Primer Dimers and Nonspecific Amplification: A Comprehensive Guide for Molecular Researchers
This article provides a thorough examination of primer dimers and nonspecific amplification, critical challenges in PCR that compromise assay accuracy and efficiency.
Primer Dimers and Nonspecific Amplification: A Comprehensive Guide for Molecular Researchers
Abstract
This article provides a thorough examination of primer dimers and nonspecific amplification, critical challenges in PCR that compromise assay accuracy and efficiency. Tailored for researchers, scientists, and drug development professionals, it explores the fundamental mechanisms behind these artifacts, from basic definitions to advanced causative models like adapter-mediated self-priming. The content delivers actionable methodological strategies for robust primer design and reaction optimization, a systematic troubleshooting framework for common experimental pitfalls, and rigorous protocols for in silico and experimental validation of assay specificity and performance. By synthesizing foundational knowledge with cutting-edge computational and experimental approaches, this guide serves as an essential resource for developing reliable, reproducible molecular diagnostics and research assays.
What Are Primer Dimers? Defining the Fundamentals of Nonspecific Amplification
A primer dimer (PD) is a common by-product in polymerase chain reaction (PCR) and other nucleic acid amplification techniques, such as Loop-Mediated Isothermal Amplification (LAMP) [1] [2]. It is formed when two primer molecules hybridize to each other via complementary base sequences instead of binding to the intended target template DNA [1] [2]. This unintended structure is then amplified by the DNA polymerase, consuming reaction reagents and potentially outcompeting the amplification of the desired target sequence [2] [3]. The formation of primer dimers is a significant concern in molecular diagnostics and research as it can lead to false-positive results, reduced amplification efficiency, low sensitivity, and signal loss [1] [4].
This article deconstructs the primer dimer, detailing its fundamental structure, formation mechanism, and the experimental approaches used to study it, thereby providing a foundational understanding for efforts aimed at mitigating nonspecific amplification.
Structural Composition and Formation Mechanism
Dimer Classifications and Structural Motifs
Primer dimers are primarily categorized based on the identity of the interacting primers, as shown in Table 1 [1].
Table 1: Classification of Primer Dimers
| Dimer Type | Description | Common Characteristics |
|---|---|---|
| Heterodimer | Formed by the hybridization of two different primers (e.g., forward and reverse primers) [1]. | Most common type in standard PCR; often involves complementary sequences at the 3' ends. |
| Homodimer | Formed when two identical primers bind to each other [1]. | Can occur in reactions with a single primer or when one primer has self-complementary regions. |
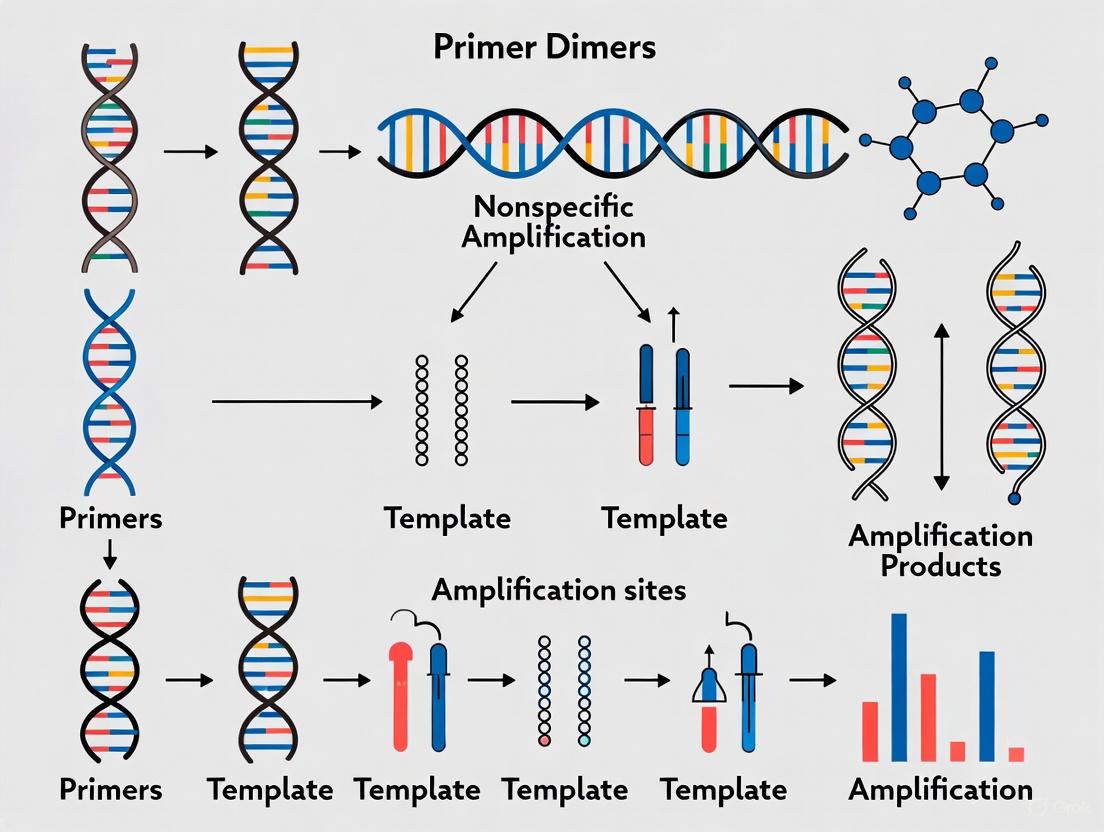
The formation of a stable, amplifiable primer dimer is a three-step process, illustrated in Figure 1 [2]:
- Annealing: Two primers anneal to each other at their 3' ends through complementary bases.
- Polymerase Extension: If this double-stranded structure is stable, DNA polymerase binds and extends both primers, synthesizing a double-stranded DNA product.
- Exponential Amplification: In subsequent cycles, the newly synthesized double-stranded dimer serves as a template for fresh primers, leading to exponential amplification of the primer dimer product [2].
A critical factor enabling this process is complementarity at the 3' ends of the primers. The DNA polymerase requires a double-stranded structure with a free 3'-OH group to initiate synthesis; thus, stable base-pairing at the 3' termini is a primary driver of dimer formation and extension [1] [2].
Figure 1: The stepwise mechanism of primer dimer formation and exponential amplification.
The Scientist's Toolkit: Key Reagents for Primer Dimer Research
Studying and mitigating primer dimers requires a specific set of reagents and tools, as detailed in Table 2.
Table 2: Essential Research Reagents and Materials
| Reagent/Material | Function in Primer Dimer Research | Specific Examples |
|---|---|---|
| DNA Polymerase | Enzyme that extends annealed primers; its activity at low temperatures can promote PDs [2]. | Standard Taq polymerase; Hot-Start variants (chemically modified, antibody-bound) [2]. |
| Primers | Synthesized oligonucleotides whose sequence dictates dimerization risk [1] [5]. | Standard DNA primers; modified primers (e.g., HANDS, chimeric, blocked-cleavable) [2]. |
| Magnesium Ions (Mg²⁺) | Essential cofactor for DNA polymerase; high concentration increases dimer formation likelihood [1] [2]. | Magnesium chloride (MgCl₂), Magnesium sulfate (MgSO₄). |
| Intercalating Dyes | Fluorescent dyes that bind double-stranded DNA, enabling real-time detection of amplification products [2] [4]. | SYBR Green I, SYTO 9, SYTO 82 [4]. |
| Capillary Electrophoresis System | Separates and analyzes DNA fragments by size and conformation to detect and quantify primer dimers [6]. | ABI 3100 system [6]. |
| Thermostable RNase HII | Enzyme used in rhPCR to cleave a blocking group from primers only at high temperature, preventing low-temperature dimer extension [2]. | Used in RNase H-dependent PCR (rhPCR) [2]. |
Experimental Detection and Quantification
Accurately detecting and quantifying primer dimers is crucial for assay optimization. The choice of method depends on the application and required sensitivity.
Standard Detection Methodologies
- Gel Electrophoresis: The most common method for endpoint detection. Primer dimers appear as a diffuse band or smear of 30-50 base pairs (bp), which is distinguishable from longer target amplicons [2] [3]. Unincorporated primers form a hazy band at the very bottom of the gel (~20-30 bp), while primer multimers can create a ladder-like pattern [3].
- Melting Curve Analysis: Used in quantitative PCR (qPCR) with intercalating dyes. Since primer dimers are short and have low GC content, they denature at a lower temperature than the specific target amplicon. This generates a distinct peak in the melting curve, allowing for discrimination between specific and nonspecific products [2].
- Sequence-Specific Probes: Technologies like TaqMan probes or molecular beacons generate a fluorescent signal only upon binding to the specific target sequence. This method prevents signal acquisition from primer dimers, although the dimers can still form and consume reaction components [2].
An Advanced Quantitative Protocol: Free-Solution Conjugate Electrophoresis (FSCE)
To gain a deeper, quantitative understanding of the biophysical parameters driving dimerization, researchers have developed sophisticated capillary electrophoresis methods. One such protocol is detailed below, adapted from a study aiming to parameterize computational models of dimerization risk [6].
Objective: To precisely quantify heterodimerization between primer-barcode pairs as a function of temperature and complementary region length [6]. Experimental Workflow:
- Sample Design: A set of fluorescently labeled 30-mer oligonucleotides (mimicking primer-barcodes) are designed with complementary regions of differing lengths and patterns.
- Drag-Tag Conjugation: One oligonucleotide in each pair is conjugated to a synthetic, neutral "drag-tag" (e.g., a poly-N-methoxyethylglycine chain). This tag increases the hydrodynamic drag of the molecule, altering its electrophoretic mobility and allowing clear separation of single-stranded and double-stranded conformations [6].
- Annealing: The drag-tagged and non-drag-tagged DNA primers are mixed, heat-denatured, and annealed at a defined temperature.
- Electrophoresis & Quantification: The annealed mixtures are separated by free-solution capillary electrophoresis (no sieving matrix) at various temperatures (e.g., 18°C to 62°C). The drag-tag creates a mobility shift, enabling precise separation and quantification of the ssDNA and ds primer-dimer peaks via laser-induced fluorescence detection [6].
Figure 2: Workflow for quantifying primer dimer formation using Free-Solution Conjugate Electrophoresis (FSCE).
Key Quantitative Findings from FSCE: This rigorous experimental approach yielded precise biophysical insights:
- Complementary Length: Dimerization occurred reliably only when more than 15 consecutive base pairs could form between primers [6].
- Stability Requirement: Non-consecutive base pairs did not create stable dimers, even when 20 out of 30 possible base pairs were bonded, highlighting the importance of contiguous homology for stable duplex formation [6].
- Temperature Dependence: For partially complementary primers (less than 30/30 bp), dimerization was inversely correlated with temperature [6].
Thermodynamics and Primer Design Implications
The formation of a primer dimer is governed by the laws of thermodynamics. The nearest-neighbor model is the most accurate method for predicting the stability of nucleic acid duplexes, including primer dimers [4]. This model calculates the change in Gibbs free energy (ΔG) for hybridization, considering the identity and orientation of adjacent base pairs. A more negative ΔG indicates a more stable, and therefore more problematic, potential dimer [4].
The quantitative data from FSCE and thermodynamic modeling directly inform primer design rules to minimize dimerization risk, as summarized in Table 3.
Table 3: Key Parameters for Preventing Primer Dimer Formation
| Parameter | Optimal or Target Value | Rationale |
|---|---|---|
| 3'-End Complementarity | Avoid ≥ 3 contiguous complementary bases, especially at the 3' ends [5]. | Prevents stable annealing and extension by DNA polymerase [2] [5]. |
| Self-Complementarity | Avoid hairpins and internal loops with ≤ 3 contiguous bases [5]. | Reduces the chance of primers folding on themselves or annealing to each other. |
| GC Content | Maintain between 40–60% [5]. | Prevents overly stable (high GC) or overly weak (low GC) primers that can promote mishybridization. |
| Consecutive Bases | Avoid long runs (≥ 4) of the same base [5]. | Reduces the chance of slippage and misalignment. |
A primer dimer is not merely an artifact but a specifically structured, polymerase-amplifiable nucleic acid product arising from the unintended hybridization of primers. Its formation is a quantifiable thermodynamic process driven by complementary sequences, particularly at the 3' ends of primers. Advanced experimental methods like FSCE provide critical quantitative data on the thresholds of complementarity required for dimerization, which in turn refines computational prediction and primer design. A deep understanding of the structure and biophysics of primer dimers, as outlined in this article, is therefore fundamental to designing robust and reliable molecular assays and advancing research in diagnostics and drug development.
In polymerase chain reaction (PCR) and related amplification technologies, the precise binding of primers to their specific target sequences is fundamental to successful DNA amplification. However, a common artifact known as primer-dimer (PD) formation can occur when primers anneal to each other instead of the intended target DNA [2]. Primer dimers are small, unintended DNA fragments that form when primers hybridize to one another through complementary base regions, creating substrates that DNA polymerase can extend [7]. This nonspecific amplification presents a significant challenge in molecular biology, particularly in quantitative PCR and diagnostic applications where it competes for precious reaction reagents and can lead to false positives or inaccurate quantification [2] [1]. Understanding the precise molecular mechanisms behind this aberrant priming is crucial for researchers and drug development professionals seeking to optimize amplification assays and ensure result reliability.
The formation of primer dimers represents a fundamental failure of the specific primer-template recognition process that PCR depends upon. Instead of binding to flanking sequences of the target DNA, primers interact with each other through complementary regions, leading to the synthesis of short, unwanted products that typically range from 30 to 100 base pairs in length [7]. This phenomenon is not merely an inconvenience—it can drastically reduce amplification efficiency, sensitivity, and specificity, particularly when working with low-template samples or in multiplex reactions where multiple primer sets are employed simultaneously [1]. Within the broader context of primer-dimer and nonspecific amplification research, this technical guide examines the molecular mechanisms governing how and why primers anneal to each other rather than their intended targets, providing experimental approaches for detection and prevention.
Molecular Mechanisms of Primer-Dimer Formation
The Stepwise Formation Process
The formation of primer dimers follows a defined three-step mechanism that exploits the inherent biochemical properties of DNA primers and polymerase activity [2]:
Step I - Initial Annealing: Two primers anneal at their 3' ends through complementary base pairing. This initial hybridization is typically facilitated by short regions of complementarity between primers, often involving only 2-3 complementary bases, though longer stretches significantly increase stability and likelihood of dimer formation [2] [8].
Step II - Polymerase Extension: Once a stable primer-primer duplex forms, DNA polymerase binds and extends both primers according to their complementary sequences. Even though DNA polymerases used in PCR are most active around 70°C, they retain some polymerizing activity at lower temperatures, which can facilitate this extension process when primer dimers form during reaction setup [2].
Step III - Template Amplification: In subsequent PCR cycles, the extended primer-dimer products from Step II serve as templates for fresh primers to anneal, leading to exponential amplification of the primer-dimer artifacts alongside the target sequence [2].
The following diagram illustrates this stepwise formation process:
Structural Determinants Facilitating Aberrant Annealing
Several structural features of primers predispose them to form dimers rather than anneal to the intended target:
3' End Complementarity: Complementarity at the 3' ends of two primers is a major factor in dimer formation. Even 2-3 complementary bases at the 3' end can provide sufficient stability for polymerase binding and extension [8]. The 3' end is particularly critical because this is where DNA polymerase initiates synthesis.
High GC Content: Regions with high GC-content, particularly at the 3' ends, contribute significantly to dimer stability due to the stronger triple hydrogen bonds between G and C bases compared to the double bonds between A and T [2]. This enhanced stability increases the likelihood that the primer-primer construct will survive long enough for polymerase binding.
Self-Complementarity: Internal complementary regions within a single primer can lead to self-dimerization or hairpin structures that alter primer conformation and facilitate aberrant interactions with other primers [1].
Primer Length and Concentration: Excessively short primers (<20 nucleotides) have reduced specificity and are more likely to form dimers, while high primer concentrations increase collision frequency and opportunities for primer-primer interactions [7] [8].
The stability of the initial primer-primer hybrid is a critical determinant in whether dimer formation proceeds to extension. A stable construct with high GC-content and longer overlap regions significantly increases the probability that DNA polymerase will bind and extend the primers, committing the reaction to dimer amplification [2].
Detection and Characterization Methods
Electrophoretic and Melting Curve Analysis
Researchers employ multiple methods to detect and characterize primer dimers, each with distinct advantages and applications:
Table 1: Detection Methods for Primer Dimers
| Method | Principle | Key Characteristics | Applications |
|---|---|---|---|
| Gel Electrophoresis [7] | Separation by molecular weight in agarose gel | 30-50 bp band/smear; fuzzy appearance; runs ahead of target amplicon | Conventional PCR; quality control check |
| Melting Curve Analysis [2] | Monitoring dsDNA dissociation with temperature changes using intercalating dyes | Lower melting temperature (Tm) than target amplicon | Quantitative PCR with SYBR Green I |
| No-Template Control (NTC) [7] | PCR reaction without DNA template | Primer dimers appear as only amplification product | Distinguishing true target amplification from artifacts |
After electrophoresis, primer dimers typically appear as a moderate to high intensity band or smear between 30-50 base pairs, distinguishable from the target sequence which is generally longer than 50 bp [2] [7]. The smeary appearance reflects the heterogeneous nature of primer-dimer products, which can form through various complementary regions between primers [7]. In quantitative PCR using intercalating dyes like SYBR Green I, melting curve analysis allows distinction between specific products and primer dimers based on their different denaturation temperatures—PDs melt at lower temperatures due to their shorter length and potentially lower GC content [2].
Experimental Protocol: Gel-Based Detection
Objective: To identify primer dimer formation in PCR samples using agarose gel electrophoresis.
Materials:
- Standard PCR reaction components [9]
- DNA ladder (e.g., 100 bp ladder)
- Agarose
- Electrophoresis buffer (TAE or TBE)
- Nucleic acid stain (ethidium bromide or SYBR Safe)
- Gel documentation system
Methodology:
- Prepare a 2-4% agarose gel in appropriate buffer with nucleic acid stain [10].
- Mix PCR products with loading dye and load alongside appropriate DNA size markers.
- Run gel at constant voltage (5-10 V/cm) until sufficient separation is achieved.
- Visualize under UV light and document results.
Interpretation: Primer dimers appear as a bright, fuzzy band or smear below 100 bp, typically well separated from the target amplicon [7]. For better resolution of small primer dimers, run the gel longer to ensure these fragments migrate past the desired PCR products [7].
Experimental Protocol: Melting Curve Analysis
Objective: To distinguish primer dimers from specific amplification products in quantitative PCR.
Materials:
- qPCR reaction mixture with intercalating dye (e.g., SYBR Green I) [2] [10]
- Real-time PCR instrument with melting curve capability
Methodology:
- Perform qPCR amplification with standard cycling conditions.
- After amplification, heat products to 95°C for 15 seconds to denature all double-stranded DNA.
- Cool to lowest temperature (e.g., 60°C) and gradually heat to 95°C while continuously monitoring fluorescence.
- Analyze derivative plot (-dF/dT vs. Temperature) to identify melting peaks.
Interpretation: Specific amplicons display a distinct, sharp peak at higher melting temperatures, while primer dimers produce a peak at lower temperatures due to their shorter length and different sequence composition [2]. The four-step PCR protocol can be employed where signal is acquired below the melting temperature of the target sequence but above that of PDs to minimize their contribution to fluorescence signals [2].
Prevention Strategies and Experimental Solutions
Primer Design and Reaction Optimization
Preventing primer dimer formation requires a multi-faceted approach addressing both primer design and reaction conditions:
Table 2: Strategies to Prevent Primer-Dimer Formation
| Strategy | Mechanism | Implementation |
|---|---|---|
| Optimized Primer Design [2] [8] | Minimizes complementary regions between primers | Use design software to avoid 3' complementarity; target GC content 40-60% |
| Hot-Start PCR [2] | Inhibits polymerase activity during reaction setup until high temperatures | Use chemically modified or antibody-inhibited polymerases |
| Lower Primer Concentration [7] [8] | Reduces primer-primer collision frequency | Titrate primers (0.1-0.5 µM typically); maintain primer:template balance |
| Increased Annealing Temperature [7] | Enhances stringency of primer binding | Test gradient PCR (55-70°C) to find optimal temperature |
| Magnesium Concentration Optimization [2] [1] | Reduces non-specific priming | Titrate MgCl₂ (1.5-3.0 mM typically) |
| Structural Modifications [2] | Prevents extension of primer-duplexes | Use blocked-cleavable primers or HANDS system |
Primer design represents the most crucial factor in preventing dimer formation. Bioinformatics tools like OligoAnalyzer and Multiple Primer Analyzer can predict potential self-dimers and heterodimers before primer synthesis [11] [12]. These tools evaluate complementarity, particularly at the 3' ends, and help select primer pairs with minimal interaction potential. The HANDS (Homo-Tag Assisted Non-Dimer System) represents an innovative structural approach where a nucleotide tail complementary to the 3' end of the primer is added to the 5' end, creating a stem-loop structure that prevents dimer formation while permitting target annealing [2].
Advanced Molecular Approaches
Several advanced molecular biology techniques provide specialized solutions for challenging amplification scenarios:
RNase H-dependent PCR (rhPCR): This method utilizes blocked-cleavable primers that only become active after processing by a thermostable RNase HII enzyme at high temperatures. The RNase HII displays minimal activity at low temperatures, preventing primer dimer formation during reaction setup, and also provides inherent primer:template mismatch discrimination [2].
Self-Avoiding Molecular Recognition Systems (SAMRS): SAMRS incorporates nucleotide analogues (T, A, G, C) into primers that can bind to natural DNA but not to other SAMRS-containing primers. This approach essentially creates primers that "ignore" each other while maintaining ability to hybridize to natural DNA templates, effectively eliminating primer-primer interactions [2].
Blocking Primers: In applications like DNA metabarcoding for dietary analysis, blocking primers can be designed to suppress amplification of predator DNA while allowing prey DNA amplification. These primers anneal to specific non-target sequences and prevent their amplification through either annealing inhibition or elongation arrest mechanisms [13].
The Scientist's Toolkit: Essential Reagents and Solutions
Table 3: Research Reagent Solutions for Primer-Dimer Prevention
| Reagent/Solution | Function | Specific Examples |
|---|---|---|
| Hot-Start DNA Polymerases [2] [7] | Inhibits polymerase activity at low temperatures during reaction setup | Antibody-inhibited, chemically modified, or cold-sensitive Taq polymerases |
| Universal Annealing Buffers [14] | Provides consistent annealing conditions for diverse primers | Platinum SuperFi II DNA Polymerase with isostabilizing components |
| Primer Design Software [11] [12] | Predicts potential dimer formation before synthesis | OligoAnalyzer, Multiple Primer Analyzer, NCBI BLAST |
| Modified Primer Chemistries [2] | Prevents extension of primer-duplexes | HANDS primers, chimeric RNA-DNA primers, blocked-cleavable primers |
| Magnesium Regulation Systems [2] | Controls magnesium availability to reduce non-specific amplification | Slow-release magnesium compounds bound to chemical matrices |
The following workflow diagram illustrates how these solutions integrate into an experimental approach for preventing and troubleshooting primer dimers:
The phenomenon of primers annealing to each other instead of their intended targets represents a significant challenge in molecular biology with implications for research accuracy, diagnostic reliability, and therapeutic development. The mechanisms underlying primer-dimer formation—initiated by complementary regions between primers, stabilized by favorable thermodynamic conditions, and amplified through polymerase activity—highlight the importance of careful experimental design and optimization. Within the broader context of nonspecific amplification research, understanding these mechanisms provides insights not only for PCR optimization but also for the development of novel amplification technologies with enhanced specificity.
As molecular techniques continue to evolve, incorporating more multiplex reactions, point-of-care applications, and complex diagnostic panels, the prevention and management of primer-dimers becomes increasingly critical. Emerging solutions such as SAMRS nucleotides, rhPCR systems, and sophisticated bioinformatics tools offer promising avenues for eliminating these artifacts. For researchers and drug development professionals, implementing systematic approaches to primer design, reaction optimization, and artifact detection ensures the reliability of molecular data and supports the advancement of precision medicine initiatives. Through continued research into the fundamental mechanisms of primer annealing and recognition, the scientific community can develop increasingly sophisticated solutions to the challenge of nonspecific amplification.
Non-specific amplification and primer dimer formation represent significant challenges in polymerase chain reaction (PCR) fidelity, particularly in diagnostic and drug development applications where precision is paramount. This technical guide examines the fundamental mechanisms through which primer complementarity, excessive primer concentration, and suboptimal annealing temperatures compromise PCR specificity. Through systematic analysis of experimental data and biochemical principles, we establish that these factors collectively promote unintended amplification artifacts by facilitating primer-primer interactions and off-target binding. The insights provided herein form a critical foundation for developing robust PCR-based assays in research and clinical settings, enabling researchers to preemptively address the root causes of amplification artifacts through informed primer design and reaction optimization.
Polymerase chain reaction (PCR) serves as a cornerstone technology in molecular biology, with applications spanning pathogen detection, genetic testing, and fundamental research. Despite its widespread utility, PCR is susceptible to amplification artifacts that compromise result interpretation and experimental reproducibility. Non-specific amplification refers to the generation of unintended DNA products beyond the targeted amplicon, while primer dimers are short, artifactual fragments formed by the hybridization and extension of primers on themselves or each other [3] [1]. These artifacts compete with target amplification for reaction components, reduce overall efficiency, and can lead to false-positive results in quantitative applications [15] [7].
Understanding the primary causes of these artifacts is essential for researchers and drug development professionals who rely on precise DNA amplification. This guide examines three interconnected factors that drive non-specific amplification: primer complementarity, primer concentration, and annealing temperature. By framing these issues within a biochemical context and providing structured experimental data, we aim to equip practitioners with the knowledge to troubleshoot existing protocols and design more specific amplification assays.
The Biochemical Basis of Primer Dimer Formation
Primer dimers form through two primary mechanisms: self-dimerization and cross-dimerization. In self-dimerization, a single primer contains regions of self-complementarity that allow it to fold and create a free 3' end accessible for polymerase extension [7]. Cross-dimerization occurs when forward and reverse primers contain complementary sequences, enabling them to hybridize to each other instead of the target template [16]. Both mechanisms create short, amplifiable duplexes that DNA polymerase can extend, generating artifacts typically between 20-100 bp in length [3] [7].
The formation of these dimers is governed by the same principles that facilitate specific primer-template binding: hydrogen bonding between complementary bases and stabilization by reaction components. Guanine-cytosine (GC) base pairs form three hydrogen bonds, making them more stable than adenine-thymine (AT) pairs, which form only two [16]. Consequently, primers with complementary GC-rich regions, particularly at the 3' ends, demonstrate higher propensity for dimer formation due to stronger binding energies. The negative Gibbs free energy (ΔG) value associated with these interactions indicates spontaneous reaction favorability, with ΔG values more negative than -9 kcal/mol representing significant dimerization risk [17].
Figure 1: Biochemical pathways of primer dimer formation. Self-complementarity or inter-primer complementarity enables dimerization, which polymerase extends into amplifiable artifacts.
Primary Cause 1: Primer Complementarity
Mechanisms and Consequences
Primer complementarity issues manifest primarily through three mechanisms: self-dimers, cross-dimers, and hairpin structures. Self-dimers occur when identical primers anneal to each other, while cross-dimers form between forward and reverse primers due to inter-primer homology [16]. Hairpins (or self-3'-complementarity) result from intramolecular interactions within a single primer when regions of three or more nucleotides complement each other, causing the primer to fold back on itself [16]. These secondary structures prevent proper annealing to the target template and provide aberrant substrates for polymerase extension.
The practical consequences of primer complementarity are significant. Studies have demonstrated that primers with strong complementary regions can generate primer dimers that effectively outcompete target amplification, particularly in later PCR cycles [3] [1]. This competition reduces the yield of desired products and can lead to complete amplification failure in severe cases. In quantitative PCR (qPCR), these artifacts are particularly problematic as they generate false fluorescence signals, compromising quantification accuracy [15].
Experimental Evidence
Research by Kim et al. highlights the impact of primer dimers in loop-mediated isothermal amplification (LAMP), where multiple primers at high concentrations increase dimerization risk [1]. Their findings demonstrate that nonspecific amplification resulting from primer dimer formation directly causes false-positive results in detection assays for Listeria monocytogenes. Similarly, studies on Scorpion primer-probes show that intramolecular structures can interfere with target annealing, though properly designed hairpin structures can actually prevent dimer formation by making the primer unavailable for intermolecular interactions [1].
Table 1: Experimental Studies Demonstrating Complementarity Effects
| Study | System | Key Finding | Impact |
|---|---|---|---|
| Kim et al. [1] | LAMP | Multiple primers at high concentrations facilitate dimer formation | False positives in pathogen detection |
| Whitcombe et al. [1] | Scorpion primers | Intramolecular hairpins prevent primer-dimer formation | Reduced non-specific amplification |
| IDT Design Guidelines [17] | PCR/qPCR | ΔG > -9 kcal/mol prevents significant dimerization | Improved amplification specificity |
Primary Cause 2: Primer Concentration
Concentration-Dependent Artifact Formation
Excessive primer concentration directly promotes non-specific amplification by altering the primer-template binding kinetics. At high concentrations (typically >1μM), the probability of primer-primer interactions increases exponentially compared to primer-template binding [18] [17]. This imbalance favors dimer formation, as primers encounter each other more frequently than they encounter the target sequence, particularly in early PCR cycles when template concentration is minimal.
The mechanistic explanation involves mass action principles and the stoichiometry of PCR components. In a standard reaction, primers are typically included in substantial molar excess relative to the template DNA (e.g., 10-1000× higher). While this ensures efficient target amplification, excessive primer concentrations exceed the polymerase's extension capacity, leaving unincorporated primers available for dimerization [3] [7]. This effect is compounded by the fact that primer dimers, once formed, are typically shorter than target amplicons and can be amplified more efficiently, allowing them to dominate the reaction in later cycles.
Empirical Optimization Data
Manufacturer recommendations for various DNA polymerases provide specific guidance on optimal primer concentrations. For Phusion and Phire DNA polymerases, Thermo Fisher Scientific recommends working concentrations between 200-1000 nM, with 500 nM as the ideal starting point [18]. Integrated DNA Technologies (IDT) similarly suggests that primer concentrations should be optimized to achieve the ideal primer-to-template ratio, noting that excessive concentrations directly promote nonspecific binding [17].
Experimental data demonstrates that reducing primer concentration from 1μM to 200nM can decrease primer dimer formation by up to 80% without significantly impacting target amplification yield, provided the template concentration is sufficient [18] [7]. This optimization is particularly crucial in multiplex PCR applications, where multiple primer pairs compete for reaction components, and in qPCR, where accurate quantification depends on specific amplification.
Table 2: Primer Concentration Guidelines by Polymerase System
| Polymerase System | Recommended Concentration | Adjustment Range | Key Considerations |
|---|---|---|---|
| Phusion High-Fidelity [18] | 500 nM | 200 nM - 1,000 nM | Concentration affects calculated Tm |
| Phire Hot Start [18] | 500 nM | 200 nM - 1,000 nM | Primers should have Tm ≥60°C |
| Standard Taq [17] | 200-500 nM | 100-900 nM | Lower end reduces dimer risk |
| Q5 Polymerase [19] | 500 nM | 200-900 nM | Use NEB Tm calculator for accuracy |
Primary Cause 3: Low Annealing Temperatures
Temperature Effects on Specificity
Annealing temperature (Ta) serves as a critical determinant of PCR stringency, directly controlling the binding specificity of primers to their target sequences. When Ta is set too low, typically 5°C or more below the primer melting temperature (Tm), primers tolerate mismatches and bind to non-complementary regions through partial hybridization [19] [17]. This permissive binding enables amplification from off-target sites and facilitates primer dimer formation by stabilizing otherwise transient primer-primer interactions.
The relationship between annealing temperature and amplification bias was systematically investigated in a study examining templates with perfect matches versus single mismatches. This research demonstrated that lower annealing temperatures (45°C) significantly reduced amplification bias between matched and mismatched templates compared to higher temperatures (60°C) [20]. While this may be desirable in some applications like degenerate PCR, it generally increases non-specific amplification in standard target-specific assays.
Melting Temperature Calculations and Optimization
Melting temperature (Tm) represents the temperature at which 50% of primer-template duplexes dissociate into single strands, and serves as the reference point for annealing temperature optimization [19]. Tm calculation methods vary, with simpler formulas considering only base composition (Tm = 4(G+C) + 2(A+T)), while more sophisticated nearest-neighbor models incorporated in modern calculators (e.g., IDT OligoAnalyzer, NEB Tm Calculator) account for buffer composition, magnesium concentration, and other reaction components that significantly impact duplex stability [17] [16].
Table 3: Annealing Temperature Optimization Strategies
| Method | Protocol | Applications | Considerations |
|---|---|---|---|
| Gradient PCR | Run reactions across a temperature range (e.g., 45-65°C) | Initial assay optimization | Identifies optimal Ta for specific primer-template pairs |
| Formula-Based | Ta = Tm - 5°C (standard) or Tm + 3°C (high stringency) | Routine applications | Requires accurate Tm calculation |
| Universal Annealing | Set single Ta (e.g., 60°C) for all assays | High-throughput settings | Limited to primers with similar Tms |
| Touchdown PCR | Incrementally decrease Ta during initial cycles | Complex templates | Improves specificity for difficult amplicons |
Figure 2: Impact of annealing temperature on amplification outcomes. Low temperatures promote permissive binding and artifacts, while higher temperatures enforce specificity.
Manufacturer guidelines consistently recommend annealing temperatures approximately 3-5°C below the calculated Tm of the primers [18] [17]. For primers ≤20 nucleotides, using the lower Tm value provided by calculators is advised, while for longer primers (>20 nt), an annealing temperature 3°C higher than the lower Tm is recommended [18]. These guidelines require adjustment when using additives like DMSO, which decreases Tm by approximately 5.5-6.0°C per 10% concentration [18].
Integrated Experimental Approaches
Comprehensive Troubleshooting Protocol
Addressing non-specific amplification requires a systematic experimental approach that simultaneously evaluates multiple parameters. The following integrated protocol combines assessment of all three primary causes:
Initial Primer Analysis: Screen primer sequences using tools such as IDT OligoAnalyzer or PrimerQuest to evaluate self-complementarity, hairpin formation, and heterodimer risk. Acceptable parameters include ΔG > -9 kcal/mol for dimers and minimal self 3'-complementarity [17] [16].
Concentration Titration: Prepare a primer matrix with concentrations ranging from 100-900 nM in 100 nM increments while maintaining constant template concentration. Identify the lowest concentration that provides robust target amplification without artifacts [7].
Temperature Gradient PCR: Run reactions across an annealing temperature gradient spanning from 5°C below to 5°C above the calculated Tm. Use a no-template control (NTC) to identify primer-derived artifacts [19] [7].
Hot-Start Polymerase Implementation: Employ hot-start enzymes (antibody-mediated or chemical modification) to prevent pre-PCR polymerization during reaction setup, significantly reducing early primer dimer formation [7] [21].
This protocol should be executed sequentially, with each optimization step informing the next. For example, primer concentration findings should be incorporated into temperature gradient experiments, and both should be validated with appropriate positive and negative controls.
Research Reagent Solutions
Table 4: Essential Reagents for Troubleshooting Non-Specific Amplification
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Hot-Start Polymerases | Phire Hot Start, Hot Start Taq | Prevents enzymatic activity during setup | Critical for low-temperature protocols |
| Tm Calculation Tools | NEB Tm Calculator, IDT OligoAnalyzer | Determines precise melting temperatures | Must input specific buffer conditions |
| Buffer Additives | BSA, Betaine, DMSO | Reduces secondary structure, inhibits artifacts | Concentration-dependent effects |
| Primer Design Software | Primer-BLAST, OligoAnalyzer | Identifies complementary regions | ΔG cutoff of -9 kcal/mol recommended |
| Gradient Thermal Cyclers | Various commercial systems | Empirical Ta optimization | Essential for assay development |
The interrelated factors of primer complementarity, excessive primer concentration, and suboptimal annealing temperatures collectively represent the primary drivers of non-specific amplification and primer dimer formation in PCR. Primer complementarity enables the initial aberrant interactions that DNA polymerase subsequently extends, while excessive concentration provides the stoichiometric conditions that favor these non-productive interactions. Low annealing temperatures further exacerbate the problem by reducing reaction stringency and permitting stabilization of these off-target complexes.
Addressing these issues requires a integrated optimization strategy that begins with in silico primer design, incorporates empirical testing of reagent concentrations, and implements precise thermal cycling parameters. The experimental approaches and reagent solutions outlined in this guide provide researchers with a systematic framework for developing highly specific amplification assays. As PCR continues to evolve as a fundamental tool in research and diagnostics, understanding and controlling these fundamental variables remains essential for generating reliable, reproducible results in molecular biology applications.
In the realm of molecular biology, the polymerase chain reaction (PCR) stands as a cornerstone technique for DNA amplification, yet its efficiency is frequently compromised by a pervasive artifact known as primer dimer (PD). Primer dimers are short, unintended DNA fragments that form when PCR primers anneal to each other through complementary base sequences rather than binding to their intended target DNA template [2] [7]. This nonspecific amplification product typically ranges between 30-50 base pairs in length—significantly shorter than most target amplicons—and manifests as a moderate to high-intensity smear or band in gel electrophoresis analysis [2] [7]. As a common by-product in both conventional and quantitative PCR (qPCR), primer dimers present a formidable challenge to experimental accuracy, particularly in sensitive applications such as diagnostic testing, gene expression studies, and drug development research [15] [22].
The formation of primer dimers initiates a cascade of molecular inefficiencies that systematically undermine PCR performance. These aberrant structures compete with legitimate amplification targets for essential reaction components, including primers, DNA polymerase, nucleotides, and magnesium ions [2] [23]. This competition establishes a resource allocation conflict wherein primer dimers deplete reagents that would otherwise support amplification of the desired DNA sequence, ultimately reducing amplification efficiency and potentially leading to false negative results [22]. In quantitative applications, the consequences are particularly severe as primer dimers can generate false positive signals, especially when using intercalating dyes like SYBR Green I, thereby skewing quantification data and compromising experimental conclusions [2] [15]. Understanding the mechanisms behind primer dimer formation and their multifaceted impact on PCR outcomes is thus fundamental to producing reliable, reproducible scientific data in molecular research and diagnostic applications.
Mechanisms of Primer Dimer Formation
The molecular genesis of primer dimers follows a sequential three-step pathway that initiates with primer-primer hybridization and culminates in the amplification of these aberrant structures. Understanding this mechanism is crucial for developing effective prevention strategies.
Molecular Pathways to Dimerization
Primer dimer formation initiates when two primers anneal at their 3' ends due to complementary base sequences (Step I) [2]. This complementarity need not be extensive; even a few complementary nucleotides can facilitate this interaction, particularly at low temperatures where molecular interactions are more permissive [22]. The stability of this initial hybridization construct is significantly enhanced by high GC-content at the 3' termini and extended overlap regions between primers, as GC base pairs form three hydrogen bonds compared to the two formed by AT base pairs [2] [16]. Once this primer-primer duplex forms, DNA polymerase recognizes it as a legitimate substrate and extends both primers according to their complementary sequences (Step II) [2]. The reaction culminates in subsequent PCR cycles when single-stranded products from Step II serve as templates for fresh primers, leading to exponential amplification of the primer dimer product (Step III) [2].
Two distinct structural configurations characterize primer dimer formation: self-dimers and cross-dimers. Self-dimerization occurs when a single primer contains regions complementary to itself, enabling intramolecular hybridization that creates a free 3' end for polymerase extension [7] [22]. Cross-dimerization arises when forward and reverse primers bear complementary regions, allowing intermolecular annealing that generates dual extension sites for DNA polymerase [7] [22]. Both pathways ultimately yield the same detrimental outcome—amplification of non-target artifacts that compete with the desired amplicon.
Contributing Factors and Conditions
Several experimental conditions predispose PCR reactions to primer dimer formation. Excessive primer concentration creates a high local density of primer molecules, increasing the probability of chance encounters and nonspecific interactions [7] [15]. Low annealing temperatures permit stable hybridization of primers with minimal complementarity, allowing weak primer-primer interactions that would be disrupted at higher, more stringent temperatures [7] [16]. The presence of complementary regions within or between primers—particularly at the 3' ends—establishes the structural foundation for dimerization [24] [22].
Perhaps most critically, primer dimer formation frequently occurs during reaction setup before thermal cycling commences [7] [22]. At room temperature, primers have maximal opportunity to interact nonspecifically, and if DNA polymerase possesses any activity at these lower temperatures (as non-hot-start enzymes do), extension can occur before the first denaturation step [2] [7]. This pre-PCR amplification establishes a pool of primer dimer templates that undergo exponential amplification during subsequent cycles, cementing their competition with the target amplicon.
Quantitative Impact of Primer Dimers on PCR Results
The presence of primer dimers exerts multifaceted detrimental effects on PCR efficiency and accuracy through resource competition and signal interference. These impacts manifest differently in conventional PCR versus quantitative real-time PCR, with particularly severe consequences in the latter.
Resource Depletion and Amplification Efficiency
Primer dimers directly compete with the target amplicon for essential reaction components, creating a resource allocation crisis that diminishes amplification efficiency. This competition occurs across multiple critical reagents:
- Polymerase Engagement: DNA polymerase molecules engaged in extending primer dimers become unavailable for target amplification [22].
- dNTP Consumption: Deoxynucleotide triphosphates (dNTDs) are incorporated into primer dimer products, depleting the pool available for legitimate target amplification [22].
- Primer Sequestration: Primers bound in dimer complexes cannot participate in target-directed amplification [23].
- Magnesium Ion Binding: Dimer structures chelate magnesium ions, potentially reducing the availability of this essential cofactor for enzymatic activity [2].
The cumulative effect of this resource competition is reduced amplification efficiency of the desired target sequence, particularly problematic for low-abundance targets where reaction components are already limiting [22]. This can manifest as diminished yield in conventional PCR or increased cycle threshold (Ct) values in qPCR, potentially leading to false negative results in detection applications [22].
Interference with Accurate Quantification
In quantitative PCR, primer dimers introduce substantial errors in quantification through multiple mechanisms. When using intercalating dyes like SYBR Green I, the dye binds nonspecifically to any double-stranded DNA, including primer dimers, generating fluorescence signal unrelated to target amplification [2] [15]. This signal contamination leads to overestimation of initial template concentration and consequently inaccurate quantification [15].
The severity of this interference depends on the relative abundance of primer dimers versus specific amplicon. At low template concentrations, primer dimers may dominate the amplification profile, generating false positive signals in no-template controls and potentially leading to erroneous conclusions about template presence [15] [22]. Experimental data demonstrates that primers with high dimerization tendencies can produce significantly higher Ct values compared to optimized primers with minimal dimer formation—a critical concern for sensitive diagnostic applications [22].
Table 1: Quantitative Impacts of Primer Dimers on PCR Performance
| Parameter Affected | Impact of Primer Dimers | Consequence |
|---|---|---|
| Amplification Yield | Decreased target product | Reduced downstream application efficiency |
| Reaction Efficiency | Increased Ct values | Diminished detection sensitivity |
| Quantification Accuracy | Skewed quantification curves | Overestimation of template concentration |
| Detection Specificity | False positive/false negative results | Compromised diagnostic reliability |
Multiplex PCR Complications
In multiplex PCR reactions where multiple targets are amplified simultaneously, primer dimer formation becomes exponentially more problematic [22]. The increased primer concentration necessary for multiple amplifications elevates the probability of intermolecular interactions between different primer pairs [22]. Furthermore, the complex network of potential interactions among numerous primers creates challenges for in silico prediction and prevention [22]. Consequently, primer optimization becomes particularly critical in multiplex applications, often requiring extensive empirical testing to identify primer combinations that minimize cross-dimer formation while maintaining efficient target amplification [22].
Detection and Identification Methods
Accurate detection of primer dimers is essential for troubleshooting PCR experiments and validating assay specificity. Multiple complementary techniques facilitate identification of these artifacts across different PCR formats.
Gel Electrophoresis Analysis
In conventional PCR, gel electrophoresis remains a fundamental method for visualizing primer dimers. Following amplification, PCR products are separated by size using agarose or polyacrylamide gel electrophoresis, with primer dimers exhibiting characteristic properties:
- Short Fragment Size: Typically appearing as bands or smears between 30-50 base pairs, migrating rapidly through the gel matrix [2] [7].
- Distinct Banding Pattern: Often manifesting as a diffuse, fuzzy smear rather than a sharp, well-defined band due to heterogeneity in the dimer products [7].
- Size Discrimination: Easily distinguishable from most target amplicons, which generally exceed 50 bp in length [2].
To enhance resolution of primer dimers from target amplicons, extended gel run times are recommended to provide sufficient separation between the fast-migrating dimers and slower target bands [7]. This approach allows visual confirmation of primer dimer presence and semi-quantitative assessment of their abundance relative to the desired product.
Melting Curve Analysis
In quantitative PCR utilizing intercalating dyes, melting curve analysis provides a powerful tool for discriminating specific amplicons from primer dimers. This technique exploits differences in melting temperature (Tm) between target products and primer dimers:
- Temperature Ramping: Following amplification, the reaction temperature is gradually increased while continuously monitoring fluorescence [2].
- Tm Discrepancy: Primer dimers, being shorter and often having lower GC content, denature at lower temperatures (typically below 80°C) compared to most specific amplicons [2] [15].
- Peak Differentiation: The resulting derivative plot (-dF/dT versus temperature) displays distinct peaks for each product species, allowing identification and quantification of primer dimer contribution to the total signal [2].
This method enables researchers to confirm reaction specificity and identify problematic amplification without additional post-processing steps. The "four steps PCR" protocol leverages this principle by acquiring fluorescence signals at temperatures between the Tm of primer dimers and the specific product, effectively excluding dimer-derived signal from quantification [2].
No-Template Controls (NTC)
Incorporating no-template controls (NTCs) represents a critical experimental practice for identifying primer dimer formation [7] [15]. NTC reactions contain all PCR components except the template DNA, thus any amplification signal detected must derive from nonspecific interactions, most commonly primer dimers [7]. The cycle threshold (Ct) value observed in the NTC provides a quantitative measure of primer dimer propensity, with lower Ct values indicating more pronounced dimer formation [22]. Reactions where NTCs amplify with Ct values within a few cycles of experimental samples indicate significant primer dimer interference that compromises result reliability [15].
Advanced Detection Tools
Recent computational advances have produced specialized tools for detecting primer dimers in sequencing data. URAdime (Universal Read Analysis of DIMErs) represents one such tool that analyzes primer sequences within sequencing reads to identify dimers and other unwanted amplicons like super-amplicons [25]. This approach enables researchers to attribute artifact generation to specific primers in multiplex reactions, facilitating targeted optimization of problematic primer pairs [25].
Experimental Strategies for Prevention and Minimization
Systematic approach combining computational design, reaction optimization, and enzymatic control provides the most effective defense against primer dimer formation. The following evidence-based strategies address the problem at multiple stages of the experimental workflow.
Primer Design Optimization
Judicious primer design represents the first and most crucial line of defense against primer dimer formation. Computational tools employing sophisticated algorithms significantly enhance primer quality by evaluating multiple parameters simultaneously:
- Minimize Complementarity: Primer design software checks for self-complementarity (hairpins) and inter-primer complementarity, especially at the 3' ends where extension initiates [2] [24]. Tools like Oligoanalyzer calculate dimer strength (ΔG), with values ≤ -9 kcal/mol indicating problematic interactions [15].
- Optimal Length and Tm: Design primers between 18-30 nucleotides with melting temperatures of 60-75°C, ensuring forward and reverse primers have Tm values within 2-5°C of each other [5] [24] [16].
- GC Content Management: Maintain GC content between 40-60%, with a GC clamp (G or C bases) at the 3' end to promote specific binding, but avoid more than 3 consecutive G/C residues which promote nonspecific interactions [24] [16].
- Avoid Repetitive Sequences: Eliminate runs of 4 or more identical bases and dinucleotide repeats (e.g., ACCCC or ATATATAT) that increase mispriming potential [24].
Advanced tools like varVAMP address additional challenges in specialized applications by designing degenerate primers for highly variable viral pathogens, incorporating degenerate nucleotides while maintaining specificity and minimizing dimer formation [26].
Reaction Condition Optimization
Fine-tuning PCR conditions establishes experimental parameters that favor specific amplification while suppressing dimer formation:
- Annealing Temperature Optimization: Implement temperature gradients to identify the highest possible annealing temperature that maintains efficient target amplification while disrupting weaker primer-primer interactions [7].
- Primer Concentration Titration: Reduce primer concentrations to decrease interaction probability while maintaining sufficient amplification efficiency, typically testing concentrations between 0.1-0.5 μM [7] [15].
- Magnesium Concentration Adjustment: Optimize Mg²⁺ concentration, as excess magnesium stabilizes nonspecific interactions and promotes dimer formation [5].
- Template Quality Assurance: Use high-quality template DNA to maximize specific amplification competitiveness against dimer formation [23].
Table 2: Research Reagent Solutions for Primer Dimer Prevention
| Reagent Category | Specific Examples | Function in Prevention |
|---|---|---|
| Hot-Start Polymerases | Antibody-inhibited, chemically modified, or aptamer-blocked enzymes | Prevent primer extension during reaction setup at low temperatures |
| Optimized Buffer Systems | Magnesium-separated formulations, additive-enhanced buffers | Control cation availability and stabilize specific interactions |
| dNTP Mixtures | Balanced dNTP solutions at optimal concentrations | Ensure nucleotide availability for specific amplification |
| Specific Detection Chemistries | TaqMan probes, Molecular Beacons, Dual-Labeled Probes | Generate signal only from specific amplicons, ignoring primer dimers |
Hot-Start PCR Techniques
Hot-start methodologies physically or chemically separate reaction components until high temperatures are reached, preventing enzymatic activity during reaction setup:
- Chemical Modification: A small molecule covalently bound to the DNA polymerase active site is released only after extended incubation at 95°C, rendering the enzyme inactive until activated by heat [2].
- Antibody Inhibition: DNA polymerase is non-covalently bound by a neutralizing antibody that denatures at high temperatures, releasing active enzyme [2].
- Physical Separation: Reaction components are physically separated by wax barriers that melt at high temperatures, mixing components only after the reaction reaches denaturing temperatures [2].
- Magnesium Sequestering: Magnesium ions are chemically complexed and released only at elevated temperatures, depriving the polymerase of essential cofactors during setup [2].
These approaches collectively prevent the pre-PCR primer extension that establishes the initial primer dimer templates, significantly reducing their subsequent amplification [7] [22].
Structural Primer Modifications
Innovative primer engineering strategies create structural barriers to dimer formation while maintaining amplification efficiency:
- HANDS (Homo-Tag Assisted Non-Dimer System): Incorporation of a nucleotide tail complementary to the 3' end of the primer creates a stem-loop structure that prevents intermolecular annealing while permitting target binding [2].
- Chimeric Primers: Strategic replacement of DNA bases with RNA bases creates sequences with lower melting temperatures in primer-primer interactions than in primer-target interactions, enabling temperature-based specificity [2].
- SAMRS (Self-Avoiding Molecular Recognition Systems): Incorporation of nucleotide analogues that bind to natural DNA but not to other SAMRS-containing primers, effectively eliminating primer-primer interactions [2].
- Blocked-Cleavable Primers: Use of primers with removable 3' end blocks that are only removed at high temperatures by specialized enzymes, preventing extension at low temperatures [2].
Primer dimers represent a multifactorial challenge with significant implications for PCR efficiency and accuracy, particularly in quantitative applications and diagnostic assays. Their formation depletes critical reaction resources, competes with specific amplification targets, and generates misleading signals that compromise experimental conclusions. Through comprehensive understanding of dimerization mechanisms and implementation of integrated prevention strategies—including sophisticated primer design, reaction optimization, hot-start methodologies, and structural modifications—researchers can significantly mitigate these detrimental effects. As PCR technologies continue to evolve toward greater sensitivity and multiplexing capacity, vigilant attention to primer dimer prevention remains fundamental to generating reliable, reproducible molecular data that advances both basic research and clinical applications.
In the realm of molecular biology, particularly within polymerase chain reaction (PCR) and primer design research, the accurate interpretation of gel electrophoresis results is paramount. Non-specific amplification artifacts represent a significant challenge, compromising experimental integrity, reducing amplification efficiency, and skewing quantitative results. These artifacts—including primer dimers, smears, and unexpected bands—directly impact the reliability of data in fields ranging from diagnostic assay development to drug discovery. This technical guide provides a comprehensive framework for identifying these artifacts within the broader context of primer dimer and nonspecific amplification research, equipping scientists with the diagnostic tools necessary for troubleshooting and optimizing molecular assays.
Understanding Non-Specific Amplification
Non-specific amplification occurs when PCR processes deviate from their intended target, generating unwanted DNA products that compete with target amplicons for reaction components. This phenomenon is fundamentally rooted in the biochemical principles of DNA polymerization, where primers anneal to non-target sequences or to each other under suboptimal conditions. The exponential nature of PCR means that even minor mis-priming events occurring early in the cycling process can become significantly amplified, leading to substantial artifacts that obscure experimental results [3].
The clinical and research implications of these artifacts are substantial. In diagnostic applications, non-specific amplification can lead to false positives or inaccurate quantification. In preparative applications like cloning or sequencing, these artifacts can reduce the yield of the desired product or necessitate additional purification steps. Understanding the mechanisms behind these artifacts is therefore critical for both troubleshooting failed experiments and designing robust, reliable assays from the outset [3].
A Systematic Guide to Common Gel Electrophoresis Artifacts
Systematic identification of gel electrophoresis artifacts requires understanding their distinct visual characteristics, underlying causes, and diagnostic features. The following table provides a consolidated reference for the most commonly encountered artifacts.
Table 1: Characteristics of Common Gel Electrophoresis Artifacts
| Artifact Type | Visual Appearance on Gel | Typical Size Range | Primary Causes | Impact on Downstream Applications |
|---|---|---|---|---|
| Primer Dimers | Single bright band at the very bottom of the gel | 20-60 base pairs [3] | Primer-primer hybridization due to complementary sequences, especially at 3' ends [27] | Competes with target amplification; usually removed in clean-up processes [3] |
| Primer Multimers | Ladder-like pattern of multiple bands at the bottom of the gel [3] | ~100 bp, 200 bp, and larger multiples [3] | Joining of multiple primer dimers into amplifiable complexes; high primer concentration [3] | Interferes with interpretation and sequencing; difficult to remove [3] |
| PCR Smears | Continuous, diffuse spread of DNA across a size range [3] | Variable (often appears as a background smear) | Highly fragmented DNA template; degraded primers; too low annealing temperature; excessive template DNA [3] | Obscures specific bands; makes amplicons unsequenceable; typically requires PCR repetition [3] |
| Unexpected Discrete Bands | One or more distinct bands at unanticipated sizes [3] | Variable | Non-target amplification; mis-priming on genomic DNA; secondary primer binding sites [3] | Skews quantitative results; may lead to false conclusions in diagnostic assays |
| DNA Stuck in Wells | DNA failing to enter the gel, visible as bright material in wells [3] | N/A | Malformed wells; carryover of impurities from DNA extraction; overloaded PCR product; extremely large DNA complexes [3] | Prevents analysis of PCR products; indicates issues with sample quality or gel integrity [3] |
Primer Dimers and Multimers
Primer dimers represent one of the most frequently encountered artifacts in PCR. These structures form when two primers hybridize to each other rather than to the template DNA, creating a short, amplifiable duplex. The resulting amplicon typically consists of the two primer sequences joined end-to-end, sometimes with additional nucleotides in between [3]. The stability of 3' end complements is a critical factor in dimer formation, as this region facilitates polymerase binding and elongation [27].
It is crucial to distinguish true primer dimers from residual unincorporated primers, which also appear at the gel bottom but form a more diffuse, hazy band approximately 21-30 bp in size (the exact length of the primers used) [3]. While primer dimers themselves may not always interfere with interpreting larger amplicons, they compete for PCR reagents and can significantly reduce amplification efficiency of the target sequence.
When primer dimers join together, they form higher-order structures known as primer multimers, which create a characteristic laddering pattern on gels. These multimers are particularly problematic because they cannot be easily removed through standard clean-up methods and may co-migrate with target amplicons of interest, complicating gel interpretation and downstream applications [3].
PCR Smears and DNA in Wells
Smears represent a fundamentally different class of artifact characterized by continuous DNA distribution across a range of sizes rather than discrete bands. This pattern typically indicates random, non-specific DNA amplification occurring throughout the PCR process. Common causes include using heavily fragmented template DNA (which provides numerous unintended priming sites), degraded primers that behave unpredictably, or excessively low annealing temperatures that permit priming at non-specific sites [3].
The phenomenon of DNA becoming stuck in wells, while not always resulting from non-specific amplification, frequently co-occurs with smearing and indicates potential issues with sample quality or gel integrity. This artifact can result from carryover of impurities from DNA extraction (such as proteins or salts), overloading of PCR product, or the formation of exceptionally large DNA complexes that physically cannot enter the gel matrix [3]. When encountered, this issue necessitates troubleshooting of both the PCR conditions and the electrophoresis setup.
Diagnostic Workflow and Troubleshooting Approaches
A systematic approach to diagnosing and addressing gel artifacts significantly improves troubleshooting efficiency. The following workflow provides a logical progression for identifying and resolving common issues.
Diagram 1: Diagnostic workflow for troubleshooting common gel electrophoresis artifacts. The chart provides a systematic approach to identifying and addressing specific artifact types based on their visual characteristics.
Experimental Protocols for Artifact Identification
Standardized protocols ensure consistent and accurate identification of gel artifacts. The following methodology details the process from gel preparation through analysis.
Agarose Gel Electrophoresis Protocol
Equipment and Reagents:
- Casting tray and well combs
- Gel box and voltage source
- UV light source or blue light transilluminator
- Microwave or heating plate
- Agarose powder
- TAE or TBE buffer
- DNA intercalating dye (e.g., ethidium bromide, GelRed, or SYBR Safe)
- DNA ladder (size range appropriate for expected amplicons)
- Gel loading buffer [28]
Procedure:
- Gel Preparation: Prepare a 1-2% agarose gel by mixing agarose powder with buffer. Microwave until completely dissolved, then cool to approximately 50°C before adding DNA intercalating dye according to manufacturer's instructions. Pour into casting tray with well comb and allow to solidify completely [28].
- Sample Preparation: Mix DNA samples with 6X loading buffer (containing density agent and tracking dyes). The loading buffer increases sample density for proper well loading and provides visual markers to monitor electrophoresis progress [28].
- Electrophoresis: Place solidified gel in electrophoresis chamber and cover with running buffer. Load DNA ladder in first lane, followed by experimental samples. Run gel at 80-150V until the dye front has migrated 75-80% of the gel length [28].
- Visualization: Image gel using UV or blue light transmission. For subsequent purification, minimize DNA exposure to UV light to prevent damage [28].
Troubleshooting Notes:
- For better resolution of similarly sized bands, increase agarose percentage or run gel at lower voltage for longer duration [28].
- If bands appear fuzzy, ensure gel was poured and run uniformly, and that appropriate voltage was applied.
- Always include appropriate controls (no-template control, positive control) to distinguish true artifacts from contamination or other issues.
Advanced Troubleshooting Strategies
Beyond the basic diagnostic approach, several advanced strategies can address persistent artifact problems:
For Persistent Primer Dimers:
- Computational Prediction: Utilize tools like PrimerROC or other dimer prediction software during primer design phase to identify sequences with high dimerization potential before experimental validation [27].
- Primer Redesign: Avoid complementary sequences at 3' ends, particularly in the last 3-5 bases, as these are critical for polymerase binding and extension [27].
- Temperature Optimization: Implement touchdown PCR or two-step PCR protocols that use higher annealing temperatures to increase stringency.
For Smearing Issues:
- Template Quality Assessment: Verify template DNA integrity by running an aliquot on a gel before PCR. High-quality genomic DNA should appear as a single high-molecular-weight band with minimal smearing below.
- Cycle Number Optimization: Reduce PCR cycle number to minimize amplification of non-specific products that tend to appear in later cycles.
- Enzyme Selection: Use polymerases with enhanced fidelity and hot-start capabilities to prevent mis-priming during reaction setup.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful troubleshooting requires appropriate laboratory tools and reagents. The following table outlines essential components for investigating and addressing amplification artifacts.
Table 2: Essential Research Reagents and Solutions for Artifact Investigation
| Reagent/Solution | Primary Function | Application Notes |
|---|---|---|
| Hot-Start Polymerase | Reduces non-specific amplification by maintaining inactivity until high temperatures are reached [3] | Essential for preventing primer dimer formation during reaction setup; available in antibody-based or chemical modification formats |
| Gel Staining Dyes (e.g., EtBr, GelRed, SYBR Safe) | Enables visualization of DNA fragments under specific light sources [28] | EtBr is a known mutagen requiring careful handling; alternative dyes offer improved safety profiles with comparable sensitivity |
| DNA Ladder/Marker | Provides size reference for interpreting experimental bands [29] | Select ladders with size ranges appropriate for expected amplicons; essential for quantifying unexpected bands |
| PCR Optimization Kits | Provide pre-formulated mixtures of buffers, additives, and enhancers | Often include proprietary components that improve specificity, particularly for challenging templates like GC-rich regions |
| Primer Design Software (e.g., tools with dimer prediction algorithms) | Identifies potential primer-primer interactions and secondary structures before synthesis [27] | Computational prediction significantly reduces experimental troubleshooting time; tools like PrimerROC use ΔG calculations for dimer likelihood [27] |
Future Directions in Primer Dimer and Non-Specific Amplification Research
The field of amplification artifact research is evolving rapidly, with several promising avenues emerging. Deep learning approaches are now being applied to predict sequence-specific amplification efficiency, offering the potential to identify problematic sequences before experimental validation. Recent studies using convolutional neural networks (CNNs) have demonstrated high predictive accuracy for amplification efficiency based solely on sequence information, potentially revolutionizing primer and amplicon design processes [30].
Additionally, novel amplification methods that circumvent traditional primer-based approaches are under development. Recent research has explored primer-less amplification techniques using padlock probes and engineered polymerases capable of using RNA targets as primers, potentially bypassing many issues associated with conventional primer design [31]. While these methodologies remain in development, they represent promising alternatives that may reduce or eliminate certain classes of amplification artifacts in future applications.
The integration of these advanced computational and biochemical approaches with traditional troubleshooting methods will continue to enhance researchers' ability to design specific, efficient amplification assays, ultimately improving the reliability and reproducibility of molecular biology research across diverse applications.
The accurate identification and interpretation of gel electrophoresis artifacts—particularly primer dimers, smears, and unexpected bands—represents a critical skill set for researchers engaged in PCR-based methodologies. Through systematic observation, application of targeted troubleshooting strategies, and utilization of appropriate computational and biochemical tools, scientists can effectively diagnose and address the underlying causes of non-specific amplification. As research in this field advances, incorporating both improved predictive algorithms and novel amplification methodologies, the molecular biology community moves closer to the goal of eliminating these confounding artifacts, thereby enhancing the precision and reliability of genetic analysis across basic research, diagnostic, and therapeutic applications.
In polymerase chain reaction (PCR) research, nonspecific amplification represents a significant challenge that extends far beyond the well-characterized phenomenon of primer dimers. While primer dimers are a common focus of troubleshooting efforts, the broader landscape of amplification artifacts includes high-melting-temperature artifacts, smearing, and amplicons of unexpected sizes that can severely compromise experimental reproducibility and data interpretation [15] [3]. Within the context of primer dimer and nonspecific amplification research, it is crucial to recognize that these artifacts are not merely isolated inconveniences but rather symptoms of complex biochemical interactions involving template, non-template, and primer concentrations in the reaction mixture [15].
The reliability and reproducibility of quantitative PCR (qPCR) experiments depend heavily on standardized technical aspects and minimized on-bench times, as these factors directly influence the frequency and severity of nonspecific amplification [15]. This technical guide examines the full spectrum of amplification artifacts, provides detailed methodologies for their identification and prevention, and offers a scientific toolkit for researchers and drug development professionals seeking to optimize their amplification workflows. Through a comprehensive understanding of these phenomena, scientists can design more robust experiments and improve the accuracy of their genetic analyses across diverse applications from basic research to clinical diagnostics.
Defining the Problem: A Spectrum of Amplification Artifacts
Nonspecific amplification encompasses a range of unintended PCR products that compete with the target amplicon for reaction components and can lead to false positives, reduced amplification efficiency, and compromised data interpretation. These artifacts manifest in several distinct forms, each with characteristic causes and visual signatures in analytical methods such as gel electrophoresis.
Primer Dimers and Multimers
Primer dimers are short, unintended DNA fragments that form when primers anneal to each other rather than to the target template DNA [7] [32]. These artifacts typically range from 20-60 base pairs in length and appear as bright bands at the bottom of an electrophoresis gel, often with a smeary appearance rather than a well-defined band [7]. Two primary mechanisms govern primer dimer formation: self-dimerization (homodimer), where a single primer contains regions complementary to itself, and cross-primer dimerization (heterodimer), where forward and reverse primers share complementary regions [32]. When primer dimers join with other dimers, they can form larger primer multimers of 100 bp, 200 bp, or more, creating a laddering effect that further interferes with result interpretation [3].
PCR Smearing
PCR smearing appears as a continuous spread of DNA fragments of varying sizes on an electrophoresis gel, rather than discrete bands [3]. This phenomenon occurs when DNA is randomly amplified, producing a wide range of fragment lengths. Smearing can result from multiple factors including high levels of DNA fragmentation that produce fragments capable of self-priming, excessive template DNA concentration that increases the chance of self-priming, degraded primers, or excessively low annealing temperatures [3]. Unlike discrete nonspecific bands, smearing represents a more generalized amplification failure that can completely obscure target amplicons and render results uninterpretable.
High and Low Melting Temperature Artifacts
In qPCR applications, nonspecific amplification can produce artifacts with different melting temperatures than the target amplicon. Research has demonstrated that the occurrence of both low and high melting temperature artifacts is determined by annealing temperature, primer concentration, and cDNA input [15]. These artifacts are particularly problematic because they can go undetected in standard amplification curves but contribute significantly to fluorescence measurements, leading to inaccurate quantification [15]. The measurement of artifact-associated fluorescence can be minimized through careful primer design and modifications to the qPCR protocol, such as including a small heating step after the elongation phase [15].
Amplicons of Unexpected Sizes
Non-specific amplification can produce one or more discrete amplicons that differ in size from the expected target [3]. These artifacts may be either smaller or larger than the intended product and typically result from primers binding to non-target sequences with sufficient complementarity to allow extension. The presence of these unexpected amplicons indicates issues with primer specificity, annealing conditions, or template quality, and they can be particularly problematic when they co-migrate with legitimate products in gel electrophoresis or when they generate false positive signals in qPCR applications.
Table 1: Classification of Nonspecific Amplification Artifacts and Their Characteristics
| Artifact Type | Size Range | Gel Electrophoresis Appearance | Primary Causes |
|---|---|---|---|
| Primer Dimers | 20-60 bp | Fuzzy bands at gel bottom; smeary appearance | Primer complementarity; high primer concentration; low annealing temperature [3] [7] |
| Primer Multimers | 100+ bp | Ladder-like pattern at bottom of gel | Joining of multiple primer dimers; extendable 3' ends in dimer structures [3] |
| PCR Smearing | Continuous range | Continuous spread of DNA across lane | Highly fragmented DNA; too much template DNA; degraded primers; low annealing temperature [3] |
| High Tm Artifacts | Variable | Not always visible on standard gels | High cDNA input; specific primer/template interactions [15] |
| Unexpected Amplicons | Variable | Discrete bands at unexpected positions | Primers binding to non-target sequences; mispriming; repetitive elements in template [3] |
Quantitative Analysis: Factors Governing Artifact Formation
The formation of nonspecific amplification products follows predictable patterns based on reaction components and conditions. Understanding the quantitative relationships between these factors is essential for designing experiments that minimize artifacts while maintaining efficient target amplification.
The Template/Non-Template Concentration Balance
Research has demonstrated that the frequency of correct product amplification versus artifact formation depends significantly on the concentration of non-template cDNA in the reaction [15]. This finding challenges the conventional use of dilution series in which both template and non-template concentrations decrease simultaneously, as this approach alters the fundamental balance between these components. Titration experiments with plasmid DNA and mouse cDNA have revealed that valid quantification of the correct product is directly influenced by non-template cDNA concentration, suggesting that the ratio of template to non-template DNA may be more critical than absolute template concentration alone [15]. This phenomenon may be related to "jumping" PCR, where extended primers share homology with sequences elsewhere in the genome, leading to completely new amplification products [15].
Primer and Template Concentration Effects
The occurrence of PCR artifacts shows a clear dependence on both template and primer concentrations in the reaction mixture [15]. Excessive primer concentrations promote primer-dimer formation by increasing the likelihood of primer-primer interactions, while insufficient template concentration reduces competition for primer binding, further favoring artifact formation. Optimization experiments typically recommend primer concentrations in the range of 0.1-1 μM, with adjustments based on specific reaction conditions [33]. For conventional PCR, a primer-to-template ratio favoring specific amplification must be empirically determined for each primer-template system.
Thermal Cycling Parameters
Annealing temperature represents one of the most critical factors in controlling amplification specificity. The optimal annealing temperature is generally 3-5°C below the lowest primer Tm [33], but often needs adjustment based on empirical results. Annealing time also significantly impacts specificity, with longer annealing times increasing the opportunity for primers to bind to non-target sequences [33]. Similarly, extension times that are too long can promote the amplification of nonspecific products that may have formed during earlier cycles.
Table 2: Quantitative Parameters Influencing Nonspecific Amplification
| Parameter | Optimal Range | Effect on Specificity | Experimental Optimization Approach |
|---|---|---|---|
| Primer Concentration | 0.1-1 μM | High concentrations promote primer-dimer formation; low concentrations reduce amplification efficiency [33] | Checkerboard titration with varying primer concentrations |
| Template Quantity | 1-100 ng (genomic DNA) | Excessive DNA increases mispriming; insufficient DNA reduces competitive specific amplification [33] | Dilution series with fixed primer concentration |
| Annealing Temperature | 3-5°C below Tm | Increased temperature enhances specificity but may reduce yield; decreased temperature promotes artifacts [33] | Temperature gradient PCR |
| Mg2+ Concentration | 1.5-2.5 mM (varies by polymerase) | Excess Mg2+ promotes non-specific priming; insufficient Mg2+ reduces polymerase efficiency [33] | Mg2+ concentration gradient |
| Cycle Number | 25-35 | Excessive cycles amplify early-formed artifacts; too few cycles yield insufficient product [33] | Monitor amplification in real-time or with endpoint analysis |
Experimental Protocols for Identification and Troubleshooting
Melting Curve Analysis for qPCR Artifact Detection
Melting curve analysis represents a powerful method for identifying nonspecific amplification in qPCR applications. This protocol enables discrimination between the target amplicon and artifacts based on their distinct melting temperatures.
Protocol Steps:
- Following qPCR amplification, slowly heat the product from 65°C to 95°C while continuously monitoring fluorescence.
- Plot the negative derivative of fluorescence versus temperature (-dF/dT) to identify peaks corresponding to different amplification products.
- Identify the target amplicon peak based on its expected melting temperature and compare with control reactions.
- Investigate additional peaks as potential artifacts, particularly those with melting temperatures lower than the target amplicon, which often represent primer dimers [15].
- For validation, compare melting curves from reactions with different template concentrations, as artifact prevalence often increases with dilution [15].
Gradient PCR for Annealing Temperature Optimization
Systematic optimization of annealing temperature is crucial for minimizing nonspecific amplification. Gradient PCR allows simultaneous testing of multiple annealing temperatures in a single run.
Protocol Steps:
- Set up identical PCR reactions with standardized template and primer concentrations.
- Program the thermal cycler to use a temperature gradient across the block during the annealing step, typically spanning 5-10°C above and below the calculated Tm of the primers.
- After amplification, analyze products by gel electrophoresis or other appropriate methods.
- Identify the annealing temperature that produces the strongest target signal with minimal nonspecific products.
- For qPCR applications, additionally consider amplification efficiency and Cq values when selecting the optimal temperature.
Checkerboard Titration for Primer and Template Optimization
This protocol systematically evaluates interactions between primer and template concentrations to identify conditions that favor specific amplification.
Protocol Steps:
- Prepare a matrix of reactions with varying primer concentrations (e.g., 0.1, 0.25, 0.5, 0.75, 1.0 μM) and template concentrations (e.g., 1:10 serial dilutions).
- Include appropriate positive and negative controls in the experimental design.
- Perform amplification using the optimized annealing temperature determined from gradient PCR.
- Analyze results to identify primer:template combinations that yield specific amplification with minimal artifacts.
- For qPCR applications, also consider amplification efficiency when selecting optimal conditions.
Diagram 1: Decision pathway for troubleshooting nonspecific amplification
The Scientist's Toolkit: Research Reagent Solutions
Successful prevention and minimization of nonspecific amplification requires careful selection of reagents and implementation of specialized techniques. The following toolkit provides essential solutions for researchers addressing amplification artifacts.
Table 3: Research Reagent Solutions for Nonspecific Amplification
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| Hot-Start DNA Polymerases | Remain inactive until high-temperature activation, preventing primer-dimer formation during reaction setup [33] | Essential for high-sensitivity applications; reduces pre-amplification artifacts |
| PCR Additives (DMSO, Betaine) | Reduce secondary structure, improve specificity especially for GC-rich templates [33] | Concentration must be optimized; typically used at 1-10% final concentration |
| Mg2+ Optimization Solutions | Adjust co-factor concentration to balance specificity and efficiency [33] | Excessive Mg2+ promotes nonspecific binding; insufficient reduces efficiency |
| Primer Design Software (Primer-BLAST, OligoAnalyzer) | Identify primers with minimal self-complementarity and high specificity [15] [34] | Critical for minimizing 3' complementarity that leads to primer dimers |
| Gradient Thermal Cyclers | Enable empirical optimization of annealing temperature across a range [33] | More efficient than sequential testing of individual temperatures |
| No-Template Control (NTC) | Detects contamination and primer-dimer formation in absence of template [7] | Essential control for every PCR experiment |
| Unified Annealing Temperature Master Mixes | Allow multiple targets to be amplified at a single annealing temperature [35] | Particularly valuable for multiplex PCR applications |
Advanced Computational Approaches for Multiplex PCR
As PCR panels scale to higher multiplex levels, computational approaches for primer design become increasingly important for minimizing nonspecific interactions. Traditional primer design tools struggle with highly multiplexed panels due to the quadratic increase in potential primer dimer interactions with each additional primer [35].
The SADDLE Algorithm for Highly Multiplexed Primer Design
Simulated Annealing Design using Dimer Likelihood Estimation (SADDLE) represents an advanced computational framework specifically developed to address the challenges of highly multiplexed PCR primer design [35]. This algorithm employs a stochastic optimization approach to navigate the vast sequence space and identify primer combinations with minimal dimer formation potential.
Key Algorithm Steps:
- Primer candidate generation: Systematic generation of potential primers with optimal binding energies (ΔG° ≈ -11.5 kcal/mol) and appropriate length constraints [35].
- Initial primer set selection: Random selection of primer pair candidates for each target amplicon.
- Loss function evaluation: Calculation of a "Badness" score representing potential primer dimer interactions between all primer pairs in the set [35].
- Iterative optimization: Repeated random modification of the primer set with probabilistic acceptance based on improved Loss function scores.
- Convergence to optimal set: Continuation until an acceptable primer set with minimal dimer potential is identified.
Experimental validation of SADDLE-designed primer sets demonstrated a dramatic reduction in primer dimer formation, from 90.7% in naively designed primer sets to just 4.9% in optimized 96-plex sets (192 primers) [35]. Even when scaling to 384-plex reactions (768 primers), the optimized primer sets maintained low dimer formation, enabling highly multiplexed detection applications such as the identification of 56 distinct gene fusions in lung cancer from a single-tube assay [35].
Diagram 2: SADDLE workflow for multiplex primer design optimization
Nonspecific amplification and smearing represent multidimensional challenges in PCR that extend well beyond simple primer dimer formation. Successful mitigation requires a comprehensive approach addressing primer design, reaction component balance, thermal cycling parameters, and specialized biochemical additives. The quantitative relationships between template, non-template, and primer concentrations play a particularly crucial role in determining whether specific amplification or artifacts predominate [15].
For researchers and drug development professionals, implementation of systematic troubleshooting workflows and advanced computational design tools like SADDLE can dramatically improve amplification specificity, even in highly multiplexed reactions [35]. As PCR technologies continue to evolve toward higher sensitivity and greater multiplexing capabilities, the principles and protocols outlined in this technical guide provide a foundation for achieving reliable, reproducible results across diverse applications from basic research to clinical diagnostics.
By recognizing nonspecific amplification as a predictable consequence of reaction component interactions rather than random experimental failure, researchers can adopt proactive design and optimization strategies that minimize artifacts before they compromise experimental outcomes. This systematic approach to understanding and addressing amplification artifacts represents an essential component of rigorous molecular research practice.
Strategic Assay Design: Proactive Methods to Prevent Primer Dimers and Nonspecific Binding
Polymerase chain reaction (PCR) stands as a cornerstone technology in molecular biology, with its success fundamentally reliant on the precise design of oligonucleotide primers. A critical challenge in PCR assay development involves avoiding artifacts such as nonspecific amplification and primer-dimer formation, which predominantly originate from primer self-complementarity and hairpin structures. These artifacts compete for reagents, reduce amplification efficiency, and can lead to false-positive or false-negative results, particularly in sensitive applications like diagnostic PCR and quantitative PCR (qPCR) [22].
The advent of in silico primer design tools has revolutionized primer development by enabling researchers to identify and eliminate problematic primers before costly laboratory experimentation. This technical guide explores how computational approaches leverage thermodynamic parameters and algorithmic checking to predict and prevent self-complementarity and hairpin formation, thereby enhancing PCR specificity and efficiency within the broader context of primer-dimer and nonspecific amplification research.
Fundamental Primer Design Parameters
Effective primer design requires balancing multiple thermodynamic and sequence-based parameters to ensure optimal binding specificity and amplification efficiency while minimizing secondary structures.
Core Design Criteria
The table below summarizes the critical parameters for optimal primer design:
| Parameter | Optimal Range | Biological Significance | Consequence of Deviation |
|---|---|---|---|
| Length | 18-24 nucleotides [16] | Balances specificity with binding efficiency | Short primers: reduced specificity; Long primers: slower hybridization [16] |
| Melting Temperature (Tm) | 54°C-65°C [16]; 50°C-72°C [36] | Temperature at which 50% of primer-template duplex dissociates | Low Tm: non-specific binding; High Tm: secondary annealing [16] |
| GC Content | 40%-60% [16] [36] | Affects duplex stability (GC bonds have 3 H-bonds vs. AT's 2) [16] | Low GC: weak binding; High GC: non-specific binding & primer-dimer formation [16] |
| GC Clamp | 1-2 G/C in last 5 bases at 3' end [16] [37] | Promotes stable binding at critical extension point | >3 G/C at 3' end: non-specific binding & false positives [16] |
| Self-Complementarity | Minimal (particularly at 3' end) [38] | Prevents self-dimerization and hairpin formation | Primer-dimer artifacts, reduced target amplification [2] [22] |
Thermodynamic Considerations
The melting temperature (Tm) represents a critical factor in determining annealing conditions. Primer pairs should have Tm values within 2-5°C of each other to ensure synchronous binding [16] [36]. The annealing temperature (Ta) is typically set 2-5°C below the Tm of the primers [16]. Multiple formulas exist for Tm calculation, with salt concentration adjustments being necessary for accuracy [34] [16].
The presence of a GC clamp (1-2 G or C bases at the 3' end) enhances binding stability but should not exceed three G/C residues in the final five bases to prevent non-specific binding [16] [37]. GC content should be distributed uniformly throughout the primer, as clustering of G/C bases can promote mispairing [37].
Mechanisms of Primer Artifact Formation
Self-Complementarity and Primer-Dimer Formation
Self-complementarity occurs when regions within a single primer or between forward and reverse primers contain complementary sequences, leading to three primary types of artifacts:
Self-dimers: Form when two copies of the same primer anneal to each other due to intra-primer homology [16] [7]. This occurs when a single primer contains regions complementary to itself.
Cross-dimers: Result from hybridization between forward and reverse primers due to inter-primer homology [16] [7]. These are particularly problematic as they provide free 3' ends for polymerase extension.
Hairpin structures: Form through intramolecular interactions when two regions of three or more nucleotides within a primer are complementary to each other [16]. These secondary structures prevent proper binding to the target template.
The formation mechanism follows a three-step process: (1) two primers anneal at their 3' ends through complementary sequences; (2) DNA polymerase binds and extends the primers according to the complementary sequence; (3) in subsequent cycles, the extended product serves as a template for further amplification, leading to exponential accumulation of primer-dimer artifacts [2].
Impact on PCR Efficiency
Primer artifacts negatively impact PCR performance through multiple mechanisms:
- Resource competition: Primer-dimers consume dNTPs, primers, and DNA polymerase that would otherwise amplify the target sequence [22]
- Template competition: Primers bound to themselves or each other are unavailable for target binding [22]
- Detection interference: In qPCR, primer-dimers can be amplified and detected, particularly with intercalating dyes like SYBR Green, leading to false-positive results [2] [22]
- Reduced sensitivity: For low-copy targets, resource depletion may prevent detection, resulting in false negatives or increased Ct values [22]
In Silico Tools and Workflows
Computational Design Pipeline
Modern primer design employs sophisticated algorithms that integrate multiple checks for potential secondary structures. The following workflow illustrates a comprehensive in silico primer design and validation process:
Key In Silico Tools
Primer-BLAST: Integrates NCBI's Primer3 design engine with BLAST specificity checking to generate target-specific primers while avoiding off-target amplification [34] [37]. Users can define position ranges, set Tm calculations using SantaLucia 1998 parameters, and specify organism databases for specificity checking [34].
Primer3/Primer3Plus: Widely-used algorithms for designing PCR primers, hybridization probes, and sequencing primers with comprehensive parameter controls [39]. These tools provide input to Primer3 of design parameters determined by the scientist [39].
OligoAnalyzer: Thermodynamic tool for screening primer designs; ideal ΔG values for potential dimers should be weaker than approximately -9 kcal/mol (less negative) [37].
Specificity Checking Parameters
Primer-BLAST allows researchers to search primers against multiple databases to determine whether a primer pair can generate PCR products on unintended targets [34]. Key specificity parameters include:
- Database selection: RefSeq mRNA, Refseq representative genomes, or custom databases [34]
- Organism restriction: Limits specificity checking to specified organisms, improving speed and relevance [34]
- Mismatch requirements: Requires at least one primer to have a specified number of mismatches to unintended targets [34]
- Exon junction spanning: Directs the program to return primers that span exon-exon junctions for cDNA-specific amplification [34]
Experimental Validation Protocols
In Silico Validation Workflow
After initial primer design, rigorous in silico validation is essential:
Step 1: Specificity Verification
- BLAST each primer individually against the appropriate genome database
- Confirm minimal off-target matches, especially at the 3' end
- Use Primer-BLAST's specificity reports to identify potential cross-hybridization [34] [37]
Step 2: Secondary Structure Analysis
- Screen for self-complementarity and hairpin formation using thermodynamic tools
- Pay particular attention to the 3' end complementarity
- Calculate ΔG values for potential dimers; values less negative than -9 kcal/mol are preferred [37]
Step 3: In Silico PCR Simulation
- Simulate amplicons using tools like UCSC in silico PCR
- Confirm expected product size and absence of spurious products [37]
- Verify primer binding sites do not overlap known SNPs or repetitive elements [37]
Laboratory Validation Methods
No-Template Control (NTC) Assay
- Perform PCR with all components except template DNA [7]
- In SYBR Green qPCR, primer-dimers will appear as amplification in NTC [22]
- Analyze NTC products by gel electrophoresis - primer-dimers typically appear as smeary bands below 100 bp [7]
Melting Curve Analysis
- For qPCR with intercalating dyes, perform melting curve analysis post-amplification
- Primer-dimers denature at lower temperatures than specific amplicons [2]
- Distinct, consistent melting peaks indicate specific amplification [2]
Gel Electrophoresis Characterization
- Run PCR products on high-percentage agarose gels (2-3%)
- Primer-dimers appear as fuzzy smears or bands at 30-50 bp [7] [2]
- Run gels longer to separate primer-dimers from specific products [7]
Optimization Strategies for Problematic Primers
Computational Redesign Approaches
When primers exhibit self-complementarity or hairpin tendencies:
- Shift binding sites: Move primers 5-10 bases upstream or downstream to avoid self-complementary regions [37]
- Adjust length: Slightly increase or decrease primer length by 1-2 bases to disrupt complementary stretches [16]
- Modify 3' ends: Terminate primers with A or T bases to reduce 3' end stability and minimize dimerization [16]
- Check alternative sequences: Target different conserved regions identified through multiple sequence alignment [38]
Experimental Optimization Techniques
PCR Condition Adjustments
- Increase annealing temperature incrementally (2-5°C) to reduce non-specific interactions [7] [36]
- Lower primer concentration (0.05-1.0 μM) to decrease primer-dimer probability [7] [36]
- Use touchdown PCR where annealing temperature starts high and gradually decreases [36]
- Increase denaturation times to ensure complete separation of secondary structures [7]
Reagent Selection
- Utilize hot-start DNA polymerases that remain inactive until high temperatures are reached [7] [2]
- Consider specialized polymerases for GC-rich templates with secondary structures [36]
- Add DMSO or other stabilizers for problematic templates with strong secondary structures [37]
Research Reagent Solutions
The following table outlines essential reagents and their functions in preventing primer artifacts:
| Reagent/Tool | Function | Application Context |
|---|---|---|
| Hot-Start DNA Polymerase | Prevents enzymatic activity at low temperatures during reaction setup | All PCR applications, particularly multiplex and qPCR [7] [2] |
| DMSO | Reduces secondary structure formation in template and primers | GC-rich targets, problematic templates with strong hairpins [37] |
| SYBR Green I | Intercalating dye for real-time detection of double-stranded DNA | qPCR optimization; enables melting curve analysis [2] [22] |
| TaqMan Probes | Sequence-specific fluorescent probes with reporter and quencher | qPCR applications requiring high specificity; prevents false positives from primer-dimers [22] |
| NCBI Primer-BLAST | Integrated primer design and specificity checking tool | Initial primer design and in silico validation [34] [37] |
| OligoAnalyzer | Thermodynamic analysis of secondary structures | Screening candidate primers for self-dimers and hairpins [37] |
Case Study: SARS-CoV-2 Primer Design
A comprehensive study designing SARS-CoV-2 detection primers demonstrates the application of these principles. Researchers analyzed 2,341 genome sequences to identify conserved regions, then designed nine primer systems targeting these areas [38]. In silico validation against 211,833 SARS-CoV-2 sequences revealed that optimized primers exhibited:
- Lower self-complementarity (both overall and 3') compared to previously described primers [38]
- Higher specificity with minimal off-target binding to human, bacterial, fungal, and other betacoronavirus genomes [38]
- Improved detection of recent viral variants compared to earlier primer sets [38]
This systematic approach highlights how in silico design targeting conserved regions can produce robust primers resistant to sequence polymorphisms while minimizing secondary structures.
In silico primer design represents a critical first step in developing specific and efficient PCR assays. By leveraging computational tools to identify and eliminate primers with self-complementarity and hairpin-forming potential, researchers can significantly reduce primer-dimer formation and nonspecific amplification. The integration of sophisticated algorithms like those in Primer-BLAST with thermodynamic analysis provides a powerful framework for predicting primer behavior before laboratory validation.
As PCR applications continue to expand into diagnostics, multiplex assays, and complex genomic targets, the importance of rigorous in silico design only increases. Future developments in machine learning and improved thermodynamic models will further enhance our ability to predict and prevent primer artifacts, ultimately leading to more reliable molecular assays across diverse research and clinical applications.
In molecular biology, the polymerase chain reaction (PCR) is a ubiquitous, fast, flexible, and cost-effective technique for amplifying DNA regions of interest. The success of PCR and its numerous derivative applications hinges almost entirely on the precise design of oligonucleotide primers. Manual primer design can be an error-prone and time-consuming process, particularly when dealing with large numbers of target sites. Primer design software has emerged as an essential solution to these challenges, integrating sophisticated algorithms that optimize physical parameters and evaluate specificity to ensure successful amplification while minimizing nonspecific byproducts such as primer-dimers. The evolution of these tools represents a critical advancement in molecular biology, enabling researchers to move from labor-intensive manual design to automated, high-throughput pipelines that dramatically improve experimental reliability and reproducibility.
The fundamental challenge in primer design lies in simultaneously optimizing multiple, often competing, parameters. Effective primers must bind specifically to their intended target sequences with high efficiency while avoiding the formation of secondary structures or off-target amplification. Within the context of broader research on primer dimers and nonspecific amplification, computational tools provide systematic approaches to address these persistent experimental problems. This technical guide explores the core principles, software capabilities, and experimental methodologies that underpin modern primer design, with particular emphasis on ensuring specificity and optimizing physical parameters for successful DNA amplification across diverse applications.
Computational Tools for Primer Design and Analysis
The landscape of primer design software encompasses both standalone tools and integrated platforms, each offering distinct capabilities tailored to different experimental needs. These tools employ sophisticated algorithms to optimize critical primer parameters and evaluate potential specificity issues before physical experiments begin.
Large-scale automated design pipelines represent the cutting edge in primer bioinformatics. CREPE (CREate Primers and Evaluate) exemplifies this category, fusing the functionality of Primer3 with In-Silico PCR (ISPCR) to create an integrated pipeline that performs both primer design and specificity analysis at scale. This system processes multiple target sites simultaneously through a custom evaluation script, generating a final output that summarizes the lead primer pair for each target alongside measures of off-target binding likelihood and additional decision-making metrics. Experimental validation of CREPE demonstrates successful amplification for more than 90% of primers deemed acceptable by the software, highlighting its reliability for targeted amplicon sequencing applications [40].
Species-specific primer design requires additional considerations to ensure amplification selectivity across closely related organisms. PrimeSpecPCR addresses this need through an open-source Python toolkit that automates the workflow of species-specific primer and probe design and validation. Its modular architecture comprises four main components: (1) automated retrieval of genetic sequences from NCBI databases based on taxonomy identifiers; (2) multiple sequence alignment using MAFFT to generate consensus sequences; (3) thermodynamically optimized primer and probe design via Primer3-py; and (4) multi-tiered specificity testing against the NCBI GenBank database. This toolkit accelerates primer development through parallel processing, automatic caching of intermediate results, and interactive HTML reports that visualize specificity profiles across taxonomic groups [41].
Commercial primer design tools offer user-friendly interfaces with optimized default parameters. Eurofins Genomics provides a web-based PCR Primer Design Tool that analyzes entered DNA sequences to select optimum primer pairs based on constraints including primer length, GC content, melting temperature, and amplicon characteristics. The tool employs thermodynamic calculations using the nearest-neighbor model of Borer and parameters determined by SantaLucia, avoiding primers with extensive self-dimer and cross-dimer formations to minimize secondary structures [42]. Similarly, VectorBuilder's Primer Design tool offers a customizable interface that optimizes primer length, GC content, melting temperature, secondary structures, and specificity for applications including cloning, sequencing, and CRISPR genome editing [43].
Comprehensive primer analysis platforms like Benchling provide integrated environments for primer design, management, and validation. Benchling's Molecular Biology application enables both manual primer design and automated wizard-based design, with features for visualizing primer binding sites, calculating thermodynamic properties (including Gibbs Free Energy values for homodimers and monomers, melting temperatures, and GC content), and building custom primer libraries for team collaboration. A particularly valuable feature is the platform's integration with in silico PCR and NCBI BLAST, allowing researchers to verify primer specificity directly within the workflow [44].
Table 1: Overview of Primer Design Software Tools
| Tool Name | Primary Application | Key Features | Specificity Validation Method |
|---|---|---|---|
| CREPE | Large-scale targeted amplicon sequencing | Integrated Primer3 and ISPCR pipeline, parallel processing | In-Silico PCR (ISPCR) against user-defined background |
| PrimeSpecPCR | Species-specific PCR/qPCR | Taxonomic sequence retrieval, consensus generation, multi-tiered testing | BLAST against NCBI GenBank with taxonomic profiling |
| NCBI Primer-Blast | Standard PCR with specificity checking | Combines Primer3 with BLAST capabilities | Automatic BLAST against selected organism databases |
| Eurofins PCR Designer | Standard PCR and RT-PCR | Physical parameter optimization, dimer prediction | Internal algorithms checking non-specific binding sites |
| BatchPrimer3 | High-throughput primer design | Batch processing of multiple templates, various primer types | No automatic BLAST; requires separate validation |
| VectorBuilder Tool | Cloning, sequencing, CRISPR | User-friendly interface, parameter customization | Basic specificity checking based on sequence uniqueness |
For specialized applications, primer design tools have diversified to support emerging methodologies. PrimerDigital.com offers tools for quantitative fluorescent PCR (QF-PCR), genotyping, loop-mediated isothermal amplification (LAMP), recombinase polymerase amplification (RPA), and Gibson assembly, addressing the unique requirements of each technique [45]. The development of cooperative primer technology represents another significant innovation, with demonstrated reductions in nonspecific amplification by up to 2.5 million-fold compared to conventional primers. This approach greatly reduces primer-dimer propagation, enabling successful amplification of 60 template copies amidst a background of 150,000,000 primer-dimers—a scenario where normal primers with or without hot start experience signal dampening and false negatives [46].
Physical Parameters for Optimal Primer Design
The biochemical properties of primers fundamentally determine their performance in amplification reactions. Primer design software optimizes a constellation of interconnected physical parameters to ensure efficient and specific binding to target sequences. Understanding these parameters and their optimal ranges provides researchers with critical insights for both automated design and manual refinement.
Primer length and melting temperature (Tm) represent two of the most critical design considerations. Primers are typically designed to be 18-25 nucleotides long, as shorter primers may lack specificity while longer primers are more prone to forming secondary structures [43]. The melting temperature, defined as the temperature at which 50% of the primer is bound to template DNA while the other half exists in single-stranded form, ideally falls between 55°C and 65°C [43]. Perhaps more importantly, forward and reverse primers should have similar Tm values, ideally within 2-3°C of each other, to ensure both primers bind and extend simultaneously during the amplification process [43]. Modern software calculates Tm using sophisticated thermodynamic models such as the nearest-neighbor method, which accounts for the sequence-dependent stability of adjacent nucleotides and provides more accurate predictions than simple GC-content-based calculations [42].
GC content and sequence composition significantly impact primer stability and binding characteristics. The proportion of guanine (G) and cytosine (C) bases in a primer should ideally be between 40-60%, as the three hydrogen bonds between G and C (versus two between A and T) contribute to binding stability [43]. Excessively high GC content can increase melting temperature potentially leading to inefficient amplification, while low GC content may result in reduced primer stability and weak binding [43]. The 3' end of the primer demands particular attention in design, as this is where DNA polymerase initiates synthesis. Most design tools avoid placing more than two G or C nucleotides at the 3' end, as this can create overly strong binding and increase the risk of non-specific amplification [43]. This design feature, often called a "GC clamp," helps ensure efficient and accurate primer extension while minimizing off-target binding.
Secondary structure formation represents a major challenge in PCR specificity. Primer design software incorporates algorithms to detect and avoid sequences prone to forming hairpins (where a primer folds back on itself) or dimers (where primers bind to each other instead of the target sequence) [43]. These secondary structures interfere with primer availability for target DNA, thereby hindering the amplification process. Advanced tools like Benchling provide Gibbs Free Energy values for homodimer and monomer formation, along with secondary structure diagrams that visually represent potential interaction problems [44].
Table 2: Optimal Ranges for Key Primer Physical Parameters
| Parameter | Optimal Range | Impact on Performance | Calculation Method |
|---|---|---|---|
| Length | 18-25 nucleotides | Specificity increases with length; secondary structures risk with longer primers | Direct sequence count |
| GC Content | 40-60% | Stability increases with GC content; very high GC increases Tm excessively | (G+C)/(G+C+A+T) × 100% |
| Melting Temperature (Tm) | 55-65°C | Must be compatible with polymerase activity and specific enough for binding | Nearest-neighbor thermodynamics [42] |
| Tm Difference | ≤2-3°C between forward and reverse | Ensures both primers hybridize simultaneously | Difference between calculated Tm values |
| 3' End Stability | Avoid >2 G/C in last 5 bases | Reduces non-specific initiation while maintaining extension efficiency | Terminal nucleotide analysis |
Reaction conditions significantly influence primer behavior and must be considered during the design process. Salt concentration (typically 50mM default) and primer concentration (typically 50nM default) affect both primer and product melting temperatures [42]. These parameters factor into the thermodynamic calculations performed by design tools, emphasizing the importance of specifying actual reaction conditions rather than relying solely on default values. The development of recombinase polymerase amplification (RPA) and other isothermal techniques has introduced additional design considerations, as these methods operate at lower temperatures (37-42°C) and employ different enzyme systems compared to conventional PCR [47] [45].
Experimental Protocols for Primer Validation
Computational primer design represents only the first step in developing robust amplification assays. Experimental validation through carefully designed protocols is essential to confirm specificity and optimize reaction conditions. The following methodologies provide systematic approaches for validating primer performance and addressing nonspecific amplification.
Specificity Testing and In Silico PCR
Before synthesizing primers, comprehensive in silico specificity analysis should be performed. The CREPE pipeline exemplifies this approach by integrating ISPCR to predict potential off-target binding sites across the relevant genome or sequence database [40]. Similarly, PrimeSpecPCR implements multi-tiered specificity testing against the NCBI GenBank database with taxonomic profiling to identify potential cross-reactivity with non-target species [41]. For researchers using standalone tools, NCBI Primer-Blast provides integrated specificity checking by automatically comparing proposed primer sequences against selected organism databases [48]. Benchling facilitates this process through direct integration with NCBI BLAST, allowing users to highlight primer sequences and submit them directly to BLASTN without manual copy-paste steps [44]. These computational specificity checks significantly reduce the risk of nonspecific amplification by identifying primers with potential for off-target binding before proceeding to laboratory testing.
The experimental workflow for primer design and validation follows a systematic path from computational design to laboratory verification, with multiple checkpoints to ensure specificity and optimal performance.
Wet Lab Validation Using Gradient PCR
Upon receiving synthesized primers, systematic experimental validation begins with gradient PCR. This protocol enables empirical determination of optimal annealing temperatures while identifying potential nonspecific amplification:
Reaction Setup: Prepare a master mix containing buffer, dNTPs, DNA polymerase, and template DNA (50-100ng genomic DNA or 1-10ng plasmid DNA). Aliquot equal volumes into PCR tubes and add forward and reverse primers to a final concentration of 0.1-0.5µM each.
Thermal Cycling with Gradient: Program a thermal cycler with a gradient annealing temperature range spanning at least 10°C (e.g., 50-60°C). Other cycling parameters include: initial denaturation at 95°C for 2-5 minutes; 30-35 cycles of denaturation at 95°C for 30 seconds, gradient annealing for 30 seconds, and extension at 72°C for 1 minute per kb; final extension at 72°C for 5-10 minutes.
Product Analysis: Separate PCR products by agarose gel electrophoresis (1.5-2% agarose in TAE or TBE buffer) alongside an appropriate DNA ladder. Visualize bands under UV transillumination after ethidium bromide or SYBR Safe staining.
Interpretation: Identify the annealing temperature that produces a single, intense band of the expected size with minimal nonspecific products or primer-dimer formation. Sequence-confirm the amplicon to verify specificity.
For quantitative applications, additional validation through standard curve analysis is necessary. Prepare serial dilutions of template DNA and amplify using the optimized conditions. PCR efficiency should fall between 90-110% (slope of -3.1 to -3.6) with correlation coefficient (R2) >0.98 [47].
RPA-LFD Assay Development for Rapid Detection
The development of recombinase polymerase amplification with lateral flow dipstick (RPA-LFD) detection represents an advanced application of primer design for isothermal amplification. The following protocol adapted from Sun et al. (2025) details primer validation for this emerging technique [47]:
Primer Design for RPA: Design primers 30-35 nucleotides in length targeting the conserved fiber-2 gene region. Modify the 5' ends with fluorescein isothiocyanate (FITC) (forward primer) and biotin (reverse primer). Validate specificity in silico against related adenoviruses.
RPA Reaction Optimization: Prepare 50µL reactions containing 29.4µL rehydration buffer, 2.4µL each forward and reverse primers (10µM), 11.8µL nuclease-free water, 2µL template DNA, and 1µL magnesium acetate (280mM). Incubate at 42°C for 30 minutes.
Lateral Flow Detection: Apply 70µL of diluted RPA product to the sample port of the LFD device containing: rabbit anti-FITC antibody conjugated to fluorescent microspheres on the conjugate pad; streptavidin at the test line (T); and goat anti-rabbit antibody at the control line (C). Interpret results after 5-10 minutes: simultaneous T and C lines indicate positive; C line only indicates negative.
Analytical Validation: Determine detection limit using serial dilutions of target DNA (e.g., 101 to 106 copies/µL). Verify specificity against related pathogens that could cause cross-reactivity. Assess clinical performance compared to reference methods (e.g., qPCR) using statistical measures including kappa (κ) values.
Advanced Techniques to Minimize Nonspecific Amplification
Despite careful conventional primer design, nonspecific amplification remains a persistent challenge in PCR-based applications, particularly in complex multiplex reactions or when amplifying low-copy targets. Advanced primer technologies and specialized design strategies have emerged to address these limitations with remarkable efficacy.
Cooperative primer technology represents a groundbreaking approach to reducing nonspecific amplification by orders of magnitude. Conventional primers, even with hot start methods, cannot prevent the propagation of primer-dimers once formed. Even small numbers of primer-dimers can compete efficiently with target amplification, leading to false negatives and/or false-positives. Cooperative primers address this fundamental limitation through structural modifications that greatly reduce primer-dimer propagation. Experimental results demonstrate that cooperative primers enable successful amplification of 60 template copies with no signal dampening even in a background of 150,000,000 primer-dimers. In contrast, normal primers with or without hot start experienced signal dampening with as few as 60 primer-dimers and false-negatives with only 600 primer-dimers. This performance represents a 2.5 million-fold improvement in the reduction of nonspecific amplification [46].
The mechanism of cooperative primer function involves preventing the extension of primer-dimers once formed, thereby stopping the cascade of nonspecific amplification that can overwhelm conventional PCR reactions. This technology maintains sensitivity while dramatically improving specificity, making it particularly valuable for applications requiring high levels of multiplexing or detection of rare targets in complex backgrounds. Additionally, cooperative primers can incorporate detection probes with 2.5 times more signal than conventional fluorescent probes, further enhancing their utility in quantitative applications [46].
Secondary structure prediction and avoidance constitute another critical strategy in advanced primer design. Modern primer design tools incorporate algorithms to detect potential self-dimers, cross-dimers, and hairpin structures based on thermodynamic calculations. Benchling's primer analysis features, for example, provide Gibbs Free Energy values for homodimer and monomer formation along with visual secondary structure diagrams [44]. The PrimerDigital platform offers specialized tools for analyzing self-dimer and cross-dimer formation in primer pairs, calculating thermodynamic parameters to predict stable secondary structures that could interfere with amplification [45]. By identifying and eliminating primers with significant secondary structure potential during the design phase, these tools prevent a common source of amplification failure and nonspecific products.
Table 3: Research Reagent Solutions for Primer Validation Experiments
| Reagent/Material | Function in Validation | Application Examples | Considerations |
|---|---|---|---|
| Highly Fluorescent\nEuropium (III)\nNanoparticles | Signal generation in lateral flow detection | RPA-LFD assays for visual amplicon detection [47] | Long fluorescence decay lifetime improves specificity and sensitivity |
| Rabbit Anti-FITC Antibody | Binds FITC-labeled amplicons in detection | Conjugated to microspheres in LFD assays [47] | Critical for capture at test line in lateral flow format |
| Streptavidin | Binds biotin-labeled amplicons | Immobilized at test line in LFD devices [47] | Forms bridge between antibody-microsphere and biotin-primer |
| Hot-Start DNA Polymerase | Reduces nonspecific amplification during reaction setup | Conventional PCR, qPCR, multiplex applications | Activates only after high-temperature incubation |
| Cooperative Primers | Prevents primer-dimer propagation | Multiplex PCR, low-copy target detection [46] | Proprietary technology with specialized design requirements |
| 1-Ethyl-3-(3-dimethylaminopropyl)\nCarbodiimide (EDC) | Crosslinking agent for antibody-microsphere conjugation | Preparation of LFD conjugate pads [47] | Activates carboxyl groups on microspheres for antibody coupling |
Multiplex tiling PCR panels represent a specialized application of primer design for next-generation sequencing target enrichment. PrimerDigital's custom multiplex tiling PCR panel design tool addresses the unique challenges of designing dozens to hundreds of primer pairs that must work efficiently under identical cycling conditions while avoiding primer-primer interactions. This tool designs primer sets optimized for amplicon sequencing across diverse organisms, assuming reference genome sequences are available. The computational pipeline eliminates primers with complementary regions that could form dimers with other primers in the multiplex reaction, a critical feature for successful large-scale multiplexing [45]. Similar capabilities are implemented in the CREPE pipeline, which is specifically optimized for targeted amplicon sequencing on Illumina platforms, with experimental validation showing >90% success rates for primers deemed acceptable by the software [40].
The integration of these advanced techniques—cooperative primers, sophisticated secondary structure prediction, and specialized multiplex design algorithms—provides researchers with powerful strategies to overcome the persistent challenge of nonspecific amplification. These approaches enable more reliable detection, particularly for demanding applications including low-abundance targets, complex multiplex reactions, and point-of-care diagnostic tests where robustness is essential.
Primer design software has evolved from simple primer suggestion tools to sophisticated computational pipelines that simultaneously optimize multiple biochemical parameters while predicting and preventing specificity problems. The integration of thermodynamic modeling, specificity checking against comprehensive databases, and advanced algorithms for avoiding secondary structures has transformed primer design from an art to a science. Tools like CREPE, PrimeSpecPCR, and commercial platforms with built-in validation represent the current state of the art, enabling researchers to design primers with >90% experimental success rates [40].
The ongoing development of specialized technologies such as cooperative primers demonstrates that innovation in primer design continues to address fundamental challenges in molecular biology. The 2.5 million-fold improvement in reducing nonspecific amplification achieved by cooperative primer technology [46] highlights the potential for continued advancement in this field. Similarly, the adaptation of primer design principles to emerging isothermal amplification methods like RPA [47] and LAMP [45] ensures that these computational tools remain relevant across evolving laboratory techniques.
For researchers investigating primer dimers and nonspecific amplification, modern primer design software provides both practical solutions and theoretical insights. The systematic approach to optimizing physical parameters including length, GC content, melting temperature, and secondary structure potential, combined with rigorous experimental validation protocols, offers a comprehensive framework for developing robust amplification assays. As these tools continue to evolve through machine learning and improved thermodynamic modeling, they will further enhance our ability to design precise molecular tools that drive advances in research, diagnostics, and therapeutic development.
The precision of quantitative PCR (qPCR) hinges on the meticulous optimization of core reaction components, a process central to mitigating artifacts like primer dimers and nonspecific amplification. This technical guide delves into the interdependent roles of primer concentration, magnesium ions (Mg2+), and deoxynucleoside triphosphates (dNTPs) in securing assay specificity and efficiency. Framed within a broader research context on PCR artifacts, we present structured quantitative data, detailed experimental protocols for optimization, and visual workflows to aid researchers and drug development professionals in achieving robust, reproducible molecular results.
The polymerase chain reaction is a cornerstone of modern molecular biology, but its success is not guaranteed. A primary challenge faced by researchers is the failure to amplify the intended target specifically, often due to the formation of primer dimers and amplification of nonspecific products [15] [23]. These artifacts compete for precious reaction components, reduce the yield of the desired amplicon, and can lead to profound inaccuracies in quantitative interpretation, especially in qPCR [49].
The occurrence of these artifacts is not random; it is governed by the delicate balance between template, non-template, primer, and cofactor concentrations [15]. This guide focuses on optimizing three levers central to this balance: primer concentration, Mg2+ concentration, and dNTP concentration. Understanding and controlling these components is not merely a technical exercise but a fundamental requirement for any rigorous research, particularly in drug development where quantitative results can influence critical decisions.
Core Component Functions and Interdependencies
Primer Concentration: The Specificity Gatekeeper
PCR primers are short, single-stranded DNA oligonucleotides designed to flank the target sequence. Their function is to provide a starting point for the DNA polymerase. However, higher primer concentrations increase the likelihood of nonspecific binding and primer-to-primer annealing, leading to spurious amplification [50] [51]. Visually, this manifests in agarose gel electrophoresis as extra, unwanted bands or a smear below the main product, and in qPCR melt curves as multiple peaks.
- Optimal Range: A final concentration of 0.1–0.5 µM for each primer is typically effective for standard PCR [52]. For qPCR, particularly with SYBR Green I, concentrations between 0.2–0.4 µM are often optimal to minimize background [49].
- Effects of Deviation:
Magnesium Ion (Mg2+) Concentration: The Polymerase Cofactor
Magnesium is an essential cofactor for Taq DNA polymerase, directly influencing its enzymatic activity [50]. It stabilizes the double-stranded DNA structure and facilitates the formation of the complex between the enzyme and the primer-template duplex.
- Optimal Range: The standard optimal concentration for Taq DNA Polymerase is 1.5–2.0 mM [52] [51].
- Effects of Deviation:
Deoxynucleoside Triphosphates (dNTPs): The Building Blocks
dNTPs (dATP, dCTP, dGTP, dTTP) are the fundamental nucleotides incorporated by the DNA polymerase to synthesize the new DNA strand. They are typically used in equimolar concentrations.
- Optimal Range: A final concentration of 0.2 mM of each dNTP is standard for most applications [52] [50].
- Effects of Deviation:
- Too High (>0.2 mM each): Can reduce specificity and, counterintuitively, inhibit the reaction. High dNTP concentrations also chelate Mg2+, effectively reducing the free Mg2+ available for the polymerase [50] [51].
- Too Low (<0.05 mM each): Reduces PCR yield and can cause premature reaction termination [50]. Lower concentrations (0.05–0.1 mM) can enhance fidelity but at the cost of yield [52].
The Interplay Between Mg2+ and dNTPs
A critical and often overlooked relationship exists between Mg2+ and dNTPs. Since dNTPs bind Mg2+, the effective concentration of free Mg2+ available to the polymerase is the total Mg2+ minus that bound by dNTPs [50]. Therefore, any change in dNTP concentration necessitates a re-evaluation of the Mg2+ concentration. If dNTP concentrations are increased, Mg2+ concentrations may need to be proportionally increased to compensate.
Quantitative Optimization Guidelines
The following tables summarize the optimal ranges and effects of varying each key component.
Table 1: Optimal Concentration Ranges and Effects of Deviation for Key PCR Components
| Component | Standard Optimal Range | Effect of High Concentration | Effect of Low Concentration |
|---|---|---|---|
| Primers | 0.1 - 0.5 µM [52] [49] | Nonspecific amplification, primer dimers [50] [51] | Low or no yield [50] |
| Mg2+ | 1.5 - 2.0 mM [52] | Nonspecific products, spurious bands [52] | No amplification [52] |
| dNTPs | 0.2 mM each [52] [50] | Reduced specificity/fidelity; chelates Mg2+ [50] | Reduced yield [50] |
Table 2: Troubleshooting Common PCR Artifacts via Component Adjustment
| Problem | Possible Causes | Optimization Strategies |
|---|---|---|
| Primer Dimers | High primer concentration [23]; Low annealing temperature [23]; Complementary 3' ends [49] | Lower primer concentration (0.1-0.3 µM) [49]; Increase annealing temperature [51]; Redesign primers [49] |
| Nonspecific Bands/Smearing | High primer/Mg2+/dNTP concentration [52] [50]; Low annealing temperature | Titrate Mg2+ down in 0.5 mM steps [52]; Use gradient PCR to find optimal Ta [49]; Use hot-start polymerase [23] |
| Low or No Yield | Low primer/Mg2+/dNTP concentration [52] [50]; High annealing temperature | Increase primer concentration (up to 0.5 µM); Titrate Mg2+ up; Increase template quality/amount [51] |
Experimental Protocols for Systematic Optimization
Primer Concentration Optimization (Checkerboard Titration)
A checkerboard titration is the most rigorous method for identifying the ideal concentration pair for forward and reverse primers, especially crucial for assays prone to dimerization.
Methodology:
- Prepare Stock Solutions: Dilute forward and reverse primers to a high concentration (e.g., 10 µM).
- Set Up Reaction Matrix: In a 96-well PCR plate, set up a series of reactions where the forward primer concentration is varied across rows (e.g., 50 nM, 100 nM, 200 nM, 400 nM) and the reverse primer concentration is varied down columns (e.g., 50 nM, 100 nM, 200 nM, 400 nM). A master mix containing buffer, Mg2+, dNTPs, polymerase, template, and water should be aliquoted first to ensure consistency.
- Run qPCR: Perform qPCR amplification using a standardized two-step or three-step cycling protocol.
- Analyze Results: Select the primer concentration combination that yields the lowest Cq value (indicating high efficiency), the highest endpoint fluorescence (indicating good yield), and a negative no-template control (NTC) [49]. The chosen combination should also show minimal primer-dimer formation in a melt curve analysis.
Mg2+ Concentration Optimization
As Mg2+ is a critical cofactor, its optimization can resolve issues with both specificity and yield.
Methodology:
- Prepare Master Mix: Create a master mix containing all components except Mg2+. Use a pre-optimized primer concentration.
- Supplement Mg2+: Aliquot the master mix into separate tubes and supplement with MgCl2 to create a concentration series (e.g., 0.5 mM, 1.0 mM, 1.5 mM, 2.0 mM, 2.5 mM, 3.0 mM, 4.0 mM). Note that the PCR buffer usually contains a baseline level of Mg2+ (often 1.5 mM), so calculate the final total concentration accordingly.
- Run PCR and Analyze: Perform PCR and analyze the products using agarose gel electrophoresis. Identify the Mg2+ concentration that produces the strongest specific band with the least background smearing or nonspecific bands [52].
Annealing Temperature (Ta) Optimization
The annealing temperature is a pivotal cycling parameter that works in concert with component concentrations.
Methodology:
- Calculate Tm: Determine the melting temperature (Tm) of your primers using a reliable calculation method (e.g.,
Tm= 2(A+T) + 4(G+C)or an online tool like OligoAnalyzer) [51]. - Set Up Gradient PCR: Using optimized primer and Mg2+ concentrations, set up identical reactions and use a thermocycler with a gradient function to test a range of annealing temperatures, typically from 55°C to 65°C or 3–5°C below the calculated Tm [51] [49].
- Identify Optimal Ta: The optimal annealing temperature is the highest temperature that still produces a robust, specific amplicon with the lowest Cq in qPCR and a single, clean band on a gel [49]. Techniques like touchdown PCR can also be employed to enhance specificity during initial cycles [51].
Visualizing the Optimization Workflow
The following diagram illustrates the logical workflow and decision-making process for optimizing PCR reactions to suppress artifacts, integrating the components and protocols discussed.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for PCR Optimization
| Item | Function & Rationale |
|---|---|
| Hot-Start DNA Polymerase | Enzyme engineered to be inactive at room temperature, preventing nonspecific priming and primer-dimer formation during reaction setup [15] [23]. |
| SYBR Green I Master Mix | Optimized commercial mixture containing buffer, salts, dNTPs, and a sensitive DNA-binding dye. Reduces pipetting error and variability [53]. |
| Nuclease-Free Water | Solvent for diluting primers and preparing master mixes. Ensures the reaction is free of RNases and DNases that could degrade components. |
| dNTP Mix (10mM each) | Provides the balanced, high-purity nucleotide building blocks necessary for efficient and accurate DNA synthesis [50]. |
| MgCl2 Solution (25mM) | Separate magnesium chloride solution allows for precise titration of this critical cofactor independent of the reaction buffer [52]. |
| Primer Design Software | Tools like Primer-BLAST or OligoAnalyzer help design primers with appropriate Tm, avoid self-complementarity, and check for specificity [15] [53]. |
Achieving specific and efficient amplification in PCR is a systematic exercise in biochemical balance. The interplay between primer concentration, Mg2+, and dNTPs forms the foundation of a robust assay. By adhering to the structured optimization protocols outlined herein—employing checkerboard titrations, Mg2+ gradients, and annealing temperature gradients—researchers can effectively suppress primer dimers and nonspecific amplification. This rigorous approach is indispensable for generating reliable, reproducible data that underpins high-quality research and accelerates drug development.
In the realm of molecular biology, the polymerase chain reaction (PCR) stands as a cornerstone technique for nucleic acid amplification. However, a pervasive challenge faced by researchers is the phenomenon of nonspecific amplification, primarily manifested through primer dimer formation and off-target products. Primer dimers are short, double-stranded DNA fragments that arise when primers anneal to each other instead of binding to the target DNA template, significantly compromising reaction efficiency and analytical accuracy [23]. Within this context, thermal cycling conditions emerge as a critical determinant of PCR success, governing the stringency of primer-template interactions and ultimately controlling the specificity and yield of amplification products. For researchers and drug development professionals, mastering these parameters is not merely technical refinement but a fundamental requirement for generating reliable, reproducible data in diagnostic assay development, gene expression studies, and genetic disorder screening.
The precision of thermal cycling directly addresses the core challenge of primer dimer formation, which can reduce the efficiency of target amplification and generate nonspecific products that complicate subsequent analysis [23] [5]. This technical guide provides an in-depth examination of how annealing temperatures and ramp rates can be systematically optimized to establish stringent reaction conditions that minimize artifacts while maximizing specific product yield, framed within the broader research context of understanding and controlling nonspecific amplification.
Theoretical Foundations: Primer-Template Interactions and Thermal Dynamics
Biochemical Basis of Primer Dimer Formation
Primer dimers represent a significant failure mode in PCR optimization, originating from biochemical interactions between primer molecules themselves rather than between primers and the target template. These artifacts form primarily through two mechanisms: self-annealing due to complementary regions within a single primer, or cross-annealing between forward and reverse primers, particularly through complementary sequences at their 3' ends [23] [5]. The 3' end complementarity is especially problematic as it provides a substrate for DNA polymerase extension, creating short duplex products that compete with the target amplicon for reaction components.
The formation of these primer-dimers is governed by thermodynamic principles, with the Gibbs free energy (ΔG) of dimerization predicting the likelihood of this undesirable interaction. Analysis using specialized software such as OligoArchitect or Oligoanalyzer reveals that primers with 3'-end dimer ΔG values below -2.0 kcal/mol, particularly those with extendable overlaps, pose significant risks for dimer formation [54] [15]. One study employing these tools demonstrated that primers with strong 3'-terminal complementarity (ΔG < -6.0 kcal/mol) consistently generated primer dimers that dominated the reaction products, underscoring the importance of thermodynamic evaluation during primer design [54].
Impact of Nonspecific Amplification on Research Outcomes
The consequences of nonspecific amplification extend beyond mere reaction inefficiency to fundamentally compromise experimental outcomes and conclusions. In quantitative applications, primer dimers consume reaction components—including primers, nucleotides, and polymerase activity—there reducing the resources available for target amplification and distorting quantification [54]. For gene expression studies using SYBR Green-based detection, the DNA-binding dye will intercalate with any double-stranded DNA product, including primer dimers and other nonspecific amplicons, generating false-positive signals and inaccurate Cq values that misrepresent true expression levels [54] [15].
The broader implications for research integrity are substantial. A survey of 93 validated assays targeting Wnt-pathway genes revealed that nonspecific amplification occurred frequently and was not reliably predicted by conventional quality metrics such as Cq values or PCR efficiency calculations [15]. This finding underscores how unoptimized thermal conditions can introduce systematic errors that undermine experimental reproducibility, particularly in low-template scenarios common in rare transcript detection, single-cell analysis, and pathogen detection at low titers.
Annealing Temperature Optimization
Theoretical Calculation and Empirical Verification
The annealing temperature (Ta) represents perhaps the most critical parameter governing PCR specificity, acting as the thermal gatekeeper that determines which primer-template hybrids remain stable enough to serve as extension initiation sites. While primer melting temperature (Tm) calculations provide a theoretical starting point, the optimal Ta must be determined empirically to account for the complex reaction environment and specific instrument characteristics [54] [55].
Theoretical Tm calculations, typically derived using nearest-neighbor algorithms, should guide initial parameter selection with the standard approach setting the Ta approximately 3-5°C below the calculated Tm of the primer pair [55]. However, this relationship requires adjustment based on specific reaction conditions. Table 1 summarizes the key relationships between annealing temperature and amplification performance that researchers must consider during optimization.
Table 1: Effects of Annealing Temperature on PCR Performance
| Temperature Range | Specificity Impact | Yield Impact | Common Observations |
|---|---|---|---|
| Too Low (<3°C below Tm) | Significant nonspecific binding | Potentially high but nonspecific | Primer dimers, multiple bands, smeared gel products |
| Optimal Range (3-5°C below Tm) | High specificity through stringent binding | Maximum specific yield | Single clear band of expected size |
| Too High (>Tm) | Excessive stringency prevents binding | Low to no amplification | Faint or absent target band |
Gradient PCR for Rapid Annealing Temperature Optimization
The gradient function of modern thermal cyclers represents an indispensable tool for efficient Ta optimization, allowing simultaneous testing of a temperature range across a single PCR block [56]. This methodology dramatically accelerates the optimization process, compressing what would otherwise require weeks of sequential testing into a single experimental run [56].
Table 2: Gradient PCR Implementation Protocol
| Step | Parameter | Specification | Technical Notes |
|---|---|---|---|
| Initial Range Selection | Temperature span | Calculated Tm ± 7°C | Based on primer Tm calculation algorithms |
| Reaction Setup | Master mix distribution | Uniform across gradient | Single preparation ensures consistency |
| Thermal Cycling | Gradient during annealing | Linear across block | Other steps (denaturation, extension) remain uniform |
| Product Analysis | Methodologies | Gel electrophoresis, melt curve analysis | Identify temperature with strongest specific product |
| Secondary Optimization | Narrowed range | ± 2°C around best result | Refine optimal temperature with higher precision |
The experimental workflow for systematic annealing temperature optimization using gradient PCR follows a logical progression from setup through analysis, as illustrated in the following workflow:
The power of gradient optimization extends beyond initial protocol establishment to troubleshooting and validation applications. When adapting established assays to different thermal cyclers, subtle differences in block temperature uniformity and accuracy may necessitate Ta adjustments that gradient PCR can rapidly identify [56]. Similarly, when working with complex templates—such as those with high GC content, secondary structure, or homologous sequences—the optimal Ta may deviate significantly from theoretical predictions, making empirical determination essential [56] [55].
Ramp Rate Considerations and Thermal Cycler Performance
The Impact of Transition Speeds on Reaction Specificity
While annealing temperature receives significant attention in PCR optimization, ramp rates—the speed at which the instrument transitions between temperature setpoints—represent an equally important but frequently overlooked parameter influencing reaction specificity. Ramp rates directly determine the time reactions spend at non-optimal temperatures during transitions, creating windows where nonspecific interactions can occur [57].
The transition between annealing and extension phases presents a particular vulnerability period for nonspecific amplification. Slower ramp rates prolong exposure to permissive temperatures where weakly matched primer-template hybrids remain stable, allowing extension of off-target products including primer dimers [57]. This phenomenon explains observations that faster ramp rates can improve amplification specificity by limiting the duration available for nonspecific interactions [57]. However, the relationship between ramp rate and performance is not universally linear, as excessively rapid transitions may cause overshooting or inadequate temperature equilibration across the block, potentially compromising reproducibility.
Thermal Cycler Performance Metrics for Stringent Protocols
The physical implementation of thermal cycling parameters through instrument hardware directly determines the practical stringency achievable in PCR protocols. Several key performance metrics distinguish thermal cyclers in their ability to maintain precise, uniform thermal conditions essential for minimizing nonspecific amplification:
Table 3: Thermal Cycler Performance Metrics Impacting Specificity
| Performance Metric | Definition | Impact on Specificity | Optimal Specification |
|---|---|---|---|
| Temperature Accuracy | Deviation from programmed setpoint | Critical for true annealing stringency | Typically ±0.2°C or better |
| Block Uniformity | Maximum temperature variance across block | Ensures consistent results across all wells | ≤0.5°C variance at 60°C |
| Ramp Rate | Speed of temperature transitions (°C/second) | Limits time at permissive temperatures | Instrument-dependent balance |
| Heated Lid | Temperature control above reactions | Prevents evaporation and concentration changes | 105°C for aqueous solutions |
The integration of these performance characteristics creates what might be termed the "thermal precision" of an instrument, determining how faithfully theoretical optimization translates to practical results. For applications requiring the highest specificity—such as diagnostic assay development, genotyping, and mutation detection—selection of thermal cyclers with demonstrated excellence across these metrics becomes a critical consideration in experimental design [57].
Integrated Experimental Protocols
Comprehensive Annealing Temperature Optimization
This protocol provides a detailed methodology for establishing optimal annealing temperature using gradient PCR, incorporating best practices for reaction setup and analysis to maximize specificity while minimizing primer dimer formation.
Materials and Reagents:
- Target DNA template (e.g., genomic DNA, cDNA)
- Primer pair (lyophilized and resuspended in sterile TE buffer or nuclease-free water)
- High-fidelity DNA polymerase with recommended buffer
- dNTP mix (typically 10mM each)
- MgCl₂ solution (if required separately)
- Nuclease-free water
- Gradient thermal cycler
- Agarose gel electrophoresis equipment or melt curve analysis capability
Procedure:
- Primer Design and Validation: Design primers following established guidelines: length of 18-30 bases, GC content 40-60%, Tm of 55-72°C with minimal difference between forward and reverse primers, and avoidance of self-complementarity particularly at the 3' ends [58] [5]. Validate primer specificity using tools such as Primer-BLAST.
Reaction Mixture Preparation: Prepare a master mix on ice according to the following formulation for a single 50μL reaction:
- 5.0 μL 10X PCR buffer
- 1.0 μL dNTP mix (10mM each)
- 1.0 μL forward primer (20μM)
- 1.0 μL reverse primer (20μM)
- 0.5-1.0 μL DNA polymerase (1-2.5 units)
- 1.0 μL template DNA (10-100ng)
- Nuclease-free water to 50μL
Mix thoroughly by gentle pipetting. For gradient optimization, multiply these volumes by the number of gradient positions plus controls.
Gradient PCR Setup: Distribute equal volumes of the master mix across the gradient block positions. Program the thermal cycler with an initial denaturation at 95°C for 2-5 minutes, followed by 30-35 cycles of:
- Denaturation: 95°C for 20-30 seconds
- Annealing: Gradient range (e.g., 55-70°C) for 20-30 seconds
- Extension: 72°C for 1 minute per kilobase of expected product
Conclude with a final extension at 72°C for 5-10 minutes.
Product Analysis: Analyze PCR products using agarose gel electrophoresis or melt curve analysis. The optimal annealing temperature produces the strongest amplification of the target product with minimal nonspecific bands or primer dimers [56].
Primer Concentration Optimization Using Checkerboard Titration
While annealing temperature represents the primary specificity governor, primer concentration optimization provides a complementary approach to fine-tuning reaction stringency. This protocol employs a checkerboard titration design to identify optimal primer concentration pairs that maximize specific amplification while minimizing dimer formation.
Procedure:
- Prepare separate master mixes varying forward and reverse primer concentrations independently across a range typically from 50nM to 600nM [54].
Set up reactions representing all possible combinations of forward and reverse primer concentrations while maintaining constant conditions for other components.
Perform amplification using the previously established optimal annealing temperature.
Identify the primer concentration combination that yields the lowest Cq value (in qPCR) or highest product yield (in conventional PCR) while maintaining negative results in no-template controls [54].
This methodology is particularly valuable for multiplex PCR applications, where multiple primer pairs must function efficiently under identical cycling conditions, or when dealing with challenging templates that require fine-balanced primer ratios for optimal amplification.
The Scientist's Toolkit: Essential Research Reagents and Equipment
Successful implementation of stringent thermal cycling protocols requires careful selection of reagents and equipment specifically designed to enhance specificity and reproducibility. The following table catalogues essential solutions for optimizing annealing conditions and controlling ramp parameters.
Table 4: Research Reagent Solutions for Thermal Cycling Optimization
| Category | Specific Product | Function in Optimization | Application Context |
|---|---|---|---|
| Polymerase Systems | Hot-start DNA polymerases | Reduce pre-extension primer dimer formation | High-specificity applications, complex templates |
| Buffer Additives | DMSO, betaine, MgCl₂ | Modify effective stringency, disrupt secondary structures | GC-rich templates, challenging amplicons |
| Thermal Cyclers | Gradient function instruments | Enable parallel annealing temperature screening | Protocol establishment, primer validation |
| Design Software | OligoAnalyzer, Primer-BLAST | Predict Tm and dimer formation potential | In silico primer evaluation and selection |
| Specificity Enhancers | Touchdown PCR programs | Automatically increase stringency during cycling | Applications requiring high specificity |
The establishment of stringent annealing temperatures through systematic optimization represents a foundational element in controlling primer dimer formation and nonspecific amplification in PCR. By integrating theoretical calculations with empirical validation using gradient PCR, researchers can identify thermal conditions that balance specificity with efficiency, creating robust protocols resistant to artifact generation. The consideration of ramp rates and thermal cycler performance characteristics further refines this optimization, addressing often-overlooked aspects of the thermal profile that impact reaction specificity.
For the research and drug development professional, mastery of these thermal cycling parameters translates directly to improved experimental reproducibility, more reliable quantification, and increased confidence in analytical results. As PCR technologies continue to evolve toward higher throughput and greater sensitivity, the principles of thermal stringency outlined in this guide will remain essential for maximizing the technical quality and scientific validity of molecular analyses across diverse applications.
Polymerase chain reaction (PCR) remains a foundational technology in molecular biology, yet its effectiveness is often compromised by nonspecific amplification and primer-dimer artifacts that originate during reaction setup. This technical review examines the critical role of hot-start polymerases in suppressing premature enzymatic activity that leads to these amplification artifacts. We explore the molecular mechanisms underlying different hot-start methodologies, including antibody-mediated, chemical, aptamer, and primer-based inhibition strategies. Through quantitative analysis of experimental data, we demonstrate how specialized enzyme formulations significantly enhance amplification specificity, sensitivity, and yield across diverse applications from genetic testing to clinical diagnostics. The implementation of hot-start technology represents an essential advancement for researchers and drug development professionals seeking to improve assay reliability while maintaining the practical flexibility required for high-throughput environments.
The exquisite sensitivity of the polymerase chain reaction presents a paradoxical challenge: the same enzymatic activity that enables exponential amplification of target sequences also promotes the generation of nonspecific byproducts that compromise assay results. Conventional DNA polymerases, including Taq DNA polymerase, exhibit significant enzymatic activity at room temperature and below [59] [60]. During the critical period when PCR reactions are assembled at ambient temperatures, these enzymes can extend primers that have hybridized to non-target sequences or to each other with partial complementarity [61] [62]. This premature extension occurs because reaction mixtures must pass through the polymerase's optimal extension temperature (typically 68-72°C) during the initial ramp to the first denaturation step at 94°C [62].
The resulting nonspecific amplification manifests primarily as two problematic artifacts:
- Mis-priming: Occurs when primers bind to regions of the nucleic acid target with partial complementarity under low-stringency conditions, leading to amplification of unintended sequences that compete with the desired target [61].
- Primer-dimer formation: Results from hybridization between primer molecules themselves, followed by polymerase extension that creates short, amplifiable artifacts [61] [4]. These structures are particularly problematic because they can be efficiently amplified in subsequent PCR cycles, depleting reagents and potentially dominating the reaction.
The consequences of these artifacts extend beyond mere inconvenience. In quantitative applications, nonspecific amplification can lead to significant underestimation of target concentration, false positives in diagnostic assays, reduced sensitivity for low-abundance targets, and compromised performance in multiplex reactions [61] [46]. As PCR-based methodologies continue to evolve toward more complex applications including multiplex assays, low-copy number detection, and next-generation sequencing library preparation, the imperative for robust solutions to nonspecific amplification has only intensified [61].
Fundamental Principles of Hot-Start PCR
Hot-start PCR represents a refined methodological approach designed to circumvent the limitations of conventional PCR by controlling polymerase activity through the initial critical phases of reaction setup and thermal cycling initiation. The core principle underlying this technology is the reversible inhibition of DNA polymerase during reaction assembly and the initial temperature ramp, with subsequent activation occurring only after the reaction mixture has reached stringent temperatures that promote specific primer-template hybridization [59] [62].
The molecular strategy employs modified DNA polymerases that remain inactive at room temperature through various inhibition mechanisms. This enzymatic blockade is maintained until a specific heat activation step, typically the initial denaturation at 95°C, denatures the inhibitory complex and releases fully active polymerase [59] [60]. By preventing enzymatic activity during non-stringent conditions, hot-start methods ensure that primer extension only initiates when the temperature has reached a point where primer binding is highly specific to the intended target sequences.
The thermodynamic rationale for this approach leverages the temperature dependence of nucleic acid hybridization specificity. At elevated temperatures (>55°C), only primer-template interactions with sufficient complementarity to form stable duplexes persist, whereas nonspecific interactions with lower melting temperatures are unstable [62]. By activating polymerase activity exclusively under these stringent conditions, hot-start methods fundamentally alter the reaction kinetics in favor of specific amplification.
The practical implementation of hot-start technology has evolved significantly from early manual methods that involved physical separation of reaction components or the manual addition of essential reagents after the reaction reached denaturation temperatures [62]. These approaches, while effective in principle, introduced workflow complexities and potential contamination risks that limited their utility in standardized and high-throughput applications. Contemporary hot-start methodologies have converged on integrated inhibition systems that require no manual intervention, broadly categorized into polymerase-targeted and primer-targeted mechanisms that will be explored in subsequent sections.
Molecular Mechanisms of Hot-Start Inhibition
The efficacy of hot-start PCR derives from specific molecular strategies that temporarily inhibit DNA polymerase activity. These approaches differ in their inhibitory mechanisms, activation kinetics, and practical implications for PCR performance. The table below summarizes the primary hot-start methodologies currently employed.
Table 1: Comparative Analysis of Hot-Start Inhibition Mechanisms
| Inhibition Method | Mechanism of Action | Activation Requirements | Advantages | Limitations |
|---|---|---|---|---|
| Antibody-mediated [63] [62] [60] | Monoclonal antibody binds polymerase active site | Brief denaturation (1-3 min at 95°C) | Rapid activation; Complete polymerase release | Animal-derived antibodies; Potential contamination |
| Chemical modification [60] | Covalent attachment of inhibitory chemical groups | Extended activation (>10 min at 95°C) | Gradual activation; High stability; Low contamination risk | Slow activation; Potential incomplete activation; DNA damage risk |
| Aptamer-based [62] [60] | Oligonucleotides bind polymerase active site | Very rapid activation (30 sec at 95°C) | Fastest activation; Non-animal origin | Less stringent binding; Potential nonspecific amplification |
| Primer modification [61] | Thermolabile groups block 3' primer extension | Heat-labile deprotection during cycling | Targeted inhibition; No polymerase modification required | Specialized primer synthesis; Additional cost |
Antibody-Mediated Inhibition
Antibody-based hot-start systems employ monoclonal antibodies that specifically bind to the active site of DNA polymerases, forming an immunocomplex that sterically blocks the enzyme's catalytic activity [63] [62]. The inhibitory complex remains stable at temperatures below 70°C, effectively preventing primer extension during reaction setup. During the initial denaturation step at 95°C, the antibody undergoes irreversible protein denaturation, dissociating from the polymerase and restoring full enzymatic activity [60]. This approach typically requires only a brief activation period of 1-3 minutes at 95°C, after which the polymerase behaves identically to its unmodified counterpart.
Recent innovations have expanded the sources of inhibitory antibodies, including the development of chicken egg-derived polyclonal antibodies (IgY) that offer a cost-effective alternative to traditional monoclonal approaches [63]. Studies demonstrate that these avian antibodies effectively block Taq DNA polymerase activity at 50°C while having no negative impact on polymerase activity after heat activation or on reverse transcriptase activity in RT-PCR applications [63]. Quantitative assessments indicate that approximately 1.0 μg of hot-start IgY effectively inhibits 5 units of Taq DNA polymerase activity [63].
Chemical Modification Strategies
Chemical hot-start methods utilize covalent modification of DNA polymerase with specific chemical groups that block enzymatic activity at ambient temperatures [60]. These inhibitory groups remain attached until elevated temperatures cleave the chemical bonds, progressively restoring polymerase function. A significant advantage of this approach is the gradual activation profile, where a portion of the polymerase population remains inactive during initial cycles and becomes activated in later cycles, potentially enhancing amplification efficiency throughout the reaction [60].
The primary limitation of chemical inhibition is the extended activation time required, often exceeding 10 minutes at 95°C, which may promote DNA template damage through prolonged exposure to high temperatures [60]. Additionally, the chemical deprotection process may be incomplete, potentially leaving a fraction of polymerase molecules permanently inhibited and reducing overall reaction efficiency, particularly for longer amplicons (>3 kb) [60].
Aptamer-Based Inhibition
Aptamer-mediated hot-start employs short, single-stranded DNA or RNA molecules engineered to bind specifically to DNA polymerase with high affinity at lower temperatures [62] [60]. These oligonucleotide aptamers undergo conformational changes or dissociate from the polymerase as temperature increases, rapidly releasing active enzyme. This mechanism offers the fastest activation time among hot-start methods, typically requiring only 30 seconds at 95°C [60].
The synthetic nature of aptamers eliminates concerns regarding animal-derived components present in antibody-based systems [60]. However, the binding affinity of aptamers may be less stringent than antibodies, potentially permitting low-level enzymatic activity at intermediate temperatures that could contribute to nonspecific amplification in some applications [60].
Advanced Primer Modification Approaches
Beyond polymerase-targeted methods, innovative primer-based strategies have emerged as an alternative hot-start paradigm. One significant advancement utilizes primers containing thermolabile 4-oxo-1-pentyl (OXP) phosphotriester (PTE) modifications at 3'-terminal and penultimate internucleotide linkages [61]. These modifications impair DNA polymerase primer extension at lower temperatures but spontaneously convert to natural phosphodiester linkages at elevated temperatures, producing fully functional primers precisely when needed during thermal cycling [61].
This primer-directed approach demonstrates significant improvements in amplification specificity and efficiency across conventional PCR, one-step RT-PCR, and real-time PCR applications with both SYBR Green I and TaqMan detection chemistries [61]. By directly modifying the primer rather than the enzyme, this method offers a complementary strategy that can be combined with polymerase-targeted hot-start methods for enhanced specificity.
Diagram 1: Comparative Pathways of Conventional vs. Hot-Start PCR. This workflow illustrates how hot-start methods intercept the critical early steps where nonspecific amplification initiates in conventional PCR protocols.
Quantitative Performance Assessment
The theoretical advantages of hot-start methodologies must be validated through rigorous experimental assessment of performance metrics. The table below summarizes quantitative improvements observed with various hot-start polymerases compared to conventional enzymes.
Table 2: Quantitative Performance Metrics of Hot-Start DNA Polymerases
| Performance Parameter | Conventional Taq | Hot-Start Taq | Experimental Conditions | Source |
|---|---|---|---|---|
| Primer-dimer tolerance | Signal dampening with 60 primer-dimers; False negatives with 600 primer-dimers | Successful amplification with 150,000,000 primer-dimers | 60 template copies background | [46] |
| Sensitivity limit | Variable; Often >10 copies | ~5 copies reliably detected | Human genomic DNA (529 bp amplicon) | [64] |
| Amplification speed | 1 min/kb | 15 sec/kb | Standard plasmid template | [64] |
| Inhibitor tolerance | Sensitive to common inhibitors | Resists humic acid (1.3 µg/mL), hemin (6 µM), xylan (0.26 mg/mL) | 1 kb human genomic target | [64] |
| Benchtop stability | Limited (minutes) | 24 hours at room temperature | Pre-mixed reaction components | [64] |
Specificity and Sensitivity Enhancements
The most significant quantitative improvement afforded by hot-start polymerases is the dramatic reduction in nonspecific amplification, particularly primer-dimer formation. Research demonstrates that cooperative primer technology, when combined with hot-start activation, enables successful amplification of 60 template copies against a background of 150,000,000 primer-dimers [46]. In stark contrast, conventional primers experience signal dampening with as few as 60 primer-dimers and produce false negatives with only 600 primer-dimers, representing a 2.5 million-fold improvement in tolerance to nonspecific amplification [46].
Sensitivity enhancements are equally impressive, with hot-start polymerases reliably detecting approximately 5 copies of target DNA in human genomic background, a challenging matrix that often promotes nonspecific amplification [64]. This improved sensitivity stems from the preferential amplification of specific targets without competition from mis-primed products that deplete essential reaction components including dNTPs, primers, and polymerase activity [61].
Efficiency and Robustness Metrics
Beyond specificity, hot-start polymerases demonstrate superior performance in amplification efficiency and reaction robustness. Engineered hot-start enzymes such as Platinum II Taq Hot-Start DNA Polymerase achieve synthesis rates approximately four times faster than conventional Taq polymerases (15 sec/kb versus 1 min/kb), enabling complete PCR runs in as little as 30 minutes [64].
The practical advantage of this accelerated synthesis is particularly evident in co-cycling applications where multiple targets of different lengths are amplified simultaneously using a universal annealing temperature of 60°C [64]. This streamlined approach eliminates the optimization typically required for different primer pairs and enables efficient amplification of targets ranging from 132 bp to 3.9 kb in a single run [64].
Reaction robustness is another distinguishing characteristic, with hot-start formulations demonstrating exceptional tolerance to common PCR inhibitors including humic acid, hemin, and xylan, as well as compatibility with challenging sample types such as formalin-fixed, paraffin-embedded (FFPE) tissue extracts [64]. Additionally, the stabilized enzyme formulations maintain performance for up to 24 hours at room temperature after reaction assembly, providing exceptional workflow flexibility [64].
Experimental Protocols and Methodologies
Standardized Hot-Start PCR Protocol
The following protocol outlines a standardized approach for implementing hot-start PCR using commercially available enzyme systems, adaptable to various experimental needs:
Reaction Assembly:
- Prepare master mix on ice containing:
- 1× PCR buffer (supplemented with MgCl₂ or MgSO₄ to 1.5-2.5 mM final concentration)
- 200 μM of each dNTP
- 0.2-0.5 μM of each forward and reverse primer
- 0.5-2.5 units of hot-start DNA polymerase
- Template DNA (10-100 ng genomic DNA or 1-10 ng cDNA)
- Nuclease-free water to final volume
Thermal Cycling Parameters:
- Initial activation: 95°C for 1-3 minutes (antibody/aptamer-based) or 10+ minutes (chemical modification)
- Denaturation: 95°C for 15-30 seconds
- Annealing: 45-65°C for 15-30 seconds (primer-specific) or 60°C universal annealing
- Extension: 68-72°C for 15-60 seconds per kb
- Repeat steps 2-4 for 30-40 cycles
- Final extension: 68-72°C for 5 minutes
- Hold: 4°C indefinitely
Critical Considerations:
- Maintain reactions on ice during setup and transfer directly to pre-heated thermal cycler when possible
- Avoid prolonged exposure to room temperature during reaction assembly
- Include no-template controls (NTC) to detect contamination or primer-dimer formation
- For difficult templates, consider touchdown PCR or gradient annealing temperature optimization
OXP-Modified Primer Implementation Protocol
For researchers employing the novel OXP-modified primer approach, the following specialized protocol is recommended [61]:
Primer Design and Synthesis:
- Design primers according to standard criteria (18-25 bp, 40-60% GC content, Tm 55-65°C)
- Incorporate one or two OXP phosphotriester modifications at 3'-terminal and/or 3'-penultimate internucleotide linkages during oligonucleotide synthesis
- Purify primers using reverse-phase HPLC to ensure complete modification
- Verify modification efficiency by mass spectrometry
PCR Implementation:
- Prepare reaction mixture using standard unmodified DNA polymerase
- Utilize OXP-modified primers at same concentration as conventional primers (0.2-0.5 μM)
- Apply standard thermal cycling conditions without extended initial activation
- The OXP groups automatically convert to natural phosphodiester linkages during initial denaturation, activating primers specifically for the intended amplification
Validation Procedures:
- Compare amplification efficiency between OXP-modified and unmodified primers using dilution series of target template
- Assess specificity improvement by gel electrophoresis or melt curve analysis
- Quantitate reduction in primer-dimer formation using no-template controls
Performance Assessment Methodology
To quantitatively evaluate hot-start polymerase performance, implement the following experimental approaches:
Specificity Assessment:
- Perform amplification with serial template dilutions (1 ng-1 fg)
- Include no-template controls to assess primer-dimer formation
- Analyze products by agarose gel electrophoresis or capillary electrophoresis
- Quantify specific versus nonspecific product using image analysis software
Sensitivity Determination:
- Prepare limiting dilution series of target template
- Perform amplification with 8-10 replicates per dilution
- Calculate limit of detection (LOD) using probit analysis or Poisson distribution
- Compare Cq values between conventional and hot-start polymerases
Kinetic Analysis:
- Monitor real-time amplification with intercalating dyes
- Calculate amplification efficiency from standard curve slope
- Determine linear dynamic range across template concentrations
- Assess reaction robustness using inhibitor-spiked samples
Diagram 2: Molecular Inhibition Mechanisms of Hot-Start PCR. This schematic illustrates how different inhibition strategies converge on the common pathway of maintaining polymerase inactivity until heat activation enables specific amplification.
Research Reagent Solutions
The successful implementation of hot-start PCR methodologies depends on access to specialized reagents optimized for specific applications. The table below outlines essential research reagents and their functions in establishing robust hot-start protocols.
Table 3: Essential Research Reagents for Hot-Start PCR Implementation
| Reagent Category | Specific Examples | Function & Mechanism | Application Notes |
|---|---|---|---|
| Antibody-based Hot-Start Polymerases | Platinum II Taq Hot-Start, AmpliTaq Gold | Antibody binds polymerase active site until heat denaturation | Ideal for standard PCR; Balance of specificity and activation speed |
| Aptamer-based Hot-Start Polymerases | OneTaq Hot Start DNA Polymerase | Oligonucleotide aptamer inhibits polymerase until temperature increase | Rapid activation; Avoids animal-derived components |
| Chemically Modified Hot-Start Polymerases | CleanAmp Taq | Covalent modification inactivated until thermal cleavage | Gradual activation; Enhanced performance in later cycles |
| Modified Primers | OXP-modified primers | Thermolabile groups block extension until conversion to PDE | Can be combined with any polymerase; Targeted approach |
| Specialized dNTPs | CleanAmp dNTP Mix | 3' thermolabile protecting groups prevent misincorporation | Additional specificity layer; Removes dNTP-related artifacts |
| Enhancement Reagents | Platinum GC Enhancer | Improves amplification of GC-rich targets | Essential for difficult templates (>65% GC content) |
| Inhibitor-Tolerant Buffers | Platinum II PCR Buffer | Engineered buffer composition resistant to common inhibitors | Critical for direct amplification from crude samples |
Commercial System Selection Guidelines
When selecting hot-start polymerase systems for specific applications, consider the following criteria:
For high-throughput applications:
- Choose systems with room temperature setup capability
- Select formulations with 24-hour benchtop stability
- Prefer universal annealing temperature compatibility
- Consider master mix formats for pipetting efficiency
For difficult templates:
- Prioritize systems with optional GC enhancers
- Select polymerases with high inhibitor tolerance
- Choose systems with validated FFPE compatibility
- Consider graduated activation for complex backgrounds
For maximum sensitivity:
- Implement antibody or aptamer-based systems for rapid activation
- Combine with modified primers for dual specificity enhancement
- Utilize optimized buffer systems with betaine or DMSO additives
- Validate with limiting dilution experiments
For quantitative applications:
- Select systems with minimal primer-dimer formation
- Choose lots with certified low DNA contamination
- Verify consistent performance across multiple runs
- Implement rigorous no-template controls
Hot-start polymerase technology represents a fundamental advancement in PCR methodology, addressing the longstanding challenge of nonspecific amplification through sophisticated molecular inhibition strategies. The diverse approaches—including antibody-mediated, chemical modification, aptamer-based, and primer-directed methods—offer researchers multiple pathways to enhance assay specificity, sensitivity, and reliability. Quantitative assessments demonstrate remarkable improvements, including million-fold enhancements in primer-dimer tolerance and consistent detection of single-digit template copies across challenging sample types.
The implications for research and diagnostic applications are substantial. In clinical diagnostics, enhanced specificity directly translates to improved test accuracy with reduced false positives and negatives. For drug development professionals, robust PCR performance enables more reliable quantification of biomarkers and transcriptional responses. In research settings, the implementation of hot-start methodologies facilitates investigation of low-abundance targets and complex samples that were previously intractable to amplification-based analysis.
Future developments will likely focus on further integration of hot-start principles with emerging amplification technologies, including isothermal methods and point-of-care diagnostic platforms. The continued refinement of inhibitor-tolerant polymerases will expand applications to direct amplification from crude samples, while engineered enzymes with enhanced fidelity may improve accuracy for sequencing and cloning applications. As PCR remains the cornerstone technology for genetic analysis across diverse fields, the role of specialized enzymes like hot-start polymerases in ensuring reliable, reproducible results will only grow in importance.
For researchers embarking on new assay development, the implementation of hot-start methodology should be considered essential rather than optional, providing a foundational improvement that enhances performance across virtually all amplification applications while reducing optimization time and experimental variability.
Non-specific amplification is a fundamental challenge in polymerase chain reaction (PCR) that compromises the fidelity, efficiency, and reliability of molecular diagnostics and research. This phenomenon, characterized by the amplification of non-target DNA sequences, manifests primarily as primer dimers and PCR smears on electrophoretic gels [3]. Primer dimers, short amplifiable artefacts of 20-60 bp formed by two primers hybridizing, compete with target amplicons for reaction resources. In multiplex PCR reactions containing numerous primer pairs, the probability of such interactions increases dramatically, severely limiting multiplexing efficiency [65]. The root cause lies in unintended primer interactions, where rather than binding to the target DNA, primers hybridize to each other or to non-target sites, leading to elongated dimer products that consume primers, DNA polymerases, and dNTPs [65] [3]. These artefacts create unpredictable sequences that propagate through subsequent amplification cycles, potentially obscuring results and leading to false conclusions [65].
LNA and PNA: Fundamental Properties and Mechanisms
Locked Nucleic Acids (LNA) and Peptide Nucleic Acids (PNA) are synthetic nucleic acid analogues engineered to overcome the limitations of natural DNA and RNA probes. Their unique chemical structures confer superior binding affinity and specificity, making them powerful tools for combating non-specific amplification.
Locked Nucleic Acid (LNA) Chemistry and Characteristics
LNAs are third-generation antisense oligonucleotides characterized by a bridged sugar-phosphate backbone. A methylene bridge connects the 2'-oxygen to the 4'-carbon of the ribose ring, "locking" the furanose in the C3'-endo conformation [66]. This structural constraint confers exceptional properties:
- Enhanced Thermal Stability: LNA modifications significantly increase the melting temperature (T~m~) of duplexes—by 2–8°C per incorporation—towards complementary DNA or RNA [67] [68].
- Superior Binding Affinity and Specificity: The pre-organized structure reduces entropy loss upon hybridization, enabling stronger and more specific binding, which is crucial for distinguishing highly similar sequences [66] [68].
- Nuclease Resistance: LNAs exhibit remarkable resistance to enzymatic degradation, enhancing their stability in biological applications [68].
Peptide Nucleic Acid (PNA) Chemistry and Characteristics
PNAs feature a radically different N-(2-aminoethyl)glycine pseudopeptide backbone that replaces the sugar-phosphate backbone entirely. Nucleobases are attached via carbonyl methylene linkers, preserving the spacing for Watson-Crick binding [69] [67].
- Electroneutral Backbone: The lack of phosphate groups removes negative charge repulsion with complementary nucleic acids, leading to higher affinity and more stable hybrids [69] [67].
- High Biological Stability: PNAs are resistant to both nucleases and proteases, making them stable in cell extracts and serum [69].
- Sequence Discrimination: PNAs demonstrate excellent specificity, including strong single-nucleotide mismatch discrimination [69].
Table 1: Comparative Properties of DNA, LNA, and PNA
| Property | DNA | LNA | PNA |
|---|---|---|---|
| Backbone Structure | Deoxyribose-phosphate | Bridged ribose-phosphate | N-(2-aminoethyl)glycine |
| Charge | Negative | Negative | Neutral |
| Binding Affinity | Baseline | High (↑Tm 2-8°C/mod) | Very High (↑Tm >10°C/mod) |
| Nuclease Resistance | Low | High | Very High |
| Sequence Specificity | Good | Excellent | Excellent |
| Primary Application | Native PCR | Probes, ASOs | Probes, Antigene/Antisense |
Experimental Protocols for Enhanced Specificity
LNA-Mediated Specificity in Quantitative Assays
Protocol: Designing LNA-Enhanced Probes for Real-Time PCR
LNA incorporation is particularly effective in TaqMan probes and molecular beacons. The following methodology ensures maximal specificity:
- Probe Design: Substitute every 3rd-4th nucleotide in a standard DNA oligonucleotide probe with an LNA monomer. Avoid LNA modifications in the enzyme cleavage site if using TaqMan probes.
- Positioning for SNP Detection: For single-nucleotide polymorphism (SNP) discrimination, place the LNA modification directly at the polymorphic site to maximize differentiation in melting temperatures.
- Experimental Workflow:
- Step 1: Design LNA-containing probes using specialized software that calculates Tm for LNA/DNA mixes.
- Step 2: Synthesize and purify probes using standard phosphoramidite chemistry.
- Step 3: Perform qPCR with a temperature gradient to optimize annealing/extension conditions.
- Step 4: Validate specificity using melt curve analysis or sequencing of products.
Diagram 1: Experimental workflows for LNA and PNA specificity enhancement.
PNA Clamping for Suppression of Non-Target Amplification
Protocol: PNA-Mediated Clamping PCR for Allele Discrimination
PNA clamping utilizes PNAs to block the amplification of non-target sequences, such as wild-type alleles when detecting mutations:
- PNA Clamp Design: Design a PNA oligomer (typically 12-18 bases) perfectly complementary to the non-target DNA sequence. The PNA should target a region encompassing the variant site.
- PCR with PNA Clamp:
- Reaction Setup: Prepare a standard PCR master mix including template DNA, primers, and dNTPs.
- PNA Addition: Add the PNA clamp at a concentration of 0.5-2.0 µM—this requires optimization as excessive PNA can inhibit all amplification.
- Thermal Cycling: Employ a modified protocol with an initial denaturation at 95°C for 5 min, followed by 35-40 cycles of denaturation at 95°C for 30s, and a combined annealing/extension at an elevated temperature (e.g., 70-75°C) for 60s. The higher hybridization temperature favors PNA binding.
- Mechanism: The PNA clamp binds tightly to the non-target sequence during PCR annealing, physically blocking DNA polymerase access and preventing amplification. The target sequence (with mismatch to PNA) amplifies preferentially.
Quantitative Data and Performance Comparison
The enhanced performance of LNA and PNA technologies is demonstrated through measurable improvements in key parameters.
Table 2: Quantitative Performance Comparison of Specificity-Enhancing Methods
| Method | ΔTm per Modification | Specificity Gain | Optimal Length | Multiplexing Capacity |
|---|---|---|---|---|
| Standard DNA Primers | Baseline | Baseline | 18-25 bp | Limited by primer dimers [65] |
| Crosslinked Primers | Not Applicable | ~30-50% reduction in primer dimers [65] | 18-25 bp | High (34-plex demonstrated) [65] |
| LNA-Modified Probes | +2 to +8°C [68] | High (excellent SNP discrimination) [66] | 15-20 bp | Moderate to High |
| PNA Clamps | +1°C per base (vs. DNA) [69] | Very High (allele-specific suppression) | 12-18 bp | Moderate |
Research Reagent Solutions
Successful implementation of these advanced techniques requires specific reagents and materials tailored to LNA and PNA applications.
Table 3: Essential Research Reagents for LNA and PNA Applications
| Reagent / Material | Function / Application | Key Characteristics |
|---|---|---|
| LNA Phosphoramidites | Chemical synthesis of LNA oligonucleotides | Enables incorporation of LNA monomers during solid-phase synthesis [68] |
| Ionizable Lipids (e.g., Lipid 10, 15) | Formulation of lipid nanoparticles (LNPs) for LNA delivery | pH-sensitive; enhance cellular uptake and endosomal escape [66] |
| PNA Monomers | Synthesis of PNA oligomers | N-(2-aminoethyl)glycine backbone units with nucleobase protections [69] |
| Cell-Penetrating Peptides (CPPs) | Conjugation to PNA for improved cellular delivery | Cationic or amphipathic peptides that facilitate membrane translocation [69] |
| High-Affinity LNA | Applications requiring maximum binding strength | Optimized LNA chemistry for superior target affinity [68] |
| Adjustable LNA | Applications requiring tunable binding properties | Modified LNA with flexibility in thermal stability [68] |
The integration of LNA and PNA technologies represents a paradigm shift in overcoming the persistent challenge of non-specific amplification in molecular biology. Through their distinct mechanisms—LNA with its pre-organized structure enabling superior hybridization discrimination, and PNA with its neutral backbone permitting unmatched binding affinity—these synthetic nucleic acid analogues provide researchers with powerful tools to enhance assay specificity. The experimental protocols and reagent frameworks outlined in this technical guide enable direct implementation of these advanced techniques, paving the way for more reliable genetic analysis, accurate diagnostics, and robust therapeutic development. As these technologies continue to evolve and converge with advanced delivery systems such as lipid nanoparticles, their impact on reducing primer dimer formation and other amplification artefacts will undoubtedly expand, further empowering precision medicine and fundamental biological research.
Diagnosing and Resolving PCR Artifacts: A Step-by-Step Troubleshooting Guide
Gel electrophoresis is a cornerstone technique in molecular biology for separating and analyzing DNA, RNA, and proteins. However, the presence of artefacts can compromise data interpretation, leading to incorrect conclusions in critical research and diagnostic settings. Within the context of primer dimers and nonspecific amplification research, recognizing these failure modes is paramount for ensuring assay specificity, particularly in applications like pathogen detection where false positives present a major problem [70]. This guide provides an in-depth analysis of common gel electrophoresis artefacts, their root causes, and robust experimental protocols for their prevention and identification.
A Systematic Catalogue of Common Artefacts
Artefacts in gel electrophoresis can arise from issues in sample preparation, reaction composition, or the electrophoresis process itself. The table below summarizes the primary artefacts associated with research on primer dimers and nonspecific amplification.
Table 1: A Catalogue of Common Gel Electrophoresis Artefacts
| Artefact | Appearance on Gel | Primary Causes | Impact on Research |
|---|---|---|---|
| Primer Dimers [23] | A diffuse smear or sharp band, typically between 30-50 bp, much smaller than the target amplicon. | Primer self-complementarity leading to primer-primer annealing; low annealing temperatures; high primer concentration. | Consumes reagents, reduces target amplification efficiency and sensitivity, can lead to false positives [70]. |
| DNA Smearing [71] | A continuous "smear" of DNA down the lane instead of sharp, distinct bands. | Nuclease-mediated DNA degradation; excessive voltage causing localized heating and DNA denaturation; incomplete restriction enzyme digestion. | Makes band identification and quantification impossible; indicates sample or reaction failure. |
| Unexpected/Multiple Bands [72] | Additional bands at unexpected molecular weights, separate from the target band. | Protease activity degrading the protein of interest before heating; non-specific primer binding to non-target genomic sequences; presence of contaminating proteins like keratin. | Complicates interpretation of purification or amplification success; can be misidentified as specific products. |
| "Smiling" or "Frowning" Bands [71] | Bands that curve upwards ("smiling") or downwards ("frowning") at the edges of the gel. | Uneven heat distribution across the gel (Joule heating), often from high voltage; incorrect buffer concentration; high salt in samples. | Distorts apparent molecular weight and can impair accurate quantification. |
| Keratin Contamination [72] | A heterogeneous cluster of bands around 55-65 kDa on reducing SDS-protein gels. | Contamination of sample or lysis buffer with skin cells or dandruff. A common contaminant in sensitive assays like silver staining. | Can be mistaken for bona fide protein bands or interfere with western blot analysis. |
Experimental Protocols for Diagnosing Artefacts
Protocol: Diagnosing Protease Degradation vs. Asp-Pro Bond Cleavage
Unexpected protein bands can stem from different causes. This protocol helps distinguish protease activity from heat-induced peptide bond cleavage [72].
Materials:
- Protein sample
- 2x SDS-PAGE sample buffer (with β-mercaptoethanol or DTT)
- Heating block(s)
- Centrifuge
- Gel electrophoresis system
Method:
- Divide your protein sample into two equal aliquots and add each to an equal volume of SDS-PAGE sample buffer.
- Tube A: Heat immediately at 95-100°C for 5 minutes.
- Tube B: Incubate at room temperature for 2-4 hours, then heat at 95-100°C for 5 minutes.
- Briefly centrifuge both tubes to pellet any insoluble material.
- Load the supernatants onto an SDS-PAGE gel and run.
Interpretation:
- If the sample in Tube B (delayed heating) shows significant degradation (more or lower molecular weight bands) compared to Tube A, it indicates protease activity [72].
- If both samples show a specific, consistent pattern of cleavage, it may indicate Asp-Pro bond cleavage from excessive heating. Testing a lower heating temperature (e.g., 75°C) can confirm this [72].
Protocol: Testing for Primer-Dimer Formation and Specificity
This protocol evaluates primer pairs for their propensity to form dimers and amplify non-specifically.
Materials:
- Primer pairs
- Target DNA template
- Non-target DNA templates (e.g., genomic DNA from a related species)
- No-template control (NTC, nuclease-free water)
- PCR reagents (polymerase, dNTPs, buffer)
- Thermocycler
- Gel electrophoresis system and staining equipment
Method:
- Set up a series of PCR reactions:
- Reaction 1: Target template + Primer pair
- Reaction 2: Non-target template + Primer pair
- Reaction 3: No-template control (NTC) + Primer pair
- Use optimized cycling conditions, but consider a gradient of annealing temperatures to find the optimal stringency.
- Analyze all reactions on a high-percentage agarose gel (e.g., 2-3%) to better resolve small primer dimers.
- Set up a series of PCR reactions:
Interpretation:
- A clean, single band only in Reaction 1 indicates high specificity.
- Bands in Reaction 2 indicate non-specific amplification.
- A band or smear in the 30-50 bp range in Reaction 3 (NTC) confirms primer-dimer formation [23]. This necessitates primer re-design.
Visual Guide to PCR Specificity Testing
The Scientist's Toolkit: Key Reagents & Solutions
Successful experimentation requires high-quality reagents. The following table details essential materials for troubleshooting amplification and electrophoresis artefacts.
Table 2: Essential Research Reagents for Troubleshooting
| Reagent / Material | Function & Rationale | Troubleshooting Application |
|---|---|---|
| Hot-Start Polymerase [23] | Polymerase is inactive at room temperature, preventing enzymatic activity during reaction setup. | Critical for minimizing primer-dimer formation and non-specific amplification in the early PCR cycles. |
| dNTPs | Building blocks for DNA synthesis. | Imbalanced concentrations can reduce yield and specificity. Use quality, nuclease-free preparations. |
| Agarose & Polyacrylamide [73] | Gel matrices that separate molecules by size. Polyacrylamide has finer resolution. | Choose concentration based on target size: 2-3% agarose for primer dimers; higher % polyacrylamide for small DNA fragments. |
| DNA Ladder / Marker | Molecular weight standard for sizing bands. | Essential for identifying the size of unknown bands and confirming primer dimers (~30-50 bp). |
| Urea [72] | A denaturing agent for proteins and nucleic acids. | Used in sample buffers to help solubilize problematic proteins (e.g., histones). Caution: Can contain cyanate which causes protein carbamylation. |
| Benzonase Nuclease [72] | Recombinant endonuclease that degrades all forms of DNA and RNA. | Added to viscous cell extracts prior to lysis to reduce sample viscosity caused by nucleic acids, preventing streaking. |
| Ethidium Bromide / SYBR Safe [73] | Intercalating dyes for visualizing nucleic acids under UV light. | Ethidium bromide is a classic stain; SYBR Safe is a safer, less mutagenic alternative. Allows band visualization post-electrophoresis. |
Advanced Concepts: AI and Machine Learning in Gel Analysis
The field of gel image analysis is being revolutionized by artificial intelligence. Traditional software relies on algorithms to convert lanes into 1D profiles and apply peak-finding, which can miss bands or clip boundaries [74]. Modern AI-based systems, such as GelGenie, use a segmentation approach, training neural networks on hundreds of manually labeled gel images to classify each pixel as 'band' or 'background' with high accuracy, even in sub-optimal conditions like high background or warped bands [74]. Furthermore, machine learning is being applied to predict PCR success; recurrent neural networks (RNNs) can be trained on "pseudo-sentences" generated from primer-template interactions to predict amplification outcomes with high accuracy, potentially reducing reliance on preliminary experiments [70].
Workflow of AI-Based Gel Analysis
The accurate interpretation of gel electrophoresis results is a fundamental skill that requires a deep understanding of potential artefacts. Primer dimers, smears, and unexpected bands are not mere inconveniences; they are diagnostic tools that provide insight into the quality of a reaction. By systematically understanding their causes—be it suboptimal primer design, nuclease contamination, or improper electrophoresis conditions—researchers can implement effective strategies to mitigate them. The integration of robust experimental protocols, high-quality reagents, and emerging AI-powered analytical tools provides a comprehensive framework for ensuring data integrity. This is especially critical in drug development and diagnostic applications, where the cost of misinterpretation can be exceptionally high.
Within the broader research on polymerase chain reaction (PCR) artifacts, understanding and diagnosing nonspecific amplification remains a critical challenge for assay development. This phenomenon, which includes the formation of primer dimers and other non-target amplicons, directly compromises the efficiency, specificity, and reliability of PCR-based analyses in research and drug development [7] [3]. Nonspecific amplification occurs when primers anneal to non-target DNA sequences or to each other, leading to the amplification of unintended fragments [3]. This guide provides a systematic, visual framework for researchers to diagnose the root causes of these common PCR complications, enabling more robust and reproducible experimental outcomes.
Background: Primer Dimers and Nonspecific Amplification
Defining the Artifacts
Nonspecific amplification encompasses any amplification of non-target DNA during PCR. The most prevalent form is the primer dimer, a small, unintended DNA fragment that forms when primers anneal to each other instead of the target template [7]. Primer dimers typically appear as a fuzzy smear or a bright band below 100 bp on an agarose gel and form via two primary mechanisms:
- Self-dimerization: A single primer contains regions complementary to itself, creating a free 3' end for DNA polymerase extension [7].
- Cross-dimerization: Two primers with complementary regions bind together, creating amplifiable units [7].
Other forms of nonspecific amplification include primer multimers (larger complexes that produce a laddering effect), smears (indicating random amplification of DNA fragments of various lengths), and discrete amplicons of unexpected sizes [3].
Impact on Research and Assay Development
The presence of these artifacts competes with target amplicons for reaction components, reducing PCR efficiency and yield [7] [3]. This is particularly problematic in quantitative PCR (qPCR), where primer dimers can lead to false-positive signals and inaccurate quantification. For downstream applications like cloning and sequencing, nonspecific amplicons can obscure results or require additional purification steps, increasing time and resource expenditure.
A Systematic Flowchart for Diagnosis and Troubleshooting
The following diagnostic flowchart provides a structured methodology for identifying the root cause of nonspecific amplification in PCR experiments. This systematic approach guides researchers through a series of investigative steps and decision points based on gel electrophoresis results and reaction conditions.
Experimental Protocols for Verification and Optimization
Protocol 1: No-Template Control (NTC) Reaction
Purpose: To verify that amplification artifacts originate from the PCR reaction itself rather than template-specific interactions [7].
Methodology:
- Prepare a standard PCR reaction mixture containing all components (primers, polymerase, dNTPs, buffer, Mg2+) except the DNA template.
- Replace the template volume with nuclease-free water.
- Run the NTC reaction alongside experimental samples using identical thermal cycling conditions.
- Analyze all samples using agarose gel electrophoresis.
Interpretation: If primer dimers or other nonspecific products appear in the NTC lane, this confirms the issues are inherent to the primer design or reaction conditions, not the template [7].
Protocol 2: Annealing Temperature Gradient Optimization
Purpose: To empirically determine the optimal annealing temperature that maximizes specific amplification while minimizing artifacts [33].
Methodology:
- Prepare a master mix containing all PCR components and aliquot equally into multiple tubes.
- Using a thermal cycler with gradient capability, set an annealing temperature range that spans at least 5°C above and below the calculated primer Tm.
- Run the PCR with the gradient annealing step.
- Analyze results by gel electrophoresis to identify the highest temperature that still yields robust target amplification.
Interpretation: The optimal annealing temperature is typically 3–5°C below the lowest primer Tm but should be empirically verified [33]. Higher annealing temperatures generally increase specificity.
Protocol 3: Magnesium Concentration Titration
Purpose: To optimize Mg2+ concentration, a critical cofactor for DNA polymerase that significantly influences reaction specificity and efficiency [33].
Methodology:
- Prepare a series of PCR reactions with Mg2+ concentrations varying from 1.0 mM to 4.0 mM in 0.5 mM increments.
- Keep all other reaction components constant.
- Run PCR using standardized cycling conditions.
- Analyze products by gel electrophoresis for yield and specificity.
Interpretation: Excessive Mg2+ promotes nonspecific amplification, while insufficient Mg2+ reduces yield [33]. Select the concentration that provides the highest target yield with minimal background.
Quantitative Data for Systematic Optimization
Thermal Cycling Parameter Adjustments
Table 1: Optimization of Thermal Cycling Parameters to Reduce Nonspecific Amplification
| Parameter | Standard Condition | Optimization Range | Effect on Specificity | Implementation Guidance |
|---|---|---|---|---|
| Annealing Temperature | Calculated Tm - 5°C | Tm - 8°C to Tm - 2°C | Increase of 1-2°C can dramatically improve specificity | Use gradient cycler; increase in 1-2°C increments [33] |
| Denaturation Time | 15-30 seconds | 30-60 seconds | Longer times help denature complex templates | Increase for GC-rich templates (>60% GC) [33] |
| Annealing Time | 15-60 seconds | 10-30 seconds | Shorter times minimize nonspecific binding | Shorten to minimum effective time [33] |
| Extension Time | 1 min/kb | 30 sec/kb - 2 min/kb | Less impact on specificity | Adjust based on amplicon length and polymerase [33] |
| Cycle Number | 30-35 | 25-40 | Fewer cycles reduce artifact accumulation | Use minimum cycles for adequate yield [33] |
Reaction Component Optimization Ranges
Table 2: Critical Reaction Component Adjustments for Specificity Enhancement
| Component | Standard Concentration | Optimization Range | Effect of Excess | Effect of Deficiency |
|---|---|---|---|---|
| Primers | 0.5 µM | 0.1-1.0 µM | Primer dimer formation [7] [33] | Reduced yield |
| Mg2+ | 1.5 mM | 1.0-4.0 mM | Nonspecific amplification [33] | Reduced or no yield |
| DNA Polymerase | Manufacturer's recommendation | 0.5-1.5x | Nonspecific products [33] | Reduced yield |
| Template DNA | 10-100 ng | 1-200 ng | Smearing; nonspecific bands [3] | Reduced yield |
| dNTPs | 200 µM each | 50-500 µM | Increased error rate; requires more Mg2+ [33] | Reduced yield |
Research Reagent Solutions
Table 3: Essential Reagents for Troubleshooting Nonspecific Amplification
| Reagent/Category | Specific Function | Application Context |
|---|---|---|
| Hot-Start DNA Polymerase | Remains inactive at room temperature, preventing nonspecific priming during reaction setup [7] [33] | Essential for all PCR applications; critical when primer dimers persist |
| GC Enhancer/Buffer Additives | Helps denature GC-rich templates and sequences with secondary structures [33] | GC-rich targets (>60%); templates with strong secondary structure |
| Proofreading DNA Polymerases | High-fidelity enzymes with 3'→5' exonuclease activity to correct misincorporated nucleotides [33] | Cloning, sequencing, and applications requiring high accuracy |
| Gradient Thermal Cycler | Allows testing multiple annealing temperatures simultaneously in a single run [33] | Empirical optimization of annealing temperature; primer validation |
| Nuclease-Free Water | Prevents enzymatic degradation of reaction components | All molecular biology applications; essential for reproducible results |
| Mg2+ Optimization Kit | Pre-formulated Mg2+ solutions at varying concentrations for systematic titration | Fine-tuning reaction specificity; troubleshooting persistent issues |
Systematic diagnosis of nonspecific amplification requires methodical investigation of both reaction components and cycling conditions. The provided flowchart, experimental protocols, and optimization tables offer researchers a comprehensive framework for identifying and addressing the root causes of PCR artifacts. Implementation of these structured troubleshooting approaches will enhance assay specificity and reliability, ultimately strengthening the validity of research findings in molecular biology and drug development contexts. Future research directions should focus on developing more predictive algorithms for primer design and novel polymerase formulations with enhanced specificity.
The annealing temperature (Ta) stands as one of the most critical parameters determining the success of the polymerase chain reaction (PCR), exerting direct control over the stringency of primer-template binding and ultimately dictating the specificity and yield of the amplification reaction. An inappropriately low Ta permits primers to bind imperfectly to similar sequences, leading to nonspecific amplification and primer dimers, while an excessively high Ta can prevent efficient primer binding altogether, resulting in low or absent yield. This technical guide details a robust, empirical strategy for determining the optimal annealing temperature using gradient PCR, a method that systematically tests a range of temperatures in a single run. By framing this methodology within the broader context of combating nonspecific amplification artifacts, this whitepaper provides researchers and drug development professionals with a definitive protocol for enhancing the reproducibility and reliability of their PCR-based assays.
In the polymerase chain reaction, the annealing step is where primers specifically hybridize to their complementary sequences on the template DNA, a process fundamentally governed by the annealing temperature. The relationship between a primer's melting temperature (Tm)—the temperature at which 50% of the primer-template duplexes are dissociated—and the actual annealing temperature used in the protocol is pivotal [75]. For most PCR applications, the optimal annealing temperature is typically 3–5°C below the calculated Tm of the primers [76]. However, relying solely on theoretical calculations can be insufficient because Tm values can shift with changes in reagent concentration, pH, and salt conditions in the reaction buffer [75].
The consequences of a suboptimal Ta are significant to both research and diagnostic outcomes. A Ta that is too low reduces reaction stringency, allowing primers to bind to off-target sites with partial complementarity. This promotes the amplification of nonspecific products and increases the formation of primer dimers—artifacts formed when primers hybridize to each other rather than the template DNA [1] [77]. These undesired products compete with the target amplicon for reaction components (polymerase, nucleotides, and primers), thereby reducing the yield and specificity of the intended product [1]. Conversely, a Ta that is too high may prohibit stable primer-template binding, leading to inefficient amplification or complete PCR failure [76]. This delicate balance underscores the necessity for empirical determination of the optimal Ta, making it a non-negotiable step in assay development for high-impact fields like drug development and clinical diagnostics.
Theoretical Foundation: Primer-Template Interactions and PCR Efficiency
The Relationship Between Tm and Ta
The melting temperature (Tm) of a primer is a theoretical value calculated based on its length, GC content, and nucleotide sequence. A fundamental rule in PCR optimization is that the annealing temperature (Ta) must be closely matched to the Tm of the primers for successful amplification. The optimal annealing temperature for standard PCR typically falls between 55°C and 65°C, with the forward and reverse primers ideally having Tm values within 1–2°C of each other to ensure synchronous binding [76]. When the Tm values of a primer pair differ significantly, the primer with the higher Tm may bind to unintended targets at a Ta optimized for the lower-Tm primer, while the lower-Tm primer might not bind efficiently, drastically reducing yield and specificity [14].
Primer Dimers and Nonspecific Amplification
Primer dimers are formed when two primers hybridize to each other instead of the target template, facilitated by complementarity between the primer sequences, particularly at their 3' ends [1]. There are two types: homodimers (between two identical primers) and heterodimers (between the forward and reverse primers) [1]. These structures can be extended by the DNA polymerase, leading to nonspecific amplification products that consume reaction reagents and can generate false-positive signals in quantitative applications [15] [1]. The formation of primer dimers is highly dependent on reaction conditions, including primer concentration, template concentration, and critically, the annealing temperature [15] [77]. Low annealing temperatures significantly increase the likelihood of primer-dimer formation and other nonspecific hybridization events.
The Mathematical Basis of PCR Efficiency
The progression of a PCR amplification follows an exponential function described by the equation: Q(n) = Q(0) × E^n Where Q(n) is the quantity of product at cycle n, Q(0) is the initial template quantity, and E is the PCR efficiency, which ideally equals 2, representing 100% doubling of product each cycle [78]. The efficiency E is intrinsically linked to the annealing temperature. A suboptimal Ta can lower the effective efficiency, as not all primers may bind correctly and extend in each cycle. In quantitative PCR (qPCR), the quantification cycle (Cq) is the value measured to estimate the initial template quantity, and it is inversely related to the log of the initial quantity. The relationship between Cq and efficiency is described by: Cq = -log(d)/log(E) + log(T/Q(0)) / log(E) Where d is the dilution factor [78]. This underscores that accurate quantification is wholly dependent on a consistent and optimal efficiency, which is achieved through proper optimization of parameters like Ta.
Gradient PCR: A Methodology for Empirical Optimization
The Principle of Gradient PCR
Gradient PCR is a powerful technique that enables the simultaneous testing of a range of annealing temperatures across a thermal cycler's block in a single run [79] [75]. Traditional optimization methods require setting up multiple individual PCR reactions, each with a different annealing temperature—a process that is time-consuming, resource-intensive, and prone to inter-run variation. Gradient PCR circumvents these limitations by applying a temperature gradient along the plate, allowing for the direct comparison of amplification efficiency and specificity across multiple Ta values under otherwise identical reaction conditions [75]. Modern thermal cyclers can create this gradient along one axis (1D gradient), and some advanced models can create independent gradients along both the x and y axes (2D gradient), enabling the simultaneous optimization of both annealing and denaturation temperatures [79].
Experimental Protocol for Gradient PCR Optimization
The following protocol provides a detailed methodology for empirically determining the optimal annealing temperature using a gradient thermal cycler.
Preliminary Steps
- Primer Design and Tm Calculation: Design primers according to best practices (18-24 bases, 40-60% GC content, and avoidance of self-complementarity). Calculate the theoretical Tm for both forward and reverse primers using appropriate software. The goal is to have primers with Tm values within 5°C of each other [75].
- Define the Temperature Gradient: Set the gradient range to span approximately 10–15°C, centered around the average Tm of the primer pair. For example, if the average Tm is 60°C, a gradient from 55°C to 65°C is suitable [75].
Reaction Setup
- Prepare a Master Mix: To minimize pipetting errors and ensure reaction consistency, prepare a single master mix for all reactions. The table below outlines the components and their typical concentrations.
Table 1: Reaction Setup for Gradient PCR Optimization
| Component | Final Concentration | Volume per Reaction (μL) | Purpose |
|---|---|---|---|
| PCR Buffer (10X) | 1X | 2.0 | Provides optimal pH and salt conditions. |
| MgCl₂ (25 mM) | 1.5 - 2.5 mM | 0.8 - 1.2 | Essential cofactor for DNA polymerase. |
| dNTP Mix (10 mM each) | 200 μM each | 0.4 | Building blocks for DNA synthesis. |
| Forward Primer (10 μM) | 0.2 - 1.0 μM | 0.4 - 2.0 | Binds to the anti-sense strand of the target. |
| Reverse Primer (10 μM) | 0.2 - 1.0 μM | 0.4 - 2.0 | Binds to the sense strand of the target. |
| DNA Template | Variable | 1.0 - 5.0 | The target DNA to be amplified. |
| DNA Polymerase | As per manufacturer | 0.2 - 0.5 | Catalyzes the DNA synthesis. |
| Nuclease-Free Water | - | To final volume | Adjusts the total reaction volume. |
| Total Volume | 20.0 |
- Aliquot the Master Mix: Dispense equal volumes of the master mix into each tube or well of the PCR plate that will be subjected to different annealing temperatures.
Thermal Cycling Conditions
Program the thermal cycler with a gradient PCR protocol. A standard program is outlined below.
Table 2: Example Thermal Cycling Protocol with Gradient
| Step | Temperature | Time | Cycles | Notes |
|---|---|---|---|---|
| Initial Denaturation | 95°C | 2 - 5 min | 1 | Activates hot-start polymerases; fully denatures complex templates. |
| Denaturation | 95°C | 15 - 30 s | ||
| Annealing | Gradient: (e.g., 55°C - 65°C) | 20 - 30 s | 30 - 40 cycles | The critical step for optimization. |
| Extension | 72°C | 1 min/kb | ||
| Final Extension | 72°C | 5 - 10 min | 1 | Ensures all amplicons are fully extended. |
| Hold | 4° - 10°C | ∞ | 1 |
Post-Amplification Analysis
- Gel Electrophoresis: Separate the PCR products using agarose gel electrophoresis (e.g., 1.5-2% agarose).
- Visualization and Interpretation: Visualize the gel under UV light. The optimal annealing temperature is identified as the highest temperature within the gradient that produces a single, intense band of the expected amplicon size, with minimal to no nonspecific bands or primer dimers (visible as a low molecular weight smear or band) [76] [75].
Workflow and Decision Pathway
The following diagram summarizes the experimental workflow and the decision-making process for analyzing gradient PCR results.
Data Analysis and Interpretation
Systematic Analysis of Gradient PCR Results
After completing the gradient PCR run and gel electrophoresis, a systematic analysis of the results is required. The optimal annealing temperature is identified based on two key criteria: specificity (the presence of a single, clean band of the correct size) and yield (the intensity of that band). The ideal result is a balance where yield is maximized without compromising specificity. The highest temperature that still produces a strong, specific product is generally chosen, as this provides the greatest stringency against nonspecific binding and primer-dimer formation [76].
Troubleshooting Common Scenarios
The following table guides the interpretation of common results observed in a gradient PCR experiment and recommends subsequent actions.
Table 3: Troubleshooting Guide for Gradient PCR Results
| Observation | Interpretation | Recommended Action |
|---|---|---|
| A single, intense band of the expected size across a wide temperature range. | The primer pair is robust and the initial Tm calculation was accurate. The reaction is well-optimized. | Select the highest temperature within the successful range for maximum specificity in future experiments. |
| Nonspecific bands (multiple bands) and/or primer dimers at lower temperatures; clean, single band at higher temperatures. | The Ta was too low at one end of the gradient, reducing stringency. The optimal Ta is at the higher end of the range. | Proceed with the highest Ta that still produces a good yield of the specific product. |
| A clean, single band only at the lower end of the temperature gradient, with no product at higher temperatures. | The annealing temperature is too high at the upper end of the gradient, preventing efficient primer binding. The optimal Ta is at the lower end. | Use the lowest Ta that gives a specific product. Verify the primer Tm calculations and consider redesigning primers if the range is unacceptably narrow. |
| No product across the entire temperature gradient. | The gradient range may be set too high, the primers may be faulty, or the template quality may be poor. | Verify template quality and concentration. Check primer sequences and integrity. Broaden the gradient to a lower temperature range (e.g., 45°C-60°C). |
| Nonspecific amplification across the entire gradient. | The primers may have low specificity, binding to multiple off-target sites. The primer concentration may be too high, or the Mg²⁺ concentration may require optimization. | Redesign primers with stricter parameters. Titrate primer and Mg²⁺ concentrations. Consider using a hot-start polymerase to minimize mis-priming [76]. |
Advanced Applications: 2D-Gradient PCR
For challenging targets, such as GC-rich sequences or long amplicons, simultaneous optimization of both the annealing temperature and the denaturation temperature can be beneficial. A 2D-gradient thermal cycler can apply one temperature gradient along the x-axis (e.g., for denaturation temperature) and another along the y-axis (e.g., for annealing temperature), testing 96 different temperature combinations in a single run [79]. Optimizing the denaturation temperature can help fully separate DNA strands with strong secondary structures, while optimizing the annealing temperature ensures specific primer binding. This combined approach can lead to significant improvements in both specificity and product yield for difficult assays [79].
Integration with Broader Research Context
The Link to Primer Dimers and Nonspecific Amplification Research
The empirical determination of annealing temperature via gradient PCR is a foundational practice within the broader research effort to understand and mitigate primer dimers and nonspecific amplification. As highlighted in studies, the occurrence of these artifacts is not random but is determined by specific reaction conditions, with annealing temperature being a primary factor [15]. The formation of primer dimers is highly favored at low annealing temperatures, where the reduced stringency allows primers to interact with each other via short regions of complementarity. These dimers can then be extended by the DNA polymerase, generating nonspecific products that consume reagents and can lead to false-positive signals in quantitative applications like qPCR [15] [1]. Therefore, the use of gradient PCR to find the highest possible Ta that still supports efficient amplification is a direct and effective strategy to suppress this artifact formation, enhancing the validity of experimental data.
Connection to qPCR and High-Fidelity Applications
The principles of annealing temperature optimization extend directly into quantitative PCR (qPCR), where the precise measurement of DNA concentrations is critical. Unnoticed amplification of nonspecific products or primer dimers in qPCR results in inaccurate Cq values and flawed quantification [15] [80]. The MIQE guidelines (Minimum Information for publication of Quantitative real-time PCR Experiments) emphasize the importance of assay specificity and validation, for which proper Ta determination is essential [78] [80]. Furthermore, in applications requiring high fidelity, such as cloning and sequencing, the use of optimized Ta with proofreading polymerases is necessary to minimize misincorporation errors and ensure the integrity of the amplified product [76].
The Scientist's Toolkit: Essential Reagents and Materials
The following table catalogues key research reagent solutions and materials essential for successfully performing gradient PCR optimization.
Table 4: Essential Research Reagents and Materials for Gradient PCR
| Item | Function/Description | Example Notes |
|---|---|---|
| Gradient Thermal Cycler | A PCR instrument capable of generating a precise temperature gradient across its block. | Essential for the simultaneous testing of multiple annealing temperatures. Models with a 2D-gradient function allow for dual-parameter optimization [79]. |
| High-Quality DNA Polymerase | Enzyme that catalyzes the synthesis of DNA. | Hot-Start versions are recommended to minimize nonspecific amplification and primer-dimer formation during reaction setup [76]. High-Fidelity enzymes (e.g., Pfu, KOD) are used for cloning. |
| Optimized PCR Buffer | A chemical solution providing the optimal pH, ionic strength, and cofactors for polymerase activity. | Some specialized buffers contain isostabilizing agents that allow for the use of a "universal" annealing temperature (e.g., 60°C), simplifying PCR setup with multiple primer sets [14]. |
| Magnesium Chloride (MgCl₂) | An essential cofactor for DNA polymerase. Its concentration directly affects enzyme activity, specificity, and fidelity. | Typically titrated between 1.5 - 2.5 mM. A critical parameter to optimize after Ta, as high Mg²⁺ can promote nonspecific binding [76] [77]. |
| Ultrapure dNTPs | Deoxynucleotide triphosphates (dATP, dCTP, dGTP, dTTP) serving as the building blocks for DNA synthesis. | Used at 200 µM each. Quality is crucial to prevent polymerase inhibition. |
| High-Purity Primers | Oligonucleotides designed to be complementary to the flanking sequences of the target DNA. | Purified by HPLC or PAGE to ensure correct sequence and full-length synthesis, reducing failed reactions. |
| Agarose Gel Electrophoresis System | Standard method for separating and visualizing DNA fragments by size post-amplification. | Used to assess the specificity and yield of the PCR products from the gradient run. |
| Additives (e.g., DMSO, Betaine) | Reagents used to amplify difficult templates, such as GC-rich sequences. | DMSO (2-10%) can help disrupt secondary structures. Betaine (1-2 M) homogenizes base stability [76]. |
The empirical determination of optimal annealing temperature through gradient PCR is a critical, non-negotiable step in the development of robust and reliable PCR assays. While in silico calculations provide a starting point, they cannot account for the complex interplay of reagents and template in a specific reaction environment. The gradient PCR method offers a efficient and systematic experimental approach to pinpoint the Ta that delivers the ideal balance of high yield and absolute specificity. By rigorously applying this methodology, researchers and drug development professionals can effectively suppress the formation of primer dimers and nonspecific amplification artifacts, thereby ensuring the generation of reproducible, publication-quality data that accurately reflects the underlying biological question.
In quantitative polymerase chain reaction (qPCR) and related amplification technologies, the exquisite sensitivity and specificity that make these methods uniquely powerful are critically dependent on reaction chemistry. Primer dimers and other forms of nonspecific amplification represent fundamental challenges that can compromise experimental results, leading to false positives, inaccurate quantification, and reduced detection sensitivity. While primer design is universally recognized as a crucial factor, the optimization of reaction components—specifically primer concentration and master mix composition—serves as an equally critical determinant of amplification success. This technical guide examines how systematic adjustment of these chemical parameters can suppress artifact formation while maintaining robust amplification of target sequences.
The occurrence of PCR artifacts is not merely a function of poor primer design but depends profoundly on the reaction environment, including template, non-template, and primer concentrations [15]. Titration experiments have demonstrated that the frequency of both low and high melting temperature artifacts is determined by annealing temperature, primer concentration, and cDNA input [15]. Furthermore, the interpretation of dilution series is complicated by the simultaneous decrease of both template and non-template concentrations, which can unexpectedly influence artifact formation [15]. This guide synthesizes current research on reaction chemistry optimization, providing detailed methodologies and quantitative frameworks for researchers seeking to overcome nonspecific amplification in diagnostic development and fundamental research applications.
Understanding the Mechanisms of Nonspecific Amplification
Established Mechanisms and a Novel Pathway
The prevailing understanding of nonspecific amplification has primarily centered on primer-dimer formation and mispriming events. However, recent research has uncovered a more complex mechanistic landscape:
Traditional Primer-Dimer Formation: Primer-dimers typically consist of two primers hybridizing to each other, often through a few complementary bases, creating an amplifiable unit that can be exponentially amplified [3]. These artifacts generally produce amplicons of 20-60 bp in length and are visible as bright bands at the bottom of electrophoresis gels [3].
Dynamic Mismatched Primer Binding (DMPB): A recently discovered mechanism reveals that primers (~20 nt) can bind to background DNA (bgDNA) for primer extension with surprisingly few matched bases—only 6-11 fully matched (9-14 mismatched) base pairs are sufficient to initiate this process [81]. After the single-stranded DNAs attached to the first primer are produced, the other primer can bind to these ssDNAs with even shorter fully matched base pairs, ultimately producing perfect "seeds" for exponential nonspecific amplification [81].
Jumping PCR: This phenomenon involves within-genome recombination where extended primers sharing homology with sequences elsewhere in the genome create completely new amplification products [15]. The frequency of jumping depends on the ratio of template to non-template DNA rather than absolute template concentration [15].
Table 1: Mechanisms of Nonspecific Amplification
| Mechanism | Key Feature | Minimum Base Pair Requirement | Amplicon Characteristics |
|---|---|---|---|
| Primer-Dimer | Primer-primer hybridization | 3-4 complementary bases (typical) | 20-60 bp; bright band at gel bottom |
| DMPB | Primer-background DNA binding | 6-11 fully matched bases | Variable length; sequence corresponds to bgDNA regions |
| Jumping PCR | Within-genome recombination | Homology regions | Chimeric products; unexpected sizes |
| Off-Target Amplification | Primer binding to non-target sequences | 9-14 consecutive base pairs | Stable dimers; competes with target amplification |
Experimental evidence indicates that spatial arrangement of base pairs significantly impacts dimerization stability. Studies have shown that while more than 15 consecutive basepairs create stable dimers, non-consecutive basepairs do not form stable dimers even when 20 out of 30 possible basepairs are bonded [6]. This finding has profound implications for understanding how primers interact with non-target sequences under various reaction conditions.
The Impact of Nonspecific Amplification on Data Quality
The consequences of nonspecific amplification extend beyond mere presence/absence artifacts to fundamentally compromise data integrity:
False Positives in Diagnostic Applications: Nonspecific products can generate detectable signals in negative samples, leading to incorrect conclusions in clinical diagnostics [81].
Reduced Sensitivity and Dynamic Range: Normal primers experience signal dampening with as few as 60 primer-dimers and false negatives with only 600 primer-dimers, representing a significant constraint on detection limits [46].
Inaccurate Quantification: Artifact-associated fluorescence can distort quantification cycles (Cq values), leading to incorrect template quantification [15]. This effect is particularly pronounced when artifacts form early in the amplification process.
Sequencing Interference: In next-generation sequencing applications, nonspecific amplification wastes sequencing capacity on non-informative reads. One study of 16S rRNA gene sequencing found that 77.2% of Amplicon Sequence Variants from breast tumor samples aligned to the human genome rather than bacterial targets when using suboptimal primers [82].
Optimizing Primer Concentration: Quantitative Guidelines
Systematic Approaches to Primer Titration
Primer concentration represents a pivotal parameter in balancing amplification efficiency against artifact formation. Research indicates that the occurrence of nonspecific products depends significantly on primer concentrations in the reaction [15]. The following table summarizes key findings from systematic titration studies:
Table 2: Effects of Primer Concentration on Amplification Specificity
| Primer Concentration (μM) | Amplification Efficiency | Artifact Frequency | Recommended Application |
|---|---|---|---|
| 0.1 | Reduced efficiency; delayed Cq | Low | High template abundance; prevention of primer-dimer formation |
| 0.2-0.3 | Optimal for many applications | Moderate to low | Standard qPCR with well-designed primers |
| 0.5 | High efficiency | Increased risk of artifacts | Problematic templates with secondary structure |
| 1.0 | Maximum efficiency | High frequency of primer-dimers | Suboptimal conditions; not generally recommended |
Checkerboard titration experiments with MS2 phage assays have demonstrated that both excessively high and low primer concentrations can promote different types of artifacts. High primer concentrations (>0.5 μM) favor primer-dimer formation, while low concentrations (<0.1 μM) can paradoxically increase off-target amplification due to reduced competition for specific binding sites [15].
Interaction Between Primer and Template Concentrations
The optimal primer concentration is not an absolute value but depends on template abundance. Experimental evidence shows that low template concentrations increase artifact frequency, while quantitatively, low non-template concentrations lead to deviating Cq values and thus to incorrect quantification [15]. This interaction creates a complex optimization landscape:
High Template, High Primer Scenarios: When both template and primer concentrations are elevated, the specific product typically outcompetes artifacts due to kinetic advantages.
Low Template, High Primer Scenarios: This dangerous combination dramatically increases the probability of primer-dimer and off-target amplification as primers seek available binding partners.
Dilution Series Complications: The common practice of using dilution series to determine amplification efficiency is problematic because it simultaneously decreases both template and non-template concentrations [15]. This dual reduction can mask artifact formation that would occur at actual experimental concentrations.
Master Mix Composition: Strategic Formulation for Specificity
Critical Components and Their Functions
Master mix composition profoundly influences the specificity of amplification reactions by altering enzymatic behavior, hybridization kinetics, and structural constraints. The following table outlines key master mix components and their roles in suppressing nonspecific amplification:
Table 3: Master Mix Components for Controlling Nonspecific Amplification
| Component | Function | Optimal Concentration Range | Effect on Specificity |
|---|---|---|---|
| Hot-Start DNA Polymerase | Inhibits polymerase activity until high temperature | Manufacturer's recommendation | Prevents primer-dimer formation during reaction setup |
| Mg²⁺ Concentration | Cofactor for polymerase activity | 1.5-3.0 mM (requires optimization) | Higher concentrations increase non-specific binding |
| dNTPs | Building blocks for DNA synthesis | 200-400 μM each | Imbalance can increase misincorporation |
| Betaine | Destabilizes secondary structures | 0.5-1.5 M | Reduces template secondary structure competition |
| DMSO | Prevents secondary structure formation | 2-10% | Enhances specificity for GC-rich targets |
| BSA | Binds inhibitors, stabilizes enzymes | 0.1-0.5 μg/μL | Improves performance with inhibited samples |
Advanced Formulation Strategies
Beyond basic components, several specialized formulations can dramatically improve amplification specificity:
Hot-Start Mechanisms: While conventional hot-start methods reduce primer-dimer formation during reaction setup, they cannot stop the propagation of primer-dimers once formed [46]. Advanced hot-start formulations provide more stringent control of polymerase activity through chemical, antibody, or aptamer-based inhibition.
Modified Polymerase Enzymes: "Proofreading" enzymes, while valuable for high-fidelity applications, can paradoxically increase non-specific amplification caused not just by primer dimers but by physical closeness of primer pairs at mismatched sites [83].
Cooperative Primers: This innovative technology represents a breakthrough in nonspecific amplification control, demonstrating a 2.5 million-fold improvement in reduction of nonspecific amplification compared to conventional primers [46]. Successful amplification of 60 template copies has been achieved with no signal dampening in a background of 150,000,000 primer-dimers [46].
Experimental Protocols for Reaction Optimization
Primer Concentration Titration Protocol
Objective: To empirically determine the optimal primer concentration for specific target amplification while minimizing artifacts.
Materials:
- Target DNA/cDNA template
- Forward and reverse primers (100 μM stock solutions)
- qPCR master mix (2× concentration)
- Nuclease-free water
- qPCR instrument with melting curve analysis capability
Methodology:
- Prepare a series of primer dilutions in nuclease-free water to create working stocks of 10 μM, 5 μM, 2.5 μM, 1.25 μM, and 0.625 μM.
Set up 20 μL reactions containing 1× master mix, fixed template concentration, and varying primer concentrations according to the following scheme:
- Reaction 1: 0.1 μM each primer (2 μL of 1 μM working stock)
- Reaction 2: 0.2 μM each primer (2 μL of 2 μM working stock)
- Reaction 3: 0.3 μM each primer (2 μL of 3 μM working stock)
- Reaction 4: 0.5 μM each primer (2 μL of 5 μM working stock)
- Reaction 5: 1.0 μM each primer (2 μL of 10 μM working stock)
Include no-template controls for each primer concentration to assess primer-dimer formation.
Run qPCR with the following cycling parameters:
- Initial denaturation: 95°C for 5 minutes
- 40 cycles of:
- Denaturation: 95°C for 10 seconds
- Annealing/extension: 60°C for 30 seconds (with fluorescence acquisition)
- Melting curve analysis: 65°C to 95°C with 0.5°C increments
Analyze amplification curves, efficiency calculations, and melting curves to identify the primer concentration that provides the lowest Cq, highest efficiency, and clean melting peak.
Validation: Reactions should be performed in triplicate with consistent template quality and quantity. The optimal concentration is typically the lowest that provides maximum efficiency without artifact formation in no-template controls [15] [83].
Master Mix Composition Screening Protocol
Objective: To evaluate different master mix formulations for amplification specificity with challenging templates.
Materials:
- Target DNA/cDNA template (including low-concentration samples)
- Optimized primer pair
- Three different commercial master mixes (including at least one hot-start formulation)
- Potential additives (DMSO, betaine, BSA)
- qPCR instrument
Methodology:
- Prepare master mixes according to manufacturer specifications, maintaining consistent primer and template concentrations across all reactions.
For additive screening, supplement master mixes with:
- Condition A: No additives (control)
- Condition B: 3% DMSO
- Condition C: 1 M betaine
- Condition D: 0.1 μg/μL BSA
- Condition E: Combined additives (1.5% DMSO + 0.5 M betaine + 0.05 μg/μL BSA)
Run qPCR with cycling conditions optimized for each master mix according to manufacturer recommendations.
Include melting curve analysis to assess product specificity.
Analyze results based on:
- Amplification efficiency (from dilution series)
- Cq values for low-concentration templates
- Melt curve profile complexity
- Signal intensity in no-template controls
Interpretation: The optimal master mix should provide efficiency between 90-110%, a single peak in melt curve analysis, and minimal amplification in no-template controls [84] [3].
Integrated Workflow for Comprehensive Optimization
The relationship between different optimization parameters and experimental steps can be visualized through the following workflow:
Diagram 1: Experimental optimization workflow for qPCR assays
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Reagents for Controlling Nonspecific Amplification
| Reagent/Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Hot-Start Polymerases | Antibody-mediated hot-start, Chemical modification | Prevents enzymatic activity during setup | Critical for low-template applications; reduces primer-dimer formation [15] |
| Master Mix Additives | DMSO, Betaine, Formamide, BSA | Modifies hybridization stringency, reduces secondary structure | Concentrations require optimization; betaine particularly effective for GC-rich targets [3] |
| Specialized Primer Formats | Cooperative primers, Mini-hairpin primers | Prevents unintended primer interactions | Cooperative primers show 2.5 million-fold improvement in reducing nonspecific amplification [46] |
| qPCR Reagents with Modified Detection | SYBR Green, EvaGreen, Probe-based chemistries | Enables real-time monitoring of amplification | SYBR Green requires melt curve analysis; probe chemistries increase specificity [15] |
| Inhibition Resistance Additives | BSA, T4 gene 32 protein | Counteracts common polymerase inhibitors | Essential for difficult samples (e.g., blood, soil); improves robustness [84] |
Optimizing primer concentration and master mix composition represents a powerful approach for controlling nonspecific amplification in qPCR and related applications. The experimental evidence presented demonstrates that strategic adjustment of these parameters can suppress artifact formation by several orders of magnitude while maintaining robust amplification of intended targets. The most effective optimization strategies acknowledge the interconnected nature of reaction components—recognizing that primer concentration, template quality, and master mix formulation must be optimized in concert rather than isolation.
For researchers confronting challenging amplification scenarios, the integrated workflow presented here provides a systematic path toward robust, specific detection. Particularly for applications involving low-template samples, difficult templates with high secondary structure, or multiplexed detection systems, investment in comprehensive reaction chemistry optimization delivers substantial returns in data quality and experimental reproducibility. As amplification technologies continue to evolve, the fundamental principles of balanced reaction chemistry will remain essential for extracting biologically meaningful signals from molecular complexity.
In molecular biology, the quality of the nucleic acid template is a foundational determinant for the success of any amplification reaction, from routine PCR to advanced diagnostic assays. Contaminants and impurities present in a sample can co-purify with the target DNA or RNA, directly interfering with the enzymatic processes that drive specific amplification. This inhibition leads to a spectrum of issues, including reduced sensitivity, false-negative results, and the generation of non-specific products like primer-dimers that compete for precious reaction reagents. For researchers and drug development professionals, understanding these inhibitory mechanisms is not merely a technical concern but a critical component of assay validation and reliability. The pursuit of specific amplification is a battle against a silent and often invisible enemy—contaminants that can sabotage experimental integrity, diagnostic accuracy, and the development of sensitive therapeutic products.
This guide provides an in-depth technical examination of how various contaminants impede amplification, supported by quantitative data and detailed protocols for preventing, identifying, and overcoming these challenges within the broader context of primer-dimer and non-specific amplification research.
Mechanisms of Contaminant Interference in Amplification Reactions
Contaminants exert their inhibitory effects through several distinct molecular mechanisms, each disrupting a different part of the amplification workflow. Understanding these mechanisms is the first step toward developing effective countermeasures.
Direct Enzymatic Inhibition
Many contaminants directly impair the activity of polymerase enzymes. This category of interference includes:
- Metal Ions: The polymerase enzyme is highly sensitive to metal ions. While magnesium (Mg²⁺) is an essential cofactor, its concentration must be carefully optimized and controlled. Conversely, heavy metals such as cadmium (Cd) and zinc (Zn), as well as transition metals like iron (Fe), cobalt (Co), copper (Cu), and nickel (Ni), can bind to the enzyme's active site and severely impact its catalytic activity [85]. These foreign metals compete with the necessary Mg²⁺, leading to reduced processivity and yield.
- Organic Compounds: Negatively charged organic molecules, such as fulvic and humic acids which are common in environmental samples, mimic the charge profile of DNA. These molecules act as non-competitive inhibitors, meaning they do not bind to the enzyme's active site but instead interfere with the catalytic cycle, reducing the polymerase's turnover rate and overall efficiency [85].
- Nucleases: Contaminating DNases and RNases are fast-acting enzymes that can cleave and degrade the genomic template or the primers themselves. This degradation leads to false negatives, reduced sensitivity, and a general loss of amplification signal [85].
Template Degradation and Masking
Beyond affecting the enzyme, contaminants can target the template itself:
- Nuclease Degradation: As mentioned, DNases and RNases present in the sample can directly destroy the target nucleic acids before amplification even begins.
- Template Masking: Certain impurities, such as charged organics or proteins, can bind to the template DNA. This binding physically blocks the access of primers and polymerase to their target sequences, preventing efficient annealing and extension [85].
Facilitation of Non-Specific Amplification
Perhaps the most insidious effect of contaminants is their role in promoting non-specific amplification:
- Altered Reaction Kinetics: Impurities can disrupt the precise reaction equilibrium, lowering the effective stringency of the annealing step. This creates an environment where primers are more likely to bind to non-target sequences or to each other.
- Primer-Dimer Propagation: In multiplexed reactions, where large numbers of primers are present, the probability of primer-dimer formation increases significantly. Contaminants can exacerbate this by facilitating transient, non-specific interactions between primers, which are then extended by the polymerase [86]. These primer-dimers consume dNTPs, primers, and enzyme activity, thereby competing with and inhibiting the desired specific amplification.
Quantifying the Impact: A Compendium of Contaminant Effects
The following tables summarize the key classes of contaminants, their sources, and their specific impacts on amplification reactions, providing a quick reference for troubleshooting.
Table 1: Major Classes of PCR Contaminants and Their Effects
| Contaminant Class | Specific Examples | Primary Source | Impact on Amplification |
|---|---|---|---|
| Ionic Impurities | Cd²⁺, Zn²⁺, Fe²⁺, Co²⁺ [85] | Reagents, labware, water | Binds to polymerase, inhibiting catalytic activity [85] |
| Organic Compounds | Fulvic acids, humic acids [85] | Environmental samples, impurities | Non-competitive inhibition of polymerase [85] |
| Nucleases | DNase, RNase [85] | Bacterial contamination, human handling | Degradation of template DNA/RNA and primers [85] |
| Foreign DNA | Host cell DNA (hcDNA) [87], plasmid DNA impurities [88], bacterial genomic DNA [85] | Production cell lines, starting materials, kit reagents | Non-target amplification, background noise, false positives [87] [88] [85] |
| Additives & Residuals | Phthalates (e.g., DEHP), UV stabilizers (e.g., UV-328) [89] | Plastic labware, environmental contamination | Unknown interference with enzyme kinetics; potential health risk [89] |
Table 2: Quantitative Impact of Water Quality on RT-PCR Reproducibility This data demonstrates the critical need for ultrapure water in achieving quantitative and consistent results in sensitive applications like RT-PCR [85].
| Parameter | Result with Ultrapure Water | Implication |
|---|---|---|
| Quantitation Accuracy | Amount of DNA obtained matched theoretically expected amount [85] | Reactions are quantitative under ideal conditions |
| Inter-experiment Reproducibility | Standard deviation was small across 30 experiments [85] | High purity enables reliable, repeatable results |
| Intra-experiment Reproducibility | No significant difference in DNA amplification across 20 parallel runs [85] | Eliminates run-to-run variability due to water contaminants |
Advanced Detection and Analytical Methodologies
Accurately detecting and quantifying impurities is essential for developing control strategies, particularly in the manufacture of viral-based therapeutics where host cell DNA (hcDNA) is a key impurity.
Table 3: Analytical Methods for Quantifying Residual DNA Impurities This comparison is based on industry survey data for monitoring hcDNA in viral vector products [87].
| Methodology | qPCR | Digital PCR (dPCR/ddPCR) | Next-Generation Sequencing (NGS) |
|---|---|---|---|
| % of Survey Responses Using | 70% | 21% | 9% [87] |
| Principle | Quantitative (relative); targeted; data collected during exponential phase [87] | Quantitative (absolute); targeted; data collected post-amplification [87] | Quantitative (relative); global sequencing of DNA [87] |
| Key Advantages | High throughput; large dynamic range [87] | High reproducibility; less affected by PCR inhibitors; no standard curve needed [87] | Confirms long, complex sequences; no prior target knowledge needed [87] |
| Key Disadvantages / | A priori target knowledge required; lower sensitivity; prone to PCR inhibitors [87] | A priori target knowledge required [87] | Low throughput; high cost; complex data analysis [87] |
| Common Targets | E1A, E1B, 18S, Alu sequences [87] | E1A, E1B, 18S, Alu sequences [87] | Full spectrum of DNA impurities |
For non-specific amplification and primer-dimer detection, methods like gel electrophoresis and melting curve analysis are standard. Furthermore, novel primer technologies like Co-Primers have been developed to directly combat primer-dimer formation. This technology divides the PCR primer into two segments separated by a PEG linker. Both segments must cooperate to bind the target, preventing the propagation of primer dimers because they cannot hybridize to the required capture region [86].
Experimental Protocols for Contamination Control
Implementing rigorous, systematic protocols is paramount for obtaining reliable amplification results, especially when working with low-biomass samples or sensitive assays.
Protocol for Ultra-Clean Sample Collection in Low-Biomass Studies
Based on consensus guidelines for low-biomass microbiome research [90] [91].
- Pre-Sampling Decontamination: Decontaminate all reusable equipment, tools, and surfaces with 80% ethanol to kill microorganisms, followed by a nucleic acid degrading solution (e.g., sodium hypochlorite, UV-C light, hydrogen peroxide) to remove residual DNA. Note that autoclaving and ethanol alone do not reliably remove persistent DNA [90].
- Use of Physical Barriers: Personnel should wear appropriate personal protective equipment (PPE)—including gloves, masks, cleansuits, and shoe covers—to limit the introduction of human-associated contaminants from skin, hair, or aerosols [90].
- Employ Single-Use, DNA-Free Materials: Wherever possible, use single-use, pre-sterilized plasticware or glassware for sample collection and storage. These items should remain sealed until the moment of use [90].
- Incorporate Comprehensive Controls: A robust experimental design must include multiple negative controls collected alongside samples:
- Field/Collection Blanks: An empty collection vessel or a swab exposed to the air during sampling.
- Equipment Blanks: A swab of the PPE or surfaces the sample may contact.
- Reagent Blanks: An aliquot of the preservation or extraction solution.
- Extraction Blanks: A tube containing only water taken through the DNA extraction process. These controls are essential for identifying the source and extent of contamination and for informing downstream bioinformatic filtering [90].
Protocol for Establishing a Rapid RPA-LFD Assay
This protocol outlines the development of a rapid, contamination-aware isothermal amplification assay, as demonstrated for duck adenovirus type-3 (DAdV-3) [47].
- Primer and Probe Design: Design RPA primers targeting a conserved region of the pathogen's genome (e.g., the fiber-2 gene for DAdV-3). The 5' ends of the forward and reverse primers must be labeled with FITC and biotin, respectively [47].
- Assay Assembly and Optimization:
- Prepare the RPA reaction mix according to the manufacturer's instructions, incorporating the labeled primers.
- Perform amplification at a constant temperature of 37–42°C for 20–40 minutes. No thermal cycler is required.
- Optimize reaction conditions (time, temperature, primer concentration) using a positive control of known concentration and include a no-template control (NTC) to check for contamination or non-specific amplification.
- Lateral Flow Detection:
- Dilute the RPA amplification product and apply it to the sample pad of the lateral flow dipstick (LFD).
- The LFD contains: i) fluorescent microspheres conjugated to an anti-FITC antibody on the conjugate pad, ii) streptavidin at the test line (T), and iii) an anti-species antibody at the control line (C).
- As the product migrates, double-labeled amplicons are captured at the T line, producing a visible signal. The appearance of only the C line indicates a negative result. The simultaneous appearance of both T and C lines indicates a positive result [47].
- Validation: Test the assay's specificity against a panel of related but distinct pathogens (e.g., other avian viruses) and determine its limit of detection using serial dilutions of the target. Finally, validate its clinical performance against a gold-standard method like qPCR [47].
Diagram 1: RPA-LFD assay workflow for rapid pathogen detection.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table lists key reagents and materials critical for preventing contamination and ensuring the integrity of amplification experiments.
Table 4: Essential Reagents and Materials for Contamination Control
| Tool/Reagent | Key Function & Rationale | Application Notes |
|---|---|---|
| Ultrapure Nuclease-Free Water (Resistivity 18.2 MΩ·cm, TOC <5 ppb) [85] | Provides a contamination-free reaction medium; removes nucleases, ions, and organics that inhibit polymerase. | Use for preparing all buffers, reagents, and dilutions. A final UF polishing step is recommended [85]. |
| Single-Use, DNA-Free Plasticware | Prevents introduction of contaminants from manufacturing or cross-contamination between uses. | Use pre-sterilized, PCR-certified tubes and tips. |
| Ultrapure Nucleotides (dNTPs) | Ensures optimal base incorporation; contaminants can directly inhibit polymerase. | Use high-quality, nuclease-free dNTPs at a balanced concentration. |
| Contamination-Control Additives | Can help overcome residual inhibitors in complex samples (e.g., soil, blood). | BSA, betaine, or proprietary commercial blends. |
| Advanced Primer Systems (e.g., Co-Primers) | Novel architecture reduces primer-dimer propagation, enhancing specificity in multiplex reactions [86]. | Ideal for complex assays where non-specific amplification is a persistent issue [86]. |
| Sterile Connectors & Closed Systems | Minimizes bioburden and environmental contamination during fluid transfer in ATMP manufacturing [92]. | Critical for aseptic processing in therapeutic production; encouraged by Annex 1 [92]. |
The fidelity of nucleic acid amplification is inextricably linked to the purity of the template and reaction environment. Contaminants—ranging from ionic impurities and nucleases to foreign DNA and organic compounds—pose a constant threat to assay specificity and sensitivity by directly inhibiting enzymes, degrading templates, and promoting non-specific artifacts like primer-dimers. Addressing these challenges requires a holistic strategy that integrates a deep understanding of inhibitory mechanisms, rigorous experimental protocols for contamination prevention, and the adoption of advanced reagents and technologies designed to safeguard reaction integrity. For scientists and drug developers, mastering template quality is not a peripheral task but a central pillar of producing reliable, reproducible, and meaningful molecular data.
High-Resolution Melting (HRM) analysis is a powerful, post-PCR, closed-tube method for identifying genetic variations and assessing amplicon purity by analyzing the thermal denaturation behavior of double-stranded DNA [93] [94]. Its exceptional sensitivity to sequence variations—down to a single base pair—makes it an indispensable tool for differentiating specific PCR products from nonspecific artifacts like primer dimers, which is a critical challenge in primer dimer and nonspecific amplification research [93] [95]. When optimized, HRM provides a more reproducible and less time-consuming alternative to older mutation scanning technologies such as denaturing HPLC (dHPLC) and denaturing gradient gel electrophoresis (DGGE) [93]. The technique relies on three key technological advancements: saturating DNA dyes, precise temperature-control instrumentation, and sophisticated analysis software that together enable the detection of subtle melt curve differences invisible to classical melt curve analysis [94] [96]. This guide details the experimental protocols and analytical frameworks for leveraging HRM to ensure amplicon specificity, a cornerstone of reliable genetic data.
Fundamental Principles of HRM for Specificity Assessment
How HRM Distinguishes Sequence Variations
DNA melting is a fundamental physical process where double-stranded DNA denatures into single strands as temperature increases. In HRM, this process is monitored in real-time using fluorescent dyes that bind preferentially to double-stranded DNA [93]. The resulting melt curve, which plots fluorescence against temperature, serves as a unique fingerprint for the amplicon. The shape and position of this curve are determined by the amplicon's nucleotide sequence, length, GC content, and strand complementarity [95]. A specific, pure PCR product will typically melt as a single, coherent domain, producing a smooth, predictable curve. In contrast, a reaction contaminated with nonspecific artifacts like primer dimers will contain multiple DNA species with different sequences and lengths, each with its own melting characteristic. This mixture results in a complex, atypical melt curve whose shape differs noticeably from that of the pure product [97]. This ability to detect heterogeneity is the foundation for using HRM in artifact identification.
HRM vs. Classical Melt Curve Analysis
While classical melt curve analysis can flag the presence of gross nonspecific amplification, HRM provides a superior level of detail and information, making it the preferred method for rigorous specificity assessment and genotyping [94].
Table: Comparison of Classical Melt Curve and HRM Analysis
| Feature | Classical Melt Curve Analysis | High-Resolution Melting (HRM) Analysis |
|---|---|---|
| Temperature Resolution | Monitors differences >1 °C [94] | Monitors differences <0.5 °C [94] |
| Data Point Density | Moderate number of data points per °C [94] | High density of data points per °C [94] |
| Primary Application | Finding nonspecific amplicons (e.g., primer dimers) [94] | Identification of DNA differences (genotyping, methylation) and detailed specificity analysis [94] |
| Instrument & Chemistry | Can be done with any real-time cycler with nonsaturating dyes (e.g., SYBR Green) [94] | Requires special instruments and optimized chemistry with saturating dyes (e.g., LCGreen, EvaGreen) [94] [96] |
| Heteroduplex Detection | Poor ability to detect heteroduplexes formed by heterozygous variants or mixtures [96] | High sensitivity for detecting heteroduplexes, enabling identification of heterozygotes and impure samples [96] |
The following diagram illustrates the core workflow and decision-making process for using HRM to differentiate specific products from artifacts:
Experimental Design for Optimal Specificity
PCR Primer Design Guidelines
Careful primer design is the first and most critical step in minimizing artifacts and ensuring successful HRM analysis. Primers must be designed to amplify a single, specific product and to avoid interactions that lead to primer-dimer formation [93].
Table: PCR Primer Design Guidelines for HRM
| Parameter | Recommendation | Rationale |
|---|---|---|
| Amplicon Size | 70–350 bp, ideally 70–150 bp for SNP analysis [93] [97] | Smaller amplicons maximize the melting temperature (Tm) shift from a single base change. Products <70 bp risk co-melting with primer dimers [97]. |
| Primer Length | ~20 nucleotides [93] | Standard length for providing sufficient specificity. |
| Melting Temperature (Tm) | 58–62°C, optimal 60°C [93] | Ensures efficient and specific annealing during PCR. |
| Forward/Reverse Primer Tm Difference | <2°C [93] | Promotes balanced amplification and reduces mispriming. |
| GC Content | 30–80% [93] | Avoids extremes that can hinder amplification or create complex melt domains. |
| GC Clamp | Maximum of 2 G or C nucleotides in the last 5 nt at 3' end [93] | Reduces mispriming and stabilizes the primer-template hybrid. |
| Complementarity | Avoid complementarity within or between primers [93] | Minimizes primer-dimer and hairpin formation. |
| Genomic Coverage | Design overlapping amplicons (25 bp overlap) to avoid missing variants [93] | Ensures complete coverage of the genomic region of interest. |
Template DNA and Reaction Component Quality
The sensitivity of HRM means that inconsistent DNA quality or reaction components can lead to irreproducible melt curves and false positives in artifact detection.
- DNA Quality and Quantity: Use a standardized DNA isolation method (e.g., QIAGEN kits) to minimize contaminant carryover like salts or alcohols, which can affect Tm [93] [97]. Normalize all DNA samples to the same concentration (e.g., 1–50 ng/μL for genomic DNA) using the same dilution buffer [93] [97]. Using comparable amounts of DNA ensures CT values differ by no more than three cycles, promoting uniform amplification [97].
- PCR Reagents: Use a master mix specifically formulated for HRM, such as the MeltDoctor HRM Master Mix or Type-it HRM PCR Kit [93] [97]. These kits contain saturating dyes (e.g., MeltDoctor HRM dye, a stabilized SYTO 9) and hot-start DNA polymerase, which minimize nonspecific amplification and primer-dimer formation from the outset [93]. It is recommended to use 200 nM of each PCR primer in a 20 μL reaction [93].
Detailed HRM Experimental Protocol
Step-by-Step Workflow
This protocol outlines the process from PCR setup to data analysis for specificity screening.
PCR Setup
- Prepare a master mix containing all HRM-optimized reagents, primers, and water. Aliquot the master mix into the reaction wells.
- Add a consistent mass of template DNA (e.g., 20 ng) to each reaction [93]. Always include a no-template control (NTC) to detect contamination and primer-dimer formation, and a known wild-type control for curve comparison [93] [97].
- Run the PCR for 40 cycles to ensure all reactions reach the plateau phase, generating comparable amounts of product for a reliable melt analysis [93] [97].
High-Resolution Melting
- After amplification, the plate is transferred to the HRM instrument (if a separate instrument is used).
- The protocol involves a gradual denaturation of the amplified products. The instrument heats the samples with a very fine temperature increment (e.g., 0.1–0.2°C) while continuously collecting fluorescence data [94]. This high data density is key to resolving subtle curve differences.
Data Analysis
- The instrument software first normalizes the raw fluorescence data, setting pre-melt fluorescence to 100% and post-melt fluorescence to 0% [93] [95].
- Analyze the normalized melt curves and their derivative plots (the negative derivative of fluorescence over temperature, -dF/dT). A single, sharp peak in the derivative plot indicates a single, pure PCR product. Multiple peaks or a broad, shifted peak suggest a mixture of products or the presence of artifacts [97].
- Use the software's difference plot, which displays the fluorescence difference between each sample and a reference (usually wild-type), to easily cluster samples with identical melt curves and flag outliers caused by artifacts or sequence variants [93].
The Scientist's Toolkit: Essential Research Reagents
Table: Key Reagent Solutions for HRM Specificity Analysis
| Item | Function | Examples & Notes |
|---|---|---|
| HRM-Optimized Master Mix | Provides saturating dye, hot-start polymerase, dNTPs, and optimized buffer in a single solution for robust, specific amplification. | MeltDoctor HRM Master Mix [93], Type-it HRM PCR Kit [97]. Simplifies setup and ensures component compatibility. |
| Saturating DNA Dye | Binds dsDNA without inhibiting PCR and without dye redistribution during melting, which is critical for high-resolution data. | MeltDoctor HRM dye (SYTO 9) [93], LCGreen Plus [96], EvaGreen [94]. Preferable to SYBR Green I for heteroduplex detection [96]. |
| High-Quality DNA Purification Kit | Isulates pure, consistent template DNA free of contaminants (salts, alcohols) that can alter melting temperatures. | QIAamp or DNeasy Kits [97]. Standardization of the extraction method across all samples is crucial. |
| Optimized PCR Primers | Amplify the specific target of interest with high efficiency while minimizing primer-dimer and nonspecific amplification. | Designed with ~20 nt length, 60°C Tm, and checked for homodimers/heterodimers [93]. |
| Positive Control DNA | Serves as a known reference for a specific amplicon's melt profile, essential for identifying aberrant curves. | A validated wild-type DNA sample for the target region [93] [97]. |
The following workflow diagram summarizes the entire HRM process, from preparation to final analysis, highlighting critical control points:
Data Interpretation and Troubleshooting
Identifying Primer Dimers and Nonspecific Products
The primary method for identifying artifacts is the visual inspection of normalized and derivative melt curves.
- Primer Dimers: Typically appear as low-temperature melt peaks, often below 80°C, due to their short length and low Tm. The no-template control (NTC) is vital here; a peak in the NTC confirms primer-dimer formation. If the same low-temperature peak appears in samples, it indicates co-amplification of primer dimers alongside the specific product [97].
- Nonspecific Products: Appear as additional, unexpected peaks in the derivative plot or cause a broadening and distortion of the primary melt curve. Complex melt curves with multiple domains can also be caused by very long amplicons or sequences with biased GC/AT distribution, underscoring the importance of proper amplicon design [97].
Troubleshooting Common HRM Issues
Even with careful design, issues can arise. The following logic tree helps diagnose and solve common problems related to specificity and data quality:
High-Resolution Melting analysis transcends its role as a simple genotyping tool, emerging as a critical methodology for diagnosing PCR fidelity within the broader context of primer dimer and nonspecific amplification research. By integrating rigorous experimental design—featuring optimized primers and controlled template quality—with a detailed understanding of melt curve interpretation, researchers can confidently differentiate specific amplicons from confounding artifacts. The structured protocols, reagent guidelines, and troubleshooting logic provided in this technical guide establish a reliable framework for implementing HRM as a robust, closed-tube quality control step. This application not only streamlines the workflow by eliminating the need for gel electrophoresis but also enhances the accuracy and reproducibility of genetic analyses, from mutation scanning prior to sequencing to the precise characterization of genomic variants.
Ensuring Assay Robustness: Validation, Comparative Analysis, and Emerging Computational Tools
Quantitative PCR (qPCR) is a cornerstone of modern molecular biology, yet its reliability is entirely dependent on rigorous assay validation. Within the broader context of investigating primer dimers and nonspecific amplification, establishing clear benchmarks for key parameters is not merely a recommendation but a fundamental requirement for data integrity. This technical guide provides an in-depth examination of the core validation parameters—amplification efficiency, correlation coefficient (R²), and dynamic range—detailing their theoretical basis, acceptable benchmarks, and step-by-step experimental determination. By framing these technical benchmarks as essential defenses against artifacts, this whitepaper equips researchers and drug development professionals with the protocols and criteria necessary to ensure the reproducibility and accuracy of their qPCR results.
The exquisite sensitivity and specificity of qPCR make it uniquely powerful, but these same attributes also render it vulnerable to subtle imperfections in assay design and execution. Amplification artifacts, such as primer dimers and off-target products, are a pervasive threat to data credibility, leading to false positives and inaccurate quantification [15]. The occurrence of these artifacts is not random; it is systematically influenced by reaction conditions, including the delicate balance between template, non-template, and primer concentrations [15].
Therefore, the process of assay validation is fundamentally about establishing a controlled operational window where the desired specific product is amplified reliably and efficiently, while the formation of artifacts is suppressed. The key parameters of amplification efficiency, the correlation coefficient (R²), and linear dynamic range serve as the primary indicators of this optimal performance. Compliance with the MIQE guidelines ensures that all aspects of the qPCR experiment, from sample preparation to data analysis, are documented and standardized, thereby promoting reproducibility and transparency [98] [99]. This guide delves into the establishment of these critical benchmarks, providing a rigorous framework for validation that is essential for both fundamental research and clinical diagnostic applications.
Theoretical Foundations of Key qPCR Parameters
Amplification Efficiency
Amplification efficiency (E) is a measure of how effectively a qPCR assay duplicates the target DNA during each cycle of the amplification process. The ideal, or 100% efficient, reaction results in a perfect doubling of the target amplicon with every cycle, corresponding to a twofold increase [100]. Efficiency is intrinsically linked to the slope of the standard curve generated from a serial dilution series. The relationship is defined by the equation: E = [10^(-1/slope)] - 1 [84] [100].
An ideal efficiency of 100% (E=1.0) corresponds to a slope of -3.32. Deviations from this ideal value have direct implications for data accuracy, particularly when using relative quantification models that assume optimal efficiency. Efficiencies significantly below 90% suggest issues such as poor primer design, suboptimal reaction conditions, or the presence of inhibitors that prevent the polymerase from functioning optimally [101]. Conversely, efficiencies consistently exceeding 110% often indicate the presence of polymerase inhibitors in concentrated samples, pipetting errors, or the amplification of nonspecific products like primer dimers, especially when using intercalating dyes [84]. In such cases, the apparent higher efficiency is an artifact; inhibitors in the more concentrated samples cause a delay in the Cq value, flattening the standard curve slope and inflating the calculated efficiency [84].
Correlation Coefficient (R²)
The correlation coefficient (R²) is a statistical measure of the linearity of the standard curve. It quantifies how well the data points of the serial dilutions fit a straight line. An R² value close to 1.0 (typically ≥0.985-0.990) indicates a strong, linear relationship between the logarithm of the template concentration and the resulting Cq value across the dilution series [102] [101]. A high R² value is a prerequisite for confident quantification, as it confirms that the assay responds predictably to changes in template input. Values below the accepted threshold suggest technical issues such as inaccurate pipetting during dilution series preparation, sample degradation, or inconsistent reaction performance across the dynamic range.
Linear Dynamic Range
The linear dynamic range is the span of template concentrations over which the qPCR assay can detect and quantify the target with consistent accuracy and precision [102]. Within this range, the Cq values maintain a linear relationship with the log of the starting concentration. The lower limit of this range is defined by the limit of detection (LOD), while the upper limit is often constrained by the presence of polymerase inhibitors in highly concentrated samples [84] [102]. A well-optimized assay should have a broad dynamic range, typically spanning 6 to 8 orders of magnitude (e.g., from 10^1 to 10^7 copies), allowing for the accurate quantification of targets that vary widely in abundance within a single run [102]. Establishing this range is critical to ensure that unknown samples fall within the quantitative scope of the assay.
Establishing and Validating Benchmarks
The following section provides a detailed experimental roadmap for determining the critical validation parameters for any qPCR assay.
Experimental Protocol for a Standard Dilution Series
A serial dilution of a known template is the foundational experiment for determining efficiency, R², and dynamic range.
- Template Preparation: The template can be a PCR product of the target gene (purified and diluted to approximately 0.01 ng/μL), a synthetic oligonucleotide, or cDNA derived from a sample with known high expression of the target [101].
- Serial Dilution: Prepare a minimum of five, and preferably more, serial dilutions. Standard dilution factors are 2-fold, 5-fold, or 10-fold [100] [101]. A 10-fold dilution series is most common and is recommended to span 6 to 8 orders of magnitude to adequately define the dynamic range [102].
- qPCR Run: Analyze each dilution in triplicate using the qPCR assay to be validated. The reaction conditions (primer concentration, annealing temperature, etc.) should be identical to those planned for experimental samples.
- Data Analysis:
- Plot the mean Cq value for each dilution against the logarithm of its concentration (or relative dilution factor).
- Generate a linear regression trendline through the data points.
- Record the slope and correlation coefficient (R²) of this line.
- Calculate the amplification efficiency using the formula: E = [10^(-1/slope)] - 1 [100].
Benchmark Criteria and Troubleshooting
The table below summarizes the target benchmarks for a validated qPCR assay and outlines common causes for failure.
Table 1: Validation Benchmarks and Troubleshooting Guide for qPCR Assays
| Parameter | Target Benchmark | Common Causes of Deviation | Troubleshooting Strategies |
|---|---|---|---|
| Amplification Efficiency | 90–110% [101] (Ideal: 100%, slope = -3.32 [100]) | Low Efficiency (<90%): Poor primer design, primer-dimer formation, suboptimal Mg²⁺ concentration, reaction inhibitors [84].High Efficiency (>110%): PCR inhibitors in concentrated samples, inaccurate dilution series, nonspecific amplification [84]. | Redesign primers following MIQE principles [103]. Optimize annealing temperature and Mg²⁺ concentration. Dilute sample to reduce inhibitor effects [84]. |
| Correlation Coefficient (R²) | ≥ 0.985 – 0.990 [102] [101] | Imprecise pipetting during dilution series, degraded or variable quality template, inconsistent reaction performance. | Practice precise pipetting techniques. Check template purity (A260/280 ratios: ~1.8 for DNA, ~2.0 for RNA) [84]. Ensure homogeneous reaction mix. |
| Linear Dynamic Range | 6–8 orders of magnitude [102] | Upper Limit: Polymerase inhibition from high template concentration [84].Lower Limit: Stochastic effects from very low template concentration. | Exclude the most concentrated and most diluted points from the standard curve if they show signs of inhibition or high variability [84]. |
Successful qPCR validation relies on a suite of key reagents and bioinformatic tools. The following table details these essential components.
Table 2: Key Research Reagent Solutions and Resources for qPCR Validation
| Item | Function / Description | Example Use in Validation |
|---|---|---|
| High-Quality Polymerase | Hot-start enzymes minimize primer-dimer formation and nonspecific amplification during reaction setup [15]. | Essential for all validation and experimental runs to ensure specificity, especially in low-template samples. |
| SYBR Green I Master Mix | Intercalating dye for detection of double-stranded DNA. Requires post-run melt curve analysis to verify specificity. | Used in validation to check for primer-dimer and nonspecific products that can inflate efficiency calculations [15] [84]. |
| TaqMan Probes | Sequence-specific fluorescent probes that hydrolyze during amplification, providing superior specificity over intercalating dyes. | Reduces risk of false positives from primer-dimers; requires careful probe design to avoid self-dimers [104]. |
| Spectrophotometer/Nanodrop | Measures nucleic acid concentration and purity (A260/280 ratio). | Critical step to assess template quality before creating a dilution series; poor purity can indicate inhibitors [84]. |
| OligoAnalyzer Tool | In-silico software for analyzing primer properties (Tm, hairpins, self-dimers, hetero-dimers) [15]. | Used during primer design to check for stable dimers (ΔG ≤ -9 kcal/mol recommended) and secondary structures [15]. |
| Primer-BLAST | NCBI tool for designing and checking the target specificity of primer pairs across the genome. | Prevents amplification of off-target genomic sequences, a key step in ensuring assay specificity [15]. |
The Critical Link Between Validation and Primer Dimers
The pursuit of robust validation benchmarks is intrinsically linked to the mitigation of primer dimers and nonspecific amplification. Primer dimers are not merely a nuisance; they are a significant source of artifact-associated fluorescence that can lead to false positive results and dramatically skew quantification, particularly in samples with low target abundance [15] [104].
The formation of these artifacts is highly dependent on reaction conditions. As demonstrated in troubleshooting experiments, the frequency of nonspecific amplification is determined by factors such as primer concentration, cDNA input, and annealing temperature [15]. Furthermore, practical workflow factors like the time taken for pipetting a qPCR plate can significantly impact artifact formation, likely due to the partial failure of hot-start procedures during extended bench times [15]. This underscores the need for standardized and efficient laboratory practices.
Validation experiments, particularly the efficiency and dynamic range tests, serve as a critical diagnostic for these issues. Anomalous results, such as efficiency far exceeding 110% or a narrow dynamic range, can be the first indicator of artifact amplification. The use of a post-elosion heating step (measuring fluorescence at a temperature above the Tm of primer dimers) can prevent the quantification of artifact-associated fluorescence, ensuring that the reported Cq values reflect only the specific product [15]. This approach, combined with rigorous in-silico checks for dimer formation using tools like OligoAnalyzer, forms a comprehensive strategy to safeguard data integrity against these common artifacts [15] [104].
Establishing and adhering to strict validation benchmarks for qPCR is a non-negotiable practice for generating credible and reproducible scientific data. The parameters of amplification efficiency (90-110%), correlation coefficient (R² ≥ 0.985), and linear dynamic range (6-8 orders of magnitude) are not arbitrary targets but are empirically derived indicators of a robust, specific, and reliable assay. As explored in the context of primer-dimer research, these validation steps are our primary defense against the pervasive threats of nonspecific amplification and artifacts. By integrating the experimental protocols and troubleshooting strategies outlined in this guide, and by consistently following the MIQE guidelines for reporting, researchers and drug development professionals can ensure that their qPCR data meets the highest standards of rigor, thereby upholding the integrity of their conclusions in both research and clinical applications.
Visual Appendix
Workflow for qPCR Assay Validation and Troubleshooting
This diagram illustrates the comprehensive workflow for validating a qPCR assay, from initial primer design through to final data interpretation and troubleshooting, highlighting the iterative nature of the optimization process.
In silico validation represents a critical first step in the development of robust polymerase chain reaction (PCR) diagnostics, enabling researchers to computationally assess primer specificity and sensitivity before costly wet-lab experiments. This process involves checking designed primers and probes against extensive nucleotide databases to ensure they amplify only the intended targets. The exponential growth of available genetic sequence data, driven by high-throughput sequencing technologies, has dramatically increased the power and necessity of in silico methods [105]. Pathogens exhibit substantial genetic variation due to genetic drift, adaptation, and evolution, making it essential to regularly re-evaluate PCR tests against newly discovered variants to prevent false negatives arising from primer-target mismatches or false positives from cross-reactivity with similar sequences [105].
Within the context of primer dimer and nonspecific amplification research, in silico validation serves as the foundational methodology for identifying potential sources of amplification artifacts during the initial design phase. The core challenge in PCR diagnostics lies in designing tests that detect all variants of a target pathogen while excluding closely related non-target organisms [105]. In silico validation addresses this challenge through a two-pronged approach: determining in silico sensitivity (the ability to detect all target variants) and in silico specificity (the selective detection only of the target group) [105]. As genetic databases continue to expand exponentially, computational validation has become both more informative and more computationally intensive, necessitating sophisticated tools and standardized methodologies.
Theoretical Foundation: Specificity Challenges in PCR
Nonspecific amplification in PCR manifests primarily as primer-dimers and off-target products, both of which compromise assay accuracy and reliability. Primer-dimers are short artifacts resulting from homology between primer sequences, while off-target products are longer amplicons containing sequences that only partially overlap with the targeted region [15]. The occurrence of these PCR artifacts depends critically on reaction conditions, including template concentration, non-template concentration, and primer concentration [15]. Even with validated assays, titration experiments have demonstrated that low template concentrations increase artifact frequency, while low non-template concentrations lead to deviating quantification cycle (Cq) values and incorrect quantification [15].
The propagation of nonspecific products presents particularly serious challenges in quantitative applications. Research has shown that conventional primers with hot-start methods can experience signal dampening with as few as 60 primer-dimers and false negatives with only 600 primer-dimers [46]. These artifacts result in false positives and fundamentally question the interpretation of dilution series where both template and non-template concentrations decrease simultaneously [15]. Even minimal amounts of nonspecific amplification can significantly impact diagnostic results in sensitive applications such as clinical pathogen detection or low-abundance gene expression analysis.
The Mismatch Tolerance Problem
A fundamental challenge in primer design lies in the inherent mismatch tolerance of DNA polymerase enzymes during amplification. Studies investigating the effects of mismatches between targets and primers have consistently demonstrated that targets can be amplified even with several mismatches to the primers [106]. While the precise relationship varies between studies, the consensus indicates that a two-base mismatch at the 3' end generally prevents amplification, whereas a single base mismatch (even at the very 3' end), as well as multiple mismatches in the middle or toward the 5' end, still allows amplification, though potentially with reduced efficiency [106].
This mismatch tolerance creates a significant challenge for specificity validation, as primers must be checked not only for perfect matches but also for partial matches that might still yield amplifiable products. The variable effects of mismatches depending on their position and composition necessitate computational tools capable of detecting several mismatches across the entire primer range while offering flexibility in specificity stringency settings [106].
Core Computational Methodologies
BLAST-Based Specificity Analysis
The Basic Local Alignment Search Tool (BLAST) serves as a fundamental algorithm for comparing primer sequences against nucleotide databases, but its application for primer validation requires specific considerations. Standard BLAST uses a local alignment algorithm that does not necessarily return complete match information between the primer and target, particularly when matches are imperfect toward the primer ends [106]. This limitation led to the development of Primer-BLAST, which combines BLAST with a global alignment algorithm to ensure complete primer-target alignment across the entire primer sequence [106].
Primer-BLAST implements sensitive search parameters capable of detecting targets with up to 35% mismatches to the primer sequence, significantly beyond the capabilities of index-based search methods [106]. The tool uses an expect value (E-value) cutoff of 30,000 for primer-only searches, which is 3,000 times higher than standard BLAST defaults, to ensure detection of potentially amplifiable targets with significant mismatch numbers [106]. For template-based primer design, the E-value threshold is automatically adjusted even higher to maintain sensitivity while searching with the entire template sequence [106].
Table 1: Key Parameters for BLAST-Based Specificity Checking
| Parameter | Default Setting | Impact on Specificity |
|---|---|---|
| Expect value (E-value) | 30,000 (primer-only) | Higher values increase sensitivity for mismatched targets |
| Max mismatch percentage | 35% | Determines how divergent targets will be detected |
| 3' end constraints | User-definable | Critical due to 3' end sensitivity in amplification |
| Word size | Adjusted for sensitivity | Smaller values increase sensitivity at computational cost |
Multiple Sequence Alignment for Conservation Analysis
Multiple Sequence Alignment (MSA) serves as the cornerstone for evaluating in silico sensitivity by determining primer binding site conservation across target variants. The process begins by downloading all available sequences of a target organism from databases like NCBI using taxonomy ID numbers [105]. A reference sequence (typically a full genome or large representative sequence containing the PCR target) is selected, and each downloaded sequence is aligned to this reference using pairwise alignment tools like ClustalW [105].
To address orientation errors in database sequences, alignments are performed in both forward and reverse complement orientations, with the orientation yielding the highest alignment score selected [105]. The scoring scheme typically awards +1 for matches, -2 for mismatches, -3 for point deletions or gaps, and -2 for every next adjacent point deletion [105]. For efficient handling of large sequences, the workflow often involves segmenting sequences into fragments of 10,000 nucleotides, aligning them individually, and then combining the results into a comprehensive MSA [105].
The regions corresponding to primers and probes are extracted from the complete MSA to construct conservation plots sorted by decreasing total mismatches [105]. The in silico sensitivity is quantitatively expressed as the percentage of hits with a cutoff value of typically one maximum mismatch per primer or probe [105]. This approach provides researchers with a quantitative assessment of how many known variants their assay will detect, enabling informed decisions about primer selection for either broad detection or specific variant targeting.
Integrated Workflow for Comprehensive Validation
A robust in silico validation protocol integrates both specificity and sensitivity analyses through a structured workflow. The software tool PCRv exemplifies this integrated approach by coordinating the use of ClustalW for MSA generation and SSEARCH for alignment searches against comprehensive nucleotide databases [105]. The process includes adding Flagged Internal Control Sequences (FICS)—randomly generated sequences containing primer and probe sequences in all possible combinations and orientations with systematically introduced mismatches (0-10 per primer) [105]. These controls enable validation of the alignment search itself and monitoring of pipeline performance.
The following diagram illustrates the complete in silico validation workflow, integrating both sensitivity and specificity analyses:
Experimental Protocols and Implementation
Primer-BLAST Specificity Checking Protocol
Objective: To determine the target-specificity of primer pairs using NCBI's Primer-BLAST tool.
Materials Needed:
- Primer sequences (forward and reverse)
- Target template sequence (accession number or FASTA format)
- Computer with internet access
Procedure:
- Access the Primer-BLAST tool at the NCBI website (https://www.ncbi.nlm.nih.gov/tools/primer-blast/).
- Input your primer sequences in the respective fields:
- Enter the forward primer sequence (5'→3' on plus strand)
- Enter the reverse primer sequence (5'→3' on minus strand)
- Always enter primer sequence only without additional characters [34].
- Under "Primer Pair Specificity Checking Parameters," select the appropriate database:
- RefSeq mRNA for transcript-specific applications
- RefSeq representative genomes for genomic applications
- core_nt for faster searches excluding eukaryotic chromosomal sequences
- Custom database for user-provided sequences [34]
- Specify the target organism to limit specificity checking and improve search speed [34].
- Adjust advanced parameters as needed:
- Set "Max. target size" to control amplicon length consideration
- Adjust "Number of mismatches" to define specificity threshold
- Modify "Primer specificity stringency" for match requirements [34]
- Submit the search and analyze results:
- Examine the predicted amplification products
- Verify intended target is present
- Check for unintended targets with permissible mismatches
- Review primer positioning relative to exon-intron boundaries if applicable
Interpretation: A specific primer pair should generate a single amplicon corresponding to your intended target. Any additional amplification products indicate potential cross-reactivity and require primer redesign or further experimental validation [106].
Multiple Sequence Alignment Sensitivity Analysis Protocol
Objective: To evaluate primer binding site conservation across genetic variants using MSA.
Materials Needed:
- Reference template sequence
- Taxonomy ID of target organism
- Computing resources (local or cloud-based)
- Software: ClustalW2.1, SSEARCH (as implemented in tools like PCRv)
Procedure:
- Download all sequences for the target organism from NCBI using the taxonomy ID number [105].
- Select an appropriate reference sequence containing the full PCR target region.
- Perform pairwise alignment of each downloaded sequence to the reference using ClustalW:
- Calculate alignment scores for both orientations
- Select the orientation with the highest alignment score
- Use scoring: match (+1), mismatch (-2), gap opening (-3), gap extension (-2) [105]
- Combine individual pairwise alignments into a comprehensive MSA.
- Extract regions corresponding to primer and probe binding sites from the MSA.
- Construct a conservation plot sorted by decreasing total mismatches.
- Calculate in silico sensitivity as the percentage of sequence hits with a maximum of one mismatch per primer or probe [105].
Interpretation: High sensitivity percentages (>90%) indicate broad detection capability across variants. Lower percentages suggest limited detection of diverse strains and may necessitate primer redesign or multiple primer sets for comprehensive coverage.
In Silico PCR Amplification Simulation
Objective: To predict amplification products from primer pairs across entire genomes or databases.
Materials Needed:
- Primer pairs to be tested
- Reference genome or database
- In silico PCR software (FastPCR, UCSC In-Silico PCR, or custom tools)
Procedure:
- Select an appropriate in silico PCR tool based on your needs:
- FastPCR for stand-alone analysis with mismatch tolerance
- UCSC In-Silico PCR for predefined genomes
- Electronic PCR for heuristic searches with limited mismatches [107]
- Input primer sequences and select the target database or genome.
- Set parameters for permissible mismatches and product size range.
- Execute the search and collect all predicted amplification products.
- Analyze results for:
- Expected product size and location
- Unexpected amplification products
- Potential primer-dimer formation
- Amplification efficiency predictions based on binding strength
Interpretation: The ideal result shows a single amplification product at the expected location. Multiple products indicate potential nonspecific amplification, while no products may suggest poor primer binding or insufficient template similarity.
Data Analysis and Interpretation
Specificity Thresholds and Mismatch Tolerance
Establishing appropriate specificity thresholds is critical for meaningful in silico validation results. Research indicates that amplification can still occur with certain mismatch patterns, particularly when mismatches are not concentrated at the 3' end [106]. The following table summarizes key evidence on mismatch tolerance and its implications for specificity thresholds:
Table 2: Experimentally Determined Mismatch Tolerance in PCR Amplification
| Mismatch Position | Amplification Outcome | Specificity Implications |
|---|---|---|
| 2 bases at 3' end | Generally prevents amplification | Critical region for specificity |
| Single base at 3' end | Allows amplification (reduced efficiency) | Permissive but concerning |
| Multiple mismatches in middle | Allows amplification | Significant cross-reactivity risk |
| Multiple mismatches at 5' end | Allows amplification | Moderate cross-reactivity risk |
| 35% overall mismatch | Detection limit of sensitive BLAST | Upper bound for concern |
Based on this evidence, recommended specificity checking should require at least 3 mismatches distributed across the primer sequence, with particular emphasis on ensuring at least one mismatch in the last 5 bases at the 3' end for reliable exclusion of amplification [106].
Quantitative Assessment of Validation Results
The quantitative output from in silico validation pipelines enables data-driven decision making for primer selection and assay design. For sensitivity analysis, the percentage of target variants with ≤1 mismatch provides a clear metric for expected detection breadth [105]. Specificity can be quantified as the number of off-target amplification products meeting various mismatch thresholds, with emphasis on products having minimal 3' end mismatches that pose the greatest amplification risk [106].
Statistical analysis of Primer-BLAST results should consider:
- Hit distribution across different organisms and genomic regions
- Mismatch patterns with emphasis on 3' end positions
- Product size variation for off-target amplifications
- Sequence similarity in flanking regions that might affect amplification efficiency
The following diagram illustrates the decision process for interpreting in silico validation results, particularly through the Primer-BLAST analysis workflow:
Successful implementation of in silico validation requires access to specific computational tools, databases, and analytical resources. The following table details key components of the in silico validation toolkit:
Table 3: Essential Research Reagents and Resources for In Silico Validation
| Resource Category | Specific Tools/Databases | Primary Function | Key Features |
|---|---|---|---|
| Primer Design Tools | Primer-BLAST [106], Primer3 [106] | Target-specific primer design | Integration of design and specificity checking |
| Sequence Databases | NCBI nt, RefSeq mRNA [34], Custom databases | Specificity checking | Comprehensive reference sequences |
| Alignment Tools | ClustalW [105], SSEARCH [105] | Multiple sequence alignment | Conservation analysis across variants |
| In Silico PCR Tools | FastPCR [107], UCSC In-Silico PCR [107] | Amplification prediction | Genome-wide product mapping |
| Validation Pipelines | PCRv [105] | Automated validation | Integrated sensitivity/specificity analysis |
| Specialized Resources | SNP database, Exon-Intron boundaries [106] | Enhanced primer placement | Avoidance of problematic regions |
Advanced Applications and Future Directions
Emerging Technologies and Methodologies
The field of in silico validation continues to evolve with emerging technologies that address current limitations. Cooperative primers represent a significant advancement, demonstrating a 2.5 million-fold improvement in reducing nonspecific amplification compared to conventional primers with hot-start methods [46]. This technology successfully amplified 60 template copies with no signal dampening despite a background of 150,000,000 primer-dimers, where normal primers failed with only 600 primer-dimers [46].
Machine learning approaches are increasingly being integrated into prediction pipelines, with recent studies achieving R² values of 0.824 for quantitative predictions of experimental endpoints like EC3 values in skin sensitization tests [108]. These approaches combine traditional parameters (melting temperature, GC content) with novel descriptors derived from chemical structures and read-across concepts to improve prediction accuracy [108].
The development of comprehensive bioinformatics tools for in silico PCR analysis continues to advance, with newer implementations offering batch processing capabilities for large-scale primer screening and enhanced handling of complex amplification scenarios including multiplexed, nested, or tiling PCR [107]. These tools are particularly valuable for DNA fingerprinting applications and repeat sequence identification where non-specific amplification risks are elevated.
Regulatory Considerations and Quality Standards
As in silico methods gain prominence in diagnostic development, regulatory frameworks are evolving to establish standards for computational validation. Recent initiatives have focused on developing White Papers and guidelines for verification and validation of in silico models, particularly for regulatory submissions [109]. The Association for Molecular Pathology, Association for Pathology Informatics, and College of American Pathologists have jointly recommended approaches for using in silico data in next-generation sequencing bioinformatic pipeline validation [110].
Key considerations for regulatory acceptance include:
- Transparent documentation of algorithms and parameters
- Comprehensive database coverage for specificity checking
- Clear applicability domains defining model limitations
- Independent validation using appropriate test datasets
- Integration with experimental data confirming predictions
The ongoing development of standards for assessment of in silico models reflects growing recognition of their value in reducing animal testing, accelerating development timelines, and enabling more sophisticated experimental design [109] [108]. As these standards mature, in silico validation is poised to become an increasingly integral component of the molecular diagnostic development pipeline.
In molecular biology research, particularly within primer dimers and nonspecific amplification studies, the reliability of any polymerase chain reaction (PCR) or quantitative PCR (qPCR) assay hinges on rigorous experimental validation. Specificity—ensuring that an assay detects only the intended target—is a cornerstone of assay credibility. This process systematically challenges the assay with non-target genomes and complex sample matrices to confirm that it remains unaffected by homologous sequences, closely related species, or inhibitory substances commonly found in real-world samples. Such validation is crucial for producing publication-quality data and developing robust diagnostic tools, as cross-reactivity and matrix effects represent significant sources of error and false positives in amplification-based detection [111] [112]. This guide provides a comprehensive technical framework for designing and executing these critical validation experiments.
Core Principles of Assay Specificity and Robustness
The validation process is designed to address two primary challenges: specificity against non-target genomes and robustness in complex sample matrices.
- Specificity ensures that the primers and probes hybridize only to their intended target sequence. This requires testing against a panel of non-target organisms, especially those that are phylogenetically closely related to the target, to rule out cross-reactivity due to sequence homology [111] [112].
- Robustness confirms that the assay maintains its performance characteristics—including sensitivity, efficiency, and dynamic range—when applied to complex, heterogeneous samples like wastewater, soil, or clinical specimens. These matrices often contain PCR inhibitors such as humic acids, heavy metals, or complex polysaccharides that can compromise amplification [112] [113].
A failure to adequately address these aspects can lead to misinterpretation of results, ultimately undermining research conclusions or diagnostic decisions.
Experimental Protocols for Key Validation Steps
In Silico Specificity Analysis
Before any wet-lab work, comprehensive in silico analysis is essential to pre-screen assay designs for potential cross-reactivity.
- Procedure: The designed primer and probe sequences must be analyzed using bioinformatics tools.
- Use the NCBI BLAST suite (Basic Local Alignment Search Tool) to query the primers against entire nucleotide databases. This identifies any homologous regions in non-target species [17].
- Perform multiple sequence alignments (e.g., using MAFFT) of the target gene across a wide range of species, including close genetic relatives, to visualize conserved and variable regions. Primers should target variable regions that are unique to the organism of interest [112].
- Validation Criterion: The assay is considered specific in silico when the primer sequences show perfect or near-perfect complementarity only to the target sequence and exhibit significant mismatches (particularly at the 3' ends) with all non-target sequences [112] [17].
Testing Against a Panel of Non-Target Genomes
This wet-lab experiment provides the definitive proof of an assay's specificity.
- Procedure:
- Panel Selection: Assemble a panel of genomic DNA from non-target strains. The panel should include phylogenetically closely related species, species sharing the same ecological niche, and common contaminants. For example, an Acinetobacter baumannii assay was validated against 27 non-target strains to confirm 100% specificity [111].
- qPCR Run: Run the qPCR assay with each non-target genomic DNA sample as a template. Use the same reaction conditions and template concentrations (e.g., 10-100 ng/μL) as would be used for a positive target sample.
- Control Inclusion: Always include a no-template control (NTC) to detect contamination and a positive target control to confirm assay functionality [113].
- Validation Criterion: The assay passes if all non-target samples and the NTC yield no amplification signal or a significantly delayed cycle threshold (Cq) value that falls outside the range of positive results (e.g., > 10 Cq later than the positive control) [111] [113].
Table 1: Example Panel for Validating a Bacterial Assay
| Category | Example Organisms | Purpose of Inclusion |
|---|---|---|
| Close Relatives | A. pittii, A. nosocomialis | Test for cross-reactivity within genus |
| Distantly Related | Pseudomonas aeruginosa, E. coli | Test for general specificity |
| Environmental | Common soil or water bacteria | Test for false positives in sample matrix |
Sensitivity and Limit of Detection in Complex Matrices
Determining the Limit of Detection (LoD) in the presence of a complex matrix confirms the assay's practical sensitivity.
- Procedure:
- Sample Preparation: Spike a known, quantified amount of the target organism (e.g., from a cultured isolate) into a matrix-free buffer and into the complex matrix of interest (e.g., wastewater, soil extract, clinical sample). The target should be quantified using a reference method, such as culture-based counting or digital PCR [114].
- Serial Dilution: Create a serial dilution series of the spiked target in both the buffer and the complex matrix.
- qPCR Quantification: Run the qPCR assay on all dilution points with multiple replicates (at least 3-5).
- Data Analysis: Compare the standard curves generated from the buffer and the matrix. The LoD is the lowest concentration at which the target is detected in ≥95% of replicates [113] [114].
- Validation Criterion: A robust assay will show a similar LoD in both the buffer and the complex matrix. A significant increase (e.g., >10-fold) in the LoD in the matrix indicates strong inhibition, requiring further optimization or DNA purification [112] [114].
Table 2: Key Performance Metrics for qPCR Validation
| Performance Metric | Target Value | Interpretation |
|---|---|---|
| Amplification Efficiency | 90–105% | Optimal reaction kinetics |
| Linearity (R²) | > 0.980 | High predictability across dilution series |
| Intra-assay Variation (Repeatability) | Cq Standard Deviation < 0.5 | High precision between replicates |
| Inter-assay Variation (Reproducibility) | Cq Standard Deviation < 1.0 | Consistency across different runs |
| Limit of Detection (LoD) | Defined concentration | Lowest reliably detectable target level |
Control Strategies to Monitor for Contamination
The high sensitivity of qPCR makes it vulnerable to contamination, which can be mistaken for non-specific amplification.
- No-Template Controls (NTCs): These are reactions containing all assay components (master mix, primers, probe, water) except the template DNA. Amplification in the NTC indicates contamination of reagents or primer-dimer formation [113].
- No-Reverse-Transcription Controls (No-RT): For RNA targets, this control contains all components but omits the reverse transcriptase enzyme. Amplification indicates contamination with genomic DNA [113].
- Inhibition Controls: Spiking a known, low quantity of a control template (e.g., a synthetic gene) into each sample can monitor for PCR inhibition. A significant delay in the Cq of this internal control in a sample indicates the presence of inhibitors [113] [115].
Validation in Complex Environmental and Clinical Matrices
Applying the assay to real-world samples is the ultimate test of its robustness.
- Wastewater Samples: Wastewater is a complex matrix rich in PCR inhibitors. One study validated ARG assays using DNA extracted from activated sludge, river sediment, and agricultural soils, ensuring the protocols were effective in these challenging environments [112]. Pre-treatment steps or the use of inhibitor-resistant polymerases may be necessary.
- Clinical Samples: For clinical diagnostics, validation must include testing on a range of patient-derived samples (e.g., sputum, blood). The consolidated TaqMan qPCR assays were validated with "metagenomic DNA from complex environmental matrices" and clinical isolates to comprehensively assess performance [116].
- Digital PCR for Absolute Quantification: When utmost precision is required, digital PCR (dPCR) can be used for validation. dPCR provides absolute quantification without a standard curve and is more tolerant to inhibitors, making it an excellent orthogonal method to confirm qPCR results in complex samples [114].
The following workflow synthesizes the key experimental and validation stages.
The Scientist's Toolkit: Essential Reagents and Materials
Successful validation relies on high-quality, specific reagents and controls.
Table 3: Research Reagent Solutions for Validation Experiments
| Reagent / Material | Function in Validation | Technical Notes |
|---|---|---|
| High-Purity Oligonucleotides | PCR primers and probes for target amplification. | Ensure manufacturer uses cleanrooms to prevent template contamination during synthesis [113]. |
| gBlocks / Synthetic Controls | Defined, clonal DNA fragments used as positive controls and for standard curves. | Essential for determining absolute quantification and LoD without culturing live pathogens [116]. |
| Inhibitor-Resistant Master Mix | Enzyme blends designed to amplify in the presence of common PCR inhibitors. | Critical for robust performance in complex matrices like soil or wastewater [112] [113]. |
| Digital PCR System | Absolute nucleic acid quantification without a standard curve. | Used as an orthogonal method to confirm qPCR results and for ultra-sensitive LoD studies [114]. |
| No-Template Control (NTC) | Critical negative control to detect reagent contamination or primer-dimer formation. | Must be included in every run; amplification indicates a contamination event [113]. |
Rigorous experimental validation against non-target genomes and in complex sample matrices is not an optional step but a fundamental requirement for generating reliable, reproducible data in PCR-based research. By adhering to the structured protocols outlined in this guide—from comprehensive in silico analysis and controlled wet-lab testing to final validation in real-world matrices—researchers can confidently deploy assays that are specific, sensitive, and robust. This diligence is the bedrock upon which scientific credibility in the fields of molecular diagnostics, environmental monitoring, and primer-dimer research is built.
In molecular diagnostics and life sciences research, the accurate amplification and detection of nucleic acids are fundamental. However, techniques such as polymerase chain reaction (PCR) are persistently challenged by the formation of primer dimers and nonspecific amplification, which can severely compromise assay efficiency, sensitivity, and reliability [23]. Primer dimers are short, artifactual double-stranded DNA fragments formed when primers anneal to each other instead of the target DNA template, often due to complementary regions within the primers themselves, high primer concentrations, or suboptimal annealing temperatures [23]. These artifacts compete with the target for reaction reagents, leading to reduced amplification yield and inaccurate quantification, which is particularly critical in applications like pathogen detection, genotyping, and gene expression analysis [23] [70].
The pursuit of overcoming these limitations has driven the development and refinement of various nucleic acid amplification technologies. This whitepaper provides a comparative analysis of three prominent methods: quantitative PCR (qPCR), Recombinase Polymerase Amplification combined with Lateral Flow Dipstick (RPA-LFD), and digital PCR (dPCR). The analysis is framed within the context of a broader thesis on understanding and mitigating primer dimers and nonspecific amplification. It evaluates each technology's multiplexing capability, sensitivity, and suitability for different application scenarios, supported by current experimental data and protocols. The intended audience includes researchers, scientists, and drug development professionals who require a deep technical understanding to select the optimal method for their specific needs.
Core Principles and Comparative Analysis
The following table summarizes the key characteristics of qPCR, RPA-LFD, and dPCR, providing a high-level overview for initial comparison.
Table 1: Core Characteristics of qPCR, RPA-LFD, and dPCR
| Feature | qPCR (TaqMan Probe-Based) | RPA-LFD | Digital PCR (dPCR) |
|---|---|---|---|
| Fundamental Principle | End-point probe hydrolysis during thermal cycling | Isothermal amplification with visual immunochromatographic detection | End-point amplification of partitioned samples for absolute quantification |
| Amplification Temperature | Requires thermal cycling (e.g., 95°C denaturation, 60°C annealing/extension) [117] | Isothermal (37–42°C) [118] [47] [119] | Requires thermal cycling [117] |
| Typical Assay Duration | 1.5 to 2 hours [117] | 10 to 30 minutes [118] [47] [120] | ~92 minutes (including droplet generation and reading) [117] |
| Primary Readout | Fluorescence (FAM, VIC) measured in real-time (Ct value) [118] | Visual band on a lateral flow strip [47] [119] | Count of positive and negative partitions for absolute quantification [117] |
| Key Strength | Gold standard for quantification; high-throughput capability; proven multiplexing | Extreme speed and portability; minimal equipment needs | Exceptional sensitivity and precision; absolute quantification without standards |
| Key Limitation | Susceptible to PCR inhibitors; requires precise thermal cycler | Prone to nonspecific amplification; lower multiplexing potential [121] | Lower throughput; higher cost per sample; complex instrumentation |
Quantitative Data Comparison
Sensitivity, specificity, and multiplexing data from recent studies further illuminate the practical performance of these techniques.
Table 2: Comparative Performance Metrics from Recent Applications
| Application Context | Technology | Reported Sensitivity | Specificity | Multiplexing Capability Demonstrated |
|---|---|---|---|---|
| Fish Species Identification [118] | TaqMan qPCR (dual-target) | 12.5 - 125 copies/reaction | 100% (171/171 samples) | Yes, dual-channel (FAM/VIC) in a single tube |
| Duck Adenovirus (DAdV-3) Detection [47] [122] | RPA-LFD | 10 copies/μL | 100% (no cross-reactivity) | No (single-plex demonstrated) |
| Tilapia Lake Virus (TiLV) Detection [119] | RT-RPA-LFD | 3.19 copies/μL | 100% (clinical samples) | No (single-plex demonstrated) |
| Salmonella Detection [123] | RAA-LFD (RPA variant) | 10¹-10² CFU/mL | 96.88-100% (clinical samples) | Yes, dual-target for two Salmonella species |
| Gardnerella vaginalis Detection [117] | dPCR | 4.4 pg/μL | 100% (no cross-reactivity with 8 controls) | No (single-plex demonstrated) |
Detailed Experimental Protocols
Dual-Target TaqMan qPCR for Species Identification
This protocol, adapted from a study on differentiating two closely related fish species, highlights a multiplexed qPCR approach to maximize information from a single reaction [118].
- Primer and Probe Design: The assay is predicated on identifying stable Single Nucleotide Polymorphisms (SNPs) differentiating the target species. Two sets of species-specific primers and corresponding minor groove binder (MGB) TaqMan probes are designed. The probes are labeled with different fluorophores (e.g., FAM and VIC) to enable multiplexed detection in a single tube [118].
- Reaction Setup: The reaction mixture typically includes:
- 1X TaqMan Fast Advanced Master Mix
- Species-specific forward and reverse primers (e.g., 900 nM each)
- FAM-labeled MGB probe (e.g., 200 nM) for Coilia brachygnathus
- VIC-labeled MGB probe (e.g., 200 nM) for Coilia nasus
- 2–5 μL of DNA template (50-100 ng total)
- Nuclease-free water to a final volume of 20 μL [118].
- Thermal Cycling:
- Initial Denaturation: 95°C for 2 minutes (activation of hot-start polymerase).
- Amplification (40 cycles):
- Denaturation: 95°C for 15 seconds.
- Annealing/Extension: 60°C for 1 minute (data collection in both FAM and VIC channels) [118].
- Data Interpretation: For C. brachygnathus, the Ct value in the FAM channel is significantly earlier than in the VIC channel (ΔCt ≥ 1). For C. nasus, amplification occurs only in the VIC channel. This differential signal allows for unambiguous species identification [118].
RPA-LFD for Rapid Pathogen Detection
This protocol outlines the steps for a rapid, isothermal RPA-LFD assay, as used for detecting Duck Adenovirus Type-3 (DAdV-3) and other pathogens [47] [122] [119].
- Primer and Probe Design for LFD: Primers are typically 28-35 nucleotides long. The forward primer is labeled with Fluorescein Isothiocyanate (FITC) at the 5' end, and the reverse primer is labeled with biotin. A specific probe, if used in more complex RPA formats, may include a FAM label and an internal abasic site (e.g., a tetrahydrofuran residue) [47] [119].
- RPA Reaction Assembly: The reaction is assembled on ice:
- 29.4 μL of rehydration buffer (provided with the kit)
- 1.6 μL of each primer (10 μM stock)
- 2 μL of DNA template
- Nuclease-free water to 47.5 μL
- The mixture is used to resuspend a pellet containing the RPA enzymes (recombinase, single-stranded DNA binding protein, strand-displacing polymerase).
- The reaction is initiated by adding 2.5 μL of magnesium acetate (280 mM) [47] [122].
- Isothermal Amplification: The tube is incubated at a constant temperature of 37–42°C for 15–20 minutes. No thermal cycler is required [47] [119].
- Lateral Flow Detection: After amplification, 5-10 μL of the product is diluted in 100 μL of assay buffer. The LFD strip is dipped into the solution. The result is read visually within 5 minutes. The appearance of both a control line (C) and a test line (T) indicates a positive result. The control line contains an anti-species antibody, while the test line contains streptavidin to capture the biotin-labeled amplicon, which is then detected by anti-FITC antibodies conjugated to colored or fluorescent microspheres [47] [123].
Droplet Digital PCR (ddPCR) for Absolute Quantification
This protocol, based on a study for detecting Gardnerella vaginalis, details the steps for a ddPCR assay that provides absolute quantification without the need for a standard curve [117].
- Primer Design and Screening: Primers are designed to target a specific gene (e.g., the 23S rRNA gene). Multiple primer pairs (e.g., six sets) are initially screened using a standard qPCR protocol to select the pair with the best amplification efficiency and a single, sharp peak in the melting curve, indicating specific amplification and minimal primer-dimer formation [117].
- ddPCR Reaction Setup: The reaction mixture is prepared:
- 12.5 μL of reaction mix (containing buffer, salts, dNTPs)
- 1 μL of primer mix (final concentration 0.2 μM each)
- 1 μL of Taq DNA Polymerase
- 6 μL of DNA template
- 4.5 μL of ddH₂O
- Total volume: 25 μL [117].
- Droplet Generation and Thermal Cycling:
- The reaction mixture is loaded into a droplet generator (e.g., DG32 Droplet Generator) along with droplet generation oil to create thousands of nanoliter-sized water-in-oil droplets, effectively partitioning the sample.
- The emulsified sample is then transferred to a PCR plate for thermal cycling:
- Initial Denaturation: 95°C for 10 minutes.
- Amplification (40 cycles): 94°C for 30 seconds (denaturation), 60°C for 60 seconds (annealing/extension). A ramp rate of 2°C/second is typically used.
- Final Hold: 98°C for 10 minutes (enzyme deactivation) [117].
- Droplet Reading and Analysis: After PCR, the plate is transferred to a droplet reader (e.g., CS7 Biochip Reader) that counts the fluorescent positive and negative droplets. The absolute concentration of the target (copies/μL) is calculated using Poisson statistics based on the fraction of positive droplets [117].
Visual Workflows and Signaling Pathways
The following diagrams illustrate the core experimental workflows and detection principles for each technology, highlighting the steps where optimization is critical to prevent nonspecific amplification.
TaqMan qPCR Workflow
RPA-LFD Principle and Workflow
ddPCR Partitioning and Quantification
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these molecular techniques relies on a suite of specialized reagents and materials. The following table details key components and their critical functions in the context of minimizing nonspecific amplification.
Table 3: Essential Research Reagents and Their Functions
| Reagent / Material | Core Function | Role in Minimizing Nonspecificity/Primer Dimers |
|---|---|---|
| Hot-Start DNA Polymerase | A modified enzyme inactive at room temperature, activated only at high temperatures (e.g., >90°C). | Prevents primer extension during reaction setup, a common period for primer-dimer formation [23]. |
| Minor Groove Binder (MGB) Probes | TaqMan hydrolysis probes with a 3' MGB moiety that increases duplex stability and melting temperature (Tm). | Allows the use of shorter, more specific probes, improving allele discrimination and reducing false positives from mispriming [118]. |
| Strand-Displacing DNA Polymerase (e.g., in RPA) | Synthesizes new DNA without requiring a denaturation step, enabling isothermal amplification. | A core enzyme in RPA; its function is coupled with recombinase and SSBs, but requires careful primer design to avoid off-target amplification [119]. |
| Recombinase Enzyme (e.g., in RPA) | Binds to primers to form filaments that scan and invade double-stranded DNA. | Facilitates target-specific initiation without thermal denaturation, though its activity can also lead to nonspecific products if primers are not optimal [121] [119]. |
| Single-Stranded DNA Binding (SSB) Proteins (e.g., in RPA) | Stabilize the displaced DNA strand after recombinase-mediated invasion. | Preerves the single-stranded template for primer binding, promoting efficient and specific amplification [119]. |
| Biotin- and FITC-labeled Primers | Primers tagged with haptens for immunochromatographic detection on LFDs. | The dual-labeling strategy (FITC and Biotin) adds a layer of specificity, as the test line on the LFD will only appear if a properly tagged, full-length amplicon is present [47] [123]. |
The choice between qPCR, RPA-LFD, and dPCR is not a matter of identifying a single superior technology, but rather of selecting the most appropriate tool for a specific research or diagnostic question, with a clear understanding of the inherent trade-offs.
- qPCR remains the workhorse for quantitative, multiplexed analysis in laboratory settings. Its robust protocols, high-throughput capability, and ability to perform multiplexing in a single tube make it ideal for applications like gene expression studies and high-throughput genotyping. However, its reliance on precise thermal cycling and calibration curves, along with its susceptibility to inhibitors, are notable limitations.
- RPA-LFD is unparalleled for speed, simplicity, and field deployment. Its isothermal nature and visual readout make it a transformative technology for point-of-care diagnostics, field surveillance, and resource-limited settings. The primary challenges lie in its susceptibility to nonspecific amplification and its currently limited multiplexing capacity compared to qPCR, necessitating rigorous primer and probe validation [121].
- dPCR offers the gold standard in sensitivity and precision by providing absolute quantification without a standard curve. Its partitioning step effectively dilutes inhibitors and competitors like primer dimers, making it exceptionally robust for detecting rare targets, quantifying minor fold changes, and analyzing complex samples. The higher cost, lower throughput, and more complex workflow are factors that currently limit its use to applications where its superior quantification is critical.
The ongoing research into primer dimer formation and nonspecific amplification, including the use of machine learning for predictive primer design [70], will continue to refine all these technologies. The future of molecular analysis lies not only in the improvement of individual methods but also in the intelligent integration of these platforms, such as using RPA-LFD for rapid initial field screening followed by confirmatory qPCR or dPCR in central laboratories [118]. This synergistic approach will ultimately provide researchers and clinicians with a powerful, flexible arsenal for precise nucleic acid analysis.
In molecular biology, the polymerase chain reaction (PCR) is a fundamental technique for amplifying specific DNA sequences. However, a significant challenge in multi-template PCR is non-homogeneous amplification, where different DNA templates amplify at varying efficiencies due to sequence-specific factors. This bias results in skewed abundance data, compromising the accuracy and sensitivity of downstream applications in quantitative molecular biology, diagnostics, and DNA data storage [30]. Traditional optimization methods, which focus on primer design and annealing temperature, are often insufficient for multi-template PCR where primers and adapters are fixed by sequencing protocols [30].
Deep learning models, particularly one-dimensional Convolutional Neural Networks (1D-CNNs), have emerged as powerful tools for predicting sequence-specific amplification efficiency directly from DNA sequence information. This technical guide explores the application of these models to address amplification bias, providing a detailed framework for researchers aiming to integrate machine learning into their molecular biology workflows.
The Problem of Amplification Bias
In multi-template PCR, even small differences in amplification efficiency between templates can lead to dramatic representation biases due to the exponential nature of PCR. A template with an amplification efficiency just 5% below the average will be underrepresented by a factor of approximately two after only 12 cycles [30]. This bias persists even when sequences are constrained to optimal GC content, suggesting the existence of additional, sequence-specific factors beyond traditional culprits like GC content and amplicon length [30].
Experimental data from synthetic oligonucleotide pools demonstrates that PCR amplification progressively skews coverage distributions, with a small subset of sequences (approximately 2%) showing very poor amplification efficiency as low as 80% relative to the population mean [30]. This poor amplification is reproducible and independent of pool diversity, indicating an intrinsic property of the sequence itself [30].
Limitations of Conventional Approaches
Traditional primer design tools like Primer3 and Primer-BLAST rely on thermodynamic principles and empirical rules to optimize primer specificity and annealing properties [70] [34] [37]. While valuable for single-template PCR, these approaches have limitations in multi-template scenarios:
- Inability to predict relative amplification efficiencies between different templates
- Limited capacity to identify sequence motifs associated with poor amplification
- Dependence on manual optimization of reaction conditions
These limitations highlight the need for more sophisticated, data-driven approaches to predict and mitigate amplification bias.
Deep Learning Approaches
Model Architectures for Sequence Analysis
1D Convolutional Neural Networks (1D-CNNs)
1D-CNNs are particularly well-suited for DNA sequence analysis due to their ability to detect local sequence patterns and motifs. When applied to amplification efficiency prediction, 1D-CNNs process DNA sequences as one-dimensional data, using sliding filters to identify sequence motifs that correlate with amplification efficiency [30] [124].
Table 1: Performance of 1D-CNN in Predicting Amplification Efficiency
| Metric | Performance | Description |
|---|---|---|
| AUROC | 0.88 | Area Under the Receiver Operating Characteristic curve |
| AUPRC | 0.44 | Area Under the Precision-Recall Curve |
| Training Data | 12,000 random sequences with common terminal primer binding sites | Synthetic DNA pools with tracked coverage over 90 PCR cycles |
| Key Finding | Identification of adapter-mediated self-priming as a major mechanism for low amplification efficiency | Challenges long-standing PCR design assumptions |
The architecture typically includes:
- Input layer for DNA sequence representation
- Convolutional layers with multiple filter sizes to detect motifs of varying lengths
- Pooling layers to reduce dimensionality while retaining important features
- Fully connected layers for final prediction
These models achieve high predictive performance (AUROC: 0.88, AUPRC: 0.44) when trained on reliably annotated datasets derived from synthetic DNA pools [30].
Recurrent Neural Networks (RNNs)
An alternative approach uses Recurrent Neural Networks (RNNs), which process sequence data sequentially and can capture long-range dependencies. One implementation expressed primer-template relationships as pseudo-sentences of five-letter codes, successfully predicting PCR results with 70% accuracy [70]. However, RNNs generally have higher computational complexity than CNNs for capturing local dependencies critical for primer efficiency [124].
Model Selection Considerations
The choice between 1D-CNNs and RNNs depends on specific research needs:
- 1D-CNNs are preferred for identifying local sequence motifs and offer superior computational efficiency
- RNNs may be more effective for modeling complex interactions between distant sequence elements
- Hybrid models can potentially leverage the strengths of both architectures
Data Preparation and Feature Engineering
Sequence Representation
DNA sequences must be converted to numerical representations suitable for deep learning:
- One-hot encoding: Represents each nucleotide (A, T, C, G) as a binary vector
- K-mer encoding: Breaks sequences into overlapping subsequences of length k, preserving contextual information [124]
For example, the sequence "ATCGAAT" with 3-mer encoding becomes "ATC", "TCG", "CGA", "GAA", "AAT". This approach helps the model recognize biologically meaningful patterns beyond individual nucleotides.
Primer Interaction Features
To minimize false positives from non-specific amplification, computational approaches assess primer interaction potential by evaluating:
- Complementarity: How well primers bind to each other instead of the target
- Secondary structure formation: Hairpins or internal loops that interfere with binding
- Thermodynamic stability: Binding strength of unwanted interactions [124]
These features help flag problematic primer sets before experimental validation, reducing false positives in diagnostic applications [124].
Model Interpretation and Motif Discovery
A significant advantage of deep learning approaches is their ability to identify biologically relevant sequence motifs. The CluMo (Motif Discovery via Attribution and Clustering) framework interprets 1D-CNN models to identify specific motifs adjacent to adapter priming sites that associate with poor amplification [30].
This approach revealed adapter-mediated self-priming as a major mechanism causing low amplification efficiency, challenging conventional PCR design assumptions [30]. Such insights enable more rational primer design and sequence optimization.
Experimental Protocols
Data Generation for Model Training
Synthetic DNA Pool Preparation
- Design: Synthesize a diverse pool of DNA sequences (e.g., 12,000 random sequences) with common terminal primer binding sites (truncated TruSeq adapters)
- GC Control: Include a separate pool with sequences constrained to 50% GC content to control for GC bias
- Quality Control: Verify pool quality and representation through sequencing
Serial Amplification Protocol
- Perform six consecutive PCR reactions with 15 cycles each
- Sequence the amplification products after each round to track changes in amplicon composition
- Use consistent reaction conditions: template concentration, polymerase choice, and thermal cycling parameters [30]
Amplification Efficiency Calculation
For each sequence, fit sequencing coverage data to an exponential PCR amplification model with two parameters:
- Initial bias: Uneven coverage after synthesis
- Amplification efficiency (εᵢ): Sequence-specific amplification efficiency per cycle [30]
Model Training and Validation
Data Splitting and Augmentation
- Split data into training (70%), validation (15%), and test (15%) sets
- Apply data augmentation techniques by creating slightly modified sequence versions to improve model robustness
- Perform statistical checks to ensure augmentations remain within biological plausibility [124]
Training Protocol
- Implement batch training with appropriate batch sizes (32-128)
- Use early stopping based on validation performance to prevent overfitting
- Apply regularization techniques (dropout, L2 regularization) to improve generalization
Validation Experiments
- qPCR Validation: Select sequences with predicted high and low efficiencies and validate using dilution curves in single-template qPCR [30]
- Cross-pool Validation: Synthesize a new oligo pool containing sequences from original experiments and measure their amplification efficiency in a new context [30]
Table 2: Key Experimental Reagents and Solutions
| Reagent/Solution | Function/Application | Example/Specification |
|---|---|---|
| Synthetic Oligo Pools | Provide training data with known sequences | 12,000 random sequences with common adapters |
| High-Fidelity PCR Master Mix | Ensure accurate amplification with minimal polymerase errors | NEBNext High Fidelity 2X PCR Master Mix |
| Sequence-Specific Primers | Validate model predictions for specific sequences | Designed based on efficiency predictions |
| Nuclease-Free Water | Maintain reaction integrity without enzymatic degradation | PCR-grade, sterile-filtered |
| DNA Purification Kits | Clean up samples between amplification rounds | Molzym Ultra Deep Microbiome Prep |
Implementation Workflow
The following diagram illustrates the complete experimental and computational workflow for predicting amplification efficiency using deep learning:
Deep Learning Workflow for Amplification Efficiency Prediction
Applications and Benefits
Homogeneous Amplicon Library Design
By predicting sequences with poor amplification efficiency, researchers can design inherently more homogeneous amplicon libraries. This approach reduces the skew in abundance data critical for quantitative applications like metagenomics and DNA data storage [30].
Reduced Sequencing Requirements
Deep learning-guided design reduces the required sequencing depth to recover 99% of amplicon sequences by fourfold, significantly lowering sequencing costs and computational overhead for data analysis [30].
Improved Diagnostic Accuracy
In diagnostic applications, predicting and avoiding primer sequences prone to non-specific amplification reduces false positives and improves assay reliability [124]. This is particularly important for pathogen detection and clinical diagnostics.
Integration with Traditional Methods
Complementary Approaches
Deep learning methods should complement rather than replace traditional primer design principles:
- Thermodynamic Optimization: Continue to apply melting temperature (Tₘ) calculations and GC content optimization
- Specificity Checking: Use BLAST-based tools like Primer-BLAST to verify primer specificity against relevant genomes [34]
- Experimental Validation: Always validate computational predictions with controlled experiments
Hybrid Workflows
Implement hybrid workflows that combine:
- Traditional primer design using established tools
- Efficiency prediction using deep learning models
- Experimental validation through small-scale tests
This integrated approach leverages the strengths of both computational and empirical methods.
Deep learning models, particularly 1D-CNNs, offer a powerful approach for predicting sequence-specific amplification efficiency in multi-template PCR. By identifying sequence motifs associated with poor amplification and enabling the design of more homogeneous amplicon libraries, these methods address a fundamental challenge in molecular biology.
The integration of deep learning with traditional primer design principles creates new opportunities to improve the accuracy and efficiency of PCR-based applications across genomics, diagnostics, and synthetic biology. As these models continue to evolve, they will likely become standard tools in the molecular biologist's toolkit, enabling more precise control over DNA amplification and more reliable research outcomes.
The development of multiplex Polymerase Chain Reaction (PCR) assays represents a significant frontier in molecular diagnostics, enabling the simultaneous detection of multiple pathogens or genetic markers in a single reaction. However, this process is fraught with experimental challenges, primarily due to the exponential increase in optimization complexity with each additional target. For (N{\rm{t}}) multiplexed targets, if (N{\rm{Ps}}) candidate primer sets are designed for each, the total number of possible multiplex assay combinations is (N{\rm{c}} = {N{\rm{Ps}}}^{N_{\rm{t}}}). This exponential relationship means that with just 4 candidate primer sets for each of 7 targets, a researcher faces 16,384 potential combinations to test empirically [125]. This biological problem is further compounded by the persistent issue of primer-dimer formation and nonspecific amplification, where primers anneal to each other or to non-target sequences instead of the intended target, significantly reducing assay efficiency and accuracy [23]. These interactions are particularly problematic in multiplexed environments where multiple primer sets compete for shared reagents, often leading to imbalanced amplification, reduced sensitivity, and false results [126].
In response to these challenges, a new paradigm of data-driven multiplexing (DDM) has emerged, leveraging computational power to simulate and optimize assay conditions before wet-lab testing. Central to this approach is the Smart-Plexer framework, which combines empirical singleplex testing with computer simulation to develop optimized multiplex combinations [125]. This hybrid workflow analyzes kinetic inter-target distances between amplification curves to generate optimal multiplex PCR primer sets, enabling accurate multi-pathogen identification even in single-channel assays where fluorescent detection channels are limited. By addressing the fundamental issues of primer dimer formation and nonspecific amplification through computational prediction, Smart-Plexer represents a transformative methodology in assay development, particularly for diagnostic applications requiring detection of numerous pathogens with overlapping symptoms, such as respiratory infections [125] [127].
The Smart-Plexer Computational Framework
Core Architecture and Workflow
The Smart-Plexer framework operates on a fundamental hypothesis: that the kinetic information encoded in amplification curves from singleplex reactions can predict their behavior in multiplex environments. This computational approach reinvents the traditional process of multiplex assay development by coupling limited empirical testing with extensive computer simulation [125]. The framework begins with experimental application of candidate singleplex assays, collecting their real-time amplification data. These singleplex amplification curves then serve as a "card deck" from which the algorithm simulates thousands of possible multiplex combinations in silico, dramatically reducing the wet-lab testing burden [125] [127].
The computational core of Smart-Plexer relies on analyzing amplification curve similarities and differences through distance measurements. The framework processes raw amplification data through a filtering pipeline that: (i) subtracts curve background to remove fluorescence signal noise at starting cycles, (ii) removes late amplification curves to exclude non-plateau reactions, and (iii) eliminates noisy curves resulting from operator or instrumentation faults [125]. The cleaned data then undergoes sigmoidal fitting using a five-parameter model proposed by Spiess et al.:
[f\left(t\right)=\frac{a}{{\left(1+{\exp}^{-c\left(t-d\right)}\right)}^{\rm{e}}}+b]
where (t) is the amplification time (PCR cycle), (f\left(t\right)) is the fluorescence at time (t), (a) is the maximum fluorescence, (b) is the baseline of the sigmoid, (c) is related to the slope of the curve, (d) is the fractional cycle of the inflection point, and (e) allows for an asymmetric shape (Richard's coefficient) [125]. This mathematical representation enables quantitative comparison of curve kinetics beyond visual inspection.
Distance Metrics and Ranking System
Smart-Plexer employs sophisticated distance metrics to quantify the distinguishability between amplification curves from different targets. The framework calculates distances between two distinct curves using either the entire amplification curve as a time series or extracted sigmoidal features [125]. These distances form the basis for two critical scoring metrics:
- Average Distance Score (ADS): The mean of computed distances among all targets in a simulated multiplex combination.
- Minimum Distance Score (MDS): The distance between the two most similar targets, ensuring that high ADS values aren't dependent on large differences among only a subset of curves [125].
The ranking system prioritizes combinations with both high ADS and MDS values, as larger inter-curve distances enable more accurate classification using Amplification Curve Analysis (ACA) methodology. When amplification curves have high similarity (small distance values), ACA classifiers struggle to differentiate targets, reducing assay reliability [125]. This dual-metric approach ensures selected primer sets maintain sufficient kinetic distinction for robust multiplex detection.
Evolution to Smart-Plexer 2.0: Enhanced Feature Extraction and Distance Measurement
The original Smart-Plexer framework (retroactively termed Smart-Plexer 1.0) primarily utilized a single kinetic feature—parameter 'c' from the sigmoidal fitting, related to curve slope—for distance calculations [127]. While effective in controlled conditions, this single-feature approach showed limitations under complex scenarios involving PCR inhibition, variable primer efficiency, and fluctuating target concentrations [127].
Smart-Plexer 2.0 introduces significant enhancements to address these limitations:
Enhanced Feature Extraction: Smart-Plexer 2.0 incorporates twelve novel kinetic features derived from biological insights into PCR fluorescent readouts, selected through statistical evaluations for optimal distinguishability across targets [127]. These parameters complement the slope-related 'c' parameter, significantly enriching the feature space for assay selection.
Advanced Distance Measurement: Instead of median-based distance calculations, Smart-Plexer 2.0 implements clustering-based distance measures that evaluate relationships between target distributions, more effectively capturing intra-target variability [127].
These advancements translate to measurable performance improvements. In comparative evaluations, Smart-Plexer 2.0 reduced accuracy variance by an order of magnitude and improved ACA classification by 1.5% and 1% in retrospective 3-plex and 7-plex assays, respectively. In a multi-experiment, cross-concentration evaluation of a newly developed 7-plex assay, it achieved 97.6% ACA accuracy, confirming robustness across complex scenarios [127].
Table 1: Comparison of Smart-Plexer Versions
| Feature | Smart-Plexer 1.0 | Smart-Plexer 2.0 |
|---|---|---|
| Primary Feature Source | Single parameter ('c') from 5-parameter sigmoidal fitting [125] | Twelve novel kinetic features with statistical selection [127] |
| Distance Calculation | Median-based distance metrics [127] | Clustering-based distance measures [127] |
| Key Metrics | Average Distance Score (ADS), Minimum Distance Score (MDS) [125] | Enhanced ADS and MDS with distribution awareness [127] |
| Performance in Variable Conditions | Reliable in controlled reactions [127] | Robust across concentration variations and efficiency fluctuations [127] |
| Reported ACA Accuracy | Not explicitly reported for 7-plex | 97.6% in cross-concentration 7-plex evaluation [127] |
Figure 1: Smart-Plexer Workflow - The hybrid computational and experimental pipeline for optimal multiplex assay development
Experimental Protocols and Implementation
Primer and Probe Design Considerations
The foundation of successful multiplexing begins with careful primer and probe design to minimize dimerization and nonspecific amplification. Following established guidelines ensures higher success rates before computational optimization:
- Melting Temperature (Tm): Optimal primer Tm should be 60–64°C, with ideally 62°C for typical cycling conditions. The melting temperatures of the two primers should not differ by more than 2°C to ensure simultaneous binding [17].
- GC Content: Design assays with GC content of 35–65%, ideally 50%, to balance sequence complexity and uniqueness. Avoid regions of 4 or more consecutive G residues [17].
- Complementarity and Secondary Structure: Screen designs for self-dimers, heterodimers, and hairpins using tools like OligoAnalyzer. The ΔG value of any secondary structures should be weaker (more positive) than –9.0 kcal/mol [17].
- Amplicon Length: Typically target 70–150 bp for efficient amplification with standard cycling conditions. Longer amplicons up to 500 bases can be generated but require extended extension times [17].
- Probe Design (for qPCR): For hydrolysis probes like TaqMan, design probes with Tm 5–10°C higher than primers. Ideally use double-quenched probes with internal ZEN or TAO quenchers for lower background and higher signal-to-noise ratios [17].
Amplification Curve Analysis (ACA) Methodology
Amplification Curve Analysis (ACA) serves as the classification engine that enables single-channel multiplexing by leveraging unique kinetic signatures of amplification curves to identify multiple targets [127]. The ACA methodology works by extracting hand-crafted or network features from amplification curves and employing statistical learning or deep learning approaches to classify targets based on their unique curve characteristics [127].
In practice, ACA recognizes clusters from different amplification shapes which represent different targets. The method can utilize either the entire raw amplification curve as a time series or various normalized representations, such as Final Fluorescence Intensity (FFI) normalized curves [125]. For the 7-plex respiratory infection assay developed using Smart-Plexer, single-channel multiplex assays coupled with ACA demonstrated capable classification of all seven common respiratory infection pathogens in a single test [125].
The recent Smart-Plexer 2.0 enhancement improves ACA performance through more stable feature sets. The twelve newly introduced kinetic features remain consistent across different template concentrations, maintaining distinguishable patterns even when reaction efficiency varies [127]. This stability is crucial for reliable performance in clinical samples where target concentrations may differ significantly.
Wet-Lab Validation and Optimization
While Smart-Plexer dramatically reduces the experimental burden, wet-lab validation remains essential for confirming computational predictions. The validation process involves:
- Testing Top-Ranked Combinations: Select a set of singleplex assays from the top Smart-Plexer ranks for empirical multiplex testing [125].
- Empirical Multiplex Evaluation: Run actual multiplex assays with the selected primer sets to assess changes in curve shape during the transition from singleplex to multiplex environments [125].
- ACA Performance Assessment: Evaluate the classification accuracy of the validated multiplex assays using the ACA methodology [125].
- Cross-Validation: Test selected assays across different target concentrations and in the presence of potential inhibitors to ensure robustness [127].
For the 7-plex respiratory tract infection assay development, researchers applied both original and enhanced Smart-Plexer strategies, selecting six optimal multiplex assays for wet-lab validation. These were tested in qdPCR with a range of different target concentrations, confirming the superior performance of Smart-Plexer 2.0 selections in complex experimental conditions [127].
Table 2: Key Research Reagent Solutions for Smart-Plexer Implementation
| Reagent/Component | Function/Role | Specifications/Considerations |
|---|---|---|
| Bst DNA Polymerase | Strand-displacing polymerase for isothermal amplification | Bst 2.0 and Bst 3.0 variants offer improved polymerization speed, thermal stability, and reverse transcriptase activity [128] |
| Double-Quenched Probes | Target-specific detection in qPCR | Incorporate ZEN or TAO as internal quenchers for lower background and higher signal-to-noise ratios [17] |
| Hot-Start Polymerases | Reduce primer dimer formation | Remain inactive until elevated temperatures are reached, minimizing nonspecific amplification during reaction setup [23] |
| Sigmoidal Fitting Algorithms | Mathematical representation of amplification curves | Five-parameter model provides optimal fit for real-time PCR sigmoids [125] |
| Intercalating Dyes (SYBR Green, EvaGreen) | Non-specific DNA detection for ACA | Enable monitoring of amplification curves without sequence-specific probes; selection affects background fluorescence [128] |
| Primer Design Tools (OligoAnalyzer, PrimerQuest) | In-silico assay design and optimization | Screen for self-dimers, heterodimers, hairpins; calculate Tm under specific reaction conditions [17] |
Applications and Performance in Diagnostic Assays
Respiratory Pathogen Detection
Respiratory infection diagnostics represent an ideal application for Smart-Plexer optimized assays, as numerous pathogens cause similar symptoms but require different treatment approaches. The framework was applied and evaluated for seven respiratory infection targets using an optimized multiplexed PCR assay [125]. The developed panel targeted common respiratory pathogens, enabling specific and sensitive identification in a single test [125].
This application demonstrates the clinical value of data-driven multiplexing, particularly during epidemics where rapid, accurate pathogen identification is crucial for patient management and infection control. Early and accurate identification of respiratory pathogens enables targeted clinical treatment, improving patient outcomes and supporting effective epidemic surveillance, particularly during pandemics [127]. The ability to detect seven targets in a single-channel assay makes this approach accessible even with standard PCR instrumentation.
Comparative Performance Analysis
The performance of Smart-Plexer-optimized assays demonstrates significant advantages over conventional multiplexing approaches. In validation studies, assays developed using the Smart-Plexer framework have shown:
- High Classification Accuracy: Smart-Plexer 2.0 achieved 97.6% ACA accuracy in a cross-concentration evaluation of a 7-plex assay [127].
- Robustness to Concentration Variations: The enhanced feature set in Smart-Plexer 2.0 maintains stability across different template concentrations, a critical advantage for clinical samples with variable target loads [127].
- Reduced Validation Burden: By simulating thousands of potential combinations in silico, the framework reduces wet-lab testing to a manageable number of top candidates [125].
These performance characteristics make Smart-Plexer-optimized assays particularly valuable for clinical diagnostics, where reliability across diverse sample types and concentrations is essential.
Figure 2: Problem-Solution Framework - Data-driven multiplexing addresses fundamental challenges in conventional multiplex PCR development
Future Directions and Integration Opportunities
The Smart-Plexer framework continues to evolve, with several promising directions for enhancement and integration. Future developments may include:
Integration with Alternative Amplification Technologies: While initially developed for PCR-based assays, the principles of data-driven multiplexing could extend to isothermal amplification methods like LAMP (Loop-Mediated Isothermal Amplification) or HDA (Helicase-Dependent Amplification) [129] [128]. These techniques offer advantages for point-of-care testing but face similar multiplexing challenges.
Machine Learning Enhancements: As amplification datasets grow, incorporating more sophisticated machine learning algorithms for pattern recognition could further improve classification accuracy, particularly as the number of multiplexed targets increases.
Automated Platform Integration: Combining Smart-Plexer with automated liquid handling and experimental platforms could create end-to-end workflows from assay design to validation, further reducing development time and resource requirements.
Expanded Clinical Applications: Beyond respiratory pathogens, the framework shows promise for applications in sepsis diagnostics, cancer biomarker detection, and antimicrobial resistance testing—all areas where multiplex detection provides significant clinical value [126].
The ongoing development of data-driven multiplexing frameworks like Smart-Plexer represents a paradigm shift in molecular assay development, moving away from purely empirical optimization toward computationally guided design. This approach not only addresses the persistent challenges of primer dimer formation and nonspecific amplification but also makes sophisticated multiplex assays more accessible and reliable for clinical diagnostics and research applications.
Data-driven multiplexing frameworks like Smart-Plexer represent a transformative approach to overcoming the historical challenges of primer dimer formation and nonspecific amplification in multiplex PCR assay development. By leveraging computational power to simulate thousands of potential primer combinations and identify those with optimal kinetic distinctions, Smart-Plexer dramatically reduces the experimental burden while improving assay performance. The evolution from Smart-Plexer 1.0 to 2.0, with enhanced feature extraction and clustering-based distance measurements, demonstrates the continuous improvement potential of this methodology.
The integration of Smart-Plexer with Amplification Curve Analysis enables unprecedented single-channel multiplexing capabilities, allowing detection of numerous targets without instrument modifications. This combination addresses both the computational challenge of optimal primer selection and the detection challenge of target classification, providing a comprehensive solution for developing robust multiplex assays. As molecular diagnostics continues to demand higher multiplexing capabilities for comprehensive pathogen detection and genetic analysis, data-driven frameworks like Smart-Plexer will play an increasingly vital role in making these assays practical, reliable, and accessible across diverse laboratory settings.
Conclusion
Primer dimers and nonspecific amplification are not merely nuisances but fundamental challenges that dictate the success of molecular assays. Mastering their control requires a holistic approach, integrating a deep understanding of their foundational mechanisms with rigorous primer design, systematic troubleshooting, and comprehensive validation. The future of overcoming these obstacles lies in the adoption of advanced computational tools, including machine learning models for predicting amplification efficiency and data-driven frameworks for designing robust multiplex assays. For biomedical and clinical research, embracing these strategies is paramount for developing the next generation of highly sensitive, specific, and reliable diagnostic tests, ultimately accelerating drug discovery and improving patient outcomes through more accurate molecular data.
References
- 1. Primer Dimer - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/primer-dimer]
- 2. Primer dimer [https://en.wikipedia.org/wiki/Primer_dimer]
- 3. Troubleshooting Non-Specific Amplification [https://bento.bio/resources/bento-lab-advice/troubleshooting-non-specific-amplification-with-bento-lab/]
- 4. Impact of Primer Dimers and Self-Amplifying Hairpins on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5922443/]
- 5. Primer Dimer - an overview [https://www.sciencedirect.com/topics/neuroscience/primer-dimer]
- 6. Quantitative Experimental Determination of Primer-Dimer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3416024/]
- 7. Troubleshooting primer dimer in PCR [https://www.minipcr.com/primer-dimer-pcr/]
- 8. How can I avoid primer-dimer formation during PCR ... [https://www.qiagen.com/us/resources/faq/544]
- 9. Module 4.2: Denaturation, Annealing, and Primer Extension [https://pressbooks.umn.edu/cvdl/chapter/module-4-4-denaturation-annealing-and-primer-extension/]
- 10. Primer dimer – Knowledge and References [https://taylorandfrancis.com/knowledge/Medicine_and_healthcare/Physiology/Primer_dimer/]
- 11. Primer Analysis Tool and Tm Calculator [https://eu.idtdna.com/pages/tools/oligoanalyzer]
- 12. Multiple Primer Analyzer | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/multiple-primer-analyzer.html]
- 13. Development of PCR Blocking Primers Enabling DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12367867/]
- 14. How to Simplify PCR Optimization Steps for Primer Annealing [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/pcr-annealing-optimization-universal-annealing.html]
- 15. Amplification of nonspecific products in quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5727009/]
- 16. Primer design guide - 5 tips for best PCR results [https://the-dna-universe.com/2022/09/05/primer-design-guide-the-top-5-factors-to-consider-for-optimum-performance/]
- 17. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 18. Optimizing PCR Primer's T m and Annealing Temperatures [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/optimizing-tm-and-annealing.html]
- 19. FAQ: How should I determine the appropriate annealing ... [https://www.neb.com/en-us/faqs/2023/07/27/how-should-i-determine-the-appropriate-annealing-temperature-for-my-reaction?srsltid=AfmBOopf_8cU_4_08hChx2XeMVxsNl6_eZFHK5Yp0mk_owe_7xh-hj7p]
- 20. Optimization of Annealing Temperature To Reduce Bias ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC93085/]
- 21. PCR Troubleshooting: Common Problems and Solutions [https://www.mybiosource.com/learn/pcr-troubleshooting-common-problems-and-solutions/]
- 22. The Pain of Primer Dimer [https://kilobaser.com/post/the-pain-of-primer-dimer]
- 23. PCR Optimization: Strategies To Minimize Primer Dimer [https://genemod.net/blog/pcr-optimization]
- 24. PCR Primer Design Tips - Behind the Bench [https://www.thermofisher.com/blog/behindthebench/pcr-primer-design-tips/]
- 25. URAdime – a tool for analyzing primer sequences in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11838186/]
- 26. varVAMP: degenerate primer design for tiled full genome ... [https://www.nature.com/articles/s41467-025-60175-9]
- 27. PrimerROC: accurate condition-independent dimer ... [https://www.nature.com/articles/s41598-018-36612-9]
- 28. Protocols Agarose Gel Electrophoresis [https://www.addgene.org/protocols/gel-electrophoresis/]
- 29. How to Read Gel Electrophoresis Bands: Interpretation Guide [https://www.wikihow.com/Read-Gel-Electrophoresis-Bands]
- 30. Predicting sequence-specific amplification efficiency in ... [https://www.nature.com/articles/s41467-025-64221-4]
- 31. Novel primer-less amplification method for detection of ... [https://www.sciencedirect.com/science/article/pii/S1046202325002154]
- 32. What are Primer Dimers? - A Beginner's Guide [https://geneticeducation.co.in/what-are-primer-dimers-a-beginners-guide/]
- 33. PCR Troubleshooting Guide | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-troubleshooting.html]
- 34. Primer designing tool - NCBI - NIH [https://www.ncbi.nlm.nih.gov/tools/primer-blast/]
- 35. Designing highly multiplex PCR primer sets with Simulated ... [https://www.nature.com/articles/s41467-022-29500-4]
- 36. Proven tips for PCR primer design [https://www.neb.com/en-us/nebinspired-blog/proven-tips-for-pcr-primer-design?srsltid=AfmBOoqz0BSP-HjtNTJed1lDf8nBq3ZY1fKOo9TEKVV_IoYBSxajXFEK]
- 37. How to Design Primers for DNA Sequencing | Practical Lab ... [https://www.cd-genomics.com/resource-how-to-design-primers-for-dna-sequencing.html]
- 38. Design and in silico validation of polymerase chain ... [https://www.nature.com/articles/s41598-021-91817-9]
- 39. In Silico Primer Design Tools [https://www.benchling.com/in-silco-primer-design-tools]
- 40. A Computational Tool for Large-Scale Primer Design and ... [https://pubmed.ncbi.nlm.nih.gov/41010007/]
- 41. PrimeSpecPCR: Python toolkit for species-specific DNA ... [https://www.sciencedirect.com/science/article/pii/S235271102500216X]
- 42. PCR Primer Design Tool [https://eurofinsgenomics.eu/en/ecom/tools/pcr-primer-design/]
- 43. Primer Design Tool [https://en.vectorbuilder.com/tool/primer-design.html]
- 44. Primer Design Using Benchling's Molecular Biology Tools [https://www.benchling.com/primer-design-using-benchlings-molecular-biology-tools]
- 45. Web Tools for Advanced PCR Design, Genotyping, LAMP ... [https://primerdigital.com/tools/]
- 46. Cooperative Primers: 2.5 Million–Fold Improvement in the ... [https://www.sciencedirect.com/science/article/pii/S1525157813002493]
- 47. Development of a rapid recombinase polymerase ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1638182/full]
- 48. Four Free and Easy-To-Use Online Primer Design Tools [https://bitesizebio.com/42836/four-free-and-easy-to-use-online-primer-design-tools/]
- 49. PCR Assay Optimization and Validation [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/assay-optimization-and-validation?srsltid=AfmBOorAiDHZJCZBKJdQ6rGIdCVr4Ki0tItsVKeOxly1AMrzYKsR8A6Z]
- 50. PCR Setup—Six Critical Components to Consider [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-component-considerations.html]
- 51. Faster PCR Optimization [https://bitesizebio.com/31937/optimizing-your-pcr-faster/]
- 52. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOoomToBORcpjDvXQTIdNjFontfcpf-tuTxWxRf_FriTgYR96vo_8]
- 53. Improving qPCR Efficiency: Primer Design and Reaction ... [https://synapse.patsnap.com/article/improving-qpcr-efficiency-primer-design-and-reaction-optimization]
- 54. PCR Assay Optimization and Validation [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/assay-optimization-and-validation?srsltid=AfmBOoobts1hB1RZKCyKoCeuslSHvxJtOc83JcWG1Oy3GZkSLgx_1T5a]
- 55. Optimising PCR: How Thermal Cycling Conditions ... [https://www.psychreg.org/optimising-pcr-how-thermal-cycling-conditions-influence-pcr-protocol-efficiency/]
- 56. Gradient Thermal Cyclers: When and How to Use Them [https://www.labx.com/resources/gradient-thermal-cyclers-when-and-how-to-use-them/5580]
- 57. PCR and Thermal Cycler Technology: Fundamentals, ... [https://www.labx.com/resources/pcr-and-thermal-cycler-technology-fundamentals-applications-and-emerging-trends-in-dna/5577]
- 58. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 59. What is Hot Start PCR? [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/what-is-hot-start-pcr?srsltid=AfmBOooLcpUxnE_xzGIYs_oFMV89ONQ83vTvoXvbH6C3KNJdLeGVJnPE]
- 60. Hot-Start PCR: Higher specificity, higher yield, ... [https://www.genaxxon.com/shop/en/blog/hot-start-pcr-higher-specificity-higher-yield-higher-sensitivity]
- 61. Hot Start PCR with heat-activatable primers [https://pmc.ncbi.nlm.nih.gov/articles/PMC2582603/]
- 62. Hot Start PCR - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/hot-start-pcr]
- 63. Production and evaluation of egg derived hot start antibodies [https://www.sciencedirect.com/science/article/pii/S0717345820300051]
- 64. Platinum II Taq Hot-Start DNA Polymerase [https://www.thermofisher.com/us/en/home/life-science/pcr/pcr-enzymes-master-mixes/platinum-hot-start-pcr-enzyme.html]
- 65. Crosslinking of PCR primers reduces unspecific ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701220307673]
- 66. Targeting intestinal inflammation using locked nucleic ... [https://www.nature.com/articles/s41467-025-63037-6]
- 67. PNA and LNA throw light on DNA [https://www.sciencedirect.com/science/article/abs/pii/S0167779902000082]
- 68. Locked Nucleic Acids (LNA) Market Disruption and Future ... [https://www.marketreportanalytics.com/reports/locked-nucleic-acids-lna-155797]
- 69. Recent Cutting‐Edge Technologies for the Delivery of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12172603/]
- 70. Prediction of PCR amplification from primer and template ... [https://www.nature.com/articles/s41598-021-86357-1]
- 71. Troubleshooting Common Electrophoresis Problems and ... [https://www.labx.com/resources/troubleshooting-common-electrophoresis-problems-and-artifacts/5539]
- 72. Common artifacts and mistakes made in electrophoresis [https://pmc.ncbi.nlm.nih.gov/articles/PMC7295095/]
- 73. Gel Electrophoresis - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/neuroscience/gel-electrophoresis]
- 74. GelGenie: an AI-powered framework for gel ... [https://www.nature.com/articles/s41467-025-59189-0]
- 75. Gradient PCR for Optimization [https://www.eppendorf.com/us-en/lab-academy/life-science/molecular-biology/gradient-pcr-for-optimization/]
- 76. Optimizing PCR Reaction Conditions for High Fidelity and ... [https://www.labx.com/resources/optimizing-pcr-reaction-conditions-for-high-fidelity-and-yield/5579]
- 77. PCR Optimization: Factors Affecting Reaction Specificity ... [https://www.mybiosource.com/learn/pcr-optimization-factors-affecting-reaction-specificity-and-efficien/]
- 78. Efficient experimental design and analysis of real-time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3710343/]
- 79. Improve Yield and Specificity of Your PCR Using the 2D- ... [https://www.eppendorf.com/fj-en/beyond-science/method-applications/molecular-biology/improve-yield-and-specificity-of-your-pcr-using-the-2d-gradient-function/]
- 80. The Ultimate qPCR Experiment: Producing Publication ... [https://www.sciencedirect.com/science/article/pii/S0167779918303421]
- 81. Unexpected Mechanism and Inhibition Effect for ... [https://pubmed.ncbi.nlm.nih.gov/37922263/]
- 82. Non-specific amplification of human DNA is a major ... [https://www.nature.com/articles/s41598-020-73403-7]
- 83. qPCR primer design revisited - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC5702850/]
- 84. Understanding qPCR Efficiency and Why It Exceeds 100% [https://biosistemika.com/blog/qpcr-efficiency-over-100/]
- 85. Water for PCR Techniques [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/water-pcr-techniques?srsltid=AfmBOorzMnYKGOesooRxdmYxMg8o3aKKusCGPfIW8tRJ8q4hgsF6G1Uw]
- 86. Co-Primers [https://co-dx.com/co-primers/]
- 87. Impurities in viral-based therapeutics: mastering residual ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/3632/impurities-in-viralbased-therapeutics-mastering-residual-hcdna-a-harmonized-approach-for-aav-gene-therapies]
- 88. pDNA Impurities in mRNA Vaccines [https://www.mdpi.com/2076-2607/13/9/1975]
- 89. A comprehensive quantitative study of organic ... [https://www.sciencedirect.com/science/article/pii/S026974912501200X]
- 90. Guidelines for preventing and reporting contamination in ... [https://www.nature.com/articles/s41564-025-02035-2]
- 91. Guidelines for preventing and reporting contamination in ... [https://pubmed.ncbi.nlm.nih.gov/40542287/]
- 92. Regulatory Landscape and the Evolution of Contamination ... [https://progress-lifesciences.nl/publicaties/regulatory-landscape-and-the-evolution-of-contamination-control-in-2025/]
- 93. HRM Analysis for Mutation Scanning Prior to Sequencing [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/genotyping-analysis-real-time-pcr-information/high-resolution-melt-analysis-mutation-scanning-sequencing.html]
- 94. HRM vs Classical melt curve analysis [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/technology-and-research/hrm-technology/hrm-vs-classical-melt-curve-analysis]
- 95. HRM - High Resolution Melt [https://www.gene-quantification.de/hrm.html]
- 96. High Resolution Melting Analysis for Gene Scanning - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2836412/]
- 97. HRM Technology - FAQs [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/technology-and-research/hrm-technology/hrm-technology-faqs]
- 98. MIQE Guidelines | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/why-choose-taqman-assays/publish-real-time-pcr-results.html]
- 99. MIQE 2.0: Revision of the Minimum Information for ... [https://pubmed.ncbi.nlm.nih.gov/40272429/]
- 100. How do I determine the amplification efficiency of my qPCR ... [https://www.qiagen.com/us/resources/faq/2694]
- 101. Primer Efficiency qPCR [https://barricklab.org/twiki/bin/view/Lab/PrimerEfficiencyqPCR]
- 102. Quantitative PCR validation for research scientists: Part 1 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 103. qPCR primer design revisited [https://www.sciencedirect.com/science/article/pii/S221475351730181X]
- 104. Adjusting RT-qPCR conditions to avoid unspecific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7598553/]
- 105. PCR diagnostics: In silico validation by an automated tool ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7113775/]
- 106. Primer-BLAST: A tool to design target-specific primers for ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-13-134]
- 107. In silico PCR analysis: a comprehensive bioinformatics tool ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2024.1464197/full]
- 108. Development of an in silico evaluation system that ... [https://www.sciencedirect.com/science/article/abs/pii/S0278691524000103]
- 109. Current Needs, Gaps, and Challenges - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7179957/]
- 110. Recommendations for the Use of in Silico Approaches ... [https://www.sciencedirect.com/science/article/pii/S1525157822002872]
- 111. Rapid identification of Acinetobacter baumannii using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12605139/]
- 112. Design and Validation of Primer Sets for the Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11096201/]
- 113. Controlling for contamination in qPCR testing - LGC [https://blog.biosearchtech.com/how-to-control-for-contamination-in-qpcr-testing]
- 114. Crystal Digital PCR™ Enables Precise Quantification of ... [https://www.mdpi.com/2076-2607/13/11/2592]
- 115. Multiplexing qPCR [https://oligos.biosearchtech.com/support/education/multiplex-qpcr]
- 116. Optimization and validation of a consolidated Set ... [https://www.sciencedirect.com/science/article/pii/S2215016125004443]
- 117. Quantitative diagnostic method to detect Gardnerella ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12410466/]
- 118. Integrating RPA-LFD and TaqMan qPCR for Rapid On-Site ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12564251/]
- 119. Development of Reverse Transcriptase Recombinase ... [https://www.mdpi.com/2306-7381/12/9/845]
- 120. Recombinase polymerase amplification combined with ... [https://www.sciencedirect.com/science/article/pii/S2405844024049740]
- 121. Recombinase polymerase amplification technology for ... [https://www.sciencedirect.com/science/article/pii/S1201971225000554]
- 122. Development of a rapid recombinase polymerase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12380705/]
- 123. Development of a dual-target RAA-LFD assay for point-of- ... [https://www.frontiersin.org/journals/veterinary-science/articles/10.3389/fvets.2025.1684537/full]
- 124. How AI Can Help Us Spot the Fakes in DNA Testing [https://medium.com/@frederickhayes3/lets-talk-about-the-work-de01d056aa33]
- 125. Smart-Plexer: a breakthrough workflow for hybrid ... [https://www.nature.com/articles/s42003-023-05235-w]
- 126. Evolution of Multiplex PCR: Benefits, Challenges & Key ... [https://www.eurogentec.com/en/news/111_evolution-of-multiplex-pcr-benefits-challenges-key-applications]
- 127. Smart-Plexer 2.0: Leveraging New Features of Amplification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12268814/]
- 128. Advancements and applications of loop-mediated ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1406632/full]
- 129. Helicase-dependent isothermal amplification turns ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25021198]