runs; requires use of uracil-tolerant polymerases (e.g., engineered KOD) as standard archaeal polymerases stall at uracil [116]. |
| GC-Rich Enhancers | Chemical additives like DMSO or betaine that reduce secondary structure formation. Critical for amplifying difficult templates with high GC content, often used in conjunction with highly processive polymerases like KOD for optimal results [74] [116]. |
| Autoinduction Media | Chemically defined growth media containing glucose, glycerol, and lactose for recombinant protein expression. Critical for cost-effective, high-yield production of recombinant polymerases like Taq in E. coli fermenters, eliminating the need for expensive IPTG inducer [118]. |
The journey from the discovery of Taq polymerase in the hot springs of Yellowstone to the modern suite of engineered enzymes exemplifies how fundamental research drives technological innovation [24]. While Taq polymerase remains a workhorse for routine PCR, the demands of modern molecular biology, drug development, and clinical diagnostics require tools with specialized capabilities. The benchmarking data presented here clearly differentiates polymerases like the high-fidelity, high-processivity KOD and engineered enzymes from the foundational Taq.
Understanding the quantitative and functional relationships between processivity, speed, and accuracy allows researchers to make informed decisions. The choice of polymerase is no longer a compromise but a strategic selection based on application-specific needs, whether for fast diagnostics, ultra-accurate sequencing library preparation, or challenging long-range PCR. As enzyme engineering continues to advance, the performance boundaries of these critical molecular tools will continue to expand, enabling the next generation of genetic analysis and therapeutic discovery.
The discovery of DNA polymerase from Thermus aquaticus, a thermophilic bacterium isolated from the thermal springs of Yellowstone National Park, revolutionized molecular biology by providing a thermostable enzyme essential for the polymerase chain reaction (PCR) [20] [12]. Unlike the Klenow fragment of Escherichia coli DNA Polymerase I originally used in PCR, which required replenishment after each denaturation cycle, Taq polymerase's stability at high temperatures allowed for automated, efficient DNA amplification [20] [12]. This robustness made Taq the cornerstone enzyme for routine PCR, forming the foundation for countless applications in research, diagnostics, and biotechnology.
However, as scientific inquiry progressed, the limitations of Taq polymerase, particularly its relatively low replication fidelity, became apparent for applications demanding high sequence accuracy. This spurred the search for and engineering of a new class of enzymes: high-fidelity DNA polymerases. These enzymes, often derived from hyperthermophilic archaea such as Pyrococcus furiosus (source of Pfu polymerase), possess intrinsic proofreading capabilities that significantly reduce error rates during amplification [12] [73]. The evolution from Taq to high-fidelity enzymes represents a critical advancement in molecular biology, enabling precise genetic manipulation essential for modern genomics, cloning, and therapeutic development. This guide provides a structured framework for researchers to select the appropriate DNA polymerase based on the specific requirements of their experimental goals.
Fundamental Properties and Mechanisms of DNA Polymerases
Key Characteristics for Selection
The choice between Taq and a high-fidelity polymerase hinges on understanding their distinct biochemical properties, which directly impact PCR outcomes. The table below summarizes the core differences.
Table 1: Fundamental Properties of Taq vs. High-Fidelity DNA Polymerases
| Property | Taq & Family A Polymerases | High-Fidelity & Family B Polymerases |
|---|---|---|
| 3'→5' Exonuclease (Proofreading) | No [119] [105] | Yes [119] [105] |
| 5'→3' Exonuclease Activity | Yes [120] [119] | No [120] [105] |
| Fidelity (Error Rate) | ~1 × 10⁻⁵ errors/base [73] [105] | ~1 × 10⁻⁶ errors/base [73] [105] |
| Fidelity Relative to Taq | 1x (Baseline) [120] [121] | 6x to >280x higher [120] [121] [73] |
| Extension Rate | High (~150 nucleotides/sec) [105] | Slower (~25 nucleotides/sec) [105] |
| Resulting PCR Ends | 3´ 'A-overhangs' [120] [119] | Blunt ends [120] [119] |
| Primary Applications | Routine PCR, genotyping, qPCR [120] [122] | Cloning, sequencing, mutagenesis [120] [122] [105] |
The Mechanism of Fidelity: Proofreading Explained
Polymerase fidelity refers to the accuracy with which a DNA polymerase copies a template sequence, measured as the number of errors per base incorporated per doubling event [121]. Taq polymerase lacks 3'→5' exonuclease (proofreading) activity, meaning it cannot remove a misincorporated nucleotide from the growing 3' end of the DNA chain. Its accuracy relies solely on the geometry of its active site to select the correct nucleotide, resulting in a higher intrinsic error rate [121] [20].
In contrast, high-fidelity enzymes like Q5 and Pfu possess a dedicated proofreading domain. When a mismatched nucleotide is incorporated, it causes a perturbation that slows polymerization. This delay allows the polymerase to backtrack, moving the incorrect nucleotide into the exonuclease site where it is excised. The correct nucleotide is then inserted, and synthesis resumes [121]. This mechanism provides a dramatic increase in replication accuracy, as shown by the 125-fold decrease in error rate observed when comparing the proofreading-deficient Deep Vent (exo-) to the proofreading-proficient Deep Vent polymerase [121].
Quantitative Fidelity Comparison of DNA Polymerases
The fidelity of DNA polymerases is quantified using advanced sequencing methods. Next-generation sequencing platforms, particularly PacBio Single-Molecule Real-Time (SMRT) sequencing, have enabled highly accurate measurements by sequencing the same molecule multiple times to generate a consensus sequence with a very low background error rate (~9.6 × 10⁻⁸ errors/base) [121]. The following data, largely derived from such methods, allows for direct comparison.
Table 2: Experimentally Determined Error Rates of Common DNA Polymerases [121]
| DNA Polymerase | Substitution Rate (per base per doubling) | Accuracy (1 base error per X bases) | Fidelity Relative to Taq |
|---|---|---|---|
| Taq | 1.5 × 10⁻⁴ | 6,456 | 1x |
| Deep Vent (exo-) | 5.0 × 10⁻⁴ | 2,020 | 0.3x |
| KOD | 1.2 × 10⁻⁵ | 82,303 | 12x |
| Pfu | 5.1 × 10⁻⁶ | 195,275 | 30x |
| Deep Vent | 4.0 × 10⁻⁶ | 251,129 | 44x |
| Phusion | 3.9 × 10⁻⁶ | 255,118 | 39x |
| Q5 | 5.3 × 10⁻⁷ | 1,870,763 | 280x |
Other studies using direct sequencing of cloned PCR products have confirmed this hierarchy, reporting error rates for Taq in the 10⁻⁵ range, while high-fidelity enzymes like Pfu, Pwo, and Phusion exhibit error rates in the 10⁻⁶ range, representing a more than 10-fold improvement [73].
Application-Based Selection Guide
Choosing the correct polymerase is paramount to experimental success. The following section provides a detailed guide, with decision workflows and reagent recommendations, for common application scenarios.
When to Use Taq DNA Polymerase
Taq polymerase is the ideal choice for applications where the primary goal is to detect the presence, size, or approximate quantity of a DNA fragment, and where perfect sequence integrity is not critical [122].
- Routine PCR and Genotyping: For colony PCR, genotype screening, or checking the size of an insert, Taq's speed and reliability are sufficient [120] [119].
- Quantitative PCR (qPCR): Taq's inherent 5'→3' exonuclease activity enables its use in hydrolysis probe-based (TaqMan) qPCR assays, where signal generation is coupled to primer extension [20] [119].
- Rapid Amplification: For simple, fast PCR where turnaround time is a priority, Taq's high extension rate is advantageous.
A critical technical consideration for standard Taq is the risk of nonspecific amplification during room-temperature reaction setup. To mitigate this, Hot-Start Taq versions are recommended. These are engineered to remain inactive until the first high-temperature denaturation step, preventing primer-dimer formation and off-target synthesis [20] [119] [105]. This is achieved through antibody-mediated inhibition or chemical modification of the enzyme [105].
When to Choose a High-Fidelity Enzyme
High-fidelity polymerases are non-negotiable for any application where the DNA sequence of the amplified product must be identical to the original template.
- Cloning and Gene Expression: Errors in the cloned insert can lead to non-functional proteins, invalidating functional studies or rendering a protein expression clone useless [120] [121].
- Site-Directed Mutagenesis: These techniques rely on introducing specific, planned mutations. A high-error background from the polymerase can obscure results and create unintended functional changes [120] [119].
- Next-Generation Sequencing (NGS) Library Prep: Errors introduced during amplification for library construction can be misinterpreted as genetic variants (e.g., SNPs), leading to false-positive results in variant calling [120] [122].
- SNP Analysis and Sequencing: Any direct sequencing of PCR products requires high fidelity to ensure the detected sequence variants are real and not polymerase-induced artifacts [120].
Specialized Polymerases and Formats
- Long-Range PCR: Amplifying fragments >5 kb, and especially >20 kb, requires a blend of high fidelity and high processivity. Specialized polymerases or mixes (e.g., LongAmp Taq, GoTaq Long PCR Master Mix) often combine a proofreading enzyme with a highly processive one to achieve both accuracy and the ability to synthesize long stretches of DNA [120] [119].
- Rapid or High-Throughput Workflows: Pre-mixed Master Mixes are available for both Taq and high-fidelity enzymes. These 2X concentrates contain the polymerase, dNTPs, and optimized buffer, simplifying setup, reducing pipetting steps, and improving reproducibility [120] [119] [122].
The Scientist's Toolkit: Essential Reagent Solutions
Table 3: Key Research Reagents for PCR-Based Workflows
| Reagent / Solution | Function / Description | Example Use-Cases |
|---|---|---|
| Standard Taq Polymerase | Core enzyme for basic amplification; lacks proofreading but is robust and fast. | Colony PCR, educational labs, genotyping, presence/absence checks. |
| Hot-Start Taq | Taq polymerase chemically or antibody-inactivated until initial denaturation. | Multiplex PCR, high-sensitivity assays, any reaction where specificity is problematic. |
| High-Fidelity Polymerase (e.g., Q5, Pfu, Phusion) | Engineered or native polymerase with 3'→5' proofreading exonuclease activity. | Cloning, NGS, mutagenesis, any application where sequence integrity is paramount. |
| Long-Range PCR Mix | Blended enzyme system optimized for high processivity and fidelity over long templates. | Amplification of large genomic regions, full-length cDNA amplification. |
| dNTP Mix | Equimolar solution of deoxyribonucleotide triphosphates (dATP, dCTP, dGTP, dTTP). | Essential building blocks for DNA synthesis in all PCR reactions. |
| MgCl₂ / MgSO₄ Solution | Divalent cation cofactor essential for polymerase activity; concentration is a key optimization parameter. | Adjusting stringency and yield in PCR; typically included with polymerase. |
| Optimized Reaction Buffer | Buffering agent (e.g., Tris-HCl), salts (e.g., KCl), and sometimes enhancers to stabilize pH and reaction components. | Providing the correct chemical environment for efficient amplification. |
The journey from the discovery of Taq polymerase in the thermal springs of Yellowstone to the sophisticated high-fidelity enzymes of today mirrors the evolving demands of precision biology. The choice between Taq and a high-fidelity enzyme is not a matter of which is superior in absolute terms, but which is optimal for the specific scientific question at hand.
For detection, sizing, and quantification where sequence perfection is secondary, Taq polymerase remains an unparalleled and cost-effective tool. However, for the rigorous demands of modern molecular cloning, next-generation sequencing, and functional genomics—where every base counts—the investment in a high-fidelity enzyme is indispensable. By applying the structured framework and experimental considerations outlined in this guide, researchers and drug developers can make informed decisions, ensuring the integrity and success of their genetic analyses.
The discovery of Taq DNA polymerase from the thermophilic bacterium Thermus aquaticus marked a revolutionary turning point in molecular biology, enabling the automation and widespread adoption of the polymerase chain reaction (PCR) [20] [12]. This thermostable enzyme, isolated from the thermal springs of Yellowstone National Park, could withstand the high temperatures required for DNA denaturation, eliminating the need to add fresh enzyme after each cycle [20] [5]. Its implementation transformed PCR from a cumbersome technique into a robust, high-throughput method fundamental to genetic research, clinical diagnostics, and forensics [5]. However, as applications diversified, the inherent limitations of wild-type Taq polymerase became apparent. Its lack of 3′ to 5′ exonuclease (proofreading) activity results in a relatively low replication fidelity, with an error rate of approximately 1 in 10,000 nucleotides [20] [123] [5]. Furthermore, its moderate processivity—adding about 50 nucleotides per binding event—and sensitivity to common PCR inhibitors constrained its utility for complex applications [123] [9].
These limitations spurred a wave of innovation aimed at creating next-generation enzymes with enhanced capabilities. Two primary strategies emerged: the creation of optimized enzyme blends and the rational design of chimeric DNA polymerases [124] [123] [125]. Enzyme blends, such as mixtures of Taq with a proofreading polymerase like Deep Vent, combine the strengths of distinct native enzymes in a single tube [123]. In parallel, advanced protein engineering techniques have enabled the creation of novel chimeric polymerases. These chimera are constructed by fusing amino acid sequences from different proteins into a single DNA polymerase, thereby combining beneficial properties such as high fidelity, thermal stability, and processivity that are not found together in nature [124] [125]. This whitepaper explores the design, creation, and application of these advanced enzymatic tools, framing them as the direct descendants of the seminal discovery of Taq polymerase.
The Building Blocks: Key Enzymatic Properties and the Structure-Function Relationship
To appreciate the engineering of chimeric polymerases, one must first understand the core enzymatic properties and the structural domains that govern them. DNA polymerases are often described as having a right-handed structure composed of palm, thumb, and fingers domains [124] [125]. The palm domain contains the active site for catalysis, the thumb domain binds the replicated DNA, and the fingers domain interacts with incoming nucleotides [125]. Additionally, some polymerases possess specialized exonuclease domains; a 5′ to 3′ exonuclease domain is involved in nick translation, while a 3′ to 5′ exonuclease domain provides proofreading activity [124] [5].
Table 1: Key Enzymatic Properties of DNA Polymerases and Their Structural Determinants
| Enzymatic Property | Description | Domain(s) Related |
|---|---|---|
| Activity | The general rate of elongating new DNA strands from 5’ to 3’ | Palm |
| Processivity | Number of nucleotides added per single binding event | Palm, Thumb, Fingers |
| Fidelity | Accuracy of base insertion | Fingers |
| Thermal Stability | Ability to resist high temperature without losing activity | All domains |
| Proofreading Activity | 3’ to 5’ exonuclease activity that excises misincorporated nucleotides | Exonuclease domain |
| Inhibitor Tolerance | Ability to remain functional against inhibitors (e.g., dyes, ions) | All domains |
These properties are critically important for specific applications. Fidelity, for example, is paramount in sequencing and cloning, where errors can lead to misinterpretations or faulty constructs. Taq polymerase's fidelity is considered low, while high-fidelity enzymes like Q5 DNA Polymerase demonstrate an error rate 280-fold lower than Taq [123]. Processivity determines how efficiently a polymerase can amplify long DNA fragments; the low processivity of Taq's Stoffel fragment (~5-10 nucleotides/binding event) limits its use for long-range PCR, a task where more processive enzymes excel [9]. Finally, thermostability is a prerequisite for modern PCR, but the degree of stability varies; Taq has a half-life of ~40 minutes at 95°C, whereas polymerases from hyperthermophiles like Pyrococcus furiosus (Pfu) are significantly more stable [124] [12].
Engineering Superior Enzymes: Strategies for Creating Chimeric DNA Polymerases
The rational design of chimeric DNA polymerases employs a Design-Build-Test-Learn (DBTL) cycle to systematically create and optimize new enzymes [124] [125]. This process begins with the selection of a parent polymerase and the identification of domains associated with desired traits, followed by the design and construction of the chimeric gene. The resulting enzyme is then expressed, purified, and rigorously tested, with performance data feeding back into the cycle for further refinement [124]. The two most prevalent design strategies are homologous domain exchange and the integration of novel functional domains.
Homologous Domain Exchange
This strategy capitalizes on the significant structural similarity among DNA polymerases, particularly in the conserved palm domain. It involves replacing a specific domain in one polymerase with the homologous domain from another polymerase to transfer a desirable characteristic [124] [125].
- Adding Proofreading to Taq: A segment of Taq polymerase (amino acids 292-423) was replaced with the corresponding 3’-5’ exonuclease domain from E. coli Pol I, successfully endowing the chimeric Taq with proofreading capability [124] [125].
- Combining Strengths of Two Polymerases: A chimeric Tth-Taq DNA polymerase was created by fusing the N-terminal of Thermus thermophilus polymerase with the C-terminal of Taq. This enzyme exhibited a fivefold increase in activity over Taq and much greater amplification specificity than Tth, effectively combining the advantages of both parents [124] [125].
- Engineering High Fidelity: By replacing the finger and palm domains of KOD DNA polymerase with those from Pfu polymerase, researchers created a chimeric enzyme that retained KOD's high processivity and thermal stability but also acquired the high fidelity of Pfu [124].
Integration of Functional Domains
This approach involves fusing an entire additional protein or functional domain to a DNA polymerase to confer a completely new function or enhance an existing one [124] [123].
- Enhancing Processivity: A highly effective method is fusing a DNA binding protein, such as Sso7d or helix-hairpin-helix (HhH) motifs, to a polymerase. This fusion increases the enzyme's affinity for DNA, significantly boosting its processivity and enabling faster amplification of long templates [124] [123]. This strategy is the foundation of commercial high-performance polymerases like Q5 and Phusion [123].
- Creating Novel Reverse Transcriptases: A chimeric reverse transcriptase was constructed by fusing the 5’-3’ nuclease domain of Thermus species Z05 polymerase with the polymerase domains of Thermotoga maritima (Tma) polymerase. This single enzyme could perform reverse transcription and DNA amplification, simplifying RT-PCR for challenging templates [124].
The following diagram illustrates the logical workflow and primary strategies used in the rational design of chimeric DNA polymerases:
Experimental Spotlight: Detailed Protocol for Engineering a Novel Reverse Transcriptase-Active Taq Variant
A recent study exemplifies the power of combinatorial engineering to create multifunctional chimeric polymerases. The goal was to develop novel Taq polymerase variants capable of catalyzing both reverse transcription (RT) and DNA amplification in a single tube without needing a separate viral reverse transcriptase [108]. This section details the key experimental workflow and methodology.
Experimental Workflow
The overall process, from library construction to final application, is visualized below:
Detailed Methodology
Library Design and Construction: Researchers started with two independently discovered mutation pools known to enhance the RT activity of Taq's truncated form, KlenTaq: RT-KTq (4 mutations: L459M, S515R, I638F, M747K) and Mut_RT (5 mutations: N483K, E507K, K540Y, V586G, I614K) [108]. A comprehensive combinatorial library was designed in the full-length Taq backbone, including all possible combinations of these mutations (a total of 256 variants). The gene library was synthesized, cloned into an expression vector, and transformed into E. coli [108].
High-Throughput Screening: A total of 2,660 individual colonies were picked to ensure >99% coverage of the library. Expression cultures were grown, and the cells were lysed. The lysates were heat-inactivated (a crucial step to denature E. coli host proteins without affecting the thermostable Taq variants) and used directly in the screening assay [108].
Functional Screening via Real-Time RT-PCR: The library screens employed a real-time RT-PCR assay previously established for SARS-CoV-2 detection [108]. This assay used either the intercalating dye SYBR Green I or hydrolytic TaqMan probes to simultaneously evaluate the RT and DNA amplification capabilities of each variant directly from RNA templates. Promising candidates demonstrating robust activity in both steps were selected.
Validation and Application: The lead chimeric variants were further validated and shown to perform quantitative multiplex RT-PCR, simultaneously detecting up to four different RNA targets in a single tube with a sensitivity of 20 copies, using a single enzyme and without requiring manganese ions [108].
The Scientist's Toolkit: Essential Reagents for Polymerase Engineering
The research and development of engineered polymerases rely on a suite of specialized reagents and tools. The following table details key components essential for this field.
Table 2: Essential Research Reagents for Polymerase Engineering and Analysis
| Reagent / Tool | Function and Utility in Polymerase Engineering |
|---|---|
| High-Copy Number Expression Vectors | Plasmids like pD451-SR_Taqpol (PCN ~78/cell) enable high-yield recombinant production of polymerase variants in E. coli [126]. |
| Autoinduction Media | Chemically defined media using lactose as a low-cost inducer facilitates high-density fermentation and scalable production without monitoring [126]. |
| Thermostable DNA Polymerases | Native enzymes from thermophiles (e.g., Taq, Pfu, Tth, KOD) serve as the foundational scaffolds and domain donors for chimeric designs [124] [125] [12]. |
| Real-Time qPCR Instruments | Critical for high-throughput screening of mutant libraries for desired functions (e.g., RT activity) and for assessing performance metrics like sensitivity and multiplexing capability [108] [126]. |
| Fluorogenic Hydrolysis Probes (TaqMan) | Used in real-time assays to confirm the 5' nuclease activity of engineered polymerases and to enable specific target quantification in multiplexed applications [20] [108]. |
The journey from the discovery of wild-type Taq polymerase to the rational design of sophisticated chimeric enzymes illustrates a paradigm shift in biotechnology. The initial focus on utilizing natural enzymes has evolved into a precision engineering discipline, where polymerases are tailored to overcome specific application bottlenecks. By employing strategies like homologous domain exchange and functional domain integration, scientists can now generate novel enzymes that combine the thermal stability of archaeal polymerases, the processivity of bacterial enzymes, and the fidelity of proofreading polymerases, all within a single polypeptide chain [124] [125].
The creation of a single enzyme capable of quantitative, multiplex reverse transcription PCR exemplifies the power of this approach, promising simplified workflows, reduced costs, and enhanced robustness for molecular diagnostics [108]. As the DBTL cycle continues, fueled by deeper structural insights and more advanced screening technologies, the next generation of engineered polymerases will further expand the boundaries of what is possible in genomics, synthetic biology, and molecular medicine, solidifying the legacy of Taq polymerase as the foundation for decades of innovation.
Conclusion
The discovery of Taq polymerase stands as a paradigm of how fundamental, curiosity-driven research on extremophiles can yield tools that transform science and society. Its integration into PCR democratized DNA manipulation, creating foundational capabilities for modern molecular biology, clinical diagnostics, and drug development. While Taq polymerase remains the robust, versatile workhorse for routine amplification, the evolution of high-fidelity proofreading enzymes addresses its key limitation of replication accuracy for advanced applications. The future of DNA polymerase technology lies in continued protein engineering, creating next-generation enzymes with enhanced speed, tolerance to inhibitors, and ultra-high fidelity. For researchers in biomedicine and drug development, a deep understanding of Taq's properties, optimal use cases, and limitations, as detailed in this review, is essential for designing robust experiments and driving innovation in genomics, personalized medicine, and molecular diagnostics.
References
- 1. The Taq behind PCR: The Story of a Heat-tolerant Enzyme [https://bitesizebio.com/1953/the-taq-behind-pcr/]
- 2. Thomas D. Brock [https://en.wikipedia.org/wiki/Thomas_D._Brock]
- 3. Discovering Life in Yellowstone Where Nobody Thought it ... [https://www.nps.gov/articles/thermophile-yell.htm]
- 4. The Extremophile Protein That Revolutionized DNA Sequencing [https://astrobiology.com/2025/01/the-extremophile-protein-that-revolutionized-dna-sequencing.html]
- 5. Taq polymerase [https://en.wikipedia.org/wiki/Taq_polymerase]
- 6. The polymerase chain reaction: An overview and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3327995/]
- 7. Fine Structure of Thermus aquaticus, an Extreme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC248237/]
- 8. Thermus aquaticus - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/thermus-aquaticus]
- 9. Taq Polymerase - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/taq-polymerase]
- 10. The History of PCR [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/history-pcr.html]
- 11. The World's Second Best Molecule of the Year [https://www.mcgill.ca/oss/article/covid-19-general-science/worlds-second-best-molecule-year]
- 12. DNA polymerases as useful reagents for biotechnology [https://pmc.ncbi.nlm.nih.gov/articles/PMC4148896/]
- 13. The scientist who made PCR possible - Research Communities [https://communities.springernature.com/posts/the-scientist-who-made-pcr-possible]
- 14. Decades on, bacterium's discovery feted as paragon of basic ... [https://news.wisc.edu/decades-on-bacteriums-discovery-feted-as-paragon-of-basic-science/]
- 15. Basic science: a key ingredient in innovation [https://www.drugdiscoverynews.com/basic-science-a-key-ingredient-in-innovation-15756]
- 16. Bio Break: The Story of PCR and Taq Polymerase [https://starfishmedical.com/resource/bio-break-the-story-of-pcr-and-taq-polymerase/]
- 17. 5 reasons why the story behind Taq polymerase is better ... [https://solmeglas.com/5-reasons-why-the-story-behind-taq-polymerase-is-better-than-fiction/]
- 18. Kary Mullis [https://en.wikipedia.org/wiki/Kary_Mullis]
- 19. The Discovery of PCR: ProCuRement of Divine Power - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4501591/]
- 20. Taq Polymerase: what is it and its applications - LGC [https://blog.biosearchtech.com/thebiosearchtechblog/bid/48174/taq-facts]
- 21. Quantitative PCR Basics [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/quantitative-pcr?srsltid=AfmBOoqEQtWIXZyc34jyNOTxGoASlbxqQJiMI07BXFWRnyKvFBPx-PhZ]
- 22. The Nobel Prize in Chemistry 1993 - NobelPrize.org [https://www.nobelprize.org/prizes/chemistry/1993/summary/]
- 23. From discovery to innovation in physiological research - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11868022/]
- 24. Science Is Easy to Make Fun of and Hard to Live Without [https://www.atkinson.cornell.edu/blog/science-is-easy-to-make-fun-of-and-hard-to-live-without/]
- 25. Polymerase Chain Reaction (PCR) - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK589663/]
- 26. The Importance of Taq Polymerase in PCR [https://aelabgroup.com/the-importance-of-taq-polymerase-in-pcr/]
- 27. One enzyme reverse transcription qPCR using Taq DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7757722/]
- 28. Anatomy of a Polymerase - How Function and Structure are ... [https://www.neb.com/en-us/tools-and-resources/feature-articles/anatomy-of-a-polymerase-how-structure-effects-function?srsltid=AfmBOooIfK26Zw24mZpukFRvi5SmSZ8msYLEUSiRJrDOL7sVy87ACIKk]
- 29. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOorCffwxyHdVOVbImcM0kGOoQSV7bJAxZaqKr0CyYxGS4b_UQhbk]
- 30. Kary B. Mullis – Facts - NobelPrize.org [https://www.nobelprize.org/prizes/chemistry/1993/mullis/facts/]
- 31. DNA Polymerase Market Size Forecasts USD 420 Million ... [https://www.biospace.com/press-releases/dna-polymerase-market-size-forecasts-usd-420-million-in-2025]
- 32. DNA Polymerase Market Growth, Trends & Forecast 2035 [https://www.futuremarketinsights.com/reports/dna-polymerase-market]
- 33. DNA Polymerase Market Size, Share & Growth Report 2035 [https://www.researchnester.com/reports/dna-polymerase-market/6423]
- 34. PCR Basics | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-basics.html]
- 35. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOorWYfl73M0RG9xbtFXUdCk9W5Bbcq_i4H9tSkprSVh00V7CkXv-]
- 36. PCR Setup—Six Critical Components to Consider [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-component-considerations.html]
- 37. Frontiers | A Single Amino Acid Change to Taq DNA Polymerase ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2020.553474/full]
- 38. Comparison of Analytic Methods for Quantitative Real-Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4642823/]
- 39. Optimization of the real-time PCR platform method using ... [https://www.nature.com/articles/s41598-025-16972-9]
- 40. Chain Reaction: A Toolbox for Molecular Polymerase Discovery [https://pubmed.ncbi.nlm.nih.gov/39955471/]
- 41. Persistent RNA SARS-CoV-2 Detection in a HIV-Infected ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9222592/]
- 42. TaqPath COVID-19 Diagnostic PCR Kit [https://www.thermofisher.com/us/en/home/clinical/clinical-genomics/pathogen-detection-solutions/real-time-pcr-respiratory-tract-microbiota-detection/clinical-products/taqpath-covid-19-ivd.html]
- 43. A novel high-fidelity DNA polymerase-driven or Taq ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400525013358]
- 44. Advances and Challenges in SARS-CoV-2 Detection [https://www.mdpi.com/2075-4418/14/5/519]
- 45. Advancing quantitative PCR with color cycle multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417387/]
- 46. How To Interpret RT-qPCR Results [https://www.goldbio.com/blogs/articles/how-to-interpret-rt-qpcr-results?srsltid=AfmBOoq3z4nvE02BiGTZMSj-U4P7i7hAaQ2HpfA862LoG9r5w9i6KAFu]
- 47. Integrative machine learning and RT-qPCR analysis ... [https://pubmed.ncbi.nlm.nih.gov/40833705/]
- 48. Biomarker discovery for early breast cancer diagnosis ... [https://www.sciencedirect.com/science/article/pii/S0010482525009357]
- 49. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOoofcKeOwwuZBFWSaSgU1q1WQsnXYYbTl-thU-vDGfTnkmiM2xSc]
- 50. DNA Polymerase, Taq - Worthington Enzyme Manual [https://www.worthington-biochem.com/products/dna-polymerase-taq/manual]
- 51. Single-molecule Taq DNA polymerase dynamics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8916733/]
- 52. DNA Profiling in Forensic Science: A Review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8635824/]
- 53. Discovery, development, and current applications of DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1200713/]
- 54. DNA Evidence: How It's Done [https://www.forensicsciencesimplified.org/dna/how.html]
- 55. Universal TA cloning [https://pubmed.ncbi.nlm.nih.gov/11464915/]
- 56. TA cloning [https://en.wikipedia.org/wiki/TA_cloning]
- 57. TA Cloning - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/ta-cloning]
- 58. TA cloning protocol [https://science.umd.edu/biology/cichlid/protocols/Basic/Cltaclone.html]
- 59. DNA Polymerase Market Size and Forecast 2025 to 2034 [https://www.precedenceresearch.com/dna-polymerase-market]
- 60. TaqMan [https://en.wikipedia.org/wiki/TaqMan]
- 61. Advances in quantitative PCR technology: 5' nuclease ... [https://pubmed.ncbi.nlm.nih.gov/9503586/]
- 62. TaqMan Assays - 5 Interesting Facts - Taq Polymerase [https://www.thermofisher.com/blog/behindthebench/5-interesting-facts-about-taqman-assays/]
- 63. A 5′ Nuclease PCR (TaqMan) High-Throughput Assay for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC86956/]
- 64. TaqMan - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/taqman]
- 65. How TaqMan Assays Work [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/how-taqman-assays-work.html]
- 66. How TaqMan Assays Work [https://www.thermofisher.com/blog/behindthebench/how-taqman-assays-work/]
- 67. TaqMan Real-Time PCR Assays [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays.html]
- 68. The Discovery of Thermus aquaticus [https://resources.biomol.com/biomol-blog/the-discovery-of-thermus-aquaticus]
- 69. Anatomy of a Polymerase - How Function and Structure are ... [https://www.neb.com/en-us/tools-and-resources/feature-articles/anatomy-of-a-polymerase-how-structure-effects-function?srsltid=AfmBOoq_by2aKKQmRMiDAWDmYZq4xAzKkQOZUlItck8kpOQU6Weo06sl]
- 70. What does DNA polymerase fidelity mean? [https://www.neb.com/en-us/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr?srsltid=AfmBOoqnjVjL-J-I68CJu8NLxngOq7Oes2MhInWVnfeOJfyo3PY-VQ8N]
- 71. FAQ: What does exonuclease activity mean for a DNA ... [https://www.neb.com/en-us/faqs/2023/07/25/what-does-exonuclease-activity-mean-for-a-dna-polymerase?srsltid=AfmBOopXhSwBo_pdOZt4dwI3Y4cF3Ojk2Ifcw--WUubT10maXyhtj5z7]
- 72. So Many DNA Polymerases, So Little Time [https://www.goldbio.com/blogs/articles/somanypolymerasessolittletime?srsltid=AfmBOoqtb5uP4zlc435jnR9HiniyR15ZQ0vI_1Sreg-ly69esbjfKQlh]
- 73. Error Rate Comparison during Polymerase Chain Reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4150459/]
- 74. DNA Polymerase—Four Key Characteristics for PCR [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/dna-polymerase-characteristics.html]
- 75. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOopLMNbbwa7aSPTAPwscOKRHQ2mD8GH2Y4FEu2t-oxZoMYpmKMSO]
- 76. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOop1ZA0bU4fuH8GXeWsCXo-N59pRW5pKa6LX2C_kY88RZObg-diQ]
- 77. What Is the Role of MgCl2 in PCR Amplification Reactions? [https://www.excedr.com/resources/what-is-the-role-of-mgcl2-in-pcr]
- 78. Temperature Dependence and Thermodynamics of Klenow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1367323/]
- 79. Optimizing PCR Reaction Conditions for High Fidelity and ... [https://www.labx.com/resources/optimizing-pcr-reaction-conditions-for-high-fidelity-and-yield/5579]
- 80. How does HotStart PCR help minimize nonspecific ... [https://www.qiagen.com/us/resources/faq/2676]
- 81. Hot Start PCR with heat-activatable primers [https://pmc.ncbi.nlm.nih.gov/articles/PMC2582603/]
- 82. Thermostable Mismatch-Recognizing Protein MutS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3634412/]
- 83. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 84. Taq DNA Polymerase Soars to XXX million , witnessing a ... [https://www.archivemarketresearch.com/reports/taq-dna-polymerase-142535]
- 85. A robust strategy for overexpression of DNA polymerase ... [https://pubmed.ncbi.nlm.nih.gov/39966433/]
- 86. Elimination of Bacterial DNA from Taq DNA Polymerases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC85583/]
- 87. Exogenous contaminating DNA in Taq polymerases: A ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701219300314]
- 88. Optimizing Taq Polymerase Concentration for Improved ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0007010]
- 89. Enhancing Bacterial DNA Detection in Blood Samples through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10886392/]
- 90. Choosing the best sample prep method for your detection ... [https://www.takarabio.com/applications/pathogen-detection/sample-prep?srsltid=AfmBOopaTp4HRtwy__TK4ivs8_g-GT_cnHVz7k_lj4919WjUN5CQfyxQ]
- 91. Taq DNA Polymerase [https://www.qiagen.com/us/products/discovery-and-translational-research/enzymes-for-molecular-biology/taq-dna-polymerase]
- 92. Four tips for PCR amplification of GC-rich sequences [https://www.neb.com/en-us/nebinspired-blog/four-tips-for-pcr-amplification-of-gc-rich-sequences?srsltid=AfmBOoqtmkHS_q5D0DtmHyGzQhrJ8-aX6titlIQoQlA7Q32nI4LM1Wtc]
- 93. Having trouble when amplifying GC-rich sequences? [https://bionordika.no/science-hub/blog-articles/having-trouble-when-amplifying-gc-rich-sequences-here-are-four-tips-on-how-to-troubleshoot]
- 94. 5 easy tips to address problems amplifying GC-rich regions [https://bitesizebio.com/24002/problems-amplifying-gc-rich-regions-problem-solved/]
- 95. Working with specific applications and conditions [https://www.takarabio.com/learning-centers/pcr/faq/applications-and-conditions?srsltid=AfmBOoo3o8_G6NSaT2IeL7IyX-zy2h0yM5Q2-o7mIs-ijCMdjdxDk2Sv]
- 96. of History Chain Reaction (PCR) Polymerase [https://www.news-medical.net/life-sciences/History-of-Polymerase-Chain-Reaction-(PCR).aspx]
- 97. The History of PCR | Thermo Fisher Scientific - ID [https://www.thermofisher.com/id/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/history-pcr.html]
- 98. A simple and efficient method for Taq DNA polymerase ... [https://jksus.org/a-simple-and-efficient-method-for-taq-dna-polymerase-purification-based-on-heat-denaturation-and-affinity-chromatography/]
- 99. Temperature effect on polymerase fidelity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8592868/]
- 100. Engineering functional thermostable proteins using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9525910/]
- 101. Protein thermostability engineering [https://pubs.rsc.org/en/content/articlelanding/2016/ra/c6ra16992a]
- 102. What does DNA polymerase fidelity mean? [https://www.neb.com/en-us/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr?srsltid=AfmBOopac31EWJmWnO8WwwQfx1TpeGRpX_b21oES3K1GE2Q4RER48VIW]
- 103. What is Taq Polymerase? [https://www.news-medical.net/life-sciences/What-is-Taq-Polymerase.aspx]
- 104. Exploring the molecular aspect and updating evolutionary ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813024047299]
- 105. DNA polymerases used in PCR [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/introduction/enzymes-used-in-pcr]
- 106. Live culture-based qPCR screening of Taq DNA ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1624735/full]
- 107. Novel thermostable Y-family polymerases - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC1373694/]
- 108. Engineering of novel DNA polymerase variants for single ... [https://www.nature.com/articles/s41598-025-10211-x]
- 109. Fidelity of DNA replication—a matter of proofreading - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6153641/]
- 110. What does DNA polymerase fidelity mean? [https://www.neb.com/en-us/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr?srsltid=AfmBOopWRSpHHJKdh8RlXAsrHYeQ5SGSgTrTMPKBbpPd4LFX8-vJUvFx]
- 111. FAQ: What does exonuclease activity mean for a DNA ... [https://www.neb.com/en/faqs/2023/07/25/what-does-exonuclease-activity-mean-for-a-dna-polymerase?srsltid=AfmBOorEfQSDeESPT3DNHHeOSetuNTXs3uwGRJy49XeL6VQ1IDHGmARd]
- 112. “Proofreading” DNA | Biology for Majors I [https://courses.lumenlearning.com/wm-biology1/chapter/reading-proofreading-dna/]
- 113. 5' exonuclease activity of DNA polymerases: a kinetic ... [https://pubmed.ncbi.nlm.nih.gov/12459442/]
- 114. DNA polymerase δ proofreads errors made by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7084062/]
- 115. A nuclear 3′–5′ exonuclease proofreads for the ... [https://www.sciencedirect.com/science/article/abs/pii/S1568786402001155]
- 116. KOD DNA polymerase: a custom fit for emergent ... [https://blog.sekisuidiagnostics.com/dxdialogue/kod-dna-polymerase-a-custom-fit-for-emergent-technology-driven-applications]
- 117. DNA polymerases engineered by directed evolution to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4215692/]
- 118. A robust strategy for overexpression of DNA polymerase ... [https://www.nature.com/articles/s41598-025-89902-4]
- 119. Selection Guide for Taq DNA Polymerase [https://www.promega.com/products/pcr/taq-polymerase/taq-dna-polymerase-selection-guide/]
- 120. DNA Polymerase Selection Chart [https://www.neb.com/en-us/tools-and-resources/selection-charts/dna-polymerase-selection-chart?srsltid=AfmBOopgP1nkEGSi7I5nsaCtJxNJJDKSmO3FTW0VbQP7ZHNyiQ4YykMc]
- 121. What does DNA polymerase fidelity mean? [https://www.neb.com/en-us/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr?srsltid=AfmBOopPWTvXAp2uckDOBIYSdz4qHSaoZcZZ3f_A4Doq-W38_YOgUSSW]
- 122. Three Questions to Ask When Choosing a PCR Enzyme [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/choosing-pcr-enzymes.html]
- 123. Anatomy of a Polymerase - How Function and Structure are ... [https://www.neb.com/en-us/tools-and-resources/feature-articles/anatomy-of-a-polymerase-how-structure-effects-function?srsltid=AfmBOooHW4aPYWVY2fehQbjEeU8dGe6Q5ZxbLtgTyuNXP8-tlHJi0ePe]
- 124. Strategies and procedures to generate chimeric DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11339088/]
- 125. Strategies and procedures to generate chimeric DNA ... [https://link.springer.com/article/10.1007/s00253-024-13276-2]
- 126. A robust strategy for overexpression of DNA polymerase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11836315/]