Sequencing Platform Showdown: Evaluating Illumina, PacBio, and Oxford Nanopore for Advanced Microbial Ecology
This article provides a comprehensive evaluation of modern DNA sequencing platforms—Illumina, Pacific Biosciences (PacBio), and Oxford Nanopore Technologies (ONT)—for microbial ecology research.
Sequencing Platform Showdown: Evaluating Illumina, PacBio, and Oxford Nanopore for Advanced Microbial Ecology
Abstract
This article provides a comprehensive evaluation of modern DNA sequencing platforms—Illumina, Pacific Biosciences (PacBio), and Oxford Nanopore Technologies (ONT)—for microbial ecology research. Tailored for researchers and drug development professionals, it explores the foundational principles of short- and long-read technologies, their methodological applications in 16S rRNA and metagenomic studies, and strategies for troubleshooting and optimization. Through a critical validation of recent comparative studies on soil, respiratory, and aquatic microbiomes, we synthesize key performance metrics on taxonomic resolution, error rates, and diversity assessments. The review concludes with a forward-looking perspective on integrating artificial intelligence and hybrid sequencing approaches to overcome current limitations and unlock novel discoveries in clinical and environmental microbiology.
From Short to Long Reads: Understanding Sequencing Technologies for Microbial Diversity
The field of DNA sequencing has undergone a revolutionary transformation, evolving from a laborious, low-throughput process to a powerful, high-throughput technology that has become a cornerstone of modern biological research. This evolution is categorized into distinct generations, each marked by significant technological leaps. First-generation sequencing, dominated by the Sanger method, enabled the decoding of initial genomes but was limited by its scalability [1] [2]. The advent of second-generation sequencing (NGS), or next-generation sequencing, introduced massively parallel sequencing, dramatically reducing cost and time while increasing output, thus enabling large-scale genome studies [1]. Most recently, third-generation sequencing (TGS) technologies have emerged, characterized by their ability to sequence single molecules in real-time and generate exceptionally long reads, overcoming some of the fundamental limitations of previous generations [3] [1].
This progression is particularly impactful for microbial ecology research. The ability to rapidly and cost-effectively sequence complex microbial communities from environmental samples has revolutionized our understanding of microbial diversity, function, and dynamics [4] [5]. While NGS platforms like Illumina provide high accuracy for profiling microbial composition, TGS platforms from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) are proving invaluable for assembling complete genomes and resolving complex genomic regions directly from metagenomic samples [6] [5]. This guide provides an objective comparison of these sequencing platforms, framing their performance within the specific context of microbial ecology.
The core distinction between sequencing generations lies not just in output, but in their underlying biochemistry and data characteristics. The table below summarizes the fundamental properties of each major platform type.
Table 1: Fundamental Characteristics of Sequencing Platform Generations
| Platform Type | Example Technologies | Key Sequencing Principle | Read Length | Key Advantages | Main Limitations |
|---|---|---|---|---|---|
| First-Generation | Sanger Sequencing | Dideoxy chain-termination with capillary electrophoresis [1] [2] | 500-1000 bp [2] | Very high accuracy (~99.99%); long reads for its era [2] | Very low throughput; high cost per base |
| Second-Generation (NGS) | Illumina MiSeq | Sequencing-by-synthesis with reversible dye-terminators [1] [6] | 36-300 bp [1] | High throughput; low cost per base; high accuracy (error rate 0.1-1%) [4] | Short reads; PCR amplification bias [4] [3] |
| Third-Generation (TGS) | PacBio SMRT | Real-time sequencing of single molecules via fluorescence in zero-mode waveguides (ZMWs) [3] [1] | Average 10,000-25,000 bp [1] | Very long reads; detects epigenetic modifications [3] | Higher cost; historically higher error rates (addressed with HiFi mode) [3] [7] |
| Third-Generation (TGS) | Oxford Nanopore | Real-time detection of electrical current changes as DNA strands pass through protein nanopores [1] [7] | Average 10,000-30,000 bp [1] | Extremely long reads; portability; direct epigenetic detection [3] [7] | Variable error rates, though improving with new chemistries (e.g., R10, Q20+) [8] [7] |
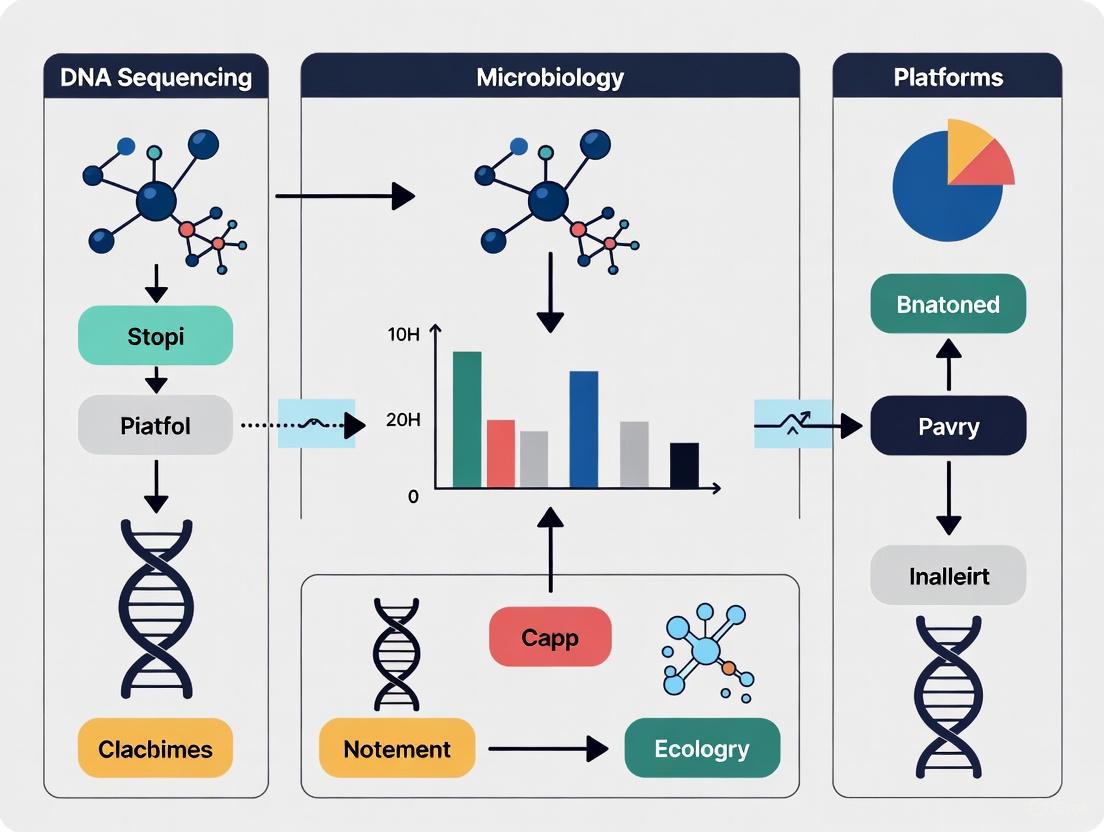
The following diagram illustrates the core workflow and logical relationship of the different sequencing technologies within a research context.
Performance Comparison in Microbial Ecology Applications
Accuracy and Read Length
Accuracy and read length are often a trade-off. Sanger sequencing remains the gold standard for accuracy for single, targeted sequences, making it ideal for validating genetic variants discovered by other methods [9] [2]. NGS platforms like Illumina provide high per-base accuracy, which is excellent for detecting single-nucleotide variations in amplicon studies (e.g., 16S rRNA sequencing) [4]. However, their short reads struggle to resolve repetitive regions or distinguish between closely related species [3] [6].
TGS platforms have historically had higher error rates, but recent chemistry improvements have been substantial. PacBio's HiFi mode generates circular consensus sequences (CCS) with accuracies exceeding 99.8% by sequencing the same molecule multiple times [3] [7]. ONT's latest R10.4.1 flow cell and Q20+ chemistry have also significantly improved raw read accuracy, with one study finding ONT R10 & Q20+ achieved the highest sample success rate for DNA barcoding [8] [7]. The defining feature of TGS is its long read length, which is transformative for metagenome-assembled genomes (MAGs), enabling the recovery of near-complete genomes from complex environments like soil [5].
Throughput, Cost, and Efficiency
The cost-effectiveness of a platform depends heavily on the project's scale and goals. Sanger sequencing is cost-prohibitive for sequencing entire genomes or many samples [8] [2]. NGS drastically reduced the cost per base, making large-scale projects like whole-genome sequencing feasible. However, for targeted sequencing of hundreds of samples, benchtop NGS sequencers can be efficient [9].
A direct comparison study estimated the cost-effectiveness of DNA barcoding relative to Sanger sequencing. It found that TGS platforms become more cost-effective when a study requires barcoding more than 61 samples for ONT Flongle, 183 for ONT MinION, or 356 for PacBio [8]. In terms of workflow, ONT protocols were noted as the quickest for library preparation [8]. For large-scale metagenomic projects, deep long-read sequencing (e.g., ~100 Gbp per sample), while a significant investment, has proven capable of recovering thousands of novel microbial genomes from complex terrestrial habitats, a task that is exceptionally challenging with short-read technologies alone [5].
Table 2: Performance Comparison in Key Microbial Ecology Applications
| Application | Best-Suited Platform | Experimental Support & Performance Data |
|---|---|---|
| 16S rRNA Amplicon Sequencing | Illumina MiSeq (for high-throughput, cost-effective profiling) [4] | Standard for microbiome studies; provides high-depth, accurate short reads suitable for amplicons [4]. |
| Whole-Genome Sequencing of Isolates | PacBio HiFi/Revio (for complete, closed genomes) [3] [6] | PacBio generated two contigs covering the entire 5-Mb, two-chromosome Vibrio parahaemolyticus genome, while NGS produced dozens of fragmented contigs [6]. |
| Metagenome-Assembled Genomes (MAGs) from Complex Samples | Oxford Nanopore (for high-quality MAG recovery) [5] | Deep Nanopore sequencing of 154 soil/sediment samples yielded 15,314 novel microbial species genomes, expanding the prokaryotic tree of life by 8% [5]. |
| Detection of DNA Modifications (e.g., 6mA) | PacBio SMRT & Oxford Nanopore (for direct epigenetic detection) [7] | Both platforms can natively detect DNA modifications. A 2025 study found SMRT and ONT's Dorado tool consistently delivered strong performance for bacterial 6mA profiling [7]. |
| Rapid In-Field Pathogen Surveillance | Oxford Nanopore (for portability and real-time analysis) [7] | ONT's portability enables sequencing outside traditional labs. Used for rapid sequencing of SARS-CoV-2 and norovirus [2] [7]. |
Detailed Experimental Protocols and Workflows
Protocol 1: Metagenomic Sequencing for MAG Recovery Using Long-Reads
This protocol is adapted from the large-scale soil microbiome study that recovered over 15,000 novel genomes using Nanopore sequencing [5].
- Sample Collection and DNA Extraction: Collect environmental samples (e.g., soil, sediment). Use a robust DNA extraction kit designed for complex environmental samples to obtain high-molecular-weight (HMW) genomic DNA. DNA integrity is critical for long-read sequencing.
- Library Preparation (ONT Ligation Sequencing): The MFD-LR MAG catalogue was built using this standard ONT workflow [5].
- DNA Repair and End-Prep: Repair DNA damage and blunt the ends of the HMW DNA fragments.
- Adapter Ligation: Ligate ONT-specific adapters to the prepared DNA ends. These adapters facilitate attachment to the nanopores and prime the sequencing reaction.
- Purification: Use solid-phase reversible immobilization (SPRI) beads to purify the adapter-ligated library from excess reagents.
- Sequencing: Load the library onto a Nanopore flow cell (R9.4.1 or R10.4.1). Sequence with a high-output device like the GridION or PromethION to achieve deep coverage (~100 Gbp per sample) [5]. Basecalling can be performed in real-time.
- Bioinformatic Analysis (mmlong2 workflow): The custom mmlong2 workflow was pivotal for high-quality MAG recovery [5].
- Assembly: Perform metagenome assembly using a long-read assembler (e.g., Flye).
- Binning: Recover MAGs using an ensemble of binners, incorporating differential coverage (using multi-sample data), iterative binning, and extraction of circular MAGs (cMAGs).
- Quality Assessment: Check MAG completeness and contamination using tools like CheckM.
Protocol 2: Diagnostic Gene Validation via Sanger Sequencing
This protocol outlines the use of Sanger sequencing for validating mutations in a diagnostic context, as used for primary hyperoxaluria [9].
- Target Amplification: Design primers flanking the genomic region of interest (e.g., a specific exon). Perform polymerase chain reaction (PCR) to amplify the target from purified genomic DNA.
- PCR Cleanup: Purify the PCR product to remove excess primers, nucleotides, and enzymes that could interfere with the sequencing reaction.
- Sanger Sequencing Reaction: This is a cycle sequencing reaction using dye-terminator chemistry [10] [2].
- Set up a reaction containing the purified PCR product, a sequencing primer, DNA polymerase, dNTPs, and fluorescently labeled ddNTPs.
- Run the reaction in a thermal cycler. The incorporation of a ddNTP randomly terminates the growing DNA chains, producing a ladder of fragments.
- Purification: Remove unincorporated dye terminators from the reaction.
- Capillary Electrophoresis: Load the purified products onto an automated DNA sequencer (e.g., ABI 310 Genetic Analyzer). The fragments are separated by size via capillary electrophoresis, and a laser detects the fluorescent dye at the end of each fragment [9].
- Data Analysis: Software (e.g., Phred) converts the fluorescence data into a chromatogram and a text sequence. Visual inspection of the chromatogram is crucial to verify base calls, especially around areas of poor resolution [10].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Sequencing Workflows
| Item | Function | Example Use Case |
|---|---|---|
| High-Molecular-Weight (HMW) DNA Extraction Kit | To isolate long, intact DNA strands from complex samples, minimizing shearing. | Essential for preparing libraries for TGS to maximize read lengths from soil or sediment samples [5]. |
| PacBio SMRTbell Express Template Prep Kit 2.0 | Prepares DNA fragments by ligating hairpin adapters to create circular templates for SMRT sequencing. | Used for generating HiFi reads for de novo genome assembly or isoform sequencing (Iso-Seq) [3]. |
| Oxford Nanopore Ligation Sequencing Kit (SQK-LSK109) | A standard kit for preparing genomic DNA libraries for Nanopore sequencing via ligation of adapters. | The primary kit used for large-scale metagenomic surveys like the Microflora Danica project [5]. |
| Illumina TruSeq DNA Custom Amplicon Kit | Designed for targeted sequencing of specific genomic regions by creating amplicon libraries. | Used in diagnostic validation studies to screen for mutations in multiple genes simultaneously via NGS [9]. |
| QIAamp DNA Blood Mini Kit | A reliable method for extracting high-quality DNA from small volumes of blood or cell cultures. | Used to obtain template DNA from patient blood samples for Sanger sequencing of disease-associated genes like AGXT, GRHPR, and HOGA1 [9]. |
| Agencourt AMPure XP Beads | SPRI magnetic beads used for efficient purification and size selection of DNA fragments in library prep. | A universal reagent for cleaning up enzymatic reactions and selecting appropriate fragment sizes in NGS and TGS workflows [9] [5]. |
The evolution from Sanger to third-generation sequencing has provided microbial ecologists with a powerful suite of tools, each with distinct strengths. The choice of platform is not a matter of identifying a single "best" technology, but rather of selecting the right tool for the specific biological question.
For high-throughput, low-cost profiling of microbial communities via 16S rRNA or shotgun metagenomics, Illumina-based NGS remains the workhorse. For applications where long-range genomic context is paramount—such as assembling complete genomes from complex metagenomes, resolving structural variations, or phasing haplotypes—PacBio and Oxford Nanopore TGS are unparalleled. The latest improvements in accuracy have made these technologies suitable for an ever-broadening range of applications. Sanger sequencing continues to hold value as an orthogonal method for validating key findings with its exceptional base-level accuracy.
The future of sequencing in microbial ecology lies in the intelligent integration of these technologies. Hybrid approaches, using Illumina for breadth and cost-efficiency and TGS for depth and resolution in complex regions, will become standard. Furthermore, as TGS continues to mature in accuracy, throughput, and cost-effectiveness, it is poised to become the dominant technology for comprehensive genomic and epigenomic characterization of the vast, uncultured microbial diversity on our planet.
In the field of microbial ecology research, selecting the appropriate DNA sequencing platform is a critical foundational decision. The choice primarily revolves around a central divide: the established dominance of second-generation short-read sequencing (exemplified by Illumina) and the rapidly advancing capabilities of third-generation long-read sequencing (championed by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT)). Each technology offers a distinct set of strengths and trade-offs in accuracy, read length, cost, and application suitability. This guide provides an objective, data-driven comparison of these platforms, framing their performance within the specific context of analyzing complex microbial communities, such as those found in soil and other environmental samples.
The fundamental difference between these platforms lies in their method of determining the sequence of DNA bases.
- Illumina (Short-Read): Utilizes Sequencing by Synthesis (SBS). DNA fragments are amplified on a flow cell to create clusters, and fluorescently labeled nucleotides are incorporated. As each nucleotide is added, a camera captures its specific fluorescent color, determining the base identity. This process generates vast volumes of highly accurate but short reads (50-300 bases) [11].
- PacBio (Long-Read): Employs Single Molecule, Real-Time (SMRT) sequencing. A single DNA molecule is sequenced by a polymerase enzyme fixed at the bottom of a tiny well called a Zero-Mode Waveguide (ZMW). The polymerase incorporates fluorescent nucleotides, and the light pulse from each incorporation is detected in real-time. Its key innovation is HiFi (High Fidelity) sequencing, where a circularized DNA template is read multiple times, producing long reads (15-20 kb) with exceptional accuracy (>99.9%) by generating a consensus sequence [12] [13].
- Oxford Nanopore (Long-Read): Based on the nanopore sensing system. A single strand of DNA or RNA is electrophoretically driven through a protein nanopore embedded in a membrane. As each nucleotide passes through the pore, it causes a characteristic disruption in the electric current. This change in current is decoded to determine the DNA sequence. This technology can produce the longest reads, sometimes exceeding a megabase, and can directly detect base modifications [12] [14].
The table below summarizes the core characteristics of each technology.
Table 1: Fundamental Comparison of Sequencing Technologies
| Feature | Illumina (Short-Read) | PacBio (HiFi Long-Read) | ONT (Long-Read) |
|---|---|---|---|
| Technology Basis | Sequencing by Synthesis (SBS) [11] | Single Molecule, Real-Time (SMRT) Sequencing [12] | Nanopore Sensing [12] |
| Typical Read Length | 50-300 bp [12] | 15,000-20,000 bp [12] [13] | 20 bp -> 1 Mb+ [12] |
| Typical Raw Read Accuracy | >Q30 (99.9%) [11] | ~Q30 (99.9%) [13] [15] | ~Q20 (99%) with latest chemistry [16] [17] |
| Primary Error Type | Low, predominantly substitutions | Random errors reduced via HiFi consensus [14] | Systematic indels, especially in homopolymers; improved with R10.4.1 flow cell [12] [14] |
| DNA Modification Detection | Requires bisulfite treatment | Direct detection of 5mC, 6mA without bisulfite treatment [12] | Direct detection of a wide range of DNA and RNA modifications [12] [17] |
The following diagram illustrates the core technological principles of each platform.
Performance Evaluation in Microbial Ecology
For microbial ecologists, the theoretical principles of a technology are less important than its performance in real-world applications like 16S rRNA amplicon sequencing for taxonomic profiling and shotgun metagenomics for functional insight and genome reconstruction.
Taxonomic Profiling with 16S rRNA Amplicon Sequencing
A 2025 study directly compared Illumina (V4 and V3-V4 regions), PacBio (full-length), and ONT (full-length) for sequencing bacterial diversity in soil microbiomes. After normalizing sequencing depth, the key finding was that ONT and PacBio provided comparable assessments of bacterial diversity, with PacBio showing a slight edge in detecting low-abundance taxa [16]. Crucially, the study concluded that despite ONT's inherently higher error rate, it did not significantly distort the interpretation of well-represented microbial taxa, and all technologies enabled clear clustering of samples by soil type [16].
Table 2: Performance in 16S rRNA Amplicon Sequencing for Microbial Ecology
| Metric | Illumina (V3-V4) | PacBio (Full-Length) | ONT (Full-Length) |
|---|---|---|---|
| Target Region | Hypervariable regions (e.g., V3-V4) [16] | Full-length 16S rRNA gene [16] | Full-length 16S rRNA gene [16] |
| Taxonomic Resolution | Limited to genus level, ambiguous due to short length [16] [15] | High, species- and often strain-level [16] [15] | High, species- and often strain-level [16] |
| Community Profile Accuracy | Reliable for overall structure | Comparable to ONT, slightly better for low-abundance taxa [16] | Comparable to PacBio for well-represented taxa [16] |
| Primary Advantage | Low cost per sample, high throughput | High accuracy with long read length | Real-time data, long reads, lower instrument cost |
Shotgun Metagenomics and Genome Reconstruction
Long-read technologies excel in shotgun metagenomics by producing contiguous sequences that span repetitive regions, which are a major challenge for short-read assemblers.
- Genome Assembly Quality: A comprehensive 2022 benchmarking study using complex synthetic microbial communities found that PacBio Sequel II generated the most contiguous assemblies, reconstructing 36 out of 71 full genomes from a mock community, followed by ONT MinION (22 genomes) [18]. The same study noted that PacBio provided the most accurate assemblies, while Illumina and MGI platforms had the lowest indel rates [18].
- Recovery of Novel Diversity: A landmark 2025 study leveraged deep ONT sequencing of 154 complex terrestrial samples, recovering 15,314 previously undescribed microbial species [5]. This demonstrates the unique power of long-read sequencing to access the vast "microbial dark matter" in highly complex environments like soil, a task that is exceptionally difficult with short-read technology alone [5].
- Metagenome-Assembled Genomes (MAGs): A 2022 clinical microbiome study compared Illumina and PacBio HiFi metagenomics and found that while both methods produced compositionally similar MAGs, long-read assemblies yielded a greater number and more complete MAGs [15]. The study also found approximately twice the proportion of long reads could be assigned functional annotations compared to short reads [15].
Table 3: Performance in Shotgun Metagenomics and Genome Assembly
| Metric | Illumina (Short-Read) | PacBio (HiFi Long-Read) | ONT (Long-Read) |
|---|---|---|---|
| Assembly Contiguity | Low; fragmented due to repeats [18] | High; produces contiguous assemblies [18] | High; produces contiguous assemblies [18] |
| Number of Recovered MAGs | Lower | Higher [15] | Higher (dependent on depth and workflow) [5] |
| MAG Quality (Completeness) | Lower | Higher [15] | High (e.g., >15,000 MQ/HQ MAGs recovered [5]) |
| Variant Detection | Strong for SNVs, small indels | Strong for all variant types: SNVs, indels, SVs [12] | Strong for SNVs and SVs; historically weaker for indels in homopolymers [12] [14] |
| Functional Annotation | Standard | Improved recovery of functional sequences [15] | Improved recovery of functional sequences [15] |
Experimental Protocols for Platform Comparison
To ensure a fair and reproducible comparison between sequencing platforms, standardized experimental protocols are essential. The following workflow, adapted from a 2025 soil microbiome study [16] and a 2022 benchmarking study [18], outlines a robust methodology.
Detailed Methodology:
Sample Selection and DNA Extraction:
- Use a well-characterized, complex sample. Synthetic mock communities with known strain compositions are ideal for absolute accuracy assessment [18]. Environmental samples (e.g., soil) are necessary for evaluating performance on novel diversity [16] [5].
- Perform High-Molecular-Weight (HMW) DNA extraction using kits designed to preserve long DNA fragments (e.g., Quick-DNA Fecal/Soil Microbe Microprep Kit) [16]. DNA quality and length are critical for long-read sequencing performance.
Library Preparation and Sequencing:
- For 16S rRNA amplicon sequencing, amplify the full-length 16S rRNA gene using universal primers for long-read platforms (PacBio, ONT) and the V3-V4 or V4 region for Illumina [16].
- For shotgun metagenomics, follow the manufacturer's recommended protocols for each platform without size selection where possible to avoid bias [18] [15].
- Sequence the same DNA sample across all platforms. For a fair comparison, it is critical to normalize sequencing depth during bioinformatic analysis (e.g., by subsampling all datasets to the same number of reads) [16].
Bioinformatic Analysis and Metric Comparison:
- Process data from each platform using optimized, platform-specific bioinformatic pipelines (e.g., DADA2 for Illumina 16S, EMU for ONT 16S) [16].
- For metagenomics, use assemblers designed for the respective read type (e.g., metaSPAdes for Illumina, metaFlye for long reads) and standard binning tools (e.g., MetaBAT2) [5] [18].
- Compare the following key metrics:
- Taxonomic Profiling: Alpha and beta diversity, sensitivity for low-abundance taxa, and accuracy against a mock community ground truth [16] [18].
- Assembly and Binning: Contig N50, number of high-quality MAGs recovered, genome completeness and contamination [18] [15].
- Variant Calling: Precision and recall for identifying single nucleotide variants (SNVs), insertions/deletions (indels), and structural variants (SVs) [12].
The Scientist's Toolkit: Key Research Reagents & Materials
The table below lists essential reagents and materials used in the comparative experiments cited in this guide.
Table 4: Essential Research Reagents and Materials for Comparative Sequencing Studies
| Item Name | Function / Application | Relevant Study / Context |
|---|---|---|
| Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) | HMW DNA extraction from complex environmental samples like soil. | Used for soil DNA extraction in 16S rRNA sequencing platform comparison [16]. |
| SMRTbell Prep Kit 3.0 (PacBio) | Library preparation for PacBio HiFi sequencing on Sequel IIe/Revio systems. | Used for preparing 16S and metagenomic libraries [16] [15]. |
| Ligation Sequencing Kit (Oxford Nanopore) | Standard library prep for ONT DNA sequencing on MinION/PromethION. | Used in multiple metagenomic studies for library construction [5] [18]. |
| ZymoBIOMICS Microbial Community Standard (Zymo Research) | Defined synthetic microbial community with known composition, used as a positive control and for benchmarking platform accuracy. | Used as a validation standard in multiple studies [16] [18]. |
| Native Barcoding Kit 96 (Oxford Nanopore) | Allows for multiplexing of up to 96 samples in a single ONT sequencing run. | Used for multiplexing samples in 16S sequencing study [16]. |
| MAS-ISO-seq for 10x Genomics (PacBio) | Library prep for high-throughput single-cell RNA sequencing with PacBio, enabling full-length transcriptome analysis. | Used in single-cell RNA sequencing protocol comparison [19]. |
The choice between Illumina, PacBio, and ONT is not about finding a single "best" technology, but rather selecting the right tool for the specific research question and context.
- Choose Illumina Short-Read Sequencing when: Your primary needs are high-throughput, low-cost per sample for large-scale cohort studies, and your analysis is focused on taxonomic profiling at the genus level or SNV calling where the highest base-level accuracy is required for variant detection [11] [16] [18].
- Choose PacBio HiFi Long-Read Sequencing when: Your research requires high accuracy (Q30) combined with long read lengths. This is ideal for resolving complex microbial communities to the species/strain level with 16S sequencing, generating high-quality metagenome-assembled genomes (MAGs), and comprehensively detecting all variant types (SNV, indel, SV) and base modifications in a single assay [12] [13] [15].
- Choose ONT Long-Read Sequencing when: Your priorities are maximal read length to span extremely long repeats, real-time data streaming for rapid pathogen identification or in-field sequencing, portability (MinION), or the direct detection of a broad range of DNA/RNA modifications. It is also a cost-effective option for generating long reads for assembly, though often requiring greater depth or computational polishing for highest consensus accuracy [12] [5] [17].
For many research groups, a multi-platform approach is becoming the most powerful strategy. A common paradigm is to use Illumina for broad, deep population screening and then employ long-read technology on a subset of key samples for deep investigation, genome resolution, and validation of complex genomic regions. As long-read technologies continue to reduce costs and improve throughput, they are poised to become the default choice for an increasing number of microbial genomics applications.
In microbial ecology, the 16S ribosomal RNA (rRNA) gene serves as a foundational genetic marker for profiling complex bacterial and archaeal communities. This gene, approximately 1,500 base pairs in length, contains a unique mosaic of nine hypervariable regions (V1-V9) interspersed with conserved areas [20]. The conserved regions enable the design of universal PCR primers, while the variable regions accumulate nucleotide changes over evolutionary time, providing signatures for taxonomic differentiation [21]. For decades, the selection of specific variable regions for amplification and sequencing has represented a critical methodological compromise, dictated by technological limitations and research objectives. While short-read sequencing platforms (e.g., Illumina MiSeq) have historically constrained researchers to target one or several hypervariable regions (~300-600 bp), the emergence of third-generation sequencing technologies (e.g., Pacific Biosciences (PacBio) and Oxford Nanopore) now enables routine high-throughput sequencing of the full-length 16S rRNA gene [22] [23]. This technological evolution necessitates a re-evaluation of historical practices, compelling researchers to understand precisely how the length and choice of targeted 16S regions directly impact the taxonomic resolution achievable in their microbiome studies.
The Compromise of Short Reads: Variable Region Performance
The inability of earlier high-throughput platforms to sequence the entire 16S rRNA gene forced researchers to target specific sub-regions, a practice that significantly influences downstream taxonomic results. Different variable regions possess varying degrees of discriminatory power, and their performance is not uniform across the bacterial kingdom.
In Silico Evidence of Differential Performance
In silico experiments, which extract sub-regions from full-length 16S sequences, starkly reveal the limitations of short-read approaches. One such analysis demonstrated that the V4 region performed poorest, with a striking 56% of in-silico amplicons failing to achieve confident species-level classification when matched to their correct sequence of origin. In contrast, using the full V1-V9 sequence allowed nearly all sequences to be accurately classified at the species level [21]. Furthermore, the choice of sub-region introduces taxonomic bias. For instance, the V1-V2 region performs poorly in classifying sequences from the phylum Proteobacteria, whereas the V3-V5 region struggles with classifying Actinobacteria [21]. This indicates that polymorphisms critical for distinguishing certain taxa are confined to specific variable regions.
Empirical Validation in Skin Microbiome Research
These computational findings are reinforced by empirical studies. A 2024 analysis of 141 skin microbiome samples sequenced on the PacBio platform concluded that while full-length sequencing provides superior taxonomic resolution, the V1-V3 region offers a resolution comparable to full-length 16S when compared to other common sub-regions like V3-V4 or V4 alone [22]. The study also confirmed that even full-length 16S sequencing cannot achieve 100% species-level resolution for complex skin communities, highlighting an inherent limitation of the 16S marker itself [22]. This makes the choice of the best possible sub-region all the more critical for studies using short-read technologies.
Table 1: Performance Comparison of Common 16S rRNA Sub-Regions
| Target Region | Approximate Length | Species-Level Classification Efficacy | Taxonomic Biases / Notes |
|---|---|---|---|
| V1-V3 | ~500 bp | Good, reasonable approximation of diversity [21]. | Poor for Proteobacteria [21]. Superior for Escherichia/Shigella; best compromise for skin microbiome [22] [21]. |
| V3-V4 | ~600 bp | Variable performance. | Poor for Actinobacteria [21]. Good for Klebsiella [21]. |
| V4 | ~300 bp | Poor (56% failure rate in in-silico experiment) [21]. | Worst-performing region in clustering experiments [21]. |
| V6-V9 | ~400 bp | Moderate. | Best sub-region for Clostridium and Staphylococcus [21]. |
| Full-Length (V1-V9) | ~1500 bp | Excellent (near-universal species-level classification) [21]. | Provides the highest taxonomic resolution and avoids regional biases [22] [21]. |
The Advantage of Full-Length Sequencing: Maximizing Resolution
The primary advantage of sequencing the entire 16S rRNA gene is the dramatic increase in taxonomic resolution. By capturing the entirety of the gene's sequence variation, full-length sequencing provides the maximum amount of phylogenetic information available from this marker, enabling more precise classification.
Direct Comparisons in Human Microbiome Studies
A direct comparative study on human saliva, subgingival plaque, and fecal samples demonstrated this advantage clearly. The research showed that while both Illumina (V3-V4) and PacBio (V1-V9) platforms assigned a similar proportion of reads to the genus level (~95%), PacBio full-length sequencing assigned a significantly higher proportion of reads to the species level (74.14% vs. 55.23%) [23]. This confirms that the additional sequence information in the full-length gene directly translates to improved discriminatory power at the species level, which is often crucial for understanding the functional roles of microbes in health and disease. Notably, the overall community profiles clustered by sample type rather than by sequencing platform, indicating that both methods capture similar broad-scale community structures despite the difference in resolution [23].
Resolving Intragenomic Variation
Another critical benefit of accurate full-length sequencing is the ability to resolve intragenomic 16S copy number variation. Many bacterial genomes contain multiple copies of the 16S rRNA gene, and these copies can contain subtle nucleotide polymorphisms within the same organism [21]. Modern PacBio Circular Consensus Sequencing (CCS) generates highly accurate long reads (HiFi reads) that are sufficiently precise to distinguish these subtle differences. Rather than being mere noise, these intragenomic 16S gene copy variants can provide strain-level information [21]. Appropriate bioinformatic treatment of this variation allows researchers to move beyond species-level identification to potentially discriminate between closely related strains, which can exhibit vastly different phenotypic properties [21].
Table 2: Comparison of Short-Read vs. Long-Read 16S Sequencing Platforms
| Factor | Short-Read (e.g., Illumina) | Long-Read (e.g., PacBio SMRT) |
|---|---|---|
| Target Region | Single or multiple variable regions (e.g., V4, V3-V4) [24] | Full-length 16S rRNA gene (V1-V9) [23] |
| Typical Read Length | ≤ 300 bp (paired-end) [21] | >1,500 bp [21] |
| Taxonomic Resolution | Genus-level (sometimes species) [24] | Species-level and strain-level (via copy variants) [21] [23] |
| Species-Level Assignment | Lower (e.g., 55%) [23] | Higher (e.g., 74%) [23] |
| Ability to Resolve 16S Copy Variants | Limited | Yes, with high accuracy [21] |
| Primary Limitation | Limited phylogenetic information; regional taxonomic bias [21] | Higher cost per sample for equivalent read depth [23] |
Experimental Protocols for 16S rRNA Gene Sequencing
To generate the data supporting the comparisons above, standardized but platform-specific laboratory protocols are essential.
Laboratory Workflow for Full-Length 16S Sequencing
The following workflow is adapted from studies that successfully compared full-length and partial 16S sequencing [22] [23]:
- DNA Extraction: Extract genomic DNA from samples (e.g., skin swabs, feces, saliva) using a kit designed for microbial lysis, such as the PowerSoil DNA Isolation Kit [22]. DNA quality and quantity should be rigorously assessed.
- Full-Length 16S Amplification: Amplify the ~1,500 bp full-length 16S rRNA gene using universal primers. A common primer pair is:
- Library Preparation & Sequencing: Prepare the PCR amplicons for sequencing using the platform-specific protocol. For PacBio, this involves damage repair, end repair, and adapter ligation with the SMRTbell Template Prep Kit. The library is then sequenced on a platform like the PacBio Sequel II using SMRT cell technology [22].
In Silico Extraction of Sub-Regions
To directly compare the performance of full-length sequences against sub-regions, an in silico extraction can be performed [22] [21]:
- Begin with a high-quality dataset of full-length 16S rRNA sequences.
- Identify the primer binding sites in the conserved regions that flank the target variable regions (e.g., primers for V1-V3, V3-V4, V4).
- Using bioinformatic tools (e.g., Cutadapt), extract the sequence between each pair of primer sites, thereby generating in silico amplicons for each sub-region from the full-length data.
- Analyze both the full-length and extracted sub-region sequences using the same bioinformatics pipeline (e.g., DADA2 for Amplicon Sequence Variants (ASVs) or the RDP classifier) and compare the resulting taxonomic assignments [21].
Table 3: Key Research Reagent Solutions for 16S rRNA Gene Sequencing
| Item / Reagent | Function / Application | Specific Examples / Notes |
|---|---|---|
| DNA Extraction Kit | Isolation of high-quality microbial genomic DNA from complex samples. | PowerSoil DNA Isolation Kit [22]; QIAamp Fast DNA Stool Mini Kit [25]. Designed to lyse tough microbial cell walls. |
| Universal 16S Primers | PCR amplification of the 16S rRNA gene or specific sub-regions. | Full-length: 27F/1492R [22] [23]. V3-V4: 341F/805R [25]. V4: 515F/806R [24]. |
| High-Fidelity PCR Master Mix | Accurate amplification of the 16S target with low error rates. | KOD One PCR Master Mix [22]. Critical for minimizing PCR-induced errors in amplicons. |
| Library Prep Kit | Preparation of amplicon libraries for sequencing on a specific platform. | SMRTbell Template Prep Kit (PacBio) [22] [25]; Nextera XT Kit (Illumina) [25]. |
| Curated Reference Database | Taxonomic classification of sequenced 16S reads. | Greengenes [21], SILVA, RDP [21]. Database choice and curation impact annotation accuracy [20]. |
| Bioinformatics Pipelines | Processing raw sequences, error-correction, taxonomic assignment, and diversity analysis. | DADA2 [23] (for ASVs), QIIME 2, MOTHUR. Essential for deriving biological insights from sequence data. |
The evidence is clear: the length and choice of the targeted 16S rRNA region are fundamental determinants of taxonomic resolution in microbiome studies. While targeting specific hypervariable regions with short-read platforms remains a practical choice under budget or DNA quality constraints, this approach entails significant compromises in species-level discrimination and introduces regional taxonomic biases [22] [21]. The emergence of third-generation sequencing technologies has made high-throughput, full-length 16S sequencing a reality, providing a level of resolution that begins to approach the full discriminatory potential of this genetic marker [23]. This allows researchers not only to achieve more accurate species-level classification but also to explore the implications of intragenomic 16S copy variation for strain-level ecology [21]. As long-read sequencing technologies continue to decline in cost and improve in accuracy, the routine use of full-length 16S sequencing is poised to become the new gold standard for amplicon-based microbial community profiling, enabling deeper and more precise insights into the composition and dynamics of microbial ecosystems across human health, biotechnology, and environmental sciences.
The selection of an appropriate DNA sequencing platform is a critical first step in microbial ecology research, as it directly influences the resolution, accuracy, and depth of microbial community characterization. The field now offers researchers a choice between second-generation short-read and third-generation long-read sequencing technologies, each with distinct performance profiles. This guide provides an objective comparison of current sequencing platforms based on key metrics—read depth, accuracy, and taxonomic resolution—to inform experimental design and platform selection for microbial ecology studies.
Sequencing Platform Performance Comparison
The table below summarizes the performance characteristics of major second and third-generation sequencing platforms based on benchmarking studies using complex synthetic microbial communities [18].
Table 1: Performance Comparison of Sequencing Platforms for Microbial Ecology
| Sequencing Platform | Read Length | Key Strengths | Key Limitations | Optimal Applications |
|---|---|---|---|---|
| Illumina | Short (150-300 bp) | High accuracy (>99.9%), high throughput [4] | Limited taxonomic resolution at species level [16] [26] | 16S rRNA amplicon studies, shallow shotgun metagenomics [16] [18] |
| PacBio (Sequel II) | Long (full-length 16S) | High accuracy (>99.9%) with CCS, enables species-level ID [16] | Lower throughput, requires DNA size filtering [18] | High-resolution taxonomic profiling, genome assembly [16] [18] |
| Oxford Nanopore (MinION) | Long (full-length 16S) | Real-time sequencing, portability for field use [26] | Higher error rates than other platforms [16] [18] | In-field monitoring, rapid pathogen detection [26] |
| MGI DNBSEQ | Short (100-150 bp) | High quality, low indel rates, cost-effective [18] | Similar limitations to Illumina short-read technology | Large-scale metagenomic surveys where cost is a factor [18] |
The Impact of Sequencing Depth on Microbial Community Analysis
Sequencing depth, or the number of reads generated per sample, profoundly impacts the detection and quantification of microbial diversity. The required depth varies significantly depending on the complexity of the microbial community and the specific research question.
Table 2: Recommended Sequencing Depth for Different Microbial Study Types
| Study Type | Recommended Depth | Rationale | Supporting Evidence |
|---|---|---|---|
| 16S rRNA Gene Taxonomy | 1 million reads | Sufficient for stable taxonomic composition at higher ranks [27] | Achieves <1% dissimilarity to full depth profile [27] |
| Shotgun Metagenomics for AMR Genes | 80+ million reads | Necessary to capture full richness of AMR gene families [27] | Rarefaction curves plateau at ~80M reads for diverse environments [27] |
| Shallow Shotgun Metagenomics | 100,000-500,000 reads | Accurate abundance estimation for cost-effective large studies [18] | Spearman correlations >0.9 for community composition [18] |
Deeper sequencing reveals greater microbial diversity, particularly for detecting rare taxa and specific genetic elements like antimicrobial resistance (AMR) genes. One study found that while 1 million reads per sample was sufficient to achieve a stable taxonomic profile (with less than 1% dissimilarity to the full profile), at least 80 million reads were required to recover the full richness of different AMR gene families in complex environmental samples [27]. Furthermore, additional allelic diversity was still being discovered in effluent samples even at 200 million reads, indicating that very deep sequencing is necessary to capture the complete genetic diversity of complex environments [27].
Experimental Protocols for Platform Comparison
Protocol 1: Comparative Evaluation of Sequencing Platforms for Soil Microbiomes
This protocol outlines the methodology for a direct comparison of Illumina, PacBio, and Oxford Nanopore technologies for 16S rRNA gene sequencing [16].
Sample Preparation:
- Sample Type: Three distinct soil types with three biological replicates each.
- DNA Extraction: Quick-DNA Fecal/Soil Microbe Microprep kit (Zymo Research).
- 16S Amplification:
- Illumina: Targeting V4 and V3-V4 regions.
- PacBio & ONT: Full-length 16S rRNA gene amplification with universal primers 27F and 1492R.
Sequencing & Analysis:
- Sequencing Depth: Normalized across platforms (10k, 20k, 25k, and 35k reads/sample).
- Bioinformatics: Standardized pipelines tailored to each platform.
- Analysis Metrics: Alpha and beta diversity, taxonomic resolution at different levels.
Protocol 2: Benchmarking Sequencing Platforms Using Synthetic Microbial Communities
This approach uses constructed synthetic communities of known composition to objectively evaluate platform performance [18].
Community Construction:
- Complexity: 64-87 microbial genomic DNAs per mock community.
- Diversity: Spanning 29 bacterial and archaeal phyla.
- Design: Uneven abundance distribution spanning three orders of magnitude.
Performance Evaluation:
- Taxonomic Profiling: Comparison of observed vs. theoretical abundances.
- Assembly Metrics: Contiguity and accuracy of metagenome-assembled genomes (MAGs).
- Error Analysis: Substitution and indel rates by platform.
Workflow for Sequencing Platform Selection
The following diagram illustrates the key decision points for selecting an appropriate sequencing platform based on research goals:
Taxonomic Resolution Across Sequencing Platforms
The choice of sequencing platform and approach significantly impacts the level of taxonomic classification achievable.
Table 3: Taxonomic Resolution by Sequencing Approach
| Sequencing Approach | Optimal Taxonomic Level | Key Determinants of Resolution | Recommendations |
|---|---|---|---|
| Short-Read 16S | Genus to Family Level | Hypervariable region selection, reference database quality [16] | Use for large cohort studies focusing on community structure shifts |
| Long-Read 16S | Species Level | Full-length 16S rRNA gene sequencing [16] [26] | Ideal for identifying specific pathogens or key taxa |
| Shotgun Metagenomics | Species to Strain Level | Sequencing depth, genome completeness, binning algorithms [18] [4] | Required for functional potential and strain-level differentiation |
Long-read sequencing technologies significantly improve taxonomic resolution. A study comparing full-length 16S rRNA gene sequencing with short-read approaches found that long-read sequencing "enables more robust classification at the species level" and "helps mitigate PCR biases and allows for better detection of rare or novel taxa" [26]. This enhanced resolution is particularly valuable for distinguishing between closely related microbial species that play different ecological roles.
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below details key laboratory reagents and their applications in microbial ecology sequencing studies.
Table 4: Essential Research Reagents for Microbial Sequencing Studies
| Reagent/Kit | Application | Function | Example Use Case |
|---|---|---|---|
| Quick-DNA Fecal/Soil Microbe Microprep Kit | DNA Extraction | Efficient lysis and purification of microbial DNA from complex samples [16] | Soil and fecal sample preparation for 16S sequencing [16] |
| ZymoBIOMICS Gut Microbiome Standard | Quality Control | Defined microbial community for evaluating extraction and sequencing biases [16] | Protocol validation and cross-study comparisons |
| SMRTbell Prep Kit 3.0 | Library Preparation | Preparation of SMRTbell libraries for PacBio sequencing [16] | Full-length 16S rRNA gene sequencing [16] |
| Native Barcoding Kit 96 | Library Preparation | Multiplexing samples for Oxford Nanopore sequencing [16] | High-throughput amplicon sequencing on MinION platform [16] |
| Ion Plus Fragment Library Kit | Library Preparation | Preparation of libraries for ThermoFisher sequencing platforms [18] | Shotgun metagenomic sequencing of synthetic communities [18] |
The optimal choice of sequencing platform for microbial ecology research involves careful consideration of trade-offs between read depth, accuracy, taxonomic resolution, and cost. Short-read platforms like Illumina offer high accuracy and are sufficient for community-level analyses, while long-read technologies from PacBio and Oxford Nanopore provide superior taxonomic resolution and are better suited for species-level identification and complex gene families. Sequencing depth requirements vary significantly based on the complexity of the microbial community and the specific research goals, with deeper sequencing needed for comprehensive analysis of gene families and rare taxa. By aligning platform capabilities with research objectives and following standardized experimental protocols, researchers can maximize the insights gained from microbial ecology studies.
Long-read sequencing technologies have historically been constrained by higher error rates compared to their short-read counterparts. However, recent advancements in flow cell chemistry and sophisticated basecalling algorithms are dramatically reshaping this landscape. This guide objectively compares the performance of Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) platforms, focusing on their application in microbial ecology research. Data from recent soil microbiome studies and technology evaluations demonstrate that these improvements are enabling unprecedented accuracy and resolution in profiling complex environmental samples, making long-read sequencing an increasingly powerful tool for ecologists.
For researchers in microbial ecology, accurately deciphering the immense diversity of microbial communities has been a persistent challenge, largely due to the limitations of sequencing technologies. While short-read Illumina platforms offer high base-level accuracy, their limited read length struggles to resolve complex genomic regions and often fails to provide complete genomic assemblies from metagenomic samples [5]. Long-read sequencing from ONT and PacBio overcome the length limitation but have traditionally been hampered by higher per-base error rates. This trade-off has forced researchers to choose between accuracy and context.
Recent breakthroughs are systematically dismantling this compromise. Innovations in flow cell chemistry, such as ONT's dual-reader-head R10.4.1 pore and PromethION Plus flow cells, are producing data with fundamentally higher fidelity [28] [29]. Concurrently, the development of advanced basecallers like Dorado, which leverage powerful neural networks, and specialized tools like Uncalled4 are translating raw signals into sequences with dramatically improved accuracy [30] [28]. For microbial ecologists, these advances are not incremental; they are transformative, enabling the recovery of high-quality metagenome-assembled genomes (MAGs) from highly complex environments like soil, which was once considered the 'grand challenge' of metagenomics [5].
Breakthrough 1: Next-Generation Flow Cell Chemistry
The physical hardware of sequencing—the flow cell and its chemistry—forms the foundation of data quality. Recent updates in this domain have been targeted at increasing raw data accuracy, yield, and consistency.
The Nanopore R10.4.1 Pore and PromethION Plus
A key advancement from Oxford Nanopore is the R10.4.1 flow cell, which features a dual-reader head. Unlike the single reader of the previous R9.4.1 pore, this design captures a longer stretch of DNA nucleotides simultaneously, resulting in a more distinctive electrical signal for each k-mer (a sequence of k nucleotides). This reduces ambiguity, particularly in homopolymer regions (repeats of the same base), which were a traditional source of error for nanopore sequencing [16] [28]. The enrichment of purines and pyrimidines at specific positions within the pore's reader head creates a more predictable and interpretable signal pattern [28].
Building on this, Oxford Nanopore has announced the PromethION Plus Flow Cell, an ultra-high-output flow cell incorporating improved chemistry. Designed for high-throughput applications, it promises significantly increased data output with enhanced consistency for long fragment libraries (>15 kb), without the need for wash protocols. This is a critical development for population-scale studies in microbial ecology, as it directly reduces the cost per genome while maintaining data richness, including epigenetic information [31] [29].
Comparative Performance in Microbial Ecology
The tangible impact of these chemistry improvements is evident in direct comparative studies. A 2025 study evaluating sequencing platforms for soil microbiome profiling found that ONT (using full-length 16S rRNA sequencing) and PacBio provided comparable assessments of bacterial diversity. The study, which normalized sequencing depth across platforms, concluded that despite ONT's historically higher error rate, its latest iterations produce results that closely match PacBio's efficiency in interpreting well-represented taxa in complex soil samples [16].
Table 1: Comparative Analysis of Sequencing Platforms for Soil Microbiome Profiling [16]
| Platform | Chemistry/Kit | Target Region | Key Finding | Relevance to Microbial Ecology |
|---|---|---|---|---|
| Oxford Nanopore | R10.4.1 Flow Cell | Full-length 16S rRNA | Results closely matched PacBio; community analysis showed clear clustering by soil type. | Enables accurate species-level identification and differentiation of microbial habitats. |
| PacBio | Sequel IIe System | Full-length 16S rRNA | Slightly higher efficiency in detecting low-abundance taxa; clear clustering by soil type. | Powerful for discovering rare members of the microbial community. |
| Illumina | MiSeq | V4 & V3-V4 regions | V3-V4 region enabled soil-type clustering; V4 region did not (p=0.79). | Limited taxonomic resolution with the V4 region can obscure ecological differences. |
Breakthrough 2: Advanced Basecalling Algorithms
Raw electrical signals from a sequencer are useless without sophisticated software to decode them. This process, known as basecalling, has become a frontier of innovation, primarily driven by machine learning.
From HAC to SUP: The Dorado Basecaller
ONT's production-grade basecaller, Dorado, offers multiple basecalling models that represent a trade-off between speed and accuracy: Fast, High Accuracy (HAC), and Super Accurate (SUP). The SUP model provides the highest raw read accuracy but requires the most computational resources [30]. These basecallers use bi-directional Recurrent Neural Networks (RNNs) that consider the context of the signal both before and after the current point, leading to more accurate base identification [30].
The machine learning pipeline for developing these models is rigorous. Training datasets incorporate diverse genomic samples, including the ZymoBIOMICS Microbial Community Standard, which is highly relevant for ecologists. The resulting models are validated on metrics including alignment accuracy, homopolymer sequencing, and—crucially—de novo genome assembly quality [30].
Specialized Tools and Signal Alignment with Uncalled4
Beyond generic basecallers, specialized tools are pushing the boundaries of accuracy further. Uncalled4 is a recently developed toolkit that improves the detection of nucleotide modifications via fast and accurate signal alignment [28]. Its basecaller-guided Dynamic Time Warping (bcDTW) algorithm aligns raw nanopore signals to a nucleotide reference much more efficiently than previous tools like Nanopolish or Tombo, being 1.3-2.7x faster than f5c (a GPU-accelerated tool) [28].
This accurate signal alignment is foundational for detecting DNA and RNA modifications, which can interfere with standard basecalling. By providing a more precise mapping between signal and sequence, Uncalled4 enables better modification detection, identifying 26% more RNA m6A modification sites than Nanopolish when used with the m6Anet detection tool, while maintaining equivalent precision [28].
Furthermore, for specific applications, iterative basecalling approaches have been shown to significantly improve the accuracy of reading modification-rich sequences, such as therapeutic RNAs or transfer RNAs (tRNAs). This method polishes initial basecalls by aligning them to a reference and iteratively retraining the basecaller, which has been proven to enhance mappability and alignment accuracy even for canonical RNAs [32].
Direct Comparison for Microbial Ecology Applications
The ultimate test of these technologies is their performance in real-world, complex applications like soil and sediment microbiome analysis.
MAG Recovery from Complex Terrestrial Habitats
A landmark 2025 study in Nature Microbiology demonstrated the power of deep long-read Nanopore sequencing for microbial ecology. Using a custom mmlong2 workflow featuring iterative binning on deep long-read Nanopore data (~100 Gbp per sample) from 154 soil and sediment samples, the study recovered 23,843 medium- and high-quality Metagenome-Assembled Genomes (MAGs). After dereplication, this yielded 15,314 previously undescribed microbial species, expanding the phylogenetic diversity of the prokaryotic tree of life by 8% [5]. This success was directly attributed to the long-read data, which enabled the recovery of complete ribosomal RNA operons and improved species-level classification in public databases.
Table 2: Experimental Protocol for High-Throughput MAG Recovery from Soils [5]
| Protocol Step | Detailed Methodology | Purpose & Rationale |
|---|---|---|
| Sample Collection | 125 soil and 28 sediment samples from 15 distinct habitats in Denmark (Microflora Danica project). | To capture a wide breadth of microbial diversity across different terrestrial ecosystems. |
| DNA Sequencing | Deep long-read sequencing on Nanopore platform (median ~95 Gbp/sample). Library prep with ligation kits for native sequencing. | To generate sufficient data depth for assembling genomes from highly complex communities. |
| Bioinformatic Analysis (mmlong2) | 1. Assembly & Polishing: Metagenome assembly and removal of eukaryotic contigs.2. Multi-feature Binning: Differential coverage, ensemble binning, and iterative binning. 3. Dereplication: Clustering of MAGs at species level. | To maximize the number and quality of recovered prokaryotic genomes from complex metagenomic data. Iterative binning alone recovered an additional 3,349 MAGs. |
The Emerging Case for Hybrid Sequencing
While long-read technology is advancing rapidly, a hybrid approach that leverages both long and short reads can offer a superior solution. A 2025 study showed that joint processing of Illumina and Nanopore data with a hybrid DeepVariant model could match or surpass the germline variant detection accuracy of state-of-the-art single-technology methods [33]. The motivation is clear: short reads excel at detecting small variants with high precision, while long reads resolve complex regions and structural variants. By integrating both, a "shallow hybrid sequencing" approach can yield competitive performance to deep sequencing with a single technology, potentially lowering costs for large-scale studies [33]. For microbial ecologists, this hybrid strategy could be ideal for achieving both high-fidelity single-nucleotide polymorphism (SNP) calling and complete genome assembly from metagenomes.
The following diagram illustrates the logical workflow for selecting a sequencing strategy based on common research goals in microbial ecology.
The Scientist's Toolkit: Essential Research Reagents & Materials
For researchers designing experiments based on these breakthroughs, the following reagents and materials are critical.
Table 3: Key Research Reagent Solutions for Advanced Long-Read Sequencing [5] [16] [30]
| Item Name | Function & Application | Specific Example/Product |
|---|---|---|
| R10.4.1 Flow Cells | The core consumable for Nanopore sequencing; provides improved raw accuracy via a dual-reader-head pore. | MinION & PromethION flow cells (Oxford Nanopore). |
| PromethION Plus Flow Cells | Ultra-high-output flow cells for cost-effective, large-scale genomic and epigenomic studies. | PromethION 24 device flow cells (Oxford Nanopore). |
| Native Sequencing Kits | Library preparation kits that preserve native DNA/RNA, enabling direct detection of base modifications. | Ligation Sequencing Kit (SQK-LSK114), Direct RNA Sequencing Kit (SQK-RNA004) (Oxford Nanopore). |
| Microbial Community Standards | Defined control communities used to validate sequencing protocols, basecaller training, and bioinformatic pipelines. | ZymoBIOMICS Microbial Community Standard (Zymo Research). |
| High-Accuracy Basecallers | Software that converts raw electrical signals to nucleotide sequences using advanced machine learning models. | Dorado basecaller with SUP model (Oxford Nanopore). |
| Signal Alignment & Modification Detection Tools | Specialized software for aligning raw signals to a reference, crucial for accurate modification detection. | Uncalled4 toolkit (github.com/skovaka/uncalled4). |
The paradigm that long-read sequencing is inherently less accurate than short-read sequencing is no longer tenable. Breakthroughs in flow cell chemistry, such as the ONT R10.4.1 and PromethION Plus cells, have fundamentally improved the quality of raw data. Simultaneously, advanced basecallers like Dorado and specialized algorithms like Uncalled4 are leveraging machine learning to extract unprecedented accuracy and epigenetic information from this data. For microbial ecologists, the evidence is clear: these advancements are already enabling the genomic exploration of previously intractable environments, leading to a massive expansion of the known microbial tree of life [5]. While the choice between PacBio and ONT may depend on specific project needs—with PacBio sometimes showing a slight edge in detecting low-abundance taxa [16]—the overall trajectory is toward more accurate, cheaper, and more information-rich long-read sequencing, empowering researchers to finally answer fundamental questions about the vast diversity of microbial ecosystems.
From Lab to Data: Practical Protocols for 16S rRNA and Metagenomic Sequencing
Standardized DNA Extraction Protocols for Complex Environmental Samples
In microbial ecology research, the accurate characterization of community structure and function hinges on the initial and critical step of DNA extraction. The choice of DNA extraction method can significantly influence downstream sequencing results, impacting the assessment of microbial diversity, abundance, and functional potential [34]. For complex environmental samples—such as soil, feces, feed, and water—this step presents particular challenges due to the presence of inhibitory substances, varying biomass, and the structural rigidity of different microbial cells. The move towards method standardization is therefore essential for ensuring reproducibility and comparability of data, especially in large-scale or multinational ecological studies [35]. This guide objectively compares the performance of various DNA extraction protocols and their interaction with different sequencing platforms, providing a foundation for robust experimental design in microbial ecology.
DNA Extraction Method Performance: A Comparative Analysis
Key Performance Metrics for Extraction Methods
The efficiency of a DNA extraction method is evaluated based on its DNA yield, purity, and its ability to provide an unbiased representation of the microbial community. Inhibitors co-extracted from environmental matrices can also affect subsequent molecular analyses like PCR or sequencing. Furthermore, the suitability of a method may vary depending on the sample type (e.g., soil vs. water) and the target organisms (e.g., bacteria vs. viruses).
Experimental Comparison of Extraction Methods
A 2025 study on detecting African swine fever virus (ASFV) in feed and environmental samples provides a direct comparison of four DNA extraction methods: two automated magnetic bead-based methods (taco Mini and MagMAX Pathogen RNA/DNA Kit), one column-based method (PowerSoil Pro Kit), and one point-of-care system (M1 Extraction) [36]. The results, derived from quantitative PCR (qPCR) analysis, are summarized in the table below.
Table 1: Performance Comparison of DNA Extraction Methods for ASFV Detection in Environmental Samples
| Extraction Method | Underlying Technology | Relative Performance (Cq Values) | Remarks |
|---|---|---|---|
| taco Mini | Automated Magnetic Bead-based | Best (Significantly lower Cq) | Higher sensitivity, able to detect ASFV DNA in feed mill surface samples. |
| MagMAX Pathogen | Automated Magnetic Bead-based | Best (Significantly lower Cq) | Higher sensitivity, able to detect ASFV DNA in feed mill surface samples. |
| PowerSoil Pro | Spin Column-based | Intermediate (Higher Cq) | Successfully detected ASFV DNA but with lower sensitivity. |
| M1 Extraction | Point-of-Care | Intermediate (Higher Cq) | Successfully detected ASFV DNA but with lower sensitivity. |
The study concluded that while all methods could detect the viral DNA, the magnetic bead-based extraction methods demonstrated significantly higher sensitivity (p < 0.05), as indicated by lower Cq values in qPCR. This enhanced performance was also evident in their ability to detect ASFV DNA on feed mill surface samples, where other methods struggled [36].
Another study comparing DNA extraction methods for metagenomic DNA from diverse human and environmental samples, including stool, fish gut, and soil, highlighted that while manual extraction methods can be effective for many sample types, broad-range commercial kits often provide higher purity and quality of DNA, which is crucial for sequencing [34].
The Importance of Standardization: Inter-Laboratory Ring Tests
The challenge of reproducibility across different laboratories was directly addressed in a 2025 inter-laboratory ring test for environmental DNA (eDNA) focused on marine megafauna detection [35]. Four laboratories, each using their established DNA extraction method (primarily column-based kits from Qiagen and Macherey-Nagel, albeit with lab-specific modifications), processed aliquots from the same set of eDNA samples.
Table 2: Extraction Protocols from an Inter-Laboratory Ring Test [35]
| Laboratory | Extraction Kit/Instrument | Key Protocol Modifications |
|---|---|---|
| UIBK | Qiagen BioSprint 96 Workstation | Elution with 100 µL TE buffer instead of AE buffer. |
| INRAE | Macherey-Nagel NucleoSpin Tissue Kit | Added 25 µL proteinase K at lysis; heated elution buffer and performed two elutions. |
| UCC & IMR | Qiagen DNeasy Blood and Tissue Kit | Used a vacuum system (IMR) or specific incubation times (UCC) for spin column steps. |
The findings revealed that while total DNA concentrations were similar, there was a significant reduction in targeted qPCR performance for one laboratory, leading that lab to modify its protocol for the remainder of the project. The study also found a significant interaction between the laboratory/extraction method and the target species, indicating that no single method is universally optimal and that protocol efficiency can be taxon-dependent [35]. This underscores the necessity of cross-validation in collaborative projects.
Interaction of DNA Extraction with Sequencing Platforms
The choice of DNA extraction method is intrinsically linked to the performance of downstream sequencing technologies. The quality, fragment length, and purity of the extracted DNA can influence sequencing accuracy, read depth, and the ability to perform certain analyses like metagenomic assembly.
Comparison of Second and Third-Generation Sequencing Platforms
A comprehensive 2022 benchmarking study compared seven second and third-generation sequencing platforms using complex, synthetic microbial communities [18]. The platforms included second-generation sequencers (Illumina HiSeq 3000, MGI DNBSEQ-G400, MGI DNBSEQ-T7, ThermoFisher Ion GeneStudio S5, and Ion Proton P1) and third-generation sequencers (Oxford Nanopore Technologies MinION and Pacific Biosciences Sequel II).
Key findings included:
- Read Accuracy: Second-generation platforms generally provided high base-level accuracy (>99%). Among third-generation platforms, PacBio Sequel II offered a very low substitution error rate, while ONT MinION had a lower identity rate (~89%) due to higher indel and substitution errors, though its accuracy has improved with newer chemistries [18].
- Taxonomic Profiling: All technologies showed high Spearman correlations (>0.9) between observed and theoretical genome abundances when sufficient sequencing depth was achieved. However, correlations were more affected by higher microbial richness in long-read technologies (MinION and PacBio) [18].
- Metagenomic Assembly: Third-generation sequencers, particularly PacBio, generated the most contiguous assemblies, reconstructing 36 full genomes from a 71-strain mock community, compared to 22 for MinION and fewer for short-read platforms. Hybrid assemblies, combining long and short reads, can further improve accuracy and contiguity [18].
Long-Read Sequencing for Improved Resolution
A 2025 study comparing sequencing platforms for soil microbiome profiling confirmed the advantages of long-read sequencing. Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) full-length 16S rRNA gene sequencing provide superior taxonomic resolution compared to Illumina short-read sequencing of hypervariable regions (e.g., V4 or V3-V4) [16]. Full-length sequences help resolve ambiguous taxonomic assignments common with short reads. The study noted that PacBio demonstrated slightly higher efficiency in detecting low-abundance taxa, but ONT results closely matched PacBio, indicating that its inherent error rate does not preclude robust community-level analysis [16].
The Scientist's Toolkit: Essential Reagents and Kits
The following table lists key reagents and kits commonly used in DNA extraction from complex environmental samples, as cited in the reviewed literature.
Table 3: Research Reagent Solutions for DNA Extraction from Environmental Samples
| Product Name/Type | Function | Example Use-Cases |
|---|---|---|
| Magnetic Beads | Bind nucleic acids in the presence of a binding buffer/chaotropic salt, allowing for washing and elution; amenable to automation. | Automated pathogen detection in feed and environmental samples [36]. |
| Silica Spin Columns | Nucleic acids bind to the silica membrane in the presence of high salt, are washed, and eluted in low-salt buffer or water. | Extraction from marine eDNA filters, soil, and stool samples [35] [34]. |
| AL Lysis Buffer | A chaotropic salt-based buffer that facilitates cell lysis and denatures contaminants, promoting binding of DNA to silica. | Initial lysis step for various sample types before DNA extraction [36]. |
| Proteinase K | A broad-spectrum serine protease that degrades proteins and inactivates nucleases. | Added during lysis to digest tough tissues and microbial cell walls [35]. |
| DNA/RNA Shield | A reagent that immediately stabilizes nucleic acids at the point of collection, preventing degradation. | Used to pre-moisten swabs for environmental sampling [36]. |
| Quick-DNA Fecal/Soil Microprep Kit | A commercial kit optimized for efficient lysis of difficult-to-lyse microbes in soil and fecal matter. | Standardized DNA extraction from soil for microbiome studies [16]. |
Decision Workflow for Method Selection and Validation
The following diagram outlines a logical workflow for selecting and validating a DNA extraction protocol for complex environmental samples, based on the insights from the comparative studies.
The journey towards fully standardized DNA extraction protocols for complex environmental samples is ongoing. Current evidence strongly indicates that magnetic bead-based automated methods offer superior sensitivity and efficiency for many applications, though column-based methods remain reliable and widely used. The choice of an extraction method must be guided by the sample type, the target of interest, and the downstream analytical application, particularly the choice of sequencing platform. Long-read sequencing technologies are overcoming earlier limitations and, when paired with high-quality DNA extracts, provide unparalleled resolution for microbial community analysis. For the scientific community, the path forward involves rigorous cross-validation of methods, as demonstrated by inter-laboratory ring tests, and a flexible, evidence-based approach to protocol selection to ensure data are both robust and comparable across studies.
In microbial ecology research, the 16S ribosomal RNA (rRNA) gene serves as a gold standard marker for taxonomic identification of bacterial communities due to its presence in all prokaryotes and its combination of highly conserved and variable regions [37]. The gene contains nine hypervariable regions (V1-V9) that provide the sequence diversity necessary for phylogenetic differentiation, flanked by conserved regions that enable primer binding for PCR amplification [22] [37]. Researchers face a fundamental methodological decision: whether to sequence the full-length 16S rRNA gene (~1,500 bp) using third-generation sequencing platforms or to target specific hypervariable regions (typically ~300-600 bp) using second-generation sequencing technologies [23] [38]. This guide provides an objective comparison of these two approaches, supported by recent experimental data, to inform researchers in selecting the most appropriate strategy for their specific research context.
Experimental Protocols and Methodologies
Full-Length 16S rRNA Gene Sequencing
PacBio SMRT Sequencing Protocol: Multiple studies have utilized similar methodologies for full-length 16S rRNA gene amplification and sequencing. The standard approach involves:
- Primer Selection: Primers 27F (AGRGTTTGATYNTGGCTCAG) and 1492R (TASGGHTACCTTGTTASGACTT) targeting the beginning and end of the 16S rRNA gene are most commonly used [22] [23].
- PCR Amplification: Typically 25-30 cycles of amplification using high-fidelity polymerases, with annealing temperatures around 55°C [22].
- Library Preparation: The SMRTbell template prep kit is used for library construction, followed by sequencing on the PacBio Sequel II system with a minimum of 5 passes and predicted accuracy ≥0.99 [22].
- Bioinformatic Processing: Circular consensus sequencing (CCS) reads are generated using SMRT Link Analysis software, followed by demultiplexing with lima v1.7.0 and primer removal with Cutadapt v1.9.1 [22].
Oxford Nanopore Technology Protocol: For nanopore-based full-length sequencing:
- Primer Design: Either standard primers (27F-I: AGAGTTTGATCMTGGCTCAG) or more degenerate variants (27F-II) can be used, with the latter showing improved coverage of diverse bacterial taxa [39] [40].
- Library Preparation: The 16S barcoding kit (SQK-RAB204) is typically employed with 50 ng of genomic DNA [39].
- Sequencing: Conducted on MinION Mk1C flow cells (R9.4.1 or newer) with real-time basecalling [40] [41].
- Optimization Considerations: Critical parameters include limiting PCR cycles (15-25) to reduce bias, selecting appropriate annealing temperatures (48-52°C), and using optimized polymerases like LongAmp Hot Start Taq [41].
Hypervariable Region-Targeted Sequencing
Illumina MiSeq Protocol: The most common approach for hypervariable region sequencing involves:
- Region Selection: Primers targeting V3-V4 (341F-805R), V1-V2 (27F-338R), or V4 (515F-806R) regions are selected based on the research application [42] [43] [44].
- PCR Amplification: Typically 25-30 cycles with region-specific annealing temperatures [43] [38].
- Library Preparation: Using kits such as the QIASeq 16S/ITS screening panel or Zymo Quick-16S Plus Library Prep methods [42] [44].
- Sequencing: Conducted on Illumina MiSeq with 2×250 bp or 2×300 bp paired-end reads [43] [23].
- Bioinformatic Processing: Denoising with DADA2 or Deblur in QIIME2 to generate amplicon sequence variants (ASVs), followed by taxonomic classification using databases like SILVA, Greengenes, or Greengenes2 [42] [43].
Table 1: Commonly Used Primer Sets for Hypervariable Region Amplification
| Target Region | Forward Primer | Reverse Primer | Approximate Amplicon Size | Common Applications |
|---|---|---|---|---|
| V1-V2 | 27F (AGAGTTTGATCMTGGCTCAG) | 338R (TGCTGCCTCCCGTAGGAGT) | ~510 bp | Skin, respiratory microbiomes [42] [43] |
| V3-V4 | 341F (CCTACGGGNGGCWGCAG) | 805R (GACTACHVGGGTATCTAATCC) | ~428 bp | Gut, oral microbiomes [23] [44] |
| V4 | 515F (GTGCCAGCMGCCGCGGTAA) | 806R (GGACTACHVGGGTWTCTAAT) | ~252 bp | General environmental samples [38] [37] |
| V5-V7 | 799F (AACMGGATTAGATACCCKG) | 1193R (ACGTCATCCCCACCTTCC) | ~394 bp | Respiratory samples [42] |
Performance Comparison: Taxonomic Resolution and Diversity Metrics
Species-Level Taxonomic Resolution
Multiple studies have demonstrated the superior species-level classification capability of full-length 16S rRNA sequencing compared to hypervariable region approaches. A 2024 study comparing PacBio full-length sequencing versus Illumina V3-V4 sequencing for human microbiome samples found that with both platforms, a similar percentage of reads was assigned to the genus level (94.79% and 95.06% respectively), but with PacBio, a significantly higher proportion of reads were further assigned to the species level (74.14% vs. 55.23%) [23]. This enhanced resolution is particularly valuable for distinguishing between highly similar species within genera such as Streptococcus or the Escherichia/Shigella group, which have minimal sequence differences in their 16S genes [23].
Even within full-length approaches, technical factors significantly impact outcomes. A comparative analysis of primer sets with different degrees of degeneracy found that the more degenerate 27F-II primer detected a broader range of taxa and showed stronger correlation with reference datasets (Pearson's r = 0.86) compared to the standard 27F-I primer (r = 0.49) in oropharyngeal samples [40]. Similarly, in fecal samples, the degenerate primer set revealed significantly higher biodiversity and a more balanced phylum-level distribution [39].
Region-Specific Performance Variations
The resolving power of different hypervariable regions varies substantially across sample types and microbial communities:
- Respiratory Samples: A 2023 study comparing V1-V2, V3-V4, V5-V7, and V7-V9 regions found that V1-V2 exhibited the highest resolving power for accurately identifying respiratory bacterial taxa, with a significant area under the curve (AUC) of 0.736, while other regions showed non-significant AUC values [42].
- Skin Microbiome: Research from 2024 revealed that the V1-V3 region offers resolution comparable to full-length 16S sequences, outperforming other hypervariable regions for skin microbiota analysis [22].
- Gut Microbiome: A longitudinal study of anorexia nervosa patients found that while dominant genera were consistently detected across V1V2 and V3V4 regions, alpha diversity measures varied between regions, with Chao1 index values being higher in the V1V2 region [43].
Table 2: Comparative Performance Across Hypervariable Regions in Different Sample Types
| Sample Type | Optimal Hypervariable Region | Key Findings | Reference |
|---|---|---|---|
| Respiratory (Sputum) | V1-V2 | Highest AUC (0.736) for taxonomic identification; superior sensitivity and specificity | [42] |
| Skin (Multiple Sites) | V1-V3 | Comparable resolution to full-length 16S; best balance of accuracy and efficiency | [22] |
| Gut (Anorexia Nervosa) | V1-V2 | Higher Chao1 diversity indices; better detection of key taxa | [43] |
| Mouse Intestine | Full-length | Differences in relative abundances and α-/β-diversity compared to V4 region | [38] |
| Oral/Oropharyngeal | Full-length with degenerate primers | Significantly higher alpha diversity (Shannon: 2.684 vs. 1.850); better population alignment | [40] |
Diversity Metrics and Community Composition
The choice of amplification strategy significantly influences both alpha and beta diversity measures. A 2022 mouse study comparing full-length 16S sequencing versus V4-region sequencing found that while primary and derived V4 region data indicated similar bacterial abundances and diversity, comparison with full-length data revealed significant differences in relative bacterial abundances, alpha-diversity, and beta-diversity [38]. This suggests that the sequence length itself, rather than the sequencing platform, drives these differences and may lead to different biological interpretations of intervention effects.
In gut microbiome studies, Bland-Altman analysis revealed a general lack of strong agreement between V1V2 and V3V4 regions, except for a few taxa such as Faecalibacterium, Ruminococcus, Roseburia, Turicibacter, and Anaerotruncus [43]. This indicates that most findings in microbiome studies are sensitive to the chosen region, potentially affecting the reproducibility and comparability of results across studies.
Technical Considerations and Methodological Recommendations
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for 16S rRNA Sequencing
| Reagent Category | Specific Products | Function and Application Notes |
|---|---|---|
| DNA Extraction Kits | ZymoBIOMICS DNA Isolation Kit, PowerSoil DNA Isolation Kit | Consistent mechanical lysis for tough-to-lyse cells; minimal bias in community representation [22] [44] |
| Polymerase Enzymes | LongAmp Hot Start Taq, iQ SYBR Green Supermix, KOD One PCR Master Mix | High-fidelity amplification; LongAmp recommended for ONT protocols [22] [41] |
| Primer Sets | 27F/1492R (full-length), 341F/805R (V3-V4), 27F/338R (V1-V2) | Degenerate primers (e.g., 27F-II) improve coverage; region selection depends on sample type [22] [39] [40] |
| Library Prep Kits | PacBio SMRTbell, ONT 16S Barcoding Kit (SQK-RAB204), Zymo Quick-16S Plus | Barcoding enables multiplexing; kit selection depends on platform [22] [41] [44] |
| Reference Databases | SILVA, Greengenes2, RDP | Taxonomic classification; SILVA-138 commonly used for full-length 16S [43] [44] |
Decision Framework for Method Selection
The choice between full-length and hypervariable region sequencing involves balancing multiple factors:
Figure 1: Decision framework for selecting between full-length and hypervariable region 16S rRNA sequencing approaches.
Methodological Recommendations by Sample Type
Based on current evidence, specific recommendations emerge for different research contexts:
- Skin Microbiome: The V1-V3 region provides a practical compromise, offering resolution comparable to full-length sequencing while being more cost-effective for larger studies [22].
- Respiratory Samples: V1-V2 demonstrates superior performance for taxonomic identification in sputum and other respiratory specimens [42].
- Oral/Oropharyngeal Samples: Full-length sequencing with degenerate primers (27F-II) more accurately captures community complexity and aligns better with population-level references [40].
- Gut Microbiome: While V3-V4 remains widely used, V1-V2 may provide better diversity estimates, and full-length sequencing enables superior species-level discrimination [43] [23].
- Low-Biomass/Forensic Samples: Hypervariable region sequencing is more practical when DNA quantity or quality is limited, though even full-length sequencing has limitations for such samples [22].
The choice between targeting hypervariable regions versus sequencing the full-length 16S rRNA gene represents a fundamental methodological decision in microbial ecology research with significant implications for taxonomic resolution, diversity assessments, and result interpretation. Full-length 16S rRNA sequencing provides superior species-level classification and reduces amplification biases, making it particularly valuable for applications requiring high taxonomic precision, such as clinical diagnostics or pathogen detection. Conversely, hypervariable region sequencing offers a cost-effective alternative for community-level analyses, especially when processing large sample sets or working with limited DNA quantities. The optimal hypervariable region varies across sample types, with V1-V3 performing best for skin microbiomes and V1-V2 for respiratory samples. As sequencing technologies continue to evolve and costs decrease, full-length 16S rRNA sequencing is poised to become more widely adopted; however, both approaches will likely maintain their relevance for specific research contexts. Researchers should carefully consider their experimental goals, sample characteristics, and resource constraints when selecting between these amplification strategies to ensure biologically meaningful and reproducible results.
The advancement of next-generation sequencing (NGS) has fundamentally transformed microbial ecology research, enabling unprecedented insights into complex microbial communities. Library preparation serves as the critical foundation of any NGS workflow, significantly influencing data quality, accuracy, and reliability. The evolving landscape of commercial library preparation kits offers diverse methodologies—including enzymatic fragmentation, tagmentation, and sonication—each with distinct advantages and limitations. For researchers navigating this complex field, a systematic comparison of these kits is essential for selecting the most appropriate protocol for specific research applications. This guide provides an objective, data-driven evaluation of DNA library preparation kits across multiple sequencing platforms, focusing on performance metrics relevant to microbial ecology studies. By synthesizing experimental data from controlled comparisons, we aim to equip researchers with the evidence necessary to optimize their sequencing workflows for studies ranging from metagenomic profiling to targeted amplicon sequencing.
Comparative Performance of DNA Library Preparation Kits
Key Performance Metrics and Experimental Findings
Library preparation efficiency varies substantially across commercially available kits, influencing sequencing outcomes through differences in ligation efficiency, fragmentation bias, and overall yield. A systematic comparison of nine library preparation kits using the same DNA sample revealed significant variations in performance, with kits that combine multiple preparation steps into a single reaction demonstrating 4 to 7 times higher final yields than conventional protocols [45].
The adaptor ligation step proved particularly variable, with efficiency ranging by more than a factor of 10 between kits. Some kits exhibited critically low ligation efficiencies (as low as 3.5%), potentially impairing original library complexity, while others achieved near-perfect 100% ligation efficiency [45]. These disparities significantly impact library quality but can be masked during PCR enrichment steps, where lower adaptor-ligated DNA inputs paradoxically lead to greater amplification yields.
Table 1: Comparative Performance of DNA Library Preparation Kits
| Kit Name | Fragmentation Method | Ligation Efficiency (%) | PCR Cycles Required | Insert Size (bp) | Key Characteristics |
|---|---|---|---|---|---|
| NEBNext Ultra II FS (NEB) | Enzymatic | Not specified | 7 (10 ng), 3 (100 ng) | 206 (10 ng), 188 (100 ng) | Flexible DNA input (1 ng-1 μg) [46] |
| KAPA HyperPlus (Roche) | Enzymatic (Fragmentase) | 100 | 9 (10 ng), Not specified | 240 (10 ng), 227 (100 ng) | Combined steps, minimal fragmentation bias [45] |
| Swift 2S Turbo | Enzymatic | Not specified | 6 (10 ng), Not specified | 330 (10 ng), 226 (100 ng) | Quick workflow, lower price [46] |
| SparQ (Quantabio) | Enzymatic | Not specified | 9 (10 ng), Not specified | 185 (10 ng), 244 (100 ng) | Cost-effective, flexible inputs [46] |
| Nextera DNA Flex (Illumina) | Tagmentation | 15-40 | 8 (10 ng), 5 (100 ng) | 326 (10 ng), 366 (100 ng) | Rapid protocol, fixed transposome concentration [46] [45] |
| TruSeq DNA PCR-Free | Sonication | Not specified | PCR-free | Not specified | Requires high input (1 μg), minimal bias [45] |
Insert size distributions also varied significantly between kits despite identical input DNA and cleanup conditions. These variations impact sequencing performance, with longer insert sizes (exceeding the cumulative read length) demonstrating improved coverage and variant detection sensitivity. Libraries with shorter inserts suffer from read overlap, reducing the informativeness of sequencing data [46].
Experimental data indicates that enzymatic fragmentation-based kits generally provide good alternatives to tagmentation-based approaches, offering reproducible results with flexible DNA inputs, quicker workflows, and lower costs. However, optimal performance often requires investment in protocol optimization tailored to specific sample types [46].
Experimental Protocols for Kit Comparison
The methodology for comparative evaluation of library preparation kits requires standardized conditions to ensure meaningful results. The following protocol outlines a systematic approach for cross-kit performance assessment:
Sample Preparation:
- Use a standardized DNA source such as NA12878 human genomic DNA ("genome in a bottle") or phiX174 amplicons to minimize sample-specific variability [46] [45].
- For input amount comparisons, include both low (10 ng) and high (100 ng) inputs to evaluate kit performance across different sample scenarios.
- Prepare multiple technical replicates (minimum n=4) for each kit and input condition to assess reproducibility [46].
Library Preparation:
- Follow manufacturer protocols for each kit while standardizing cleanup procedures (bead ratios) and PCR reagents/conditions to enable direct comparison [45].
- Include both PCR-free and PCR-enabled protocols where applicable to evaluate amplification bias.
- Use capillary electrophoresis (e.g., Tapestation, Bioanalyzer) to assess fragment size distribution for each library [46].
Efficiency Quantification:
- Employ droplet digital PCR (ddPCR) assays to precisely quantify DNA fragments bearing adaptors or P5/P7 sequences after ligation and PCR enrichment steps [45].
- Calculate stepwise yields (after A-tailing, adaptor ligation, and PCR) and overall preparation efficiency.
- Sequence libraries on a consistent platform (e.g., Illumina HiSeq X) with balanced multiplexing to avoid batch effects [46].
Data Analysis:
- Assess sequencing metrics including coverage uniformity, error rates, GC bias, and variant detection accuracy.
- Evaluate library complexity through duplicate read rates and unique molecular identifiers where available.
- Compare detected single nucleotide variants (SNVs) and indels against established benchmarks for accuracy assessment [46].
Cross-Platform Sequencing Comparisons
Platform-Specific Performance in Microbial Applications
Sequencing platform selection significantly influences data outcomes, particularly for specialized applications in microbial ecology. Comparative studies between established and emerging platforms reveal both consistencies and divergences in performance characteristics.
Table 2: Sequencing Platform Comparison for Microbial Applications
| Platform | Technology | Read Length | Applications in Microbial Ecology | Performance Notes |
|---|---|---|---|---|
| Illumina NovaSeq6000 | Sequencing by synthesis | Short-read | Metagenomics, 16S rRNA sequencing, WGS of pathogens | High accuracy, established benchmark [47] [48] |
| MGISEQ-2000 | DNB/cPAS | Short-read | Targeted bisulfite sequencing, metagenomics | Comparable to Illumina in sensitivity, consistency [47] |
| BGISEQ-500 | DNB/cPAS | Short-read | Transcriptome analysis, small RNA profiling | High concordance with Illumina for gene expression [49] |
| PacBio | SMRT sequencing | Long-read | Metagenomic assembly, full-length 16S sequencing | High accuracy, minimal bias, long reads [48] |
| Oxford Nanopore | Nanopore sensing | Long-read | Real-time pathogen detection, AMR gene identification | Portability, long reads, increasing accuracy [48] |
In targeted bisulfite sequencing for methylation analysis—a challenging application due to low sequence diversity—the MGISEQ-2000 platform demonstrated performance comparable to Illumina's NovaSeq6000. Both platforms showed high consistency in methylation level measurements (correlation coefficient: 0.999) and similar analytic sensitivity in detecting cancer signals from synthetic cell-free DNA samples [47].
For transcriptomic applications, the BGISEQ-500 platform showed high concordance with Illumina HiSeq4000 in gene quantification (correlation: 0.88-0.93) and identification of differentially expressed genes, though it exhibited greater variability in SNP and indel detection [49].
Long-read platforms significantly enhance metagenomic studies by enabling more complete genome assembly from complex samples. In a recent large-scale study of terrestrial habitats, Nanopore sequencing of 154 soil and sediment samples facilitated recovery of 15,314 previously undescribed microbial species, expanding phylogenetic diversity of the prokaryotic tree by 8% [5].
Visualization of Library Preparation Workflows
Comparative Workflow Diagram: Traditional vs. Modern Kits
The workflow visualization highlights key differences between traditional and modern library preparation approaches. Traditional multi-step protocols involve sequential purification steps that contribute to significant DNA loss and extended hands-on time. In contrast, modern combined-step kits integrate multiple reactions into single-tube processes, improving DNA recovery and reducing preparation time from hours to minutes [45].
DNA Library Preparation Kit Decision Framework
This decision framework provides researchers with a systematic approach for selecting appropriate library preparation methods based on experimental requirements. The pathway emphasizes critical decision points including DNA input amount, need for PCR-free protocols, strand-specificity, and insert size control, directing users toward optimal kit choices for their specific applications [46] [45] [50].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for Library Preparation
| Reagent/Solution | Function | Application Notes |
|---|---|---|
| Magnetic Beads (SPRI) | Size selection and purification | Standardized bead ratios crucial for reproducible size selection [45] |
| ddPCR Assay Reagents | Library quantification and quality control | Enables precise measurement of adaptor-ligated fragments [45] |
| Enzymatic Fragmentation Mixes | DNA shearing | Reduced bias compared to mechanical shearing; requires optimization [46] |
| Tagmentation Enzymes | Simultaneous fragmentation and adaptor tagging | Illumina Nextera; fixed size distribution based on bead coating [46] |
| Universal Control DNA | Process standardization | NA12878 or phiX174 DNA enables cross-kit comparisons [46] [45] |
| High-Fidelity DNA Polymerases | Library amplification | Reduced errors during PCR enrichment steps [46] |
The expanding landscape of library preparation methodologies offers researchers both opportunities and challenges in experimental design. Enzymatic fragmentation kits provide compelling alternatives to traditional sonication and emerging tagmentation approaches, particularly for projects requiring flexible DNA inputs, rapid workflows, and cost efficiency. Performance comparisons reveal that kits combining multiple preparation steps generally yield higher efficiency with reduced hands-on time, though ligation efficiency varies significantly between products. Cross-platform sequencing comparisons demonstrate that emerging sequencing technologies now deliver performance comparable to established platforms for many microbial ecology applications, though platform-specific strengths persist. By aligning kit capabilities with specific research requirements through the frameworks provided, researchers can optimize their sequencing strategies to maximize data quality and biological insights while efficiently utilizing resources. As library preparation technologies continue to evolve, ongoing comparative assessments will remain essential for navigating this dynamic landscape and leveraging the full potential of next-generation sequencing in microbial ecology research.
High-throughput sequencing technologies have revolutionized microbial ecology, enabling detailed characterization of complex communities. The choice of sequencing platform—Illumina, Pacific Biosciences (PacBio), or Oxford Nanopore Technologies (ONT)—is intrinsically linked to the selection of an appropriate bioinformatics pipeline for data analysis. DADA2, designed for Illumina's short reads, PacBio's Circular Consensus Sequencing (CCS) model, and Emu, developed for ONT's long reads, represent specialized tools that optimize the interpretation of data from their respective technologies. This guide provides an objective, data-driven comparison of these platform-pipeline pairs, focusing on their performance in microbial ecology research, to help researchers and drug development professionals make informed methodological decisions.
Sequencing Platforms and Corresponding Bioinformatics Pipelines
The core of effective microbial community analysis lies in matching the sequencing technology with a bioinformatics tool designed to handle its specific data characteristics. The following section details the primary pipelines for each major platform.
Illumina & DADA2: Illumina sequencing generates high-volume, short-read data (typically 100-400 bp) targeting hypervariable regions of the 16S rRNA gene [16]. DADA2 (Divisive Amplicon Denoising Algorithm 2) is a widely adopted pipeline for such data. It operates not by clustering reads into operational taxonomic units (OTUs) based on an arbitrary similarity threshold, but by modeling and correcting Illumina-specific sequencing errors to infer exact amplicon sequence variants (ASVs). This method provides higher resolution and greater reproducibility [51].
PacBio CCS & QIIME 2: PacBio's long-read technology enables the sequencing of the full-length 16S rRNA gene. Its Circular Consensus Sequencing (CCS) mode allows the same DNA molecule to be sequenced multiple times. By processing these multiple sub-reads, the platform generates a single, highly accurate long read (HiFi read) with an intrinsic quality exceeding 99.9% [16]. The QIIME 2 platform incorporates the
dada2 denoise-ccsmethod, which leverages the DADA2 algorithm to denoise these PacBio CCS reads, deduplicate them, and produce ASVs [52] [53].Oxford Nanopore & Emu: ONT sequencing provides very long reads in real-time, which is also suitable for full-length 16S rRNA gene amplicon sequencing. Historically, its higher error rates posed a challenge for accurate taxonomic profiling [16]. The Emu pipeline was developed specifically to address this. Instead of error correction, Emu uses an abundance-based, expectation-maximization algorithm to model and account for ONT-specific errors during the taxonomic assignment process, which has been shown to effectively minimize false positives and negatives [16] [51].
Table 1: Core Characteristics of Sequencing Platforms and Their Primary Bioinformatics Pipelines
| Platform | Read Length | Target Region | Primary Pipeline | Core Algorithm Principle | Key Taxonomic Advantage |
|---|---|---|---|---|---|
| Illumina | Short (100-400 bp) [16] | Hypervariable regions (e.g., V4, V3-V4) [16] | DADA2 | Error model-based denoising to infer Exact Amplicon Sequence Variants (ASVs) [51] | High-resolution ASVs from targeted regions |
| PacBio | Long (Full-length 16S) [16] | Full-length 16S rRNA gene [16] | DADA2 via QIIME 2 (denoise-ccs) |
Circular Consensus Sequencing (CCS) for high accuracy, followed by denoising [16] [52] | Species-level resolution from full-length gene |
| Oxford Nanopore | Long (Full-length 16S) [16] | Full-length 16S rRNA gene [16] | Emu | Abundance-based error modeling for taxonomic assignment without prior error correction [16] [51] | Species-level resolution; real-time sequencing capability |
Experimental Data and Performance Comparison
A direct comparative study of these platforms and pipelines provides critical insights into their performance in a real-world research context. A 2025 study offers a robust evaluation using soil microbiome samples, which are known for their high complexity and diversity [16].
Experimental Protocol
- Sample Collection and DNA Extraction: Soil samples were collected from three distinct soil types (Luvic Chernozem) with three independent biological replicates per type. DNA was extracted using the Quick-DNA Fecal/Soil Microbe Microprep kit (Zymo Research) [16].
- Sequencing: The same DNA samples were subjected to 16S rRNA gene sequencing on three platforms:
- Illumina: Targeting the V4 and V3-V4 regions.
- PacBio (Sequel IIe): Sequencing the full-length 16S rRNA gene.
- ONT (MinION): Sequencing the full-length 16S rRNA gene using R10.4.1 flow cells [16].
- Bioinformatics Processing: Sequencing depth was normalized across platforms (e.g., 10,000, 20,000 reads per sample). Standardized pipelines were used: DADA2 for Illumina, the
dada2 denoise-ccsmethod for PacBio CCS reads, and Emu for ONT data [16]. - Analysis: Researchers compared alpha diversity (within-sample diversity), beta diversity (between-sample diversity), and taxonomic composition across the platforms.
Key Comparative Findings
The study yielded several critical findings that highlight the trade-offs between each approach [16]:
- Diversity Assessments: ONT (with Emu) and PacBio (with CCS) provided comparable assessments of bacterial diversity in soil samples. Both long-read platforms ensured clear clustering of microbial communities based on the original soil type.
- Impact of Target Region: For Illumina (DADA2), the choice of hypervariable region significantly influenced results. While the V3-V4 region allowed for sample clustering by soil type, the V4 region alone did not (p=0.79), demonstrating that the region selected can impact the ecological conclusions drawn.
- Taxonomic Resolution: PacBio CCS, with its high innate accuracy, showed a slight advantage in detecting low-abundance taxa compared to ONT.
- Error Rate Handling: Despite ONT's inherently higher per-read error rate, the Emu pipeline successfully produced taxonomic profiles that closely matched those from PacBio. This indicates that Emu's error-modeling strategy effectively mitigates the impact of raw sequencing errors on the final interpretation of well-represented taxa.
Table 2: Performance Summary from a Comparative Soil Microbiome Study [16]
| Performance Metric | Illumina (V3-V4) + DADA2 | PacBio (Full-Length) + CCS | ONT (Full-Length) + Emu |
|---|---|---|---|
| Soil-Type Clustering | Yes (V3-V4), No (V4 only) | Yes | Yes |
| Detection of Low-Abundance Taxa | Good | Slightly Higher | Comparable |
| Handling of Sequencing Errors | High accuracy via denoising | Very high innate accuracy (>99.9%) | Error modeling with Emu |
| Overall Community Representation | Region-dependent | Comparable to ONT | Comparable to PacBio |
The following workflow diagram synthesizes the experimental and analytical steps involved in such a comparative study.
Practical Implementation and Considerations
Beyond pure performance, practical aspects like computational demand and experimental design are crucial for selecting a pipeline.
Computational Resources:
- DADA2 for Illumina: Generally efficient, with runtimes scaling reasonably with sample number and depth.
- DADA2 for PacBio CCS: Processing PacBio data with
dada2 denoise-ccsis computationally intensive. A user reported a runtime of approximately one week for 45 samples, each with 250,000 reads, using 40 threads [52]. This can be mitigated by splitting samples into smaller groups for denoising or adjusting the pooling method. - Emu for ONT: Designed to be efficient and does not typically require the same extreme computational overhead as denoising CCS reads.
Batch Effect Management: When samples are sequenced across multiple runs or batches, it is recommended to process each batch separately through the denoising step (DADA2 or Emu) and then merge the resulting feature tables and sequences for downstream analysis. This prevents the batch-specific error profiles from interfering with the denoising process [53].
The Scientist's Toolkit: Key Research Reagents and Materials
The reliability of metagenomic studies depends on the quality of wet-lab procedures. The following table lists essential reagents and their functions as identified in the cited studies.
Table 3: Essential Research Reagents and Kits for Metagenomic Sequencing
| Item | Specific Example | Function in Workflow |
|---|---|---|
| DNA Extraction Kit | Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) [16] | Efficiently extracts microbial DNA from complex environmental samples like soil, critical for unbiased representation. |
| High Molecular Weight DNA Kit | Quick-DNA HMW MagBead Kit (Zymo Research) [51] | Extracts long, intact DNA fragments, which is particularly important for long-read sequencing (PacBio, ONT). |
| Library Prep Kit (PacBio) | SMRTbell Prep Kit 3.0 [16] | Prepares DNA libraries for sequencing on the PacBio platform, optimized for SMRTbell adapter ligation. |
| Barcoding Kit (ONT) | Native Barcoding Kit 96 [16] | Allows for multiplexing of up to 96 samples in a single Oxford Nanopore sequencing run by adding sample-specific barcodes. |
| Positive Control | ZymoBIOMICS Gut Microbiome Standard [16] | A defined microbial community used to validate the entire workflow, from DNA extraction to bioinformatics analysis. |
The choice between DADA2 for Illumina, CCS for PacBio, and Emu for ONT is not a matter of selecting a universally superior option but rather of aligning the technology and pipeline with the specific research goals and constraints. For high-throughput, cost-effective community profiling where species-level resolution is not paramount, Illumina with DADA2 remains a powerful and reliable choice. When the highest possible taxonomic resolution from the 16S gene is required, PacBio CCS provides exceptional accuracy. Oxford Nanopore, analyzed with the Emu pipeline, offers a compelling alternative with comparable results for community structure, plus the unique advantages of real-time data streaming and minimal library preparation hardware, making it ideal for in-field or rapid-turnaround studies. This empirical data empowers researchers to make strategic decisions that best suit their experimental needs in unraveling the complexities of microbial ecosystems.
The implementation of deep, long-read DNA sequencing has enabled a monumental leap in microbial discovery, as demonstrated by a landmark study that identified 15,314 previously undescribed microbial species from complex terrestrial samples [5]. This case study examines how the use of Oxford Nanopore Technologies (ONT) long-read sequencing, combined with an optimized bioinformatic workflow (mmlong2), successfully addressed the long-standing "grand challenge" of recovering high-quality microbial genomes from highly complex environments like soil [5]. The findings mark a significant expansion of known microbial diversity, increasing the phylogenetic diversity of the prokaryotic tree of life by 8% and providing unprecedented access to complete genetic elements such as ribosomal RNA operons, biosynthetic gene clusters, and CRISPR-Cas systems [5].
Microbial ecosystems represent the planet's greatest reservoir of biological diversity, with the vast majority of microorganisms remaining undiscovered [5]. Traditional culturing methods have proven insufficient for characterizing this diversity, as most microbes resist laboratory isolation [5]. While metagenome-assembled genomes (MAGs) obtained through sequencing have expanded our knowledge, soil environments have remained particularly challenging due to their enormous microbial complexity [5]. Prior approaches using short-read sequencing technologies struggled with genome fragmentation, particularly in repetitive regions, limiting the recovery of complete genomes from these environments [54] [55].
Long-read sequencing technologies from Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) have emerged as transformative solutions by generating reads that span repetitive regions and entire genes, dramatically improving genome continuity and completeness [55]. This case study examines how the strategic implementation of long-read sequencing enabled unprecedented genomic discoveries from terrestrial habitats.
Experimental Design and Methodology
Sample Collection and Sequencing Strategy
The Microflora Danica project conducted deep long-read Nanopore sequencing of 154 soil and sediment samples collected from diverse terrestrial habitats in Denmark [5]. The experimental design incorporated:
- Sample Diversity: 125 soil, 28 sediment, and 1 water sample from 15 distinct habitat types
- Sequencing Depth: Deep sequencing yielding ~100 Gbp per sample (14.4 Tbp total)
- Read Characteristics: Median read length N50 of 6.1 kbp, enabling comprehensive genome assembly [5]
Table 1: Key Experimental Parameters for the Microflora Danica Project
| Parameter | Specification |
|---|---|
| Total Samples | 154 |
| Total Sequencing Output | 14.4 Tbp |
| Median Sequencing per Sample | 94.9 Gbp |
| Median Read N50 | 6.1 kbp |
| Assembly Contiguity | Median contig N50 of 79.8 kbp |
Bioinformatics Innovation: The mmlong2 Pipeline
The research team developed a custom metagenomic workflow, mmlong2, specifically optimized for recovering prokaryotic MAGs from extremely complex datasets [5]. Key innovations included:
- Multi-sample Binning: Incorporation of read mapping information from multiple samples to improve binning accuracy
- Ensemble Binning: Application of multiple binning tools to the same metagenome
- Iterative Binning: Repeated binning of the metagenome to recover additional genomes
- Circular MAG Extraction: Separate processing of circular MAGs as distinct genome bins [5]
This comprehensive approach significantly enhanced MAG recovery compared to standard workflows, with iterative binning alone recovering 3,349 additional MAGs (14% of the total) [5].
Comparative Performance Analysis: Long-Read vs. Short-Read Platforms
Genome Recovery and Quality Metrics
The superiority of long-read sequencing for genome recovery from complex environments is demonstrated by direct comparative studies:
Table 2: Performance Comparison of Sequencing Technologies for Metagenomic Applications
| Performance Metric | Long-Read (ONT/PacBio) | Short-Read (Illumina) |
|---|---|---|
| Assembly Contiguity | Contig N50: 79.8 kbp [5] to 255.5 kbp [54] | Contig N50: 7.8 kbp [54] |
| Prophage Recovery | ~60% of phages assembled as integrated elements [54] | ~5% of phages assembled as integrated elements [54] |
| MAG Quality | Higher completeness with CheckV [54] | Increased fragmentation [54] |
| Sensitivity (LRTI) | 71.9% [56] | 71.8% [56] |
| Specificity Range | 28.6% to 100% [56] | 42.9% to 95% [56] |
| Mycobacterium Detection | Superior sensitivity [56] | Standard sensitivity [56] |
Advantages of Long-Read Technologies
Oxford Nanopore Technologies (ONT) provides exceptional capabilities for real-time sequencing with rapid turnaround times, making it particularly valuable for time-sensitive applications [56] [55]. ONT platforms can produce ultra-long reads spanning hundreds of thousands of bases, enabling complete assembly of complex genomic regions [12]. Recent improvements in chemistry (R10.4 flow cells) have increased raw read accuracy to approximately 99.5% [55].
Pacific Biosciences (PacBio) HiFi Sequencing delivers exceptionally high accuracy (99.9%) through circular consensus sequencing, producing reads of 15-20 kb that are ideal for detecting structural variants and resolving complex genomic regions [55] [12]. This technology provides comprehensive variant calling including SNVs, indels, and structural variations [12].
Key Research Findings and Genomic Insights
Expansion of Microbial Diversity
The implementation of long-read metagenomics yielded extraordinary discoveries:
- Novel Species Recovery: 15,314 previously undescribed microbial species [5]
- Genus-Level Novelty: 1,086 previously uncharacterized genera [5]
- Phylogenetic Expansion: 8% increase in prokaryotic tree of life diversity [5]
- Catalog Statistics: 15,640 dereplicated species-level MAGs (4,894 high-quality, 10,746 medium-quality) [5]
Functional Genetic Elements Recovered
Long-read sequencing enabled the recovery of complete genetic elements that were previously fragmented with short-read approaches:
- Ribosomal RNA Operons: Thousands of complete rRNA operons recovered intact
- Biosynthetic Gene Clusters: Complete pathways for secondary metabolite production
- CRISPR-Cas Systems: Full immune system arrays in native genomic context [5]
Habitat-Specific Recovery Patterns
MAG recovery efficiency varied significantly across habitat types:
- Coastal Habitats: Highest MAG recovery due to dominant species and lower microdiversity
- Agricultural Fields: Lowest MAG recovery, influenced by higher microdiversity and absence of dominant species [5]
- Community Composition: Distinct phylum-level differences between habitats (e.g., more Firmicutes in agricultural fields, more Proteobacteria in coastal samples) [5]
Experimental Protocols and Workflows
Sample Preparation and DNA Extraction
Proper sample preparation is critical for successful long-read metagenomics:
- DNA Quality Requirements: High molecular weight DNA is essential, with minimal fragmentation
- Extraction Considerations: Kits must preserve long DNA fragments (>50 kb) and avoid shearing
- Recommended Kits: Circulomics Nanobind Big DNA Kit, QIAGEN Genomic-tip, and MagAttract HMW DNA Kit [55]
- Quality Control: Assessment of DNA integrity through pulsed-field gel electrophoresis or fragment analyzers [55]
Library Preparation and Sequencing
Library preparation methods must be optimized for long-read technologies:
- ONT Library Prep: Utilizes ligation-based approaches with specific considerations for DNA fragment size selection
- PacBio SMRTbell Prep: Involves ligation of hairpin adapters to create circular templates for sequencing [55]
- Handling Considerations: Gentle pipetting techniques to minimize DNA shearing during library preparation [55]
Bioinformatic Processing Workflow
The computational workflow for long-read metagenomics involves several critical steps:
- Quality Control: Filtering of reads based on length and quality metrics
- Assembly: Use of long-read assemblers such as metaFlye, Canu, or Hifiasm-meta [57]
- Binning: Application of specialized tools like mmlong2 that incorporate multi-sample and iterative binning [5]
- Quality Assessment: Evaluation of MAG completeness and contamination using CheckM or CheckV [54]
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Research Tools for Long-Read Metagenomics
| Tool Category | Specific Solutions | Function and Application |
|---|---|---|
| Sequencing Platforms | Oxford Nanopore PromethION, PacBio Revio | High-throughput long-read sequencing [55] [12] |
| DNA Extraction Kits | Circulomics Nanobind, QIAGEN Genomic-tip | Preservation of high molecular weight DNA [55] |
| Library Prep Kits | ONT Ligation Sequencing, PacBio SMRTbell | Preparation of DNA libraries for long-read sequencing [55] |
| Assembly Tools | metaFlye, Canu, Hifiasm-meta | Long-read metagenome assembly [57] |
| Binning Tools | mmlong2, MetaBAT2, vRhyme | Genome binning from metagenomic assemblies [5] [57] |
| Viral Identification | VirSorter2, DeepVirFinder, CheckV | Viral genome identification and quality assessment [54] [57] |
The implementation of long-read metagenomics has transformed our ability to explore microbial dark matter, successfully addressing the long-standing challenge of genome recovery from complex terrestrial environments [5]. The discovery of thousands of novel species through the Microflora Danica project demonstrates the profound impact of this technological advancement on microbial ecology [5].
Future developments in long-read sequencing will likely focus on further improving accuracy, reducing costs, and enhancing computational methods for data analysis [55]. As these technologies become more accessible, they will continue to expand our understanding of microbial diversity and function across diverse ecosystems, with significant implications for biotechnology, medicine, and environmental science.
The integration of long-read metagenomics into standard microbial ecology workflows represents a paradigm shift in our approach to studying uncultured microorganisms, opening new frontiers for discovery and application in the genomic era.
Navigating Pitfalls and Maximizing Data Quality in Microbial Sequencing
Long-read sequencing technologies, primarily represented by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), have revolutionized genomic studies by generating reads spanning thousands to millions of bases. This capability is particularly valuable for assembling complex genomic regions, resolving structural variants, and performing metagenomic analyses. However, both platforms exhibit characteristic error profiles that must be addressed through specialized bioinformatic tools and experimental strategies. Understanding the fundamental sources and types of these errors is crucial for selecting appropriate correction methodologies [14].
The error profiles of PacBio and Nanopore technologies differ significantly in both nature and origin. PacBio's Single Molecule Real-Time (SMRT) sequencing primarily produces stochastic errors resulting from limitations in fluorescence signal detection during DNA synthesis. These errors are randomly distributed across reads, with an initial error rate of approximately 13-15%. In contrast, Nanopore sequencing generates systematic errors concentrated in homopolymeric regions (stretches of consecutive identical bases), where current signal recognition biases lead to inaccurate base calling. Initial error rates for Nanopore sequencing typically range from 5% to 15%, varying by sample type and specific chemistry [14].
For microbial ecology research, these error profiles present distinct challenges. High error rates can lead to misassembly of closely related microbial genomes, inaccurate taxonomic profiling, and false positive variant calls. This technical guide provides a comprehensive comparison of bioinformatic strategies to mitigate these errors, supported by experimental data and practical protocols for researchers evaluating sequencing platforms for microbial studies.
Comparative Analysis of Sequencing Platform Error Rates
Technology-Specific Error Characteristics
Table 1: Error Profile Comparison Between PacBio and Nanopore Technologies
| Parameter | PacBio (SMRT) | Oxford Nanopore |
|---|---|---|
| Primary Error Type | Stochastic substitutions | Systematic indels in homopolymers |
| Initial Error Rate | ~13-15% | 5-15% (sample-dependent) |
| Dominant Error Mechanism | Fluorescence signal misinterpretation | Current signal recognition bias |
| Post-Correction Accuracy | <1% (with HiFi mode) | <2% (with deep learning basecalling) |
| Homopolymer Accuracy | High | Moderate (improved with R10 chip) |
| Recommended Applications | High-precision assembly, variant detection | Real-time sequencing, metagenomic profiling |
The fundamental differences in error profiles between platforms stem from their distinct biochemical principles. PacBio's SMRT technology relies on detecting fluorescently-labeled nucleotides incorporated by DNA polymerase immobilized in zero-mode waveguides (ZMWs). Errors primarily arise from stochastic variations in fluorescence detection and polymerase kinetics [14]. Nanopore technology measures changes in ionic current as DNA strands pass through protein nanopores, with errors predominantly occurring in homopolymeric regions where subtle current changes complicate base identification [14].
Recent technological advancements have substantially improved the native accuracy of both platforms. PacBio's HiFi (High Fidelity) mode employs circular consensus sequencing (CCS), where multiple passes of the same DNA molecule generate consensus reads with dramatically reduced error rates (<1%). Nanopore has addressed systematic errors through both hardware improvements (R10 chip with dual reader head design) and enhanced basecalling algorithms incorporating deep learning models such as Bonito and Guppy [14].
Performance Benchmarking in Microbial Metagenomics
Table 2: Experimental Performance Metrics for Complex Microbial Communities
| Sequencing Platform | Read Identity (%) | Genomes Fully Reconstructed | Assembly Mismatches (per 100 kbp) | Hybrid Assembly Improvement |
|---|---|---|---|---|
| PacBio Sequel II | >99% (highest) | 36/71 | Lowest | Minimal benefit |
| ONT MinION R9 | ~89% | 22/71 | Moderate | Significant improvement |
| Illumina HiSeq 3000 | >99% | N/A | Low | Beneficial for both platforms |
| DNBSEQ-G400 | >99% | N/A | Lowest | Beneficial for both platforms |
A comprehensive benchmarking study evaluating seven sequencing platforms on synthetic microbial communities containing 64-87 strains across 29 bacterial and archaeal phyla revealed critical performance differences. The PacBio Sequel II system achieved the lowest substitution error rate and produced the most contiguous assemblies, successfully reconstructing 36 out of 71 complete genomes from a mock community. Nanopore's MinION R9 platform showed higher error rates (approximately 89% read identity) due to elevated indel and substitution errors, but still enabled reconstruction of 22 full genomes and demonstrated significant improvement when used in hybrid assembly approaches combining long reads with short-read data [18].
Notably, the study found that while second-generation sequencers (Illumina, DNBSEQ) provided high per-base accuracy, third-generation long-read technologies (PacBio, Nanopore) offered superior performance for genome reconstruction despite their higher raw error rates. This advantage stems from the ability of long reads to span repetitive regions and resolve complex genomic structures that fragment short-read assemblies [18].
Bioinformatics Error Correction Strategies
Computational Error Correction Methodologies
Computational error correction methods have been developed specifically to address the distinct error profiles of long-read sequencing technologies. These can be broadly categorized into hybrid approaches (combining long reads with short-read data) and non-hybrid approaches (using long reads only). A comprehensive benchmarking study of 23 error-correction tools revealed that method performance varies substantially across different data types, with no single method performing best on all datasets [58] [59].
Hybrid correction methods leverage the high per-base accuracy of Illumina short reads to correct errors in long reads. These approaches typically map short reads to long reads, then use the consensus of mapped short reads to correct errors in the long reads. While hybrid methods can achieve high accuracy, they require additional sequencing and may struggle in genomic regions with complex repeat structures where short reads cannot be uniquely mapped [58].
Non-hybrid methods utilize only long-read data, employing strategies such as iterative correction, partial order alignment, and de Bruijn graph-based approaches. These methods are particularly valuable when short-read data is unavailable, but may require higher long-read coverage to achieve correction accuracy comparable to hybrid approaches. The benchmarking analysis identified that increasing k-mer size typically improves correction accuracy, though with diminishing returns beyond optimal sizes [59].
Figure 1: Bioinformatics workflow for long-read error correction, showing the decision points between hybrid and non-hybrid approaches.
Read-Based Versus Assembly-Based Correction
Error correction can be implemented at two primary stages: read-based correction (correcting errors in raw reads before assembly) and assembly-based correction (correcting errors in the assembly graph or consensus sequences after assembly). Read-based correction methods are typically more computationally intensive but can improve downstream assembly quality, while assembly-based correction (often called "polishing") refines the final assembly using the original read data [60] [58].
A benchmarking study evaluating 11 long-read assemblers for bacterial genomes demonstrated that preprocessing strategies significantly impact final assembly quality. Tools employing progressive error correction with consensus refinement (Notably NextDenovo and NECAT) consistently generated near-complete, single-contig assemblies with low misassemblies. Flye offered a strong balance of accuracy and contiguity, while Canu achieved high accuracy but produced more fragmented assemblies with significantly longer runtimes [60].
The study further revealed that preprocessing steps including quality filtering, adapter trimming, and read correction substantially influenced assembly outcomes. Filtering improved genome fraction and BUSCO completeness, trimming reduced low-quality artifacts, and correction benefited overlap-layout-consensus (OLC) based assemblers but occasionally increased misassemblies in graph-based tools [60].
Experimental Protocols for Error Reduction
DNA Extraction and Library Preparation Guidelines
Proper experimental design and library preparation are critical for minimizing errors before sequencing begins. For microbial ecology studies involving complex communities, the following protocols have been demonstrated to reduce error rates and improve data quality:
High Molecular Weight (HMW) DNA Extraction: The integrity of input DNA significantly impacts sequencing accuracy. For PacBio systems, use magnetic bead-based cleanups to remove short fragments without damaging HMW DNA. For Nanopore sequencing, prioritize extraction methods that maintain DNA integrity, such as CTAB-based protocols for difficult samples or commercial kits specifically validated for long-read sequencing [18].
Library Preparation Considerations: For PacBio, the circular consensus sequencing (HiFi) mode requires careful size selection to optimize read length and number of passes. For Nanopore, the transition from R9 to R10 flow cells has substantially improved accuracy in homopolymer regions, with the R10.4.1 chemistry demonstrating particularly enhanced performance. When preparing multiplexed libraries, use unique barcodes with sufficient edit distance to minimize barcode swapping or misassignment [14] [18].
Table 3: Quality Control Recommendations by Sequencing Technology
| Quality Control Aspect | PacBio Recommendations | Nanopore Recommendations |
|---|---|---|
| Primary Error Mitigation | HiFi mode with ≥3 passes | R10 chip with dual reader |
| Optimal Coverage | 20-30x for assembly | 30-50x for assembly |
| Data Filtering | Remove reads with low consensus quality | Filter by mean Q-score (>7) |
| Supplementary Data | Integrate Illumina for hybrid correction | Generate consensus sequences |
| Validation | Sanger sequencing of key variants | PCR validation of structural variants |
SPECTACLE Evaluation Framework for Error Correction
The SPECTACLE (Software Package for Error Correction Tool Assessment on nuCLEic acid sequences) framework provides a standardized methodology for evaluating error correction efficacy. This system employs both simulated and real reads to assess correction tools across diverse scenarios, including challenging cases with heterozygous sites, coverage variations, and repetitive elements [58].
Simulated Read Generation: Using tools such as pIRS for Illumina-like data and PBSIM for PacBio-like data, generate reads from reference sequences with precisely known error locations. Introduce variants to create diploid genome simulations for evaluating performance on heterozygous regions. For microbial ecology studies, include genomes with varying GC content and complexity to represent natural community diversity [58].
Performance Metrics Calculation: For each error correction tool, calculate sensitivity (proportion of true errors corrected), precision (proportion of corrections that were proper), and gain (overall performance balancing sensitivity and precision). Additionally, assess sequence similarity, NG50 length, supporting read coverage, and alignment quality of corrected reads [58] [59].
Application to Microbial Communities: When applying this framework to metagenomic data, pay particular attention to the tools' performance on low-abundance community members, as excessive correction may eliminate genuine rare species through erroneous over-correction. The benchmarking study by [18] demonstrated that while most genomes were accurately estimated across technologies, careful parameter optimization is needed to prevent systematic under-representation of specific taxonomic groups.
Table 4: Essential Research Reagent Solutions for Long-Read Sequencing
| Reagent/Category | Function | Technology Application |
|---|---|---|
| HMW DNA Extraction Kits | Preserve long DNA fragments | Both PacBio and Nanopore |
| Magnetic Bead Cleanup | Size selection and purification | Both PacBio and Nanopore |
| SMRTbell Express Template Prep Kit | Library construction for PacBio | PacBio-specific |
| Ligation Sequencing Kit | Library construction for Nanopore | Nanopore-specific |
| DNA Damage Repair Mix | Address DNA degradation artifacts | Both PacBio and Nanopore |
| Barcoding Expansion Kits | Multiplexed library preparation | Both PacBio and Nanopore |
| Sequencing Primers & Buffers | Initiate sequencing reactions | Platform-specific |
Computational Tools and Pipelines:
- Canu: Versatile assembler for noisy long reads, incorporates correction, trimming, and assembly [60]
- Flye: Graph-based assembler specifically designed for noisy long reads, balances speed and accuracy [60]
- NECAT: Efficient assembler for Nanopore data, demonstrates high continuity and completeness [60]
- NextDenovo: Scalable assembler with progressive error correction, excels on large genomes [60]
- Medaka: Nanopore-specific tool for consensus sequence improvement using neural networks
- Merfin: K-mer-based tool for polishing assemblies and evaluating consensus quality
- nf-core/eager: Community-curated pipeline for long-read data processing and quality control [61]
Each tool exhibits distinct strengths depending on the data characteristics and research objectives. For projects requiring maximum contiguity, NextDenovo and NECAT generally produce the most complete assemblies. When balancing accuracy, speed, and computational efficiency, Flye often provides optimal performance. For rapid draft assemblies, Shasta and Miniasm offer ultrafast processing but typically require subsequent polishing to achieve high completeness [60].
Long-read sequencing technologies continue to evolve, with both PacBio and Nanopore demonstrating rapid improvements in raw accuracy and throughput. The latest PacBio Revio system further enhances HiFi read yield and quality, while Nanopore's R10.4.1 chemistry and updated basecalling models have substantially reduced indel errors in homopolymer regions. These advancements, coupled with more sophisticated bioinformatic approaches, are progressively mitigating the challenge of high error rates in long-read data [14] [18].
For microbial ecology researchers, the choice between platforms and error correction strategies should be guided by specific research objectives and resource constraints. PacBio HiFi reads offer superior per-base accuracy advantageous for single-nucleotide variant detection and assembly of complex regions, while Nanopore provides advantages for real-time applications, ultra-long reads, and direct RNA sequencing. Hybrid approaches combining multiple technologies frequently provide the most comprehensive view of complex microbial communities, leveraging the complementary strengths of each platform [18] [62].
As algorithmic innovations continue to emerge, particularly in deep learning-based basecalling and assembly methods, the bioinformatics community is increasingly well-equipped to address the persistent challenge of sequencing errors. The standardized evaluation frameworks and benchmarking resources discussed in this guide provide a foundation for researchers to critically assess these evolving tools and implement robust error correction strategies in their microbial genomics workflows.
The accurate characterization of microbial communities is fundamental to advancing microbial ecology, yet researchers face significant technical challenges when working with complex sample types. Soil and respiratory microbiomes represent two such challenging environments: the former is characterized by immense microbial diversity and physical heterogeneity, while the latter often presents the difficulty of extremely low microbial biomass amid overwhelming host DNA contamination [4] [63] [64]. The choice of DNA sequencing platform and accompanying experimental workflow directly influences the resolution, accuracy, and biological validity of the resulting data.
This guide provides an objective comparison of current DNA sequencing platforms specifically for these challenging samples. It synthesizes recent comparative studies to evaluate the performance of Illumina, Pacific Biosciences (PacBio), and Oxford Nanopore Technologies (ONT) in capturing true microbial diversity and achieving species-level taxonomic resolution. Within the broader thesis of evaluating DNA sequencing platforms for microbial ecology, this review emphasizes that there is no single "best" platform; rather, the optimal choice depends on the specific research question, whether it requires comprehensive diversity assessment, high taxonomic resolution, or functional potential analysis [4] [4].
Sequencing Platform Comparison: Performance Metrics for Challenging Samples
The table below summarizes the key technical characteristics and performance metrics of the major sequencing platforms when applied to soil and respiratory microbiome samples.
Table 1: Sequencing Platform Comparison for Soil and Respiratory Microbiomes
| Feature | Illumina (e.g., NextSeq, NovaSeq) | Pacific Biosciences (Sequel IIe) | Oxford Nanopore (MinION Mk1C) |
|---|---|---|---|
| Read Technology | Short-read, paired-end | Long-read, Circular Consensus Sequencing (CCS) | Long-read, single-molecule |
| Typical 16S Amplicon | V3-V4 region (~460 bp) | Full-length 16S (~1,500 bp) | Full-length 16S (~1,500 bp) |
| Key Strength | High accuracy, well-established workflows | Very high accuracy for long reads | Longest read lengths, real-time analysis |
| Key Limitation | Limited to genus-level taxonomy | Higher DNA input requirements, cost | Historically higher error rates |
| Error Rate | < 0.1% [63] | >99.9% [16] | ~99% with latest chemistry [16] [64] |
| Species-Level Resolution | Limited [63] | Excellent [16] | Excellent [63] [64] |
| Best for Soil (Diversity) | Excellent for broad surveys [16] | Good, comparable to ONT [16] | Good, comparable to PacBio [16] |
| Best for Respiratory (Low Biomass) | Excellent, captures high richness [63] | N/A (Information not available in search results) | Effective with optimized DNA extraction [64] |
Analysis of Comparative Performance
Illumina remains the benchmark for high-throughput, high-accuracy sequencing. Its strength lies in detecting a broad range of taxa, making it ideal for initial microbial surveys where capturing overall diversity and richness is the goal [63]. However, its short-read lengths limit its ability to resolve closely related species, a significant constraint in clinical or ecological studies requiring fine-scale discrimination [63] [64].
Long-Read Platforms (PacBio and ONT) address this limitation by sequencing the entire 16S rRNA gene, which provides the phylogenetic resolution necessary for species-level identification [16] [64]. Recent advancements have significantly improved the accuracy of both platforms. PacBio's CCS mode achieves exceptional accuracy, while ONT's latest R10.4.1 flow cells and base-calling algorithms have pushed its raw accuracy over 99% [16] [64]. Comparative studies on soil samples show that PacBio and ONT provide comparable assessments of bacterial diversity, with PacBio showing a slight advantage in detecting low-abundance taxa [16].
For low-biomass respiratory samples, the choice of DNA extraction method becomes as critical as the sequencing platform itself. One study found that despite yielding lower total DNA, a host depletion protocol (Zymo HostZero kit) resulted in a sample containing 50–90% microbial DNA, whereas standard kits yielded less than 1% microbial DNA, dramatically affecting downstream community profiles [64].
Experimental Protocols from Key Comparative Studies
To ensure reproducible and comparable results, studies evaluating sequencing platforms must use standardized and clearly documented wet-lab and bioinformatics protocols. This section details the methodologies from two key comparative studies.
Protocol 1: Soil Microbiome Comparison (Illumina, PacBio, ONT)
A 2025 study provided a robust comparison of sequencing platforms for soil microbiomes using three distinct soil types with three biological replicates each [16].
Sample Collection and DNA Extraction:
- Soil samples were collected from 0–10 cm and 10–20 cm layers.
- Samples were sieved (1 mm mesh) under sterile conditions and stored at -20°C.
- DNA was extracted using the Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research).
Library Preparation and Sequencing:
- Illumina: Targeted amplification of the V3-V4 and V4 regions of the 16S rRNA gene.
- PacBio: Amplification of the full-length 16S rRNA gene using universal primers 27F and 1492R. Library preparation used the SMRTbell Prep Kit 3.0, and sequencing was performed on the Sequel IIe system.
- ONT: Amplification of the full-length 16S rRNA gene with the same primers. Libraries were prepared with the Native Barcoding Kit 96 and sequenced on MinION R10.4.1 flow cells.
- To ensure a fair comparison, sequencing depth was normalized across all platforms to 10,000, 20,000, 25,000, and 35,000 reads per sample prior to analysis.
Protocol 2: Respiratory Microbiome Comparison (Illumina vs. ONT)
A 2025 study directly compared Illumina and ONT for profiling respiratory microbial communities from human and pig samples [63].
Sample Collection and DNA Extraction:
- Human ventilator-associated pneumonia samples and samples from a swine model were collected.
- DNA was extracted from approximately 1 mL of sample using the Sputum DNA Isolation Kit (Norgen Biotek).
Library Preparation and Sequencing:
- Illumina: Libraries of the V3-V4 hypervariable region were prepared using the QIAseq 16S/ITS Region Panel (Qiagen) and sequenced on an Illumina NextSeq for 2x300 bp paired-end reads.
- ONT: Libraries were prepared using the 16S Barcoding Kit 24 V14 (SQK-16S114.24), sequenced on MinION Mk1C with R10.4.1 flow cells, and basecalled using the Dorado basecaller.
Bioinformatic Analysis:
- Illumina: Data was processed using the nf-core/ampliseq workflow, which uses DADA2 for error correction, chimera removal, and amplicon sequence variant (ASV) inference. Taxonomic classification was performed against the Silva 138.1 database.
- ONT: The EPI2ME Labs 16S Workflow and the Emu pipeline were used for analysis. Emu is specifically designed for ONT full-length 16S reads and uses a curated database to enhance taxonomic assignment accuracy at the species level [64].
Workflow Diagram: From Sample to Taxonomic Profile
The following diagram illustrates the core experimental and bioinformatic workflows for analyzing complex microbiomes, highlighting the divergent paths for short-read and long-read platforms.
The Scientist's Toolkit: Essential Reagents and Software
Successful microbiome analysis in challenging samples requires careful selection of reagents, kits, and software tools. The table below lists key solutions used in the cited studies.
Table 2: Research Reagent and Software Solutions for Microbiome Analysis
| Item | Function | Example Use-Case / Note |
|---|---|---|
| Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) | DNA extraction from complex, difficult-to-lyse matrices like soil. | Used for standardized DNA extraction from diverse soil types [16]. |
| Sputum DNA Isolation Kit (Norgen Biotek) | DNA extraction from viscous, high-host-content respiratory samples. | Used for parallel extraction of human and pig respiratory samples [63]. |
| ZymoBIOMICS HostZero Kit | Microbial DNA enrichment via host DNA depletion. | Critical for low-biomass respiratory samples; increased microbial DNA to 50-90% of total [64]. |
| QIAseq 16S/ITS Region Panel (Qiagen) | Targeted library preparation for Illumina sequencing. | Used for Illumina V3-V4 library prep from respiratory samples [63]. |
| ONT 16S Barcoding Kit (SQK-16S114.24) | Library prep for full-length 16S sequencing on Nanopore. | Allows multiplexing of up to 24 samples for ONT sequencing [63]. |
| SMRTbell Prep Kit 3.0 (PacBio) | Library preparation for PacBio SMRT sequencing. | Used for preparing full-length 16S amplicon libraries [16]. |
| nf-core/ampliseq | Bioinformatic pipeline for Illumina 16S data. | A standardized, reproducible workflow for processing V3-V4 data [63]. |
| Emu | Bioinformatic pipeline for ONT full-length 16S data. | Uses an expectation-maximization algorithm for improved taxonomic profiling with long, error-prone reads [64]. |
The landscape of sequencing technologies for complex microbiomes is no longer dominated by a single platform. While Illumina provides a cost-effective and highly accurate solution for broad diversity surveys, long-read sequencing from PacBio and ONT is now a mature and reliable approach for studies demanding species-level resolution. The choice between PacBio and ONT may come down to specific needs: PacBio offers slightly higher single-read accuracy, whereas ONT provides greater flexibility and real-time data streaming.
Future directions point toward the development of even more refined methodologies, such as the two-step metabarcoding approach that uses universal primers for an initial survey followed by taxa-specific primers for in-depth analysis of dominant groups, thereby overcoming primer bias [65]. Furthermore, the integration of machine learning with microbiome data is showing great promise for predicting soil health properties directly from 16S rRNA data, potentially creating powerful diagnostic tools [66]. As sequencing chemistries and bioinformatic tools continue to advance, the capacity to unravel the intricate workings of the most challenging microbial ecosystems will only grow, driving discoveries in human health, agriculture, and environmental science.
Batch Effect Correction and Contaminant Removal in Multi-Run Studies
In microbial ecology research, the utilization of high-throughput DNA sequencing has revolutionized our capacity to catalog and understand complex microbial communities. However, two significant technical challenges consistently arise in multi-run studies: batch effects and environmental contaminants. Batch effects are non-biological variations introduced when samples are processed in separate sequencing runs, using different technologies, or at different times. These technical artifacts can obscure true biological signals and compromise the integrity of comparative analyses. Simultaneously, the presence of laboratory contaminants and environmental inhibitors in samples can skew microbial community representations and reduce sequencing efficiency.
The integration of findings from multiple studies or sequencing platforms—a practice becoming increasingly common in meta-analyses—amplifies these challenges. This comparison guide objectively evaluates the performance of current sequencing platforms, wet-lab methods for contaminant removal, and computational tools for batch effect correction, providing researchers with a structured framework for optimizing their microbial ecology study designs.
Comparative Performance of DNA Sequencing Platforms
The selection of an appropriate sequencing platform significantly influences both the potential for batch effects and the capacity for contaminant identification. Third-generation long-read technologies have emerged as particularly valuable for complex environmental samples, offering improved resolution for strain-level differentiation and contaminant detection.
Technology Performance Metrics
Table 1: Comparative Performance of Major Sequencing Platforms in Microbial Ecology Applications
| Platform | Read Length | Accuracy | Strengths | Limitations | Best Suited For |
|---|---|---|---|---|---|
| Oxford Nanopore (e.g., MinION, PromethION) | Ultra-long (can exceed 10 kbp) [5] | >99% with latest chemistry (R10.4.1 flow cells) [16] | Real-time sequencing, portable options, detects base modifications | Higher per-base error rate than Illumina, though significantly improved [16] | Metagenome-assembled genomes (MAGs), in-field sequencing [5] |
| PacBio (Sequel IIe) | Long (HiFi reads: 10-25 kbp) | >99.9% with circular consensus sequencing (CCS) [16] | Extremely high accuracy for long reads | Lower throughput than competing platforms, higher DNA input requirements | Full-length 16S rRNA sequencing, resolving complex genomic regions [16] |
| Illumina (MiSeq, NextSeq) | Short (25-300 bp) [67] | >80% bases >Q30 at 2x150 bp [67] | High throughput, well-established protocols, low per-base cost | Short reads limit strain resolution and assembly continuity | 16S rRNA hypervariable region sequencing, high-population studies [16] |
Table 2: Experimental Performance in Soil Microbiome Profiling [16]
| Metric | Oxford Nanopore | PacBio | Illumina (V3-V4) |
|---|---|---|---|
| Species-Level Resolution | High (full-length 16S) | High (full-length 16S) | Moderate (partial 16S) |
| Community Composition Accuracy | Closely matches PacBio | Gold standard for full-length 16S | Subject to primer bias |
| Soil-Type Clustering | Clear differentiation | Clear differentiation | Variable with region |
| Required Sequencing Depth | 20,000-35,000 reads | 20,000-35,000 reads | 20,000-35,000 reads |
Impact on Batch Effect Generation
Different sequencing technologies exhibit characteristic error profiles that can manifest as systematic batch effects in multi-platform studies. Illumina platforms demonstrate highly consistent error rates within runs but show specific sequence-specific biases, particularly in GC-rich regions. Oxford Nanopore technologies have error profiles that are more evenly distributed across read lengths but have significantly improved with updated chemistries. The implementation of double reader-head R10.4.1 flow cells and improved basecalling algorithms has increased accuracy to over 99% [16]. PacBio's circular consensus sequencing achieves exceptional accuracy through multiple passes of the same DNA molecule, effectively randomizing errors and reducing batch effects stemming from sequence-specific biases [16].
Experimental Protocols for Contaminant Removal
Effective contaminant removal is essential for obtaining accurate microbial community profiles, particularly in samples with low bacterial biomass where laboratory contaminants can constitute a substantial proportion of sequences.
Laboratory Procedures for DNA Purification
Membrane Filtration Protocols: For water samples and DNA extraction eluates, membrane filtration effectively removes particulate contaminants and potential exogenous DNA. Microfiltration (0.1-10 µm pores) eliminates larger particles and eukaryotic cells, while ultrafiltration (0.01-0.1 µm) removes enzymes, inhibitors, and smaller contaminants. For comprehensive purification, nanofiltration (0.001-0.01 µm) can remove endotoxins, viruses, and fragmentary nucleic acids that may interfere with sequencing libraries [68].
Activated Carbon Filtration Methods: The implementation of activated carbon columns or beads during DNA extraction effectively adsorbs organic contaminants, including humic and fulvic acids that are prevalent in soil samples and inhibit enzymatic reactions in library preparation. The extremely porous structure of activated carbon provides a large surface area for binding these inhibitory compounds through chemical attraction [68]. However, researchers should note that carbon filters require regular replacement as the binding sites become saturated, and may themselves introduce bacterial DNA if not properly treated.
Experimental Protocol: Contaminant Removal for Soil DNA Extractions
- Step 1: Use the Quick-DNA Fecal/Soil Microbe Microprep kit (Zymo Research) or equivalent with soil-specific lysis buffers [16].
- Step 2: Incorporate a membrane filtration step (0.2 µm pore size) post-extraction to remove particulate matter and potential contaminating cells.
- Step 3: Apply DNA extract to activated carbon spin columns, incubating for 5 minutes at room temperature before centrifugation.
- Step 4: Implement a clean-up step using magnetic beads with size selection to remove short fragments that may represent degraded contaminants.
- Step 5: Quantify DNA using fluorometric methods (e.g., Qubit) and verify quality via fragment analyzer, monitoring for unusual peak patterns indicating contamination.
Bioinformatic Approaches for Contaminant Identification
Post-sequencing, computational methods enable the identification and removal of contaminant sequences. The use of blank extraction controls is critical for this process, as these controls reveal contaminants introduced during laboratory procedures. Bioinformatics pipelines should align sequences against databases of known laboratory contaminants (e.g., those identified in the Microflora Danica project) [5]. Additionally, taxonomic classification tools can flag unexpected taxa that appear across multiple samples from different habitats, which may indicate contamination rather than biological signal.
Computational Methods for Batch Effect Correction
Batch effects in multi-run sequencing studies arise from variations in library preparation, sequencing runs, and platform-specific biases. Computational correction methods aim to remove these technical artifacts while preserving biological signals.
Performance Evaluation of Batch Correction Methods
Table 3: Comparative Performance of scRNA-seq Batch Correction Methods [69]
| Method | Correction Efficacy | Artifact Introduction | Overall Recommendation |
|---|---|---|---|
| Harmony | High | Minimal (lowest artifacts) | Recommended - performs well across all tests |
| ComBat/ComBat-seq | Moderate | Moderate (detectable artifacts) | Use with caution - may alter biological signals |
| MNN | Moderate | High (considerable artifacts) | Not recommended - poorly calibrated |
| SCVI | Moderate | High (considerable artifacts) | Not recommended - poorly calibrated |
| LIGER | Moderate | High (considerable artifacts) | Not recommended - poorly calibrated |
| BBKNN | Moderate | Moderate (detectable artifacts) | Use with caution - may alter biological signals |
| Seurat | Moderate | Moderate (detectable artifacts) | Use with caution - may alter biological signals |
A rigorous evaluation of single-cell RNA sequencing batch correction methods revealed that most widely used algorithms are "poorly calibrated" and introduce measurable artifacts during the correction process [69]. According to this systematic assessment, Harmony was the only method that consistently performed well across all testing methodologies without introducing significant distortions to the data [69]. Methods including MNN, SCVI, and LIGER performed poorly, often altering the data considerably during correction [69].
Implementation Framework for Batch Correction
Protocol for Batch Effect Correction in Multi-Run Microbial Studies:
- Step 1: Perform sequencing run-specific quality control using tools like FastQC and monitor for platform-specific error profiles.
- Step 2: Generate a combined count table or feature table across all batches, ensuring consistent feature identification across runs.
- Step 3: Apply the Harmony algorithm to the combined dataset, using sequencing run or technology platform as the batch covariate.
- Step 4: Validate correction efficacy by visualizing data using Principal Component Analysis (PCA) or UMAP, coloring points by batch before and after correction.
- Step 5: Confirm biological preservation by verifying that known sample groupings (e.g., by habitat type) remain distinct post-correction.
Batch effect correction workflow for multi-run studies.
Integrated Workflow for Multi-Run Microbial Studies
Combining strategic wet-lab practices with computational correction produces the most robust results for multi-run microbial studies. The following integrated approach minimizes both contaminants and batch effects.
Comprehensive Experimental Design
Pre-Sequencing Phase:
- Standardize DNA extraction protocols across all samples, using the same kit lots when possible
- Include blank extraction controls in each processing batch to monitor contaminants
- For multi-run studies, distribute samples from each experimental group across sequencing runs to avoid confounding batch effects with biological signals
- Consider using negative selection probes to remove common contaminant DNA during library preparation
Sequencing Phase:
- When using multiple platforms, sequence overlapping samples across technologies to enable cross-platform calibration
- For Illumina sequencing, target the V3-V4 region rather than just V4 for improved taxonomic resolution [16]
- For Nanopore sequencing, utilize the latest flow cells (R10.4.1) and basecalling algorithms for improved accuracy [16]
Post-Sequencing Phase:
- Process all datasets through the same bioinformatic pipeline simultaneously
- Apply contaminant removal using control-based methods before batch correction
- Implement Harmony for batch effect correction when integrating data from multiple runs or platforms
- Validate results by confirming that biological replicates cluster together after correction
Multi-run study integrated workflow phases.
Essential Research Reagent Solutions
Table 4: Key Research Reagents and Materials for Contaminant-Free Microbial Studies
| Reagent/Kit | Primary Function | Application Notes |
|---|---|---|
| Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) | DNA extraction from complex matrices | Effectively removes humic acids and other PCR inhibitors common in soil [16] |
| Native Barcoding Kit 96 (Oxford Nanopore) | Multiplexed library preparation for Nanopore | Enables sample multiplexing while maintaining read length [16] |
| SMRTbell Prep Kit 3.0 (PacBio) | Library preparation for HiFi sequencing | Optimized for long-read circular consensus sequencing [16] |
| MiSeq Reagent Kits (Illumina) | Short-read sequencing chemistry | Various kit sizes available; v3 kits support 2x300 bp reads [67] |
| ZymoBIOMICS Gut Microbiome Standard | Positive control for microbiome studies | Validates entire workflow from extraction to analysis [16] |
| Activated Carbon Spin Columns | Removal of organic contaminants | Essential for environmental samples with high organic content [68] |
| Membrane Filtration Devices | Particulate and inhibitor removal | 0.2 µm pores effective for removing bacterial contaminants [68] |
The integration of data from multiple sequencing runs and platforms presents both challenges and opportunities in microbial ecology research. Based on current comparative data, Oxford Nanopore and PacBio platforms offer advantages for complex environmental samples through their long-read capabilities, which enhance strain-level resolution and reduce some forms of batch effects. For batch effect correction, Harmony emerges as the recommended computational approach due to its minimal introduction of artifacts during correction. When combined with robust wet-lab practices including standardized DNA extraction, blank controls, and strategic use of filtration technologies, researchers can effectively mitigate the impacts of both technical artifacts and contaminants. This multifaceted approach enables more valid integration of datasets across sequencing runs and platforms, ultimately supporting more powerful meta-analyses and reproducible results in microbial ecology studies.
For researchers in microbial ecology, selecting an appropriate DNA sequencing platform is a critical strategic decision that directly impacts the scope, depth, and cost of their investigations. The choice hinges on a careful balance between three competing factors: throughput (the volume of data generated), resolution (the taxonomic and functional detail obtained), and budget (encompassing both initial investment and ongoing costs). Next-generation sequencing (NGS) technologies have become the backbone of modern microbiome studies, enabling the profiling of complex communities from various environments. This guide provides an objective comparison of current sequencing platforms, grounded in experimental data, to help researchers align their technology selection with their specific scientific questions and financial constraints.
Platform Comparison at a Glance
The following tables summarize the core specifications and performance metrics of major sequencing platforms used in microbial ecology, based on manufacturer specifications and independent benchmarking studies.
Table 1: Key Technical Specifications and List Prices of Major Sequencing Platforms
| Platform (Model) | Technology Type | Max Throughput per Run | Read Length | Reported Accuracy | Estimated Capital Cost (USD) | Key Microbial Ecology Applications |
|---|---|---|---|---|---|---|
| Illumina MiSeq [70] | Short-read (SBS) | 15 Gb | 2 x 300 bp | >99.9% (Q30) | $20,000 - $100,000 [71] | 16S rRNA gene amplicon (e.g., V3-V4), shallow shotgun metagenomics [70] |
| Thermo Fisher Ion GeneStudio S5 Prime [72] | Short-read (Ion Semiconductor) | 50 Gb | 200-600 bp | >99% [72] | Mid-range [73] | Targeted gene panels, 16S rRNA gene amplicon sequencing [72] |
| PacBio Sequel IIe [74] | Long-read (SMRT) | 120 Gb (HiFi mode) | 10-20 kb HiFi reads | >99.9% [74] | High (>$500,000) [71] | Full-length 16S rRNA sequencing, metagenome-assembled genomes (MAGs) |
| Oxford Nanopore PromethION [74] | Long-read (Nanopore) | 1.9 Tb | Up to Mb-level | ~93.8% (raw); ~99.996% (consensus) [74] | Lower cost than PacBio [74] | Real-time pathogen monitoring, ultra-long read assembly, direct RNA sequencing |
Table 2: Experimental Performance Metrics from Comparative Studies
| Platform | Correlation with Theoretical Abundance (Mock Communities) [18] | Strengths (from Experimental Data) | Limitations (from Experimental Data) |
|---|---|---|---|
| Illumina (HiSeq 3000) | High (Spearman correlation >0.9) [18] | Low error rate, excellent for quantitative analysis [18] | Short reads limit resolution in complex regions [16] |
| PacBio (Sequel II) | Slightly decreased correlation vs. short-read [18] | Most contiguous assemblies (36/71 full genomes reconstructed) [18]; high accuracy for species-level ID [16] | Library preparation size filtering can bias abundance estimates [18] |
| Oxford Nanopore (MinION) | Slightly decreased correlation vs. short-read [18] | Capable of full-length 16S profiling; enables real-time analysis [16] [74] | Higher inherent error rate (though recent flow cells improve this) [16] [18] |
Note: Mock community studies involved synthetic samples with known compositions of 64-87 microbial strains to evaluate quantitative accuracy. [18]
Detailed Methodologies from Cited Experiments
To ensure the reproducibility of the comparative data cited, this section outlines the key experimental protocols from the benchmark studies.
- Sample Origin: Soil samples were collected from three distinct soil types (chernozem) in the Ob area forest-steppe region. Samples were sieved and stored at -20°C.
- DNA Extraction: performed using the Quick-DNA Fecal/Soil Microbe Microprep kit (Zymo Research), with DNA quantified via Qubit Fluorometer.
- 16S rRNA Amplification and Sequencing:
- PacBio (Sequel IIe): The full-length 16S rRNA gene was amplified using universal primers (27F/1492R) with barcodes. Library preparation used the SMRTbell Prep Kit 3.0.
- Oxford Nanopore (MinION): The full-length 16S rRNA gene was amplified with primers 27F and 1492R, and libraries were prepared with the Native Barcoding Kit 96 (SQK-NBD109).
- Illumina: The V4 and V3-V4 hypervariable regions were targeted for amplification and sequencing.
- Bioinformatics: Sequencing depth was normalized across platforms (10k-35k reads/sample). A standardized bioinformatics pipeline was applied, and data analysis included alpha and beta diversity metrics and taxonomic resolution assessment.
- Mock Communities: Three uneven synthetic microbial communities were constructed from 91 different bacterial and archaeal strains, comprising 64 to 87 genomes and representing 29 phyla. This created a controlled environment with known truth.
- Sequencing Platforms: The study compared seven platforms: Illumina HiSeq 3000, MGI DNBSEQ-G400, MGI DNBSEQ-T7, ThermoFisher Ion GeneStudio S5, Ion Proton P1, ONT MinION, and PacBio Sequel II.
- Library Preparation and Sequencing: Standardized library prep protocols specific to each manufacturer were followed for shotgun metagenomic sequencing, as detailed in the study.
- Data Analysis: Reads were aligned to reference genomes. Analyses included subsampling to evaluate the impact of sequencing depth on abundance correlation, de novo metagenomic assembly, and assembly quality assessment (e.g., mismatches per 100 kbp).
Sequencing Platform Selection Logic
The following diagram outlines a decision-making workflow to guide researchers in selecting a sequencing platform based on their primary research objective, required resolution, and budget.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Kits for Microbial Ecology Sequencing Workflows
| Item | Function in Experimental Workflow | Example Product & Manufacturer |
|---|---|---|
| Soil DNA Extraction Kit | Isolates high-quality microbial DNA from complex environmental matrices, critical for downstream success. | Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) [16] |
| 16S rRNA PCR Primers | Amplifies target hypervariable regions (e.g., V4, V3-V4) or the full-length gene for taxonomic profiling. | 27F/1492R (for full-length) [16]; Illumina 16S Amplicon Protocols |
| Library Preparation Kit | Prepares amplified DNA fragments for sequencing on a specific platform, often including barcoding. | SMRTbell Prep Kit 3.0 (PacBio) [16]; Native Barcoding Kit 96 (Oxford Nanopore) [16] |
| Metagenomic Standard | Serves as a positive control to evaluate sequencing accuracy, bias, and bioinformatics pipelines. | ZymoBIOMICS Gut Microbiome Standard (Zymo Research) [16] |
| Methylation Detection Kit | Enables direct study of DNA methylation and other epigenetic modifications without bisulfite conversion. | CUTANA meCUT&RUN (EpiCypher) [75] |
The optimal DNA sequencing platform for microbial ecology is not a one-size-fits-all proposition. Short-read platforms like Illumina MiSeq offer a cost-effective solution for high-throughput taxonomic profiling where species-level resolution is not paramount. [70] [18] Long-read platforms from PacBio and Oxford Nanopore are indispensable for achieving high taxonomic resolution through full-length 16S sequencing and for assembling complete genomes from complex metagenomic samples, despite their higher operational complexity and cost. [16] [18] [74] The most forward-looking strategies may involve hybrid approaches, using short-read data to enhance the accuracy of long-read assemblies, thereby maximizing both data quality and cost-efficiency. [18] Ultimately, a successful cost-benefit analysis requires a clear definition of research goals, an honest assessment of sample throughput, and a comprehensive understanding of the total cost of ownership.
Accurately identifying microbial species is a fundamental goal in microbial ecology, with profound implications for understanding ecosystem health, disease pathogenesis, and biogeochemical cycles. The choice of DNA sequencing platform significantly influences the taxonomic resolution achievable in microbiome studies. While short-read sequencing technologies like Illumina have been the workhorse for microbial community profiling for over a decade, their limited read length often restricts classification to the genus level. In contrast, third-generation sequencing platforms from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) generate long reads that span entire marker genes and genomic regions, enabling superior species-level and sometimes even strain-level resolution [76] [63].
This guide provides an objective comparison of current sequencing platforms, evaluating their performance for species-level identification in microbial ecology research. We summarize recent experimental data, detail methodological approaches, and provide practical strategies for platform selection based on specific research objectives.
Platform Comparison: Technical Capabilities and Performance
The following table summarizes the key characteristics of major sequencing platforms used in microbial ecology, based on recent comparative studies.
Table 1: Comparison of Sequencing Platforms for Microbial Species Identification
| Platform (Company) | Read Type | Typical Read Length | Key Strength | Key Limitation | Best Suited For |
|---|---|---|---|---|---|
| Illumina (e.g., MiSeq, NextSeq) [63] [1] | Short-read | 300-600 bp | High accuracy (~99.9%), low cost per sample, high throughput | Limited to hypervariable regions, hindering species-level resolution | Large-scale microbial surveys where genus-level profiling is sufficient |
| PacBio (Sequel IIe) [16] [77] [1] | Long-read (HiFi) | 10,000-25,000 bp | Very high accuracy (>99.9%) with HiFi reads, full-length 16S rRNA sequencing | Higher cost per sample, requires more input DNA | High-accuracy species-level identification for complex communities |
| Oxford Nanopore (MinION, PromethION) [63] [5] [1] | Long-read | 10,000-30,000 bp | Real-time sequencing, extreme portability, detects base modifications | Higher raw error rate, though improved with recent chemistry | Rapid, in-field species identification and large-scale metagenome-assembled genomes (MAGs) |
| Ion Torrent (GeneStudio S5) [78] [1] | Short-read | 200-400 bp | Fast run times, simple workflow | Issues with homopolymer errors, lower throughput | Targeted, rapid pathogen identification in clinical settings |
Recent comparative studies yield critical performance insights. For 16S rRNA gene sequencing, PacBio and ONT, which sequence the full-length ~1,500 bp gene, provide significantly finer taxonomic resolution than Illumina, which typically sequences only the V3-V4 hypervariable regions (~460 bp) [16] [63]. One soil microbiome study found that while Illumina captured greater apparent species richness, both PacBio and ONT enabled clear sample clustering by soil type, with PacBio showing a slight edge in detecting low-abundance taxa [16] [77]. For metagenomic analysis, long-read sequencing is transformative. A 2025 study using deep Nanopore sequencing of 154 soil samples recovered 15,314 previously unknown microbial species, expanding the phylogenetic diversity of the prokaryotic tree by 8% [5]. This demonstrates long-read sequencing's unparalleled power for discovering novel microbial diversity in complex environments.
Experimental Data and Methodologies from Recent Studies
16S rRNA Sequencing Platform Comparison for Soil Microbiomes
A 2025 study directly compared Illumina, PacBio, and ONT for 16S rRNA-based bacterial diversity profiling in three distinct soil types [16] [77].
Experimental Protocol:
- Sample Collection: Soil samples were collected from medium-humus chernozem in the forest-steppe region. The experimental design included three independent biological replicates per soil type.
- DNA Extraction: DNA was extracted using the Quick-DNA Fecal/Soil Microbe Microprep kit (Zymo Research).
- Library Preparation and Sequencing:
- PacBio: The full-length 16S rRNA gene was amplified and sequenced on a PacBio Sequel IIe system using the SMRTbell Prep Kit 3.0.
- ONT: The full-length 16S rRNA gene was amplified and sequenced on a MinION platform using the Native Barcoding Kit.
- Illumina: The V4 and V3-V4 regions were sequenced on an unspecified Illumina platform.
- Data Analysis: Sequencing depth was normalized across platforms (10,000 to 35,000 reads/sample). Data was processed using standardized bioinformatics pipelines tailored to each platform.
Key Findings:
- Taxonomic Resolution: Both long-read platforms (PacBio and ONT) provided comparable and superior taxonomic resolution to Illumina's short-read approach.
- Community Analysis: All technologies except Illumina's V4 region alone showed clear clustering of microbial communities by soil type.
- Accuracy: Despite ONT's inherently higher error rate, the use of modern algorithms (e.g., Emu) produced results closely matching the high accuracy of PacBio, suggesting errors do not significantly impact well-represented taxa [16] [77].
Respiratory Microbiome Profiling: Illumina vs. ONT
A 2025 study compared Illumina NextSeq and ONT MinION for 16S rRNA profiling of respiratory microbial communities from human and pig samples [63].
Experimental Protocol:
- Sample Collection: 34 respiratory samples were collected from ventilator-associated pneumonia patients and an experimental swine model.
- DNA Extraction: DNA was extracted in parallel for both platforms using the Sputum DNA Isolation Kit (Norgen Biotek).
- Library Preparation and Sequencing:
- Illumina: Libraries targeted the V3-V4 region and were sequenced on a NextSeq for 2x300 bp reads.
- ONT: Libraries were prepared for full-length 16S rRNA sequencing using the 16S Barcoding Kit and sequenced on a MinION Mk1C with R10.4.1 flow cells.
- Bioinformatics: Illumina data was processed with nf-core/ampliseq, while ONT data was basecalled with Dorado and analyzed with the EPI2ME Labs 16S Workflow.
Key Findings:
- Richness vs. Resolution: Illumina captured greater species richness, while ONT provided improved resolution for dominant bacterial species.
- Platform Bias: Differential abundance analysis (ANCOM-BC2) revealed significant platform-specific biases. ONT overrepresented certain taxa (e.g., Enterococcus, Klebsiella) while underrepresenting others (e.g., Prevotella, Bacteroides).
- Application Suitability: The study concluded that Illumina is ideal for broad microbial surveys, whereas ONT excels when species-level resolution and real-time analysis are required [63].
Recovering Microbial Genomes from Complex Environments
A landmark 2025 study demonstrated the power of long-read sequencing for large-scale recovery of metagenome-assembled genomes (MAGs) from terrestrial habitats [5].
Experimental Protocol:
- Sampling and Sequencing: 154 complex soil and sediment samples were selected from the >10,000-sample Microflora Danica project.
- DNA Sequencing: Deep long-read sequencing was performed on Oxford Nanopore platforms, generating a median of 94.9 Gbp per sample.
- Bioinformatic Analysis: The team developed the mmlong2 workflow, which incorporates:
- Multisample coverage binning: Using read mapping information from multiple samples to improve binning.
- Ensemble binning: Applying multiple binning algorithms to the same metagenome.
- Iterative binning: Repeatedly binning the metagenome to recover more MAGs.
Key Findings:
- The study recovered 23,843 MAGs, which were dereplicated into 15,640 species-level clusters.
- Of these, 15,314 represented previously undescribed microbial species, and 1,086 were from previously uncharacterized genera.
- This effort expanded the phylogenetic diversity of the prokaryotic tree of life by 8%, showcasing the profound ability of long-read sequencing to access the "microbial dark matter" in highly complex environments like soil [5].
Workflow Visualization
The following diagram illustrates a generalized experimental workflow for achieving species-level identification, integrating strategies from the cited studies.
Figure 1: Experimental workflow for species-level microbial identification.
Essential Research Reagent Solutions
The table below lists key reagents and kits used in the experimental studies cited, providing a practical resource for researchers designing similar experiments.
Table 2: Key Research Reagents and Kits for Sequencing-Based Microbial Identification
| Product Name | Manufacturer | Primary Function | Application Context |
|---|---|---|---|
| Quick-DNA Fecal/Soil Microbe Microprep Kit | Zymo Research | DNA extraction from complex environmental samples | Efficiently lyses diverse microbial cells and purifies inhibitor-free DNA from soil and fecal samples [16] [77]. |
| SMRTbell Prep Kit 3.0 | Pacific Biosciences | Library preparation for PacBio sequencing | Creates SMRTbell libraries for long-read sequencing on Sequel IIe systems, suitable for amplicons and genomes [77]. |
| 16S Barcoding Kit (SQK-16S114) | Oxford Nanopore | Preparation of full-length 16S rRNA libraries | Enables multiplexed, full-length 16S sequencing on MinION/GridION/PromethION platforms [63]. |
| QIAseq 16S/ITS Region Panel | Qiagen | Targeted 16S amplicon library prep | Designed for Illumina systems, targets hypervariable regions (e.g., V3-V4) for high-throughput short-read sequencing [63]. |
| ZymoBIOMICS Gut Microbiome Standard | Zymo Research | Community standard and positive control | Defined microbial community with known composition used to validate extraction, sequencing, and bioinformatics pipelines [16]. |
The strategic choice of a sequencing platform is paramount for achieving species-level identification in microbial ecology. Short-read platforms like Illumina remain a robust, cost-effective choice for large-scale studies where genus-level profiling is adequate. However, the emergence of high-accuracy long-read sequencing from PacBio and ONT has fundamentally advanced the field. PacBio's HiFi reads offer exceptional accuracy for full-length 16S sequencing and MAG recovery, while ONT provides unparalleled flexibility, real-time analysis, and the ability to sequence natively. For the most comprehensive exploration of complex environments like soil, where the majority of microbial diversity remains uncataloged, long-read sequencing is no longer just an alternative but is becoming the preferred tool for illuminating the "microbial dark matter" and achieving true species-level resolution [76] [5]. Future directions will likely involve hybrid approaches that leverage the complementary strengths of multiple platforms to obtain the most complete picture of microbial communities.
Head-to-Head Platform Comparison: Performance in Real-World Ecological Studies
In microbial ecology, the choice of DNA sequencing platform and analysis method can dramatically influence the interpretation of community structure and function. Achieving a fair, apples-to-apples comparison between technologies such as Illumina, Pacific Biosciences (PacBio), and Oxford Nanopore Technologies (ONT) requires a rigorous standardizing framework that controls for key variables like read depth and bioinformatic processing [16]. This guide outlines a standardized comparative approach, supported by recent experimental data, to help researchers objectively evaluate platform performance for their specific needs.
The Critical Need for Standardization in Sequencing Comparisons
The fundamental challenge in comparing sequencing technologies is that their inherent differences—read length, accuracy, and error profiles—can skew results. Without standardization, it is impossible to distinguish true biological signal from technological artifacts.
Third-generation long-read sequencing platforms, PacBio and ONT, offer full-length 16S rRNA gene sequencing, which provides finer taxonomic resolution, often enabling species-level identification [16] [79]. In contrast, traditional short-read methods like Illumina typically target hypervariable regions (e.g., V3-V4 or V4), which can lead to ambiguous taxonomic assignments [16]. However, long-read technologies have historically been associated with higher error rates, though recent improvements in chemistry and base-calling have substantially increased their accuracy [79] [63].
A robust comparison must therefore control for these variables through experimental replication and standardized data processing. Studies with limited biological replication can yield misleading conclusions, whereas incorporating multiple independent biological replicates, as done in recent comprehensive evaluations, minimizes random variation and enhances the reliability of diversity estimates [16] [79].
Standardizing Key Experimental Parameters
To ensure a fair and interpretable comparison, the following parameters must be carefully controlled and documented.
The Role of Read Depth and Coverage Uniformity
Sequencing coverage or depth describes the number of unique sequencing reads that align to a given region of a reference genome or amplicon. Greater depth provides higher statistical confidence that the results are accurate and not due to random sampling error [80].
Crucially, depth must be normalized across platforms for a direct comparison. For example, one recent soil microbiome study directly compared Illumina, PacBio, and ONT by analyzing all datasets at normalized depths of 10,000, 20,000, 25,000, and 35,000 reads per sample [16] [79]. This approach allows for the assessment of how each platform performs at different sequencing intensities.
It is also vital to distinguish between average coverage and coverage uniformity. Two datasets with the same average depth (e.g., 30x) can have vastly different scientific values if one has poor uniformity—leaving some genomic regions uncovered—while the other provides consistent coverage throughout [80]. Uniformity is particularly important for avoiding biases in microbial community representation.
Standardized Bioinformatics Pipelines
Using platform-specific bioinformatics tools without standardization introduces a major source of bias. A fair comparison requires a common analytical framework tailored to the strengths of each technology.
- Initial Quality Control (QC): Raw data from all platforms should undergo stringent QC. Tools like FastQC generate summaries of base quality scores, GC content, and adapter contamination. Low-quality bases and adapter sequences must be trimmed [81].
- Alignment and Mapping: Read alignment tools must be chosen based on read length.
- Variant and Taxon Calling: For microbiome studies, this involves assigning taxonomic units. It is critical to use a consistent reference database (e.g., SILVA) and classification algorithm across all platforms. For full-length 16S rRNA data from ONT, applying algorithms like Emu can help reduce error rates and generate fewer false positives [16].
The overall workflow for a standardized cross-platform comparison can be visualized as follows.
Experimental Protocols from Recent Comparative Studies
The following methodologies, derived from recent publications, provide a template for a well-controlled sequencing platform evaluation.
Protocol: Comparative 16S rRNA Sequencing for Soil Microbiome Analysis
This protocol is adapted from a 2025 study that provided a comprehensive evaluation of Illumina, PacBio, and ONT for soil microbiome profiling [16] [79].
1. Sample Collection and DNA Extraction
- Collect soil samples from multiple distinct soil types with independent biological replication (e.g., three distinct soil types, each with three replicates) [16].
- Homogenize samples and pass through a sterile sieve.
- Extract genomic DNA from all samples using the same commercial kit (e.g., Quick-DNA Fecal/Soil Microbe Microprep Kit, Zymo Research) following the manufacturer's protocol [16] [79].
- Quantify DNA concentration using a fluorometer (e.g., Qubit) and assess quality by agarose gel electrophoresis.
2. Library Preparation and Sequencing
- Illumina: Amplify the V3-V4 or V4 hypervariable regions of the 16S rRNA gene using region-specific primers and prepare libraries according to standard protocols (e.g., Illumina Nextera XT). Sequence on a platform such as Illumina NextSeq or NovaSeq to generate paired-end reads (e.g., 2 × 300 bp) [16] [63].
- PacBio: Amplify the full-length 16S rRNA gene using universal primers (e.g., 27F and 1492R) tagged with sample-specific barcodes. Prepare libraries using the SMRTbell Prep Kit and sequence on the PacBio Sequel IIe system [16] [79].
- Oxford Nanopore: Amplify the full-length 16S rRNA gene using the same or similar universal primers. Prepare libraries using the Native Barcoding Kit (e.g., SQK-NBD114.24) and sequence on a MinION Mk1C using an R10.4.1 flow cell [16] [63].
3. Bioinformatic Processing and Normalization
- Process raw data from each platform through a tailored but standardized pipeline.
- For Illumina, use a pipeline such as nf-core/ampliseq, which incorporates tools like DADA2 for error correction, paired-end read merging, and amplicon sequence variant (ASV) generation [63].
- For ONT, use the EPI2ME Labs 16S Workflow or a pipeline that incorporates the Emu algorithm for taxonomic classification to minimize false positives [16] [63].
- For PacBio, apply circular consensus sequencing (CCS) processing to generate highly accurate long reads (HiFi reads).
- Crucially, rarefy all datasets to the same sequencing depth (e.g., 10,000, 20,000, and 35,000 reads per sample) before performing downstream ecological analyses [16] [79].
Key Reagents and Research Solutions
The table below lists essential materials and tools used in the aforementioned protocol.
Table 1: Essential Research Reagents and Tools for Comparative Sequencing
| Item Name | Function in the Experiment | Specific Example |
|---|---|---|
| Soil DNA Extraction Kit | Isolates high-quality microbial genomic DNA from complex soil matrices. | Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) [16] |
| Universal 16S rRNA Primers | Amplifies the target region of the 16S rRNA gene for library construction. | 27F (AGRGTTYGATYMTGGCTCAG) / 1492R (RGYTACCTTGTTACGACTT) [16] |
| Long-Read Library Prep Kit | Prepares barcoded libraries for full-length 16S sequencing. | SMRTbell Prep Kit 3.0 (PacBio) [16]; Native Barcoding Kit (ONT) [16] |
| Fluorometer | Accurately quantifies double-stranded DNA concentration for library pooling. | Qubit Fluorometer (Thermo Fisher Scientific) [16] [79] |
| Standardized Database | Provides a consistent reference for taxonomic classification of sequences. | SILVA 138.1 prokaryotic SSU database [63] |
Performance Metrics and Comparative Data Analysis
After standardizing protocols and read depth, researchers can objectively compare platforms using key microbiological metrics.
Insights from Recent Platform Comparisons
Recent studies have yielded critical insights into the performance characteristics of different sequencing platforms.
Table 2: Comparative Performance of Sequencing Platforms in Microbiome Studies
| Platform | Read Length | Key Strengths | Key Limitations | Findings in Recent Comparative Studies |
|---|---|---|---|---|
| Illumina | Short (∼300 bp) | High per-base accuracy (<0.1%) [63]; excellent for genus-level surveys [63]. | Limited species-level resolution [63]; V4 region alone may not cluster samples by soil type (p=0.79) [16]. | Captured greater species richness in respiratory samples [63]. |
| PacBio | Long / Full-Length | Full-length 16S sequencing; high accuracy (>99.9%) with CCS [16] [79]; slightly better detection of low-abundance taxa [16]. | Lower throughput; reliance on error-correction algorithms [16]. | Provided comparable diversity to ONT; clustered samples by soil type effectively [16]. |
| Oxford Nanopore | Long / Full-Length | Real-time sequencing; full-length 16S for species-level resolution [63]. | Higher inherent error rates, though improved with R10.4.1 flow cells [16] [63]. | Results closely matched PacBio; errors did not significantly impact well-represented taxa interpretation [16]. |
The standardized workflow and the resulting data lead to a final analytical and decision-making process.
The choice of a sequencing platform is not one-size-fits-all but should be dictated by the specific research question. The move towards a balanced and flexible sequencing offering from companies, including the ability to generate both short and long-read data from a single instrument, is a promising trend for the future [82].
Based on the standardized comparative framework and recent data, the recommendations are as follows:
- Choose Illumina when the research goal is a broad, high-resolution survey of microbial diversity at the genus level across a large number of samples, and where the highest base-level accuracy is paramount for detecting very rare taxa [63].
- Choose PacBio when the study requires the highest possible taxonomic resolution (species to strain level) from full-length 16S sequencing with exceptional accuracy, particularly for detecting low-abundance organisms in complex communities like soil [16].
- Choose Oxford Nanopore when the application demands species-level resolution with real-time data streamin, in-field sequencing capability, or when long reads are essential for resolving complex regions, and when the latest bioinformatic tools can be leveraged to mitigate error rates [16] [63].
By adopting a standardized framework that controls for read depth and bioinformatics, researchers can make informed, objective decisions that maximize the return on sequencing investments and drive robust scientific discoveries in microbial ecology.
In microbial ecology, the choice of DNA sequencing platform is a critical decision that directly impacts the characterization of community diversity. Researchers commonly use alpha diversity to describe the species richness and evenness within a single sample and beta diversity to quantify the compositional differences between microbial communities from different samples. While Illumina short-read sequencing has been the longstanding workhorse for 16S rRNA gene amplicon studies, long-read platforms from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) are increasingly adopted for their ability to sequence the full-length 16S rRNA gene, promising enhanced taxonomic resolution. This guide objectively compares the performance of these three major platforms—Illumina, PacBio, and ONT—in generating alpha and beta diversity metrics, providing researchers with a data-driven foundation for selecting the most appropriate technology for their specific study aims in drug development and microbial ecology.
Platform Performance at a Glance
The table below summarizes the key performance characteristics of Illumina, PacBio, and ONT sequencing platforms for 16S rRNA amplicon sequencing, based on recent comparative studies.
Table 1: Comparative Overview of Sequencing Platforms for 16S rRNA Amplicon Sequencing
| Feature | Illumina | PacBio | Oxford Nanopore (ONT) |
|---|---|---|---|
| Typical Read Length | Short (e.g., 300-600 bp, V3-V4 region) [83] | Long (Full-length ~1,450 bp) [83] | Long (Full-length ~1,500 bp) [63] |
| Sequencing Accuracy | High (<0.1% error rate) [63] | Very High (HiFi reads ~Q27) [83] | Moderate (Historically higher, but improved with latest chemistries to >99%) [16] [63] |
| Species-Level Classification Rate | 47% [83] | 63% [83] | 76% [83] |
| Key Strength | High throughput & accuracy for genus-level surveys [63] | High-fidelity long reads for species resolution [83] | Ultra-long reads, real-time analysis, species-level resolution [83] [63] |
| Key Limitation | Limited to hypervariable regions, lower species-level resolution [83] | Lower throughput, higher DNA input requirements [83] | Higher raw error rate requires specialized bioinformatics [83] |
Experimental Evidence: A Deeper Dive into the Data
Taxonomic Resolution Across Platforms
A direct comparative study of rabbit gut microbiota, which sequenced the same DNA extracts on all three platforms, provides clear quantitative data on taxonomic resolution. The platforms showed comparable performance at the family level, classifying over 99% of sequences. However, significant differences emerged at finer taxonomic resolutions [83]:
- ONT demonstrated the highest species-level classification at 76%.
- PacBio followed with a 63% species-level classification rate.
- Illumina had the lowest species-level resolution at 47%.
It is crucial to note that a large proportion of these species-level identifications were labeled as "uncultured_bacterium" across all platforms, highlighting that database limitations remain a significant challenge for precise species-level characterization, regardless of the technology used [83].
Impact on Alpha and Beta Diversity
The effect of the sequencing platform on downstream diversity metrics is complex and can depend on the sample type.
Alpha Diversity (Within-sample Diversity): Findings on species richness (a component of alpha diversity) are not consistent across studies. One study on respiratory microbiomes reported that Illumina captured greater species richness than ONT [63]. In contrast, a comprehensive soil microbiome study found that PacBio and ONT provided comparable bacterial diversity assessments, with PacBio showing a slightly higher efficiency in detecting low-abundance taxa [16] [79]. This suggests that the performance may be influenced by the ecosystem's complexity.
Beta Diversity (Between-sample Diversity): The platform choice can significantly impact beta diversity results, which measure the dissimilarity between microbial communities. The rabbit gut study found that while relative abundances of taxa were highly correlated, diversity analysis showed significant differences between the taxonomic compositions derived from the three platforms [83]. Another study noted that beta diversity differences were more pronounced in complex pig respiratory microbiomes than in human samples, indicating that the platform effect is amplified in more diverse communities [63]. Despite these technical differences, a key finding from soil research is that all three platforms consistently ensured clear clustering of samples based on soil type, demonstrating that biological signals remain robust across technologies [16].
Experimental Protocols from Cited Studies
To ensure the comparability of data when evaluating different sequencing platforms, standardized protocols for sample processing and bioinformatic analysis are essential. The following workflow, generalized from the methodologies of the cited comparative studies, outlines the key steps from sample collection to data interpretation.
Figure 1: A generalized experimental workflow for comparative sequencing platform studies.
Sample Preparation and Sequencing
The following table details the specific reagents and kits used in the comparative studies discussed in this guide.
Table 2: Key Research Reagent Solutions for 16S rRNA Sequencing
| Reagent / Kit | Function | Platform Application |
|---|---|---|
| DNeasy PowerSoil Kit (QIAGEN) [83] | DNA extraction from complex samples (feces, soil) | Illumina, PacBio, ONT |
| Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) [16] | DNA extraction from complex samples | Illumina, PacBio, ONT |
| Nextera XT Index Kit (Illumina) [83] | Library preparation & indexing for Illumina | Illumina |
| SMRTbell Express Template Prep Kit 2.0/3.0 (PacBio) [83] [16] | Library preparation for PacBio systems | PacBio |
| 16S Barcoding Kit (SQK-RAB204 / SQK-16S024) (ONT) [83] [63] | Library preparation & barcoding for Nanopore | ONT |
| KAPA HiFi HotStart DNA Polymerase [83] | High-fidelity PCR amplification | PacBio |
| SILVA database [83] [63] | Taxonomic classification of 16S rRNA sequences | Illumina, PacBio, ONT |
Key Experimental Notes:
- Primer Choice: Illumina studies typically amplify specific hypervariable regions (e.g., V3-V4 using primers from Klindworth et al., 2013), while long-read platforms target the full-length 16S rRNA gene using universal primers 27F and 1492R [83] [16].
- Platform-Specific Bioinformatics: The choice of denoising pipeline is critical and often platform-dependent. Illumina and PacBio HiFi reads, due to their high accuracy, are typically processed with the DADA2 pipeline to generate Amplicon Sequence Variants (ASVs). For ONT, which has a higher inherent error rate, specialized pipelines like Spaghetti (which uses an OTU-based approach) or Emu are often employed for more accurate results [83] [16].
The selection of a sequencing platform for microbial ecology research involves a careful trade-off between read length, accuracy, and taxonomic resolution. Illumina remains a robust and cost-effective choice for large-scale studies where high-throughput and genus-level community profiling are the primary goals. In contrast, PacBio HiFi and ONT sequencing offer superior species-level resolution due to their long-read capabilities, which is invaluable for applications in drug development and clinical diagnostics requiring precise taxonomic assignment.
Current evidence suggests that while long-read platforms improve species-level classification, all technologies are susceptible to database limitations. Furthermore, the sequencing platform itself can be a significant source of variation in beta diversity analyses. Therefore, the optimal choice depends heavily on the research question: Illumina is ideal for broad microbial surveys, whereas PacBio and ONT are better suited for studies demanding high taxonomic precision. Future efforts should focus on improving reference databases and developing hybrid or integrated sequencing approaches to fully leverage the complementary strengths of these powerful technologies.
In microbial ecology, accurate taxonomic profiling of biofilms is crucial for understanding community dynamics, ecosystem functions, and biogeochemical processes in both soil and aquatic environments. The choice of DNA sequencing platform fundamentally influences the resolution of these profiles, creating a critical trade-off between genus-level identification and more precise species-level classification. This comparison guide objectively evaluates the performance of second and third-generation sequencing platforms for taxonomic profiling of soil and water biofilms, providing supporting experimental data to inform researchers and drug development professionals.
Methodological Approaches in Comparative Studies
Experimental Design and Sample Processing
Comparative studies typically employ standardized samples across multiple sequencing platforms to enable direct performance comparisons. For soil biofilms, researchers often utilize artificial soil communities or collected natural samples, with DNA extraction methods optimized for comprehensive lysis through chemical, enzymatic, and mechanical approaches [84] [85]. Water biofilm studies commonly collect epilithic biofilms from natural river systems, preserving samples in DNA stabilization buffers and employing centrifugation to concentrate biomass before extraction [86].
A key methodological consideration is the use of synthetic microbial communities with known composition, which serve as gold standards for evaluating platform performance. These mocks can contain 64-87 microbial strains spanning 29 bacterial and archaeal phyla, with relative abundance distributions spanning over three orders of magnitude [18]. This controlled approach enables precise measurement of accuracy in taxonomic assignment and abundance estimation.
Sequencing Platforms and Target Regions
The primary sequencing platforms compared in recent studies include:
- Short-read platforms: Illumina systems (targeting V4 or V3-V4 hypervariable regions of 16S rRNA gene)
- Long-read platforms: Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) (sequencing full-length 16S rRNA gene)
Bioinformatic processing typically involves platform-specific pipelines followed by taxonomic assignment against reference databases such as the Genome Taxonomy Database (GTDB) [87] [88].
Performance Comparison of Sequencing Platforms
Quantitative Analysis of Taxonomic Resolution
Table 1: Comparative Performance of Sequencing Platforms for Biofilm Analysis
| Platform | Read Length | Target Region | Species-Level Resolution | Genus-Level Resolution | Best Application Context |
|---|---|---|---|---|---|
| Illumina | ~250-300 bp | V4 or V3-V4 | Limited | Excellent | High-throughput community structure analysis [86] [89] |
| PacBio | ~1,500-3,000 bp | Full-length 16S | Excellent | Excellent | Studies requiring precise species identification [86] [16] |
| ONT | Varies | Full-length 16S | Good to Excellent | Excellent | Rapid analysis and field applications [16] [18] |
Table 2: Quantitative Performance Metrics Across Platforms
| Performance Metric | Illumina | PacBio | ONT |
|---|---|---|---|
| Estimated Error Rate | <1% [18] | ~0.1% [16] | ~1-11% [16] [18] |
| Community Structure Correlation | High [86] | High [86] | Moderate to High [16] |
| Detection of Low-Abundance Taxa | Limited [86] | Enhanced [16] | Moderate [16] |
| Throughput | High [86] [89] | Moderate [86] | Moderate [16] |
| Cost Efficiency | High [86] | Moderate [86] | Moderate [16] |
Platform Performance in Different Biofilm Environments
Soil Biofilms: In soil environments, PacBio and ONT platforms demonstrate superior ability to resolve species-level taxonomy compared to Illumina. A comparative evaluation of 16S rRNA gene sequencing in soil microbiomes found that despite differences in sequencing accuracy, ONT produced results that closely matched those of PacBio, suggesting that ONT's inherent sequencing errors do not significantly affect the interpretation of well-represented taxa [16]. Both long-read technologies enabled clear clustering of samples based on soil type, whereas the V4 region alone (typical of Illumina workflows) showed no soil-type clustering (p = 0.79) [16].
Water Biofilms: Similarly, in river biofilm samples, PacBio long-read sequencing provided higher taxonomic resolution, enabling classification of taxa that remained unassigned in short-read Illumina datasets [86]. This enhanced resolution is particularly beneficial for ecological monitoring as it improves species-level identification. Despite this difference in resolution, both sequencing methods produced comparable bacterial community structures regarding taxon relative abundance, suggesting that the sequencing approach does not profoundly affect the comparative assessment of community composition [86].
Experimental Workflow for Comparative Taxonomic Profiling
The following diagram illustrates the generalized experimental workflow for comparative analysis of sequencing platforms in biofilm studies:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Biofilm Taxonomic Profiling
| Reagent/Material | Function | Example Application |
|---|---|---|
| DNA Preservation Buffers | Stabilizes nucleic acids during sample transport and storage | Ammonium sulphate-based buffer for river biofilms [86] |
| Multi-component Lysis Reagents | Comprehensive cell disruption through chemical, enzymatic and mechanical action | ZymoBIOMICS kits with bead beating for soil and water biofilms [16] [85] |
| PCR Amplification Kits | Target region amplification with high-fidelity polymerases | Q5 high-fidelity DNA polymerase for 16S amplification [86] |
| Library Preparation Kits | Platform-specific library construction | SMRTbell Prep Kit for PacBio; Native Barcoding Kit for ONT [16] [18] |
| Reference Databases | Taxonomic classification of sequence data | Genome Taxonomy Database (GTDB) for standardized taxonomy [87] [88] |
Functional Implications of Taxonomic Resolution
The resolution achieved by different sequencing platforms has direct implications for understanding microbial community functioning. In soil biofilms, higher microbial diversity and evenness have been associated with enhanced metabolic activity, with biofilms sustaining 23-times more active microbes and consuming 65.4% more oxygen in topsoil compared to free-living communities [84]. Global metagenomic analyses have revealed distinct functional profiles between terrestrial and aquatic ecosystems, with soil metagenomes exhibiting higher abundance of genes associated with carbohydrate, sulfur, and potassium metabolisms, while water metagenomes harbor more genes related to nitrogen and iron metabolisms [90].
Species-level identification enables researchers to detect functionally distinct taxa that would be grouped together at genus level. For instance, in nuclear storage pool biofilms, proteotyping approaches identified three primary genera (Sphingomonas, Caulobacter, and Acidovorax) and revealed differential expression of metabolic pathways between them, highlighting their functional specialization within the extreme environment [91].
Based on comparative experimental data:
For studies requiring species-level resolution: PacBio platform is recommended due to its high accuracy (>99.9%) and ability to sequence full-length 16S rRNA genes, providing superior taxonomic resolution for both soil and water biofilms [86] [16].
For high-throughput community structure analysis: Illumina platforms offer cost-effective solutions for genus-level profiling and comparative assessment of community composition across large sample sets [86] [89].
For rapid analysis and field applications: ONT technologies provide a balanced approach with decreasing error rates and the advantage of real-time data processing, making them increasingly suitable for comprehensive taxonomic profiling [16] [18].
The choice between genus and species-level resolution ultimately depends on research objectives, with species-level identification being crucial for detecting subtle community shifts, identifying pathogenic variants, and understanding functional adaptations in specialized environments, while genus-level analysis may suffice for broader ecological patterns and community dynamics.
In microbial ecology research, the accurate measurement of taxonomic abundance is foundational to understanding community structure and function. However, the choice of DNA sequencing platform itself introduces specific, systematic biases that can significantly influence abundance measurements. Different sequencing technologies vary in their read length, accuracy, error profiles, and sensitivity to genomic features, all of which can alter the apparent composition of a microbial community. This guide provides an objective comparison of leading sequencing platforms—Illumina, Oxford Nanopore Technologies (ONT), and PacBio—evaluating their performance in microbial ecology applications based on recent experimental data. The objective is to equip researchers with the information needed to select the most appropriate technology and correctly interpret their metagenomic and 16S rRNA sequencing results.
Performance Comparison of Sequencing Platforms
The following tables summarize key performance metrics and their impact on abundance measurements for the major sequencing platforms, based on recent comparative studies.
Table 1: Technical Specifications and Associated Biases of Sequencing Platforms
| Platform | Read Length | Key Strengths | Key Limitations | Impact on Abundance Measurements |
|---|---|---|---|---|
| Illumina [16] [63] | Short-read (e.g., 2x300 bp) | High per-base accuracy (~99.9%); high sequencing depth [63]. | Inability to resolve full-length 16S rRNA; PCR amplification bias [16] [92]. | Captures greater species richness but may lack resolution for closely related species, skewing community diversity estimates [16] [63]. |
| PacBio [16] | Long-read (Full-length 16S) | High accuracy (>99.9%) with Circular Consensus Sequencing (CCS); enables species-level identification [16]. | Lower throughput than Illumina; requires error-correction algorithms [16]. | Provides comparable diversity assessments to ONT; slightly superior in detecting low-abundance taxa [16]. |
| Oxford Nanopore (ONT) [16] [63] [5] | Long-read (Full-length 16S, up to N50 of ~6.1 kbp) | Real-time sequencing; minimal PCR amplification needed; capable of recovering high-quality Metagenome-Assembled Genomes (MAGs) [16] [5]. | Historically higher error rates (5-15%), though recent flow cells (R10.4.1) and basecallers have improved accuracy to >99% [16] [63]. | Can overrepresent certain taxa (e.g., Enterococcus, Klebsiella) while underrepresenting others (e.g., Prevotella, Bacteroides) compared to Illumina [63]. |
Table 2: Comparative Performance in Microbial Community Profiling
| Performance Metric | Illumina | PacBio | Oxford Nanopore (ONT) |
|---|---|---|---|
| Species Richness | Higher [16] [63] | Comparable to ONT [16] | Lower than Illumina [63] |
| Species-Level Resolution | Limited [63] | High [16] | High [63] |
| Community Evenness | Comparable to ONT [63] | Information Not Available | Comparable to Illumina [63] |
| Detection of Low-Abundance Taxa | Effective | Slightly more efficient than ONT [16] | Effective, but slightly less than PacBio [16] |
| Beta Diversity Clustering | Significant differences in complex microbiomes (e.g., pig samples) [63] | Clear clustering by sample type (e.g., soil) [16] | Significant differences in complex microbiomes; clusters well by sample type [16] [63] |
Detailed Experimental Protocols from Cited Studies
Comparative Evaluation of Whole Exome Sequencing Platforms on DNBSEQ-T7
A 2025 study provided a robust framework for comparing four commercial Whole Exome Sequencing (WES) platforms: BOKE (TargetCap Core Exome Panel v3.0), IDT (xGen Exome Hyb Panel v2), Nad (EXome Core Panel), and Twist (Twist Exome 2.0) on a DNBSEQ-T7 sequencer [93].
- Sample Preparation: Genomic DNA from the reference sample NA12878 was physically fragmented to 200-300 bp using a Covaris E210 ultrasonicator [93].
- Library Construction: A total of 72 libraries were prepared using the MGIEasy UDB Universal Library Prep Set on an MGISP-960 automated system. Each library was uniquely dual-indexed with UDB primers [93].
- Hybridization Capture: The study employed both 1-plex and 8-plex hybridization designs. A key comparison was made between using each probe manufacturer's proprietary enrichment reagents and a unified protocol using MGIEasy Fast Hybridization and Wash Kit [93].
- Sequencing & Analysis: Post-capture libraries were pooled and sequenced on one lane of a DNBSEQ-T7 for PE150. Data was processed using the MegaBOLT pipeline, aligned to hg19, and variants were called against dbSNP build 151 [93].
Comparative Analysis of Illumina and Oxford Nanopore for 16S rRNA Profiling
A 2025 study directly compared Illumina and ONT for profiling respiratory microbiomes [63].
- Sample Collection: 34 respiratory samples (20 from human VAP patients, 14 from a swine VAP model) were collected and stored at -80°C [63].
- DNA Extraction: DNA was extracted using the Sputum DNA Isolation Kit (Norgen Biotek), with concentration and quality assessed via Nanodrop and Qubit Fluorometer [63].
- Illumina Library Prep & Sequencing: The V3-V4 hypervariable region of the 16S rRNA gene was amplified using the QIAseq 16S/ITS Region Panel (Qiagen). Libraries were sequenced on an Illumina NextSeq for 2x300 bp reads [63].
- ONT Library Prep & Sequencing: Full-length 16S rRNA gene sequencing libraries were prepared using the ONT 16S Barcoding Kit 24 V14 (SQK-16S114.24). Sequencing was performed on a MinION Mk1C with an R10.4.1 flow cell for up to 72 hours [63].
- Bioinformatics: Illumina data was processed with the nf-core/ampliseq pipeline, using DADA2 for Amplicon Sequence Variant (ASV) inference. ONT data was basecalled with Dorado and analyzed using the EPI2ME Labs 16S Workflow. Both used the SILVA 138.1 database for taxonomic classification [63].
Evaluation of Sequencing Platforms for Soil Microbiome Analysis
A 2025 study compared Illumina, PacBio, and ONT for analyzing bacterial diversity in soil microbiomes [16].
- Sample Collection: Soil samples were collected from three distinct soil types in a forest-steppe region, with three independent biological replicates per sample to ensure robustness [16].
- DNA Extraction: DNA was extracted using the Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) and quantified via Qubit Fluorometer [16].
- PacBio Sequencing: The full-length 16S rRNA gene was amplified with universal primers and sequenced on a PacBio Sequel IIe system using the SMRTbell Prep Kit 3.0 [16].
- ONT Sequencing: The full-length 16S rRNA gene was amplified with primers 27F and 1492R, and libraries were prepared with the Native Barcoding Kit 96 for sequencing on a MinION platform [16].
- Data Analysis: To ensure a fair comparison, sequencing depth was normalized across all platforms (10,000, 20,000, 25,000, and 35,000 reads per sample), and standardized bioinformatics pipelines were applied [16].
Workflow Diagram of a Comparative Platform Study
The diagram below illustrates the logical workflow of a typical study designed to compare sequencing platforms for microbiome analysis, integrating key steps from the cited protocols.
Comparative Sequencing Study Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
The following table lists essential kits and reagents used in the featured comparative studies, which are critical for ensuring reproducibility and accuracy in sequencing-based microbial ecology studies.
Table 3: Essential Research Reagents for Sequencing-Based Microbial Ecology
| Reagent / Kit Name | Primary Function | Specific Application / Note |
|---|---|---|
| Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) [16] | DNA Extraction | Optimized for efficient lysis of difficult-to-lyse microbial cells in soil and fecal samples. |
| Sputum DNA Isolation Kit (Norgen Biotek) [63] | DNA Extraction | Designed for optimal DNA yield and purity from low-biomass, mucinous respiratory samples. |
| MGIEasy UDB Universal Library Prep Set (MGI) [93] | Library Preparation | Used for high-throughput, automated library construction with unique dual indexing (UDI). |
| QIAseq 16S/ITS Region Panel (Qiagen) [63] | Target Amplification & Library Prep | For Illumina-based amplification of the 16S rRNA V3-V4 hypervariable region. |
| SMRTbell Prep Kit 3.0 (PacBio) [16] | Library Preparation | Used for preparing SMRTbell libraries for PacBio's long-read sequencing platform. |
| 16S Barcoding Kit (Oxford Nanopore) [63] | Target Amplification & Library Prep | Enables PCR-based barcoding and preparation of full-length 16S rRNA amplicons for ONT sequencing. |
| MGIEasy Fast Hybridization and Wash Kit (MGI) [93] | Target Enrichment | A unified hybridization capture protocol compatible with multiple commercial exome probe sets. |
| SILVA 138.1 SSU Database [63] | Bioinformatics | A curated, high-quality reference database for taxonomic classification of 16S rRNA gene sequences. |
The choice of sequencing platform is a critical experimental design decision that directly influences measurements of microbial abundance and diversity. Illumina remains a powerful tool for broad microbial surveys where high sequencing depth is required to capture species richness, particularly in highly complex communities. PacBio offers a compelling combination of long reads and high accuracy, providing superior resolution for species-level taxonomy and slightly better detection of low-abundance taxa. Oxford Nanopore technology provides the advantages of ultra-long reads and real-time sequencing, which are invaluable for genome-resolved metagenomics and field applications, though researchers must be mindful of its specific taxonomic biases.
No single platform is universally superior. The decision should be guided by the specific research question, whether the priority is high depth of coverage (Illumina), high taxonomic resolution with accuracy (PacBio), or long reads and portability (ONT). As the field advances, hybrid approaches that leverage the complementary strengths of multiple technologies are emerging as a powerful strategy for the most comprehensive and accurate characterization of microbial ecosystems.
The choice between full-length 16S ribosomal RNA (rRNA) gene sequencing and partial region sequencing represents a critical methodological crossroads in microbial ecology. Evidence from recent comparative studies indicates that full-length 16S rRNA sequencing provides superior taxonomic resolution, enabling more accurate species-level classification of complex microbial communities. However, the performance of specific hypervariable regions varies, with the V1-V3 region often delivering results closest to full-length sequences for certain sample types. The emergence of third-generation sequencing platforms (Pacific Biosciences and Oxford Nanopore Technologies) has made full-length sequencing increasingly accessible, though partial sequencing via second-generation platforms remains a cost-effective alternative for genus-level analyses where sequencing resources are constrained.
The 16S rRNA gene has served as the cornerstone of microbial ecology and environmental microbiology for decades, providing a universal genetic marker for bacterial identification and phylogenetic analysis. This gene contains nine hypervariable regions (V1-V9) flanked by conserved sequences, which together offer a reliable framework for differentiating bacterial taxa. The central question facing researchers today is whether to sequence the full-length gene (~1500 bp) or target specific hypervariable regions through partial sequencing. This comprehensive analysis synthesizes recent experimental evidence to determine how this choice impacts clustering efficiency, taxonomic classification accuracy, and the biological interpretation of microbial community data across diverse sample types and sequencing platforms.
Technical Comparison: Full-Length Versus Partial 16S Sequencing
Table 1: Technical Characteristics of Full-Length vs. Partial 16S Sequencing Approaches
| Feature | Full-Length 16S Sequencing | Partial 16S Sequencing |
|---|---|---|
| Sequencing Target | Complete 16S rRNA gene (V1-V9) | Specific hypervariable regions (e.g., V3-V4, V4, V1-V3) |
| Typical Read Length | 1,200-1,650 bp [22] [94] | 250-600 bp (depending on region targeted) |
| Taxonomic Resolution | Species to strain level [95] | Primarily genus level, limited species resolution [22] [95] |
| Primary Technologies | PacBio SMRT, Oxford Nanopore [40] [22] [16] | Illumina platforms [16] [94] |
| Key Advantage | Comprehensive phylogenetic information | Lower cost, established protocols |
| Major Limitation | Higher cost per sample, complex data analysis | Restricted phylogenetic resolution, primer bias |
Experimental Evidence: Performance Across Sample Types
Oropharyngeal Microbiome Profiling
A 2025 comparative analysis of human oropharyngeal swabs demonstrated that methodological choices significantly impact results. Researchers compared primer sets with different degeneracy for full-length 16S rRNA sequencing using Oxford Nanopore's MinION platform. The more degenerate primer set (27F-II) yielded significantly higher alpha diversity (Shannon index: 2.684 vs. 1.850; p < 0.001) and detected a broader range of taxa across all phyla compared to the standard primer (27F-I). Taxonomic profiles generated with 27F-II strongly correlated with a large-scale reference dataset (Pearson's r = 0.86, p < 0.0001), whereas profiles from 27F-I showed weak correlation (r = 0.49, p = 0.06). The standard primer overrepresented Proteobacteria and underrepresented key genera including Prevotella, Faecalibacterium, and Porphyromonas [40].
Experimental Protocol: Oropharyngeal swabs were collected from 80 donors with no history of acute inflammation. Swabs were applied to teeth, tongue, and buccal mucosa before pharyngeal insertion. Samples were transferred to DNA/RNA shielding buffer, and DNA was extracted using the Quick-DNA HMW MagBead kit. Two sequencing libraries were prepared using different primer sets (27F-I and 27F-II). PCR amplification was performed, followed by sequencing on the MinION Mk1C platform. Bioinformatic analysis included alpha diversity calculations and taxonomic profiling [40].
Skin Microbiome Analysis
A 2024 investigation of skin microbiota from multiple human anatomical sites provided direct comparison between full-length 16S sequences and derived sub-regions. Researchers conducted full-length 16S sequencing of 141 skin samples using the PacBio platform, then generated derived 16S sub-region data through in silico experiments. The study confirmed that full-length sequences provide superior taxonomic resolution, though even full-length sequencing cannot achieve 100% species-level resolution for skin samples. Notably, the V1-V3 region offered resolution comparable to full-length 16S sequences, outperforming other hypervariable regions studied. For high-abundance bacteria (TOP30), genus-level resolution remained generally consistent across different variable regions [22].
Experimental Protocol: Researchers collected 141 skin microbiome specimens from 22 healthy volunteers, including intraaural skin, circumaural skin, palmar skin, nasal skin, and oral epithelial skin swabs. Genomic DNA was extracted using the PowerSoil DNA Isolation kit. The complete 16S rRNA gene was amplified using primers 27F (AGRGTTTGATYNTGGCTCAG) and 1492R (TASGGHTACCTTGTTASGACTT). PCR conditions included initial denaturation at 95°C for 2 min, followed by 25 cycles of denaturation at 98°C for 10 s, annealing at 55°C for 30 s, extension at 72°C for 90 s, and final extension at 72°C for 2 min. Sequencing was performed on the PacBio Sequel II system. Sub-regions (V1-V2, V1-V3, V3-V4, V4, V5-V9) were extracted in silico from full-length sequences based on primer binding sites [22].
Soil Microbiome Study
A 2025 comparative evaluation of sequencing platforms for soil microbiome analysis examined Illumina (V4 and V3-V4 regions), PacBio (full-length and trimmed V3-V4/V4 regions), and Oxford Nanopore Technologies (full-length). The research demonstrated that despite differences in sequencing accuracy, ONT and PacBio provided comparable bacterial diversity assessments, with PacBio showing slightly higher efficiency in detecting low-abundance taxa. Importantly, regardless of sequencing technology and the choice of target region (full-length or partial), microbial community analysis ensured clear clustering of samples based on soil type. The sole exception was the V4 region, where no soil-type clustering was observed (p = 0.79) [16].
Respiratory Microbiome in Wildlife Conservation
A 2024 study on African elephant respiratory microbiota developed a novel approach to full-length 16S sequencing using Illumina's short-read iSeq 100 platform. Researchers implemented a modified 150 bp paired-end sequencing technique and assembly workflow that generated assembled amplicons averaging 869 bp in length. This approach provided taxonomic assignments consistent with the theoretical composition of a mock community and respiratory microbiota of other mammals. The study identified tentative bacterial signatures representing distinct respiratory tract compartments (trunk and lower respiratory tract), demonstrating the value of enhanced sequencing approaches even within technological constraints [94].
Quantitative Comparison of Annotation Performance
Table 2: Species-Level Annotation Rates Across Sample Types Using Full-Length 16S Sequencing [95]
| Sample Type | Species-Level Average Annotation Rate (Reads) | Genus-Level Average Annotation Rate (Reads) |
|---|---|---|
| Feces | 90% | 90% |
| Gut Content | 90% | 90% |
| Saliva/Sputum | 87% | 93% |
| Nasal/Oral Swab | 89% | 95% |
| Skin Swab | 88% | 90% |
| Soil | 75% | 82% |
| Water | 75% | 82% |
| Vaginal Swab | 85% | 90% |
| Rumen | 85% | 88% |
| Sludge | 72% | 75% |
| Fermentation | 90% | 95% |
Diagnostic Applications: Clinical Performance Comparison
A 2025 clinical diagnostic study compared Sanger sequencing with Oxford Nanopore Technologies (ONT) for 16S rRNA gene sequencing in 101 culture-negative clinical samples. The positivity rate for clinically relevant pathogens was significantly higher for ONT (72%) compared to Sanger sequencing (59%). ONT also detected more samples with polymicrobial presence (13 vs. 5). Concordance between Sanger and ONT sequencing was 80%. Notably, in one joint fluid sample, Borrelia bissettiiae was identified by ONT but not by Sanger sequencing. The researchers concluded that ONT sequencing improves detection of both monobacterial and multiple bacterial species in clinical diagnostics [96].
Experimental Protocol: Between June 2021 and August 2022, 101 clinical culture-negative samples positive in 16S rRNA gene PCR were analyzed. DNA libraries for ONT sequencing were prepared according to the SQK-SLK109 protocol with additional reagents from New England Biolabs. Sequencing was performed on a GridION with FLO-MIN104/R9.4.1 flow cells using super-accurate basecalling with read filtering (min_qscore = 10). ONT data were processed using the EPI2ME platform's Fastq 16S workflow and an in-house pipeline using the k-mer alignment (KMA) tool mapping reads to a database built from the NCBI RefSeq and SILVA 138.1 databases [96].
Sequencing Platform Performance
Table 3: Sequencing Platform Comparison for 16S rRNA Gene Sequencing [16]
| Platform | Technology Type | Read Length | Target Region | Key Strength | Key Limitation |
|---|---|---|---|---|---|
| PacBio | Third-generation | Long-read (full-length) | Full-length 16S | High accuracy (>99.9%) with CCS, species-level resolution | Higher cost, complex workflow |
| Oxford Nanopore | Third-generation | Long-read (full-length) | Full-length 16S | Real-time sequencing, portable | Historically higher error rates, though improved with R10.4.1 flow cells |
| Illumina | Second-generation | Short-read | Hypervariable regions (V4, V3-V4) | High throughput, low per-sample cost | Limited to partial gene sequencing |
Benchmarking and Validation Approaches
The critical importance of proper benchmarking for 16S rRNA analysis tools was highlighted by research utilizing a complex mock community comprising 235 bacterial strains representing 197 distinct species. This resource provides a gold-standard ground truth for testing OTU/ASV methods, addressing the fundamental limitation of real data where the true composition of microbial communities is unknown. Benchmarking studies using such mock communities have revealed that ASV algorithms (particularly DADA2) produce consistent output but suffer from over-splitting, while OTU algorithms (notably UPARSE) achieve clusters with lower errors but with more over-merging [97].
Additionally, synthetic data generation tools such as metaSPARSim and sparseDOSSA2 have enabled researchers to create calibrated synthetic datasets that mimic experimental data characteristics. This approach allows for validation of bioinformatic pipelines and differential abundance tests using data with known ground truth, addressing the challenge of unknown true compositions in experimental samples [98] [99].
Visual Guide: Experimental Workflow for 16S rRNA Sequencing Comparison
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagents and Solutions for 16S rRNA Sequencing Studies
| Reagent/Solution | Function | Example Use Case |
|---|---|---|
| DNA/RNA Shielding Buffer | Preserves nucleic acid integrity between sample collection and processing | Storage of oropharyngeal swabs prior to DNA extraction [40] |
| Magnetic Bead-Based DNA Extraction Kits | High molecular weight DNA extraction suitable for long-read sequencing | Quick-DNA HMW MagBead kit for oropharyngeal and soil samples [40] [16] |
| PowerSoil DNA Isolation Kit | Efficient DNA extraction from complex, inhibitor-rich samples | Skin microbiome and soil DNA extraction [22] [16] |
| SMRTbell Prep Kit | Library preparation for PacBio sequencing | Full-length 16S sequencing on PacBio Sequel II system [16] |
| Native Barcoding Kits | Multiplexed library preparation for Oxford Nanopore | 16S rRNA gene sequencing on MinION platform [16] [96] |
| Mock Microbial Communities | Validation and benchmarking of sequencing workflows | ZymoBIOMICS standards for pipeline validation [94] [97] |
The evidence comprehensively demonstrates that full-length 16S rRNA sequencing provides superior taxonomic resolution and more accurate microbial community profiling compared to partial gene sequencing approaches. The complete genetic information captured across all nine hypervariable regions enables species-level identification that cannot be consistently achieved with partial regions alone.
However, practical considerations remain. For researchers with limited sequencing resources or those focusing primarily on genus-level community dynamics, targeting specific hypervariable regions (particularly V1-V3) represents a viable alternative that balances cost with analytical precision. The choice between approaches should be guided by research objectives, sample type, and available resources.
Future methodological developments will likely focus on reducing the cost and computational burden of full-length sequencing while improving the accuracy of long-read technologies. As these trends continue, full-length 16S rRNA sequencing is positioned to become the gold standard for microbial community analysis, particularly for applications requiring high taxonomic resolution such as clinical diagnostics, microbial source tracking, and detailed ecological studies.
Conclusion
The choice of sequencing platform is not one-size-fits-all but must be strategically aligned with specific research goals. While Illumina remains a robust, high-throughput choice for broad microbial surveys, long-read technologies from PacBio and ONT are indispensable for achieving species-level resolution and discovering novel taxa, as evidenced by studies recovering over 15,000 previously unknown species. Recent advancements have significantly narrowed the accuracy gap for ONT, making it a powerful tool for real-time, full-length 16S sequencing. Future directions point toward hybrid sequencing approaches that leverage the strengths of multiple platforms, augmented by AI-driven bioinformatics for rapid genome annotation and functional prediction. For biomedical research, this evolving landscape promises more precise microbiome diagnostics, enhanced antimicrobial resistance detection, and a deeper understanding of host-microbe interactions in disease.
References
- 1. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 2. Sanger sequencing [https://en.wikipedia.org/wiki/Sanger_sequencing]
- 3. The Third-Generation Sequencing Challenge: Novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11117673/]
- 4. Metagenomic approaches in microbial ecology: an update ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7641418/]
- 5. Genome-resolved long-read sequencing expands known ... [https://www.nature.com/articles/s41564-025-02062-z]
- 6. Performance comparison of second- and third-generation ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-15-699]
- 7. Comprehensive comparison of the third-generation ... [https://www.nature.com/articles/s41467-025-59187-2]
- 8. Comparing the accuracy and efficiency of third generation ... [https://www.sciencedirect.com/science/article/pii/S2405985423000228]
- 9. Performance evaluation of Sanger sequencing for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4299716/]
- 10. Analyzing Sanger Sequencing Data [https://blog.genewiz.com/analyzing-sanger-sequencing-data]
- 11. Sequencing Quality Scores [https://www.illumina.com/science/technology/next-generation-sequencing/plan-experiments/quality-scores.html]
- 12. Comparing long-read sequencing technologies [https://www.pacb.com/blog/sequencing-101-comparing-long-read-sequencing-technologies/]
- 13. Long-read sequencing myths: debunked. Part 1- HiFi ... [https://www.pacb.com/blog/long-read-sequencing-myths-debunked-part-1-hifi-sequencing/]
- 14. Error Rate of PacBio vs Nanopore: How Accurate Are Long ... [https://www.cd-genomics.com/blog/pacbio-nanopore-error-rate-correction-strategies/]
- 15. Finding the right fit: evaluation of short-read and long ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9176275/]
- 16. Comparative evaluation of sequencing platforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12365774/]
- 17. Nanopore sequencing accuracy [https://nanoporetech.com/platform/accuracy]
- 18. Benchmarking second and third-generation sequencing ... [https://www.nature.com/articles/s41597-022-01762-z]
- 19. Comparison of single-cell long-read and short ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12231600/]
- 20. Performance and Application of 16S rRNA Gene Cycle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7484979/]
- 21. Evaluation of 16S rRNA gene sequencing for species and ... [https://www.nature.com/articles/s41467-019-13036-1]
- 22. Comparison of the full-length sequence and sub-regions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11264597/]
- 23. Full-length 16S rRNA gene sequencing by PacBio improves ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10213-5]
- 24. 16S rRNA Gene Sequencing vs. Shotgun Metagenomic ... [https://blog.microbiomeinsights.com/16s-rrna-sequencing-vs-shotgun-metagenomic-sequencing]
- 25. The effects of sequencing platforms on phylogenetic ... [https://www.nature.com/articles/sdata201868]
- 26. A long-read sequencing approach to high-resolution ... [https://www.nature.com/articles/s41598-025-96558-7]
- 27. The impact of sequencing depth on the inferred taxonomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8204541/]
- 28. Uncalled4 improves nanopore DNA and RNA modification ... [https://www.nature.com/articles/s41592-025-02631-4]
- 29. London Calling 2025 Technology Update [https://nanoporetech.com/news/london-calling-2025-technology-update]
- 30. Data analysis [https://nanoporetech.com/document/data-analysis]
- 31. Oxford Nanopore announces PromethION Plus Flow Cell ... [https://nanoporetech.com/news/oxford-nanopore-announces-promethion-plus-flow-cell-and-other-human-genetics-updates-at-ashg-2025]
- 32. A reference-guided iterative approach to polish the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12488850/]
- 33. Joint processing of long- and short-read sequencing data ... [https://www.sciencedirect.com/science/article/pii/S2667237525001432]
- 34. of Comparison Methods for Optimal Recovery of... DNA Extraction [https://pubmed.ncbi.nlm.nih.gov/31762512/]
- 35. Inter-laboratory ring test for environmental ... DNA extraction protocols [https://mbmg.pensoft.net/article_preview.php?id=128235]
- 36. Frontiers | Comparison of DNA methods for detecting... extraction [https://www.frontiersin.org/journals/veterinary-science/articles/10.3389/fvets.2025.1675115/full]
- 37. Introduce to 16S rRNA and 16S rRNA Sequencing [https://www.cd-genomics.com/blog/introduce-to-16s-rrna-and-16s-rrna-sequencing/]
- 38. Evaluation of Full-Length Versus V4-Region 16S rRNA ... [https://link.springer.com/article/10.1007/s00284-022-02956-9]
- 39. Comparative analysis of full-length 16s ribosomal RNA ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2023.1213829/full]
- 40. Comparative analysis of full-length 16s ribosomal RNA ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1658615/full]
- 41. Optimized bacterial community characterization through full ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-024-03208-5]
- 42. Determining the most accurate 16S rRNA hypervariable ... [https://www.nature.com/articles/s41598-023-30764-z]
- 43. Comparison of 16S ribosomal RNA hypervariable regions ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1665847/full]
- 44. 16S/ITS Sequencing [https://www.seqcenter.com/service/metagenome-sequencing/16s-its-sequencing/]
- 45. a systematic comparison of DNA library preparation kits for ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-016-2757-4]
- 46. a comparison of library preparation methods for Illumina ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8805253/]
- 47. Cross-platform comparisons for targeted bisulfite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10426093/]
- 48. Next-generation sequencing applications in food science [https://pmc.ncbi.nlm.nih.gov/articles/PMC12405390/]
- 49. Comparative performance of the BGISEQ-500 and Illumina ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-018-0337-0]
- 50. Comparative evaluation of RNA-Seq library preparation ... [https://www.nature.com/articles/s41598-019-49889-1]
- 51. Cross-comparison of gut metagenomic profiling strategies [https://www.nature.com/articles/s42003-024-07158-6]
- 52. Questions about the running time of qiime dada2 denoise-ccs [https://forum.qiime2.org/t/questions-about-the-running-time-of-qiime-dada2-denoise-ccs/32916]
- 53. Analyzing 2 batches of sequenced samples separately or ... [https://github.com/benjjneb/dada2/issues/1589]
- 54. Long-read metagenomics reveals phage dynamics in ... [https://www.nature.com/articles/s41586-025-09786-2]
- 55. Unraveling metagenomics through long-read sequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC10823668/]
- 56. Comparative Meta-Analysis of Long-Read and Short-Read ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566492/]
- 57. Complementary insights into gut viral genomes: a comparative ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-024-01981-z]
- 58. Comprehensive Evaluation of Error-Correction Methodologies ... [https://www.ncbi.nlm.nih.gov/books/NBK569557/]
- 59. Benchmarking of computational error-correction methods for ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-01988-3]
- 60. Research Article Benchmarking long-read assembly tools ... [https://www.sciencedirect.com/science/article/pii/S2215017X2500058X]
- 61. A systematic benchmark of Nanopore long-read RNA ... [https://www.nature.com/articles/s41592-025-02623-4]
- 62. Comparative evaluation of sequencing technologies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12130029/]
- 63. Comparative analysis of illumina and oxford nanopore ... [https://www.nature.com/articles/s41598-025-18768-3]
- 64. Evaluation of V3–V4 and FL-16S rRNA amplicon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12379586/]
- 65. A novel two-step metabarcoding approach improves soil ... [https://www.nature.com/articles/s41598-025-18936-5]
- 66. Predicting measures of soil health using the microbiome ... [https://www.sciencedirect.com/science/article/abs/pii/S0038071721003461]
- 67. MiSeq Sequencing System specifications [https://www.illumina.com/systems/sequencing-platforms/miseq/specifications.html]
- 68. Comparing Different Filtration Technologies in 2025 [https://www.san-lan.com/faq/comparing-different-filtration-technologies-in-2025.html]
- 69. Batch correction methods used in single-cell RNA ... [https://pubmed.ncbi.nlm.nih.gov/40623818/]
- 70. MiSeq System | Rapid and cost-effective sequencing [https://www.illumina.com/systems/sequencing-platforms/miseq.html]
- 71. How to Choose the Right DNA Sequencer for Your ... [https://www.labx.com/resources/how-to-choose-the-right-dna-sequencer-for-your-research-budget/5598]
- 72. Ion GeneStudio System Models [https://www.thermofisher.com/us/en/home/life-science/sequencing/next-generation-sequencing/instruments/ion-genestudio-system/models.html]
- 73. Top DNA Sequencing Products Companies & How to ... [https://www.linkedin.com/pulse/top-dna-sequencing-products-companies-how-compare-jllic/]
- 74. PacBio vs Oxford Nanopore: Which Long-Read ... [https://www.cd-genomics.com/resource-pacbio-oxford-nanopore-comparison.html]
- 75. dna sequencing market size and share analysis [https://www.coherentmarketinsights.com/market-insight/dna-sequencing-market-5531]
- 76. Why Are Long-Read Sequencing Methods Revolutionizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388134/]
- 77. Comparative evaluation of sequencing platforms: Pacific ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1633360/full]
- 78. DNA Sequencing: How to Choose the Right Technology [https://frontlinegenomics.com/dna-sequencing-how-to-choose-the-right-technology/]
- 79. Comparative Evaluation of Sequencing Platforms [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1633360/abstract]
- 80. Sequencing 101: Sequencing coverage [https://www.pacb.com/blog/sequencing-101-sequencing-coverage/]
- 81. Bioinformatics Tools for Analyzing DNA Sequencing Data [https://www.labx.com/resources/bioinformatics-tools-for-analyzing-dna-sequencing-data/5601]
- 82. The Latest Developments in Sequencing Technologies [https://frontlinegenomics.com/the-latest-developments-in-sequencing-technologies/]
- 83. Comparative analysis of Illumina, PacBio, and nanopore ... [https://www.frontiersin.org/journals/microbiomes/articles/10.3389/frmbi.2025.1587712/full]
- 84. Soil biofilm formation enhances microbial ... [https://www.sciencedirect.com/science/article/pii/S0160412019320392]
- 85. Performance evaluation of a new custom, multi-component ... [https://environmentalmicrobiome.biomedcentral.com/articles/10.1186/s40793-019-0349-z]
- 86. Unlocking River Biofilm Microbial Diversity: A Comparative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12627904/]
- 87. Accurate profiling of microbial communities for shotgun ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590682/]
- 88. Evaluation of the Microba Community Profiler for ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.643682/full]
- 89. High-throughput DNA sequencing technologies for water ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10424514/]
- 90. Metagenomics unravel distinct taxonomic and functional ... [https://www.sciencedirect.com/science/article/pii/S2589004224022727]
- 91. Taxonomical composition and functional analysis of ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2023.1148976/full]
- 92. Measuring sequencer size bias using REcount [https://pmc.ncbi.nlm.nih.gov/articles/PMC6489363/]
- 93. Performance comparison of four exome capture platforms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12551171/]
- 94. Short-read full-length 16S rRNA amplicon sequencing for ... [https://www.nature.com/articles/s41598-024-65841-4]
- 95. Full-Length 16S rRNA Sequencing Enhances Species ... [https://www.cd-genomics.com/resource-full-length-16s-rrna-sequencing-enhances-species-level-microbial-analysis.html]
- 96. Next Generation Sequencing Improves Diagnostic 16S ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12418051/]
- 97. Unlocking Reliable 16S rRNA Analysis: A Benchmarking Gold ... [https://communities.springernature.com/posts/unlocking-reliable-16s-rrna-analysis-a-benchmarking-gold-standard-ground-truth]
- 98. Benchmarking Differential Abundance Tests for 16S ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0321452]
- 99. Leveraging Synthetic Data to Validate a Benchmark... [https://f1000research.com/articles/14-621]