Preventing Primer-Dimer Formation in PCR: A Comprehensive Guide for Reliable Assays
Primer-dimer formation is a pervasive challenge in PCR that consumes reaction resources, reduces amplification efficiency, and can lead to inaccurate results in research and diagnostics.
Preventing Primer-Dimer Formation in PCR: A Comprehensive Guide for Reliable Assays
Abstract
Primer-dimer formation is a pervasive challenge in PCR that consumes reaction resources, reduces amplification efficiency, and can lead to inaccurate results in research and diagnostics. This article provides a complete framework for scientists, researchers, and drug development professionals to understand, prevent, and troubleshoot primer-dimer artifacts. It covers foundational concepts of how primer-dimers form, strategic primer design and methodological optimizations, advanced troubleshooting protocols, and validation techniques to confirm assay specificity. By integrating modern primer design tools with robust laboratory practices, this guide empowers professionals to achieve highly specific and efficient PCR amplification, thereby enhancing the reliability of downstream applications in biomedical and clinical research.
Understanding Primer-Dimer Formation: Mechanisms and Impacts on PCR Efficiency
What is a Primer Dimer? Defining the Unintended PCR Byproduct
In polymerase chain reaction (PCR) research, few issues are as ubiquitous and detrimental as the formation of primer dimers. These unintended amplification artifacts compete for valuable PCR reagents, inhibit target DNA amplification, and can compromise the accuracy of experimental results, particularly in quantitative applications. For researchers and drug development professionals, understanding and preventing primer dimer formation is not merely a troubleshooting exercise but a fundamental requirement for ensuring data integrity. This technical support center provides a comprehensive guide to identifying, understanding, and preventing primer dimers within the broader context of robust PCR experimental design.
What is a Primer Dimer?
A primer dimer (PD) is a small, unintended by-product formed during the polymerase chain reaction (PCR) when PCR primers anneal to each other instead of to the target DNA template [1] [2]. These artifacts are typically short, double-stranded DNA fragments, often appearing in the size range of 30 to 50 base pairs [1]. Their formation leads to the amplification of these short fragments, which competes with the amplification of the desired target sequence for essential PCR reagents like primers, nucleotides, and DNA polymerase. This competition can significantly reduce the efficiency and yield of the target PCR product [1] [3].
There are two primary types of primer dimers [4]:
- Self-dimer (Homodimer): Formed when two identical primers bind to each other due to self-complementary regions within a single primer sequence.
- Cross-dimer (Heterodimer): Formed when the forward and reverse primers, which are designed to be different, bind to each other because of complementary regions between them.
How Do Primer Dimers Form? The Mechanism
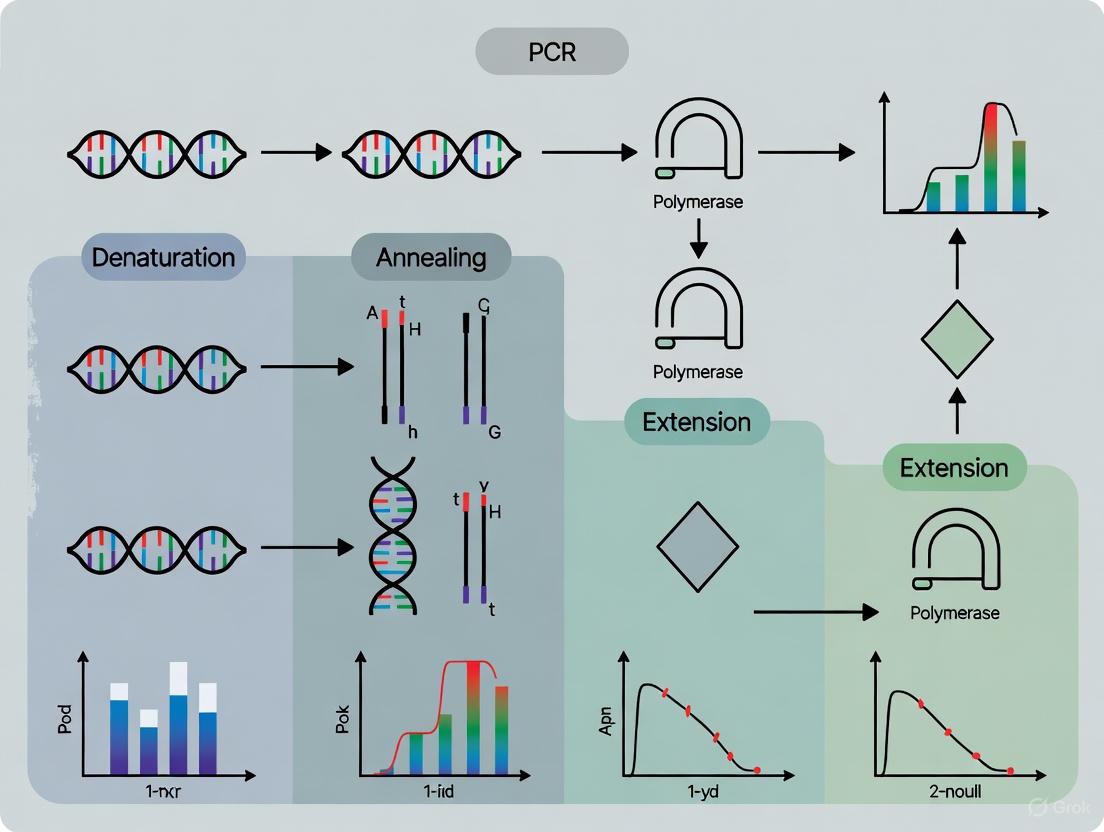
The formation and amplification of a primer dimer occur in a series of steps, as illustrated below [1]:
This process is often initiated at low temperatures, such as during reaction setup, where DNA polymerase can still exhibit some enzymatic activity [1] [5]. Primers with complementary regions, especially at their 3' ends, are particularly prone to this phenomenon [1].
How Can I Detect Primer Dimers?
Accurate detection is the first step in troubleshooting. The methods vary between conventional and quantitative PCR.
| Method | How Primer Dimers Appear | Additional Notes |
|---|---|---|
| Gel Electrophoresis (Conventional PCR) | A smeary band or fuzzy smear typically between 30-100 bp, well below the expected target amplicon [2] [4]. | Running the gel longer helps separate primer dimers from the target band. A No-Template Control (NTC) is crucial for confirmation [2]. |
| Melting Curve Analysis (qPCR with intercalating dyes) | A distinct peak at a lower temperature than the peak of the target amplicon [1]. | Primer dimers melt at lower temperatures due to their shorter length and lower GC content compared to the typically longer target product. |
| Amplification Plot (qPCR) | An amplification curve that appears earlier (lower Cq value) than the target in a No-Template Control (NTC) reaction [4]. | The short length of primer dimers allows for very efficient amplification, sometimes leading to early signal detection. |
What Causes Primer Dimer Formation?
Understanding the root causes is key to prevention. The following table summarizes the primary factors contributing to primer dimer formation.
| Category | Specific Cause | Impact on PCR |
|---|---|---|
| Primer Design & Quality | Complementarity at the 3' ends of primers (≥2 bases) [6], high primer concentration [5] [7], and poor-quality primers with truncated sequences [5] [6]. | Leads to direct initiation of the dimerization process and inefficient use of resources. |
| Reaction Conditions | Low annealing temperature [5] [8], excessive PCR cycles [5], and suboptimal Mg2+ concentration [8]. | Promotes non-specific binding and extension of primers. |
| Experimental Practice | Reaction setup at room temperature [5], early addition of non-hot-start DNA polymerase [5] [8], and contaminated reagents [8]. | Allows low-temperature activity of polymerase to extend primed dimers before PCR begins. |
How Can I Prevent Primer Dimer Formation?
A multi-faceted approach is most effective for minimizing primer dimers. The strategies below are listed from most critical and common to more advanced.
Optimize Primer Design
This is the most fundamental prevention strategy.
- Use Software: Utilize primer design software (e.g., Primer3) to check for self-complementarity, hairpins, and cross-complementarity between primers [1] [8].
- Avoid 3' Complementarity: Ensure there are no more than two to three complementary bases at the 3' ends of primer pairs [7] [6].
- Follow General Guidelines: Design primers with a length of 18-30 nucleotides, GC content of 40-60%, and closely matched melting temperatures (Tm) [9].
Apply Laboratory Best Practices
- Use Hot-Start DNA Polymerase: This is a highly effective method. Hot-start polymerases are inactive at room temperature, preventing enzymatic activity during reaction setup and the initial denaturation step, thus blocking early primer dimer extension [1] [2] [8].
- Optimize Reaction Components: Lower primer concentrations (e.g., 0.1-1 µM) to reduce the chance of primer-primer interactions [8] [7]. Optimize Mg2+ concentration, as excess Mg2+ can promote non-specific amplification [8].
- Prepare Reactions on Ice: Set up PCR master mixes and reactions on ice to maintain low temperatures and minimize enzyme activity until thermal cycling begins [5].
- Use High-Quality Reagents: Purchase HPLC-purified primers to ensure sequence fidelity and minimize truncated primers that can facilitate dimerization [5] [6].
Refine Thermal Cycling Conditions
- Increase Annealing Temperature: Use a temperature gradient to find the highest possible annealing temperature that still allows specific primer-template binding. This reduces non-specific annealing [2] [8].
- Use Touchdown PCR: This technique starts with a high annealing temperature and gradually decreases it in subsequent cycles, favoring the amplification of the specific target in early cycles when primer dimers are less likely to form [10].
- Avoid Excessive Cycles: Limit the number of PCR cycles to 25-35 where possible, as extra cycles can amplify primer dimers after the target is exhausted [5].
Explore Advanced Techniques
For persistent problems, especially in sensitive or multiplexed assays, consider:
- Structural Modifications: Techniques like the Homo-Tag Assisted Non-Dimer System (HANDS) use tailed primers to form stem-loop structures that prevent dimerization [1].
- Self-Avoiding Molecular Recognition Systems (SAMRS): SAMRS involves incorporating nucleotide analogues into primers. These analogues bind to natural DNA but not to other SAMRS-containing primers, thereby avoiding primer-primer interactions [1] [10].
- Sequence-Specific Probes: In qPCR, using TaqMan probes or molecular beacons ensures that the fluorescence signal is generated only from the specific target amplicon, not from primer dimers [1].
Research Reagent Solutions
The following table outlines key reagents and their roles in preventing primer dimer formation.
| Reagent / Tool | Function in Preventing Primer Dimers |
|---|---|
| Hot-Start DNA Polymerase | Essential. Remains inactive until a high-temperature activation step, preventing low-temperature artifacts [2] [8]. |
| HPLC-Purified Primers | Ensures high primer quality and sequence accuracy, reducing dimerization from truncated sequences [5] [6]. |
| Primer Design Software | Critical for in silico checks of self-complementarity, hairpin formation, and primer-primer interactions during the design phase [1] [3]. |
| Mg2+ Optimization Kits | Allows for fine-tuning magnesium chloride concentration, a key factor in reaction specificity [8]. |
| PCR Additives (e.g., DMSO) | Can help improve specificity in difficult reactions (e.g., high GC content), but must be used judiciously as excess can promote dimers [5] [8]. |
Frequently Asked Questions (FAQs)
Q1: Are primer dimers a sign of a failed experiment? Not necessarily. The presence of a faint primer dimer band in a gel, alongside a strong, correct target band, may not invalidate an experiment [2]. However, strong dimer formation that inhibits target amplification or leads to false positives in qPCR requires troubleshooting.
Q2: Can I still use my primers if they form dimers? It depends on the severity. If the target band is strong and the dimers are faint, you may proceed. Otherwise, you can try optimizing the reaction conditions (e.g., increasing annealing temperature, lowering primer concentration). If optimization fails, redesigning the primers is the most reliable solution [5].
Q3: Why do I see primer dimers in my negative control (NTC)? This is a classic sign of primer dimer formation. Since the NTC lacks a template DNA, any amplification product is non-specific. The presence of a band in the NTC, especially a low molecular weight one, confirms that your primers are annealing to each other and being amplified [2].
Q4: What is the most critical step in preventing primer dimers? While multiple factors are important, proper primer design is the most critical foundational step. Designing primers with minimal self- and cross-complementarity, especially at the 3' ends, prevents the initiation of the dimerization process [1] [7]. Combining well-designed primers with a hot-start polymerase is a highly effective strategy for most applications.
FAQ: What are primer dimers and how do they form?
Question: What is a primer dimer and how does its formation impact my PCR results?
A primer dimer is a small, unintended DNA fragment that can form during a polymerase chain reaction (PCR) [2]. It occurs when PCR primers anneal to each other or to themselves instead of binding to their intended target sequence in the template DNA [2]. This nonspecific amplification competes with the desired reaction, reducing the yield and efficiency of your target amplicon [3]. In severe cases, particularly in quantitative PCR (qPCR), it can lead to inaccurate quantification and misinterpretation of experimental results [3].
The following diagram illustrates the two primary mechanisms of primer dimer formation.
FAQ: What is the difference between self-dimerization and cross-dimerization?
Question: What specific sequences in my primers lead to self-dimer versus cross-dimer formation?
The distinction lies in whether one primer interacts with itself or two different primers interact with each other. The table below summarizes the key differences.
| Feature | Self-Dimerization | Cross-Dimerization |
|---|---|---|
| Definition | A single primer contains regions complementary to each other, leading to intramolecular binding [2] [11]. | Two different primers (e.g., forward and reverse) have complementary regions, leading to intermolecular binding [2] [11]. |
| Primers Involved | One primer molecule folding on itself, or two identical primers binding together [12]. | The forward primer and the reverse primer bind to each other [12]. |
| Common Cause | Regions of 3 or more bases within a single primer are complementary to another region within itself (intra-primer homology) [13]. | The forward primer sequence has homology with the reverse primer sequence, especially at the 3' ends (inter-primer homology) [13]. |
| Resulting Structure | Can form hairpin loops if the complementary regions are within the same molecule [14] [13]. | Forms a short, double-stranded duplex between two separate primers [2]. |
| Sequence Check | Compare the primer to itself for complementarity [12]. | Compare the sense primer (5'-3') with the antisense primer (3'-5') for homology [12]. |
Scientist's Toolkit: Research Reagent Solutions
The following reagents and tools are essential for preventing and troubleshooting primer dimer formation.
| Reagent / Tool | Function in Preventing Primer Dimer |
|---|---|
| Hot-Start DNA Polymerase | Remains inactive at room temperature, preventing enzyme activity during reaction setup where primer dimer formation is most likely [2] [15]. |
| PCR Additives (e.g., DMSO) | Can help denature template secondary structures and improve specificity, though may require adjustment of annealing temperature [14] [15]. |
| Magnesium Chloride (MgCl₂) | A critical cofactor for DNA polymerase; its concentration must be optimized as excess Mg²⁺ can promote nonspecific amplification and primer dimers [8]. |
| Primer Design Software | Tools like NCBI Primer-BLAST, Oligo Analyzer, and commercial software calculate complementarity to predict and avoid self- and cross-dimers during the design phase [14] [11] [16]. |
| Gradient Thermal Cycler | Allows empirical determination of the optimal annealing temperature by testing a range of temperatures simultaneously, helping to find a temperature that favors specific priming [8]. |
FAQ: How can I experimentally troubleshoot and resolve primer dimer issues?
Question: I see a primer dimer band on my gel. What are the immediate steps I can take in the lab to fix this?
If you encounter primer dimers, wet-lab optimization is required. The following workflow provides a systematic troubleshooting protocol.
Experimental Protocol for Troubleshooting
Follow this detailed methodology to diagnose and resolve primer dimer formation.
Diagnosis with a No-Template Control (NTC)
- Purpose: To confirm that the observed band is a primer dimer and not a specific product or contamination.
- Procedure: Prepare a control reaction identical to your test reactions but omitting the DNA template [2]. Replace the template volume with sterile nuclease-free water.
- Interpretation: If the same small, fuzzy band (typically below 100 bp) appears in the NTC lane, it is a primer dimer. This confirms the amplification is template-independent [2].
Wet-Lab Optimization Strategies If the NTC is positive for primer dimer, implement the following changes to your PCR protocol.
Increase Annealing Temperature
- Protocol: Increase the annealing temperature in increments of 2°C [8] [17]. Use a gradient thermal cycler if available to test a range of temperatures simultaneously.
- Rationale: Higher temperatures destabilize weak, nonspecific bonds between primers, favoring only the specific primer-template binding [2] [8].
Use a Hot-Start DNA Polymerase
- Protocol: If not already using one, switch to a hot-start enzyme. Follow the manufacturer's instructions for activation (typically a 94-95°C pre-incubation step) [2] [15].
- Rationale: This enzyme is inactive during reaction setup at room temperature, preventing the polymerase from extending primers that have bound to each other during tube preparation [2] [15].
Lower Primer Concentration
Shorten Annealing Time
- Protocol: Reduce the annealing step to 5-15 seconds [17].
- Rationale: A shorter annealing time provides less opportunity for primers to form nonspecific duplexes with each other.
Ultimate Solution: Primer Redesign If optimization fails, the primers themselves are the source of the problem and must be redesigned [8] [17].
- Protocol:
- Use primer analysis software (e.g., Oligo Analyzer) before ordering new primers [16].
- Check for self-complementarity and 3'-end complementarity [11] [13].
- Avoid runs of 4 or more of a single base (e.g., GGGG) or dinucleotide repeats (e.g., ATATAT) [14] [13].
- Ensure the 3' ends of the primer pair have fewer than 4 complementary bases, especially a G or C, as this strongly promotes dimer extension [14] [12].
- Validation: Always run an NTC with newly designed primers to validate their performance before proceeding with experimental samples.
- Protocol:
Primer-dimers are short, unintended DNA fragments that form when PCR primers anneal to each other instead of the target DNA template. Their formation and subsequent amplification compete directly with the desired reaction, leading to two major negative consequences: resource consumption and reduced sensitivity, as detailed in the table below.
Table 1: Mechanisms of Resource Consumption and Sensitivity Reduction by Primer-Dimers
| Mechanism | Impact on PCR Resources | Consequence for Assay Sensitivity |
|---|---|---|
| Consumption of Primers [3] [10] | Primers are used for off-target dimer formation instead of target amplification. | Decreased yield of the desired amplicon due to reduced primer availability for the specific reaction [3] [10]. |
| Consumption of DNA Polymerase [10] | The enzyme wastefully extends the primer-dimer complex. | Reduced efficiency of target amplification, as less polymerase is available for the intended product [10]. |
| Consumption of dNTPs [10] | Nucleotides are incorporated into the primer-dimer product. | Fewer dNTPs are available for synthesis of the target DNA sequence, limiting amplification [10]. |
| Efficient Amplification [10] | The short length of primer-dimers makes them a highly efficient amplification target. | The desired, typically longer amplicon is outcompeted, especially in later PCR cycles, leading to false negatives or inaccurate quantification [10]. |
| Interference in Multiplex Assays [18] | Multiple primer pairs increase the risk of cross-reactions and dimer formation. | Can cause false negatives by weakening the signal for the intended targets and can also lead to false positives [18]. |
The following diagram illustrates the competitive process between specific target amplification and the wasteful pathway of primer-dimer formation.
FAQ 2: What experimental data quantifies the conditions for primer-dimer formation?
Understanding the specific conditions that lead to stable primer-dimer formation is crucial for prevention. Research using Free-Solution Conjugate Electrophoresis (FSCE) has provided quantitative insights into the base-pairing requirements for dimerization.
Table 2: Experimental Conditions for Stable Primer-Dimer Formation
| Experimental Variable | Quantitative Finding | Experimental Context |
|---|---|---|
| Stable Dimer Formation | Occurs when more than 15 consecutive base pairs form between primers [19]. | A study using a mobility shift assay with drag-tagged DNA oligomers. |
| Unstable Interactions | Non-consecutive base pairs did not create stable dimers, even when 20 out of 30 possible base pairs were bonded [19]. | Same FSCE study, highlighting the importance of contiguous complementarity. |
| Temperature Correlation | Dimerization was inversely correlated with temperature for duplexes with less than 30 bonded base pairs [19]. | Electrophoresis was performed at temperatures from 18°C to 62°C. |
Experimental Protocol: Quantifying Dimerization via Free-Solution Conjugate Electrophoresis (FSCE) [19]
- Oligonucleotide Design: Synthesize two 30-mer primers with designed regions of complementarity. One primer is conjugated at the 5’-end to a neutral, hydrophilic "drag-tag" (e.g., a poly-N-methoxyethylglycine) and labeled with a fluorophore (e.g., ROX). The other primer is labeled with a different fluorophore (e.g., FAM).
- Sample Annealing: Mix the drag-tagged and non-drag-tagged primers. Heat-denature the mixture at 95°C for 5 minutes, anneal at 62°C for 10 minutes, and then cool to 25°C.
- Capillary Electrophoresis: Load the annealed samples into a capillary electrophoresis system under free-solution conditions (no sieving matrix). Use a buffer such as 1x TTE (89 mM Tris, 89 mM TAPS, 2 mM EDTA) with a dynamic capillary coating.
- Temperature-Dependent Separation: Electrophorese the samples at a range of temperatures (e.g., 18°C, 25°C, 40°C, 55°C, 62°C) with a applied voltage (e.g., 15 kV).
- Analysis: Use laser-induced fluorescence (LIF) detection to distinguish the peaks of single-stranded primers and double-stranded primer-dimers. The drag-tag causes a mobility shift, allowing for precise quantification of the proportion of dimers formed at each temperature.
FAQ 3: What advanced primer technologies can prevent dimerization?
Beyond conventional optimization, several advanced technologies have been developed to fundamentally redesign primers and avoid dimerization.
Table 3: Advanced Primer Technologies to Suppress Dimer Formation
| Technology | Mechanism of Action | Key Advantage |
|---|---|---|
| Self-Avoiding Molecular Recognition Systems (SAMRS) [10] | Uses alternative nucleobases (a, t, g, c) that pair with natural bases (T, A, C, G) but not with each other. | Dramatically reduces primer-primer interactions while maintaining binding to the DNA target, improving SNP discrimination [10]. |
| Co-Primers Technology [18] | A short primer sequence is linked to a longer "capture sequence." The primer is too short to amplify alone, but the capture sequence anchors it to the target. | The primer sequence only extends if the capture sequence binds, vastly reducing primer-dimer formation and enabling robust multiplexing [18]. |
| Hot-Start DNA Polymerases [3] [2] [15] | The polymerase is inactive until a high-temperature activation step, preventing enzymatic activity during reaction setup at low temperatures. | Suppresses nonspecific amplification and primer-dimer formation that occurs before thermal cycling begins [3] [2] [15]. |
The Scientist's Toolkit: Key Research Reagent Solutions
The following reagents and tools are essential for diagnosing and preventing primer-dimer issues in PCR research.
Table 4: Essential Reagents and Tools for Managing Primer-Dimers
| Reagent / Tool | Function | Use Case in Primer-Dimer Management |
|---|---|---|
| Hot-Start DNA Polymerase [2] [15] | A modified enzyme inactive at room temperature. | Critical for preventing dimer formation during reaction setup. Activated during the initial denaturation step [2] [15]. |
| No-Template Control (NTC) [2] | A control reaction that contains all PCR components except the DNA template. | Diagnoses primer-dimer formation. Amplification in the NTC indicates primer-dimers, as they do not require a template [2]. |
| Gradient Thermal Cycler [8] | A instrument that allows different tubes to run at slightly different temperatures simultaneously. | Empirically determines the optimal annealing temperature for a primer pair to maximize specificity and minimize dimerization [8]. |
| Primer Design Software [3] [11] | An algorithm-based tool for designing oligonucleotides. | Identifies self-complementary regions and predicts potential for dimer formation before synthesis [3] [11]. |
| DMSO [20] | A PCR additive or co-solvent. | Helps denature templates with secondary structures and can optimize reactions by lowering melting temperature, improving specificity [20]. |
| SAMRS Phosphoramidites [10] | Specialized chemical building blocks for oligonucleotide synthesis. | Used to synthesize SAMRS-containing primers that avoid primer-primer interactions [10]. |
FAQ: How can I distinguish a primer-dimer band from my target PCR product on a gel?
Primer-dimers are short, unintended byproducts of the polymerase chain reaction (PCR) that can form when primers anneal to each other instead of the target DNA template. You can distinguish them from your target amplicon based on the following characteristics [2] [21]:
- Size: Primer-dimers are typically very short, usually in the range of 30-100 base pairs (bp), and often appear around 50 bp [2] [1] [21]. They will run far below the last band of a standard 100 bp DNA ladder.
- Band Appearance: They often have a fuzzy, diffuse, or smeary appearance rather than a tight, well-defined band [2].
- Location: They migrate very quickly through the gel and are found near the bottom, close to the dye front. In contrast, your target PCR product is usually larger and will be located higher up in the gel [2] [22].
To confirm a suspicious band is a primer-dimer, you can run a No-Template Control (NTC). This reaction contains all PCR components except the DNA template. If the same fuzzy, low molecular weight band appears in the NTC lane, it is almost certainly a primer-dimer, as it formed in the absence of any target DNA [2].
FAQ: What are the main causes of primer-dimer formation, and how can I prevent it?
Primer-dimer formation is primarily caused by complementarity between primers, especially at their 3' ends, which allows them to anneal to each other and be extended by the DNA polymerase [2] [1]. The table below summarizes the root causes and corresponding preventive strategies.
Table 1: Causes and Prevention of Primer-Dimer Formation
| Cause | Prevention Strategy | Key Details |
|---|---|---|
| Complementary Primers | Careful Primer Design | Use design software (e.g., Primer3, Primer-BLAST) to avoid self-complementarity and 3'-end complementarity. Ideally, there should be ≤3 complementary bases at the 3' ends [1] [23] [7]. |
| Low Stringency Annealing | Optimize Annealing Temperature | Increase the annealing temperature in increments of 1-2°C. Use a gradient thermal cycler to find the optimal temperature [2] [8]. |
| Enzyme Activity at Low Temp | Use Hot-Start DNA Polymerase | Hot-start polymerases are inactive until a high-temperature activation step, preventing spurious amplification during reaction setup [2] [1] [8]. |
| Excess Primers | Lower Primer Concentration | Reduce primer concentration to the lowest effective amount, typically between 0.1-1 µM. Perform a primer concentration gradient test [2] [7] [8]. |
| Low-Quality Primers | Ensure High Primer Quality | Old, degraded, or poorly purified primers can increase dimer formation. Use high-purity primers and store them properly in aliquots [8] [6]. |
The following diagram illustrates the core troubleshooting workflow for addressing primer-dimer issues.
Troubleshooting Guide: Step-by-Step Protocols to Minimize Primer-Dimer
Protocol 1: Optimizing PCR Conditions
This protocol outlines a systematic, experimental approach to suppress primer-dimer formation when you are using an existing set of primers.
- Prepare a Master Mix: Create a standard PCR master mix according to your protocol, but use a hot-start DNA polymerase [2] [8].
- Test a Primer Concentration Gradient: Aliquot the master mix into several tubes. Use primer concentrations ranging from 0.1 µM to 0.5 µM in 0.1 µM increments. Keeping all other variables constant, this helps identify the lowest concentration that still provides robust amplification of your target [7] [8].
- Test an Annealing Temperature Gradient: Using the optimal primer concentration from step 2, run a PCR with an annealing temperature gradient. Start 3-5°C below the calculated Tm of your primers and increase up to the Tm itself. A gradient thermal cycler is ideal for this [8].
- Analyze and Iterate: Run the products on a gel. The ideal condition is the one that yields a strong target band with little to no primer-dimer. You may need to combine the optimal primer concentration and annealing temperature in a final verification experiment.
Protocol 2: Designing Primers to Avoid Dimerization
Prevention through careful primer design is the most effective strategy.
- Use Bioinformatics Tools: Design primers using software like Primer3 or NCBI Primer-BLAST. These tools automatically check for self-complementarity and cross-complementarity between the forward and reverse primers [1] [23].
- Check the 3' Ends Manually: Ensure there are no more than 2-3 complementary bases at the 3' ends of your primer pair, as this is a major trigger for dimerization and extension [1] [6].
- Follow General Design Rules: Design primers that are 18-25 nucleotides long, with a GC content of 40-60%, and similar melting temperatures (Tm) for each member of the pair [23].
- Validate Specificity: Use a tool like BLAST to ensure your primers are specific to your intended target sequence [23].
Research Reagent Solutions
The following table lists key reagents that are essential for preventing and troubleshooting primer-dimer formation.
Table 2: Essential Reagents for Managing Primer-Dimers
| Reagent | Function in Prevention/Troubleshooting |
|---|---|
| Hot-Start DNA Polymerase | Critical for suppressing enzymatic activity during reaction setup, dramatically reducing pre-PCR primer-dimer formation [2] [1] [8]. |
| High-Purity, Quality Primers | Primers purified (e.g., HPLC-grade) to remove truncated fragments and stored correctly in aliquots reduce nonspecific interactions and dimerization [8] [6]. |
| Gel Electrophoresis System | Required for visualizing and diagnosing primer-dimers. Includes agarose, a DNA stain (e.g., ethidium bromide, SYBR Safe), a suitable buffer (TAE or TBE), and a DNA ladder [2] [22]. |
| Gradient Thermal Cycler | Instrumental for optimizing the annealing temperature, allowing you to test multiple temperatures in a single run to find the most stringent conditions that prevent primer-dimer [8]. |
| Magnesium Chloride (MgCl₂) | A key reaction component. Its concentration can be optimized (e.g., 1.5-5.0 mM); excess Mg²⁺ can promote nonspecific amplification and primer-dimer formation [23] [8]. |
What are primer dimers and how do they form?
Primer dimers (PDs) are short, unintended DNA fragments that form as a byproduct in the polymerase chain reaction (PCR) [2] [1]. They are generated when PCR primers anneal to each other via complementary base pairs, instead of binding to their intended target sequence in the template DNA [2]. The DNA polymerase can then extend these annealed primers, leading to the amplification of a short, nonspecific product [1].
Formation occurs in several steps [1]:
- Annealing: Two primers anneal to each other at their 3' ends. This can be a self-dimer (one primer annealing to itself) or a cross-dimer (the forward primer annealing to the reverse primer) [24].
- Extension: If this double-stranded structure is stable, the DNA polymerase binds and extends the primers, synthesizing a short piece of double-stranded DNA.
- Amplification: In subsequent PCR cycles, this newly synthesized short fragment can serve as a template, leading to rapid amplification of the primer dimer product.
The following diagram illustrates the mechanism of cross-primer dimer formation:
What are the primary causes of primer dimer formation?
The causes can be divided into issues related to primer design, reaction conditions, and experimental handling. The table below summarizes the most common factors.
| Category | Specific Factor | Mechanism & Impact |
|---|---|---|
| Primer Design | Complementarity at 3' Ends [1] [5] [6] | Complementary regions, especially at the 3' ends where extension begins. As few as 2-3 complementary bases can be sufficient [7] [6]. GC-rich overlaps increase stability [5]. |
| Self-Complementarity [14] [25] | A single primer has regions that are complementary to each other, leading to hairpin loops and self-dimers [14]. | |
| Poor Overall Design [5] [14] | Primers with uneven melting temperatures (Tm), long di-nucleotide repeats, or single base runs promote nonspecific binding [14]. | |
| Reaction Conditions | Low Annealing Temperature [2] [5] [24] | Allows primers to anneal to each other via weak, nonspecific interactions despite low complementarity [24]. |
| High Primer Concentration [2] [3] [7] | Increases the likelihood of primer-primer interactions. Unused primers find each other and form dimers [3] [5]. | |
| Premature Polymerase Activity [2] [24] | Before the PCR begins, the reaction mixture is at room temperature, allowing standard polymerases to extend primers that have loosely annealed [24]. | |
| Excessive Cycle Number [5] | Once the target is amplified, excess PCR cycles promote self/cross-annealing between leftover primers [5]. | |
| Template & Reagents | Low Template Quality/Quantity [5] | With little or no target DNA available, primers are more likely to find and bind to each other [5]. |
| Poor Quality Primers [5] [6] | Impure primers (e.g., with truncated sequences) can have unpredictable binding and promote dimerization [5]. | |
| Suboptimal Mg²⁺ Concentration [5] [14] | Excess Mg²⁺ can increase non-specific binding and facilitate primer-dimer formation [5]. |
How can I detect primer dimers in my experiments?
Gel Electrophoresis
After agarose gel electrophoresis, primer dimers have distinct characteristics [2]:
- Short Length: Typically appear as a band or smear below 100 bp, often near the bottom of the gel.
- Smeary Appearance: They look like a fuzzy, diffuse band rather than a sharp, well-defined one.
Tip: Running the gel for a longer period can help separate primer dimers from your desired PCR product, which is usually larger and migrates more slowly [2].
No-Template Control (NTC)
Including an NTC is a crucial diagnostic. The NTC contains all PCR reagents except the template DNA. If amplification occurs in the NTC, it is almost certainly due to primer-dimer formation or contamination, as there is no target for the primers to bind to [2] [24].
Quantitative PCR (qPCR) Analysis
In qPCR using intercalating dyes like SYBR Green, primer dimers can be detected using melting curve analysis [1]. Because primer dimers are short, they denature (melt) at a lower temperature than the longer, specific PCR product. A secondary peak at a lower melting temperature indicates the presence of primer dimers [1].
What detailed protocols can I use to troubleshoot and prevent primer dimers?
Protocol A: Optimize Primer Design and In Silico Analysis
The most effective solution is to prevent primer dimers at the design stage [2] [14].
- Use Primer Design Software: Utilize tools like Primer-Blast or Primer3 to select primers with low self-complementarity and low 3'-end complementarity within the pair [2] [14].
- Check for Dimers Manually: Use online oligo analyzer tools to check for potential self-dimers and cross-dimers. Avoid primers with more than two complementary bases at their 3' ends [5] [7] [25].
- Follow Design Rules:
Protocol B: Optimize Thermal Cycling and Reaction Conditions
If dimers persist, wet-lab optimization is required.
- Increase Annealing Temperature: This is one of the most effective wet-lab steps. Perform a temperature gradient PCR (e.g., testing from 55°C to 68°C) to find the highest possible temperature that still allows specific amplification but discourages nonspecific primer binding [2] [5].
- Use a Hot-Start DNA Polymerase: Hot-start polymerases are inactive at room temperature. They are only activated after a high-temperature heating step (e.g., 95°C), preventing enzyme activity during reaction setup and the initial warm-up phase, where most primer dimers form [2] [1] [24].
- Lower Primer Concentration: Test a range of primer concentrations (e.g., 0.1-0.5 µM) to find the lowest concentration that still supports robust amplification of your target. This reduces the chance of primer-primer interactions [2] [3] [7].
- Prepare Reactions on Ice: Keep all reagents and the reaction tube on ice during setup. Add the polymerase last, and immediately transfer the tube to a pre-heated thermal cycler to minimize time for primer interaction at low temperatures [5].
Protocol C: Advanced and Alternative Techniques
For stubborn cases or highly multiplexed PCR, consider these advanced strategies.
- Use Modified Primers: Techniques like SAMRS incorporate alternative nucleobases that pair with natural DNA but not with other SAMRS nucleotides, thereby avoiding primer-primer interactions [10].
- Employ Probe-Based Detection: In qPCR, switch from SYBR Green to sequence-specific probes (e.g., TaqMan probes). These probes will only generate a fluorescent signal if the correct target is amplified, preventing false positives from primer dimers, though dimers may still consume reagents [1] [24].
- Try a Touchdown PCR: This protocol starts with an annealing temperature higher than the calculated Tm and gradually decreases it in subsequent cycles. This favors amplification of the specific target in the early cycles, giving it a competitive advantage over primer dimers that form at lower temperatures [10].
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function in Preventing Primer Dimers |
|---|---|
| Hot-Start DNA Polymerase | Essential. Remains inactive until a high-temperature activation step, preventing extension of primerdimers formed during reaction setup [2] [1]. |
| High-Purity (HPLC Purified) Primers | Ensures primers are full-length and free of truncated sequences that can cause nonspecific amplification and dimerization [5]. |
| Optimized PCR Buffer | Provides the correct ionic strength (e.g., K⁺) and pH. May contain additives that enhance specificity [14]. |
| Magnesium Chloride (MgCl₂) Solution | A critical cofactor for polymerase activity. Its concentration must be optimized, as too much can promote non-specific binding and dimer formation [5] [14]. |
| DMSO, Betaine, or Other Additives | Can help improve specificity and reduce secondary structures, especially for GC-rich templates. However, they must be used judiciously as they can sometimes exacerbate dimer issues [5] [14]. |
| No-Template Control (NTC) Reagents | A critical diagnostic tool. Sterile water used in place of template DNA to confirm that amplification signals are not due to contamination or primer dimers [2]. |
Strategic Primer Design and Reaction Setup to Suppress Dimerization
In polymerase chain reaction (PCR) research, the specificity and efficiency of the entire experiment hinge on the initial design of the primers. Properly designed primers are the most critical factor in preventing the formation of primer-dimers, a common cause of failed experiments and ambiguous results. Primer-dimers are short, unintended amplification artifacts that form when primers anneal to each other instead of the target DNA template, consuming reaction resources and potentially outcompeting the desired product [2]. This guide outlines the fundamental rules of primer design, framed within the context of a broader thesis on preventing primer-dimer formation, to equip researchers with the knowledge to design robust assays from the outset.
The Three Golden Rules of Primer Design
The following rules form the cornerstone of effective primer design. Adhering to them significantly reduces the risk of non-specific amplification and primer-dimer formation.
Primer Length
Primer length is the primary determinant of specificity. Excessively long primers reduce hybridization efficiency, while overly short primers compromise specificity.
Recommendation: Aim for primers between 18 and 30 nucleotides in length [26] [11]. This range provides an optimal balance, ensuring specific binding to a unique sequence within a complex genome while maintaining efficient annealing.
Melting Temperature (Tm)
The melting temperature (Tm) is the temperature at which half of the DNA duplex dissociates into single strands. For PCR, the Tm determines the appropriate annealing temperature (Ta).
Recommendations:
- Aim for a primer Tm between 54°C and 65°C, with an ideal range often cited as 65°C to 75°C [26] [11].
- The Ta is typically set 3–5°C below the lowest Tm of the primer pair [11].
- The forward and reverse primers should have Tm values within 5°C of each other to ensure synchronized binding during the annealing step [26].
GC Content
The GC content refers to the percentage of guanine (G) and cytosine (C) bases in the primer. GC base pairs form three hydrogen bonds, compared to the two formed by AT pairs, directly influencing primer stability and Tm.
Recommendation: Maintain a GC content between 40% and 60% [26] [11]. This prevents the formation of overly stable secondary structures while providing sufficient binding strength.
A GC clamp is highly recommended. This involves having a G or C base at the 3' end of the primer, which promotes stronger binding due to the additional hydrogen bond. However, avoid more than three consecutive G or C bases at the 3' end, as this can promote non-specific binding [26] [11].
The following table summarizes these core parameters for easy reference.
| Design Parameter | Optimal Range | Importance for Specificity & Preventing Primer-Dimer |
|---|---|---|
| Primer Length | 18 - 30 nucleotides [26] [11] | Short primers may bind non-specifically; long primers hybridize inefficiently. |
| Melting Temperature (Tm) | 65°C - 75°C (or 54°C - 65°C) [26] [11] | Primers with closely matched Tm (within 5°C) anneal synchronously. |
| GC Content | 40% - 60% [26] [11] | Prevents overly stable mispriming; balanced distribution of strong/weak bonds. |
| GC Clamp | G or C at the 3' end [26] | Promotes specific binding at the site of enzyme extension; avoid >3 consecutive G/C. |
The Scientist's Toolkit: Essential Reagents for PCR
Selecting the right reagents is crucial for successful amplification, especially when optimizing to prevent primer-dimer formation.
| Reagent / Material | Function & Importance |
|---|---|
| Hot-Start DNA Polymerase | A modified enzyme inactive at room temperature, preventing non-specific priming and primer-dimer formation during reaction setup. It is activated only at high denaturation temperatures [2] [8]. |
| MgCl2 Solution | A co-factor essential for DNA polymerase activity. Its concentration must be optimized, as excess Mg2+ can reduce specificity and promote non-specific amplification [8] [27]. |
| PCR Additives (e.g., DMSO, GC Enhancers) | Help denature complex templates (e.g., GC-rich sequences) and minimize secondary structures, improving primer binding specificity and yield [8] [27]. |
| Nuclease-Free Water | The solvent for all reaction components. Must be pure and free of nucleases to prevent degradation of primers, template, and PCR products. |
| Purified Primer Stocks | Primers should be resuspended in nuclease-free water or TE buffer, aliquoted to avoid repeated freeze-thaw cycles, and stored properly to maintain stability [8]. |
Experimental Protocol: A Method for Primer Design and Validation
This protocol provides a step-by-step methodology for designing and testing primers with a focus on minimizing primer-dimer artifacts.
Step 1: In Silico Primer Design
- Sequence Acquisition: Obtain the target DNA sequence from a trusted database like NCBI.
- Parameter Setting: Use a reputable primer design tool (e.g., from Eurofins Genomics, Thermo Fisher) and input the following criteria:
- Length: 18-30 nt
- Tm: 65-75°C
- GC Content: 40-60%
- Specificity Check: Use BLAST to verify primer specificity to the intended target.
- Homology Analysis: Analyze the primer sequence for self-complementarity (risk of hairpins) and inter-primer complementarity (risk of cross-dimers). The parameters "self-complementarity" and "self 3'-complementarity" should be as low as possible [11].
Step 2: Primer Ordering and Preparation
- Synthesis and Purification: Order primers with standard purification (e.g., cartridge purification). For cloning applications, a higher purification grade is recommended [26].
- Resuspension and Storage: Resuspend primers in nuclease-free water or TE buffer to create a concentrated stock (e.g., 100 µM). Aliquot and store at -20°C.
Step 3: Experimental Validation and Optimization
- Initial PCR Setup: Set up a standard PCR reaction with a positive control (known template) and a critical No-Template Control (NTC). The NTC contains all reagents except the DNA template and is essential for detecting primer-dimer formation and contamination [2].
- Annealing Temperature Gradient: Use a thermal cycler with a gradient function to test a range of annealing temperatures (e.g., from 3-5°C below the calculated Tm to 2-3°C above it).
- Analysis: Analyze the PCR products and NTC on an agarose gel. The optimal condition will show a single, sharp band of the expected size in the test reaction and a clear NTC with no smeary bands.
Troubleshooting Guide & FAQs
This section directly addresses common issues researchers encounter, with a specific focus on problems related to primer design and primer-dimer formation.
Frequently Asked Questions
Q1: What exactly is a primer dimer, and why is it problematic? A primer dimer is a small, unintended DNA fragment that forms when PCR primers anneal to each other via complementary regions instead of to the template DNA. DNA polymerase then extends these primers, creating a short product [2]. Primer dimers are problematic because:
- They compete with the desired product for reaction resources (enzyme, nucleotides, primers), reducing amplification efficiency and yield.
- In quantitative PCR (qPCR), they can lead to false positive signals, severely compromising data accuracy.
- They appear as a fuzzy smear or band below 100 bp on an agarose gel [2].
Q2: I see a fuzzy band around 50-100 bp in my PCR and my no-template control. What is this? This is almost certainly a primer dimer. The confirmation comes from its presence in the No-Template Control (NTC), as primer dimers do not require a DNA template to form. Their smeary appearance and short length are telltale signs [2].
Q3: My primers were designed with good parameters, but I still get primer dimers. What wet-lab steps can I take? If primer design is not the issue, wet-lab optimization is key:
- Increase Annealing Temperature: Raise the temperature in 1-2°C increments to discourage loose, non-specific binding [2] [8].
- Use a Hot-Start Polymerase: This enzyme is inactive during reaction setup, preventing primer-dimer formation at room temperature [2] [27].
- Lower Primer Concentration: High primer concentrations promote primer-primer interactions. Test a concentration gradient from 0.1–1 µM to find the lowest effective concentration [8] [7].
- Increase Denaturation Time: This helps ensure primers and template are fully dissociated, making primers more available for specific binding [2].
Q4: How can I identify a primer dimer in my gel results?
- Size: Typically appears below 100 bp. Run the gel long enough to separate them from your desired product.
- Appearance: Looks like a fuzzy, diffuse smear rather than a sharp, defined band.
- Control: Will be present in the No-Template Control (NTC) lane [2].
Q5: What specific sequence features in my primers should I avoid to prevent dimers? During the in silico design phase, strictly avoid:
- Complementary 3' Ends: Even 2-3 complementary bases at the 3' ends of the forward and reverse primers can lead to cross-dimer formation [7].
- Runs of Identical Bases: Avoid stretches of 4 or more of the same base (e.g., AAAA or GGGG) [26].
- Repeated Dinucleotides: Avoid repeats like ATATAT, which can misalign [26].
- Intra-primer Homology: Regions within a single primer that are complementary can form hairpin loops [26] [11].
Troubleshooting Table
| Observation | Possible Cause (Related to Design or Conditions) | Recommended Solution |
|---|---|---|
| No PCR Product | Tm calculation error; annealing temperature too high [8] [27]. | Recalculate Tm; perform an annealing temperature gradient. |
| Poor primer specificity or binding site secondary structures [8]. | Verify specificity with BLAST; redesign primers if needed; use PCR additives. | |
| Primer concentration too low [8]. | Test primer concentrations from 0.1–1 µM. | |
| Multiple Bands or Smearing | Annealing temperature too low [8] [27]. | Increase annealing temperature stepwise. |
| Primers bind to non-specific sites [8]. | Check primer design for specificity; increase annealing temperature. | |
| Excess Mg2+ or primers [8] [27]. | Optimize Mg2+ concentration; lower primer concentration. | |
| Primer Dimers (fuzzy band in NTC) | Complementary sequences between primers, especially at 3' ends [26] [7]. | Redesign primers to avoid 3' complementarity. |
| Low annealing temperature [2]. | Increase annealing temperature. | |
| High primer concentration [8] [7]. | Lower primer concentration. | |
| Use of non-hot-start polymerase [2] [27]. | Switch to a hot-start DNA polymerase. |
In polymerase chain reaction (PCR) research, the precision of your results is fundamentally dictated by the design of your primers. Among the most critical aspects of primer design is the management of the 3' end, which directly influences the specificity and efficiency of DNA amplification. Improper complementarity at the 3' end is a primary cause of primer-dimer formation, a common artifact that consumes reaction reagents and competes with the amplification of your target DNA [3] [1]. This guide provides targeted troubleshooting and FAQs to help you optimize this crucial part of your primer design, thereby preventing primer-dimer formation and ensuring the success of your experiments.
Frequently Asked Questions (FAQs)
1. Why is the 3' end of a PCR primer so critical?
The 3' end of a primer is where DNA polymerase adds new nucleotides to extend the DNA chain [1]. If this region is complementary to another primer in the reaction, the polymerase can mistakenly extend it, leading to the formation of a primer-dimer [2] [12]. These short, unintended DNA fragments reduce reaction efficiency by depleting essential reagents and can complicate the interpretation of your results, especially in quantitative PCR.
2. What is a GC clamp and how does it help?
A GC clamp refers to the presence of one or two guanine (G) or cytosine (C) bases within the last five nucleotides at the 3' end of a primer [11] [28]. Since G-C base pairs are bound by three hydrogen bonds (as opposed to the two in A-T pairs), they form stronger, more stable bonds [28]. This promotes specific and complete binding of the primer to its intended target template, enhancing the overall specificity of the amplification [28].
3. How much complementarity is too much between primers?
Primers should have fewer than 4 complementary bases at their 3' ends [12]. This is especially critical in multiplex PCR reactions, where multiple primer pairs are present, increasing the chance of intermolecular interactions. Tools for checking "self-complementarity" and "self 3'-complementarity" should be used during design, and these values should be kept as low as possible [11].
4. Can a 3' end mismatch completely block amplification?
The effect of a single mismatch at the 3' end depends on its nature. Research on the human β-globin gene has shown that a G/T mismatch may still allow efficient amplification, whereas G/A or G/G mismatches can severely reduce or even prevent the production of a specific PCR product [29]. This principle is leveraged in techniques like allele-specific PCR to distinguish between single-nucleotide variants.
Troubleshooting Guide: Primer-Dimer Formation
| Problem | Recommended Action | Protocol / Details |
|---|---|---|
| Visible primer-dimer band on gel (low molecular weight smear ~30-50 bp) [2] | 1. Optimize Primer Design: Check for 3' complementarity. Use design software to ensure low self-complementarity scores [3] [11].2. Increase Annealing Temperature: Raise temperature in 1-2°C increments to discourage nonspecific binding [2] [8].3. Use Hot-Start Polymerase: Prevents enzyme activity during reaction setup, reducing low-temperature artifacts [3] [2]. | Protocol: No-Template Control (NTC)• Include a control reaction with all PCR components except the DNA template.• If primer-dimer appears in the NTC, the issue is with primer design or reaction conditions, not the template [2]. |
| Low amplification yield (suspected reagent competition by primer-dimer) | 1. Lower Primer Concentration: Test primer concentrations in the range of 0.1–1 μM [8]. A lower ratio of primer to template can help [2].2. Optimize Mg2+ Concentration: High Mg2+ can promote nonspecific amplification. Titrate Mg2+ concentration downward [8]. | Protocol: Magnesium Titration• Set up a series of reactions with MgCl₂ concentrations varying from, for example, 0.5 mM to 3.0 mM in 0.5 mM increments.• Identify the lowest concentration that provides robust target amplification without nonspecific products [8]. |
| Persistent dimers with well-designed primers | 1. Touchdown PCR: Start with an annealing temperature above the calculated Tm and decrease it incrementally over subsequent cycles. This enriches specific targets early on [8].2. Use PCR Additives: Add co-solvents like DMSO or betaine to help disrupt secondary structures that might promote dimerization [8]. | Protocol: Hot-Start Activation• Ensure an initial prolonged denaturation step (e.g., 95°C for 5 minutes) if using a chemically modified hot-start polymerase to fully activate the enzyme [8]. |
Experimental Data and Design Parameters
Effect of 3' End Mismatches on PCR Efficiency
The following table summarizes experimental data from a study investigating the amplification of a 268 bp region of the human β-globin gene using primers with different 3' terminal mismatches [29].
| 3' End Mismatch Type | Amplification Efficiency | Key Findings |
|---|---|---|
| G/C (Match) | High | Efficient amplification across all tested annealing temperatures (45°C - 65°C). |
| G/T | High | Nearly as efficient as the matched primer at all temperatures. |
| G/A | Very Low / None | No specific PCR fragment detected at any annealing temperature. |
| G/G | Very Low | A barely detectable specific product only at lower temperatures (45°C, 50°C). |
Optimal Primer Design Parameters
Adhering to these established design parameters during the initial primer synthesis phase can preemptively avoid many common issues [11].
| Parameter | Optimal Range | Rationale |
|---|---|---|
| Primer Length | 18 - 24 nucleotides | Balances specificity with efficient hybridization [11]. |
| GC Content | 40% - 60% | Provides sufficient duplex stability without promoting mispriming [11]. |
| GC Clamp | 1-2 G/C bases in the last 5 nucleotides at the 3' end | Strengthens terminal binding; >3 can cause non-specific binding [11] [28]. |
| Melting Temperature (Tm) | 54°C - 65°C; forward and reverse primers should be within 2°C | Ensures both primers anneal efficiently at the same temperature [11]. |
Workflow and Visualization
The following diagram illustrates a systematic workflow for troubleshooting and optimizing PCR reactions to prevent primer-dimer formation, integrating the strategies discussed above.
Research Reagent Solutions
The following reagents are essential for implementing the troubleshooting and optimization strategies outlined in this guide.
| Reagent / Tool | Function in Optimization |
|---|---|
| Hot-Start DNA Polymerase | Remains inactive until high temperature is reached, preventing primer-dimer formation during reaction setup [3] [2]. |
| dNTP Mix | Deoxynucleoside triphosphates (dATP, dCTP, dGTP, dTTP) are the building blocks for DNA synthesis; unbalanced concentrations can increase error rates [8]. |
| Magnesium Chloride (MgCl₂) | A crucial co-factor for DNA polymerase; its concentration must be optimized as both low and high levels can cause amplification issues [8]. |
| Primer Design Software | Automated tools check for self-complementarity, hairpins, and calculate Tm, helping to design primers with low dimerization potential [3] [11]. |
| PCR Additives (e.g., DMSO, Betaine) | Co-solvents that help denature GC-rich templates and disrupt secondary structures, improving amplification specificity and yield [8]. |
Primer-dimer is a common yet challenging issue in polymerase chain reaction (PCR) that can significantly compromise experimental results. These short, unintended amplification artefacts form when PCR primers anneal to each other instead of the target DNA template, leading to reduced amplification efficiency, consumption of reaction reagents, and inaccurate data interpretation, particularly in quantitative applications [3] [2]. Bioinformatics tools offer powerful solutions for predicting and preventing primer-dimer formation during the experimental design phase, enabling researchers to achieve higher specificity and reliability in their PCR results.
Frequently Asked Questions (FAQs)
What is primer-dimer and how does it affect my PCR results?
Primer-dimer refers to small, double-stranded DNA fragments that form when PCR primers anneal to each other through complementary regions, creating free 3' ends that DNA polymerase can extend [3] [2]. This nonspecific amplification competes with target amplification, reducing yield, exhausting reaction components, and potentially leading to false positives or inaccurate quantification in qPCR experiments [5]. In gel electrophoresis, primer-dimers typically appear as smeary bands below 100 bp [2].
How accurate are computational tools at predicting primer-dimer formation?
The accuracy of prediction tools varies significantly. A 2019 systematic evaluation of seven publicly available dimer prediction tools found that algorithms using ROC analysis-optimized Gibbs free energy (ΔG) calculations, such as PrimerROC, achieved predictive accuracies greater than 92% [30]. This comprehensive study demonstrated that condition-independent prediction is feasible, with PrimerROC consistently outperforming other tools across different primer sets [30].
Can I completely eliminate primer-dimer through bioinformatics tools alone?
While bioinformatics tools significantly reduce primer-dimer risks, complete elimination often requires a combined approach of thoughtful primer design and optimized reaction conditions [5]. Computational tools excel at identifying problematic primer pairs during design, but laboratory techniques like hot-start PCR and annealing temperature optimization provide additional safeguards during amplification [15].
Troubleshooting Guide: Addressing Primer-Dimer Issues
Problem: Persistent primer-dimer formation despite using design tools
Possible Causes and Solutions:
- Insufficiently stringent design parameters: Ensure your primer design tool checks for 3' end complementarity, with recommended thresholds of ≤3 contiguous complementary bases at the 3' ends [23].
- Suboptimal laboratory conditions: Implement hot-start DNA polymerase to prevent nonspecific amplification during reaction setup [3] [15].
- Excessive primer concentration: Optimize primer concentrations (typically 0.1-1 μM), as high concentrations promote primer-primer interactions [5] [8].
Problem: Discrepancy between prediction and experimental results
Possible Causes and Solutions:
- Tool limitations: Use tools with demonstrated high accuracy, such as PrimerROC, which employs condition-independent prediction models [30].
- Unaccounted reaction conditions: Validate predictions empirically using a no-template control (NTC) to confirm primer-dimer formation [2].
- Primer quality issues: Use high-quality, HPLC-purified primers to minimize synthesis artifacts that might contribute to dimerization [5].
Comparison of Primer Analysis Tools
Table 1: Features of Major Primer Design and Analysis Platforms
| Tool Name | Key Features | Dimer Prediction Method | Special Considerations |
|---|---|---|---|
| Primer-BLAST [31] | Combines Primer3 with BLAST search for specificity checking | ΔG calculations with user-defined parameters | Verifies primer specificity against selected databases |
| PrimerROC [30] | Condition-independent prediction using ROC analysis | Optimized ΔG with bonus/penalty system for extensible dimers | Demonstrates >92% accuracy; particularly effective for multiplex PCR |
| PrimerQuest (IDT) [32] | Customization of ~45 parameters | Algorithm includes multiple checks for dimer formation | Provides flexible sequence entry and batch processing |
| Eurofins Primer Design [33] | Based on Prime+ of GCG Wisconsin Package | Avoids primers with extensive self-dimer and cross-dimer formation | Considers salt and primer concentration in Tm calculations |
| Oligo 7 [30] | Comprehensive primer design suite | ΔG-based calculations with condition adjustments | Performed well in comparative studies, particularly with longer primers |
Table 2: Technical Specifications for Effective Primer Design to Minimize Dimer Formation
| Parameter | Optimal Range | Rationale |
|---|---|---|
| Primer Length | 18-30 bases [23] | Balances specificity and binding energy |
| GC Content | 40-60% [23] | Prevents overly stable or unstable hybrids |
| Tm Difference | ≤5°C between primers [23] | Ensures balanced annealing kinetics |
| 3' End Complementarity | ≤3 contiguous bases [23] | Minimizes primer-dimer initiation sites |
| Self-Complementarity | ≤3 contiguous bases [23] | Reduces hairpin structure formation |
| Annealing Temperature | 3-5°C below lowest primer Tm [8] | Optimizes specificity while preventing dimer formation |
Experimental Protocols
Protocol 1: Comprehensive Primer Design Workflow Using Bioinformatics Tools
Materials Needed:
- Template DNA sequence
- Access to primer design tool (e.g., Primer-BLAST, PrimerROC)
- Computer with internet connection
Step-by-Step Methodology:
Sequence Preparation: Obtain your target DNA sequence in FASTA format. Identify the specific region you wish to amplify [33].
Parameter Setting: Configure the primer design tool with the following optimal parameters:
Specificity Checking: Enable specificity checks against appropriate databases (e.g., RefSeq mRNA for human transcripts) [31]. For gene-specific amplification, select "Primer must span an exon-exon junction" to avoid genomic DNA amplification [31].
Dimer Evaluation: Use the tool's dimer prediction function or a dedicated tool like PrimerDimer to analyze all possible primer-primer interactions (forward-forward, reverse-reverse, and forward-reverse) [30].
Primer Selection: Choose primer pairs with the most favorable scores, prioritizing those with no 3' end complementarity and high specificity [23].
Figure 1: Bioinformatics workflow for dimer-free primer design
Protocol 2: Laboratory Validation of Primer Specificity
Materials Needed:
- Designed primers
- DNA template
- PCR reagents (buffer, dNTPs, DNA polymerase)
- Thermal cycler
- Gel electrophoresis equipment
Step-by-Step Methodology:
Reaction Setup: Prepare PCR reactions with your designed primers. Always include a no-template control (NTC) to detect primer-dimer formation [2].
Thermal Cycling: Use a touchdown PCR protocol if possible: start with an annealing temperature 3-5°C above the calculated Tm, then decrease 1°C per cycle for 5-10 cycles until reaching the optimal annealing temperature [15].
Analysis: Run PCR products on a 2-3% agarose gel. Primer-dimers will appear as smeary bands below 100 bp, distinct from your specific amplicon [2].
Troubleshooting: If dimers persist, consider increasing annealing temperature, reducing primer concentration, or using hot-start DNA polymerase [5] [8].
Research Reagent Solutions
Table 3: Essential Reagents for Minimizing Primer-Dimer Formation
| Reagent/Resource | Function | Implementation Example |
|---|---|---|
| Hot-Start DNA Polymerase [3] [15] | Remains inactive until high-temperature activation, preventing nonspecific amplification during reaction setup | Use for all PCR applications, particularly multiplex and qPCR |
| PCR Additives (DMSO, GC Enhancers) [8] | Helps denature complex templates, reducing primer competition | Employ for GC-rich targets or complex templates |
| High-Quality Primer Synthesis [5] | Minimizes truncated primers that contribute to nonspecific amplification | Request HPLC purification for critical applications |
| Bioinformatics Tools [31] [30] | Predicts potential dimer formation during design phase | Integrate into standard primer design workflow |
| Gradient Thermal Cycler [8] | Enables empirical optimization of annealing temperature | Use for testing multiple annealing temperatures simultaneously |
Effective primer-dimer prediction and prevention requires a multifaceted approach combining sophisticated bioinformatics tools with optimized laboratory techniques. By leveraging condition-independent prediction algorithms like PrimerROC and adhering to established primer design parameters, researchers can significantly reduce primer-dimer formation and improve the reliability of their PCR results. Regular validation using no-template controls and gel electrophoresis remains essential for confirming computational predictions and ensuring experimental success.
Troubleshooting Guides
How do I optimize primer concentration to prevent primer-dimer formation?
The Problem: Excessive primer concentration is a common cause of primer-dimer formation. When primers are too abundant, they are more likely to anneal to each other instead of the target DNA template, leading to nonspecific amplification and reduced PCR efficiency [2] [24].
The Solution: Systematically test a range of primer concentrations to find the optimal balance that minimizes dimers while maximizing specific product yield [34].
Experimental Protocol: Primer Concentration Optimization
- Prepare a Primer Matrix: Create a series of reactions where the forward and reverse primer concentrations are varied independently. A typical testing range is between 50 nM and 600 nM for each primer [34].
- Use a Standard PCR Mix: Keep all other reaction components constant, including template DNA, Mg²⁺ concentration, polymerase, and dNTPs.
- Perform Amplification: Run the PCR using your standard cycling conditions.
- Analyze Results: Evaluate the results via gel electrophoresis or qPCR analysis. The optimal concentration combination is the one that yields the lowest Cq value (for qPCR), the strongest specific band, and a negative no-template control (NTC), indicating minimal primer-dimer formation [34].
Table 1: Guidelines for Primer Concentration Optimization
| Parameter | Recommended Range | Effect of High Concentration | Effect of Low Concentration |
|---|---|---|---|
| Primer Concentration | 0.1 - 1.0 µM [8] [20] | Increased primer-dimer formation and non-specific binding [2] [8] | Reduced amplification efficiency and yield [8] |
| Optimal for SYBR Green I | 200 - 400 nM [34] | High fluorescence background in NTC | Weak amplification signal |
How does template quality affect my PCR and how can I improve it?
The Problem: Poor template quality or the presence of PCR inhibitors can lead to failed amplification, non-specific products, smearing, or primer-dimer accumulation as the reaction falters [35] [8].
The Solution: Ensure the use of high-quality, pure template DNA and optimize its quantity in the reaction [36].
Experimental Protocol: Assessing and Optimizing Template Quality
- Assess Purity and Integrity:
- Optimize Template Quantity:
- Use Appropriate Polymerases: Select DNA polymerases with high processivity, as they are more tolerant to inhibitors commonly found in biological samples like blood, plants, or soil [8].
Table 2: Template Quality and Quantity Troubleshooting
| Issue | Cause | Solution |
|---|---|---|
| Low Purity | Contaminants like phenol, EDTA, heparin, or proteins [35] [8] | Re-purify template using spin columns or ethanol precipitation. Use inhibitor-tolerant polymerases [8]. |
| Poor Integrity | Degraded or sheared DNA [8] | Isolate fresh DNA using a gentle extraction method to minimize nicking. |
| Insufficient Quantity | Too few copies of the target sequence [8] | Increase the amount of input template or the number of PCR cycles. |
| Excessive Quantity | Too much template DNA [8] | Reduce the amount of input template to prevent non-specific amplification. |
Frequently Asked Questions (FAQs)
Why do I still get primer-dimers even when my primer concentrations are optimized?
Even with optimized concentrations, primer-dimers can form due to other factors. The most common reason is complementary sequences at the 3' ends of your primers [2] [24]. Other reasons include an annealing temperature that is too low, or polymerase activity at room temperature during reaction setup. To address this, redesign primers to avoid 3' complementarity, increase the annealing temperature, and always use a hot-start DNA polymerase to prevent pre-PCR amplification [3] [2] [15].
What is the optimal primer-to-template ratio?
There is no single universal ratio, as it depends on the complexity of the template DNA. For example, 30-100 ng of human genomic DNA is often optimal, while for plasmid or abundant genes, 10 ng may be sufficient [20]. The key is to achieve a lower primer-to-template ratio, which gives primers a higher probability of finding the target sequence rather than each other [2]. This is best determined empirically through a template dilution series.
How can I quickly identify primer-dimers in my results?
In gel electrophoresis, primer-dimers have two key characteristics [2]:
- Short length: They typically run below 100 bp.
- Smeary appearance: They look like a fuzzy, indistinct band rather than a sharp, well-defined one. Running a no-template control (NTC) is crucial. Any amplification product in the NTC is almost certainly a primer-dimer or contaminant, as it was formed without a template [2] [34].
Experimental Workflow and Relationships
The following diagram summarizes the key components and their interactions for successful PCR optimization.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for PCR Optimization
| Reagent / Tool | Function / Purpose | Optimization Consideration |
|---|---|---|
| Hot-Start DNA Polymerase | Prevents enzymatic activity at room temperature, drastically reducing primer-dimer formation before PCR begins [2] [15]. | Essential for all PCR setups to enhance specificity. |
| dNTP Mix | Building blocks for DNA synthesis. | Use balanced equimolar concentrations (typically 20-200 µM each); unbalanced dNTPs can increase error rate [20]. |
| Magnesium Ions (Mg²⁺) | Essential cofactor for DNA polymerase activity [35] [20]. | Critical parameter; titrate between 1.5 - 5.0 mM. Too little reduces yield; too much promotes non-specific binding [35] [8]. |
| PCR Additives (DMSO, BSA) | DMSO helps denature GC-rich templates; BSA can bind and neutralize inhibitors in the reaction [35] [20]. | Use sparingly (e.g., DMSO at 2-10%); may require lowering annealing temperature [35] [20]. |
| No-Template Control (NTC) | A control reaction containing all components except template DNA. | Critical for diagnosing contamination and primer-dimer formation. Any amplification in the NTC indicates a problem [2] [34]. |
FAQs: Understanding and Preventing Primer-Dimer
What is a primer-dimer and why is it a problem in PCR? A primer-dimer is a short, double-stranded DNA artifact formed when PCR primers anneal to each other instead of to the target DNA template. This occurs due to complementary regions within or between the primers, leading to nonspecific amplification [3]. Primer-dimers negatively impact PCR results by reducing the efficiency of target amplification, decreasing the yield of the desired product, consuming reaction reagents, and complicating the interpretation of results, especially in quantitative applications [3].
What are the primary causes of primer-dimer formation? The common causes include [3]:
- Primer Design: Presence of complementary sequences, especially at the 3' ends of primers.
- Reaction Conditions: Using excessively high primer concentrations or suboptimal (too low) annealing temperatures.
- Enzyme Activity: Nonspecific polymerase activity during reaction setup at room temperature.
How can primer design minimize primer-dimer formation? Follow these key design principles [11] [37]:
- Length: Design primers between 18 and 24 nucleotides.
- GC Content: Maintain a GC content between 40% and 60%.
- 3' End Stability: Avoid stretches of 3 or more G or C bases at the 3' end (a GC clamp of 1-2 bases is beneficial, but more can promote non-specific binding).
- Self-Complementarity: Use design software to check and minimize parameters for "self-complementarity" and "self 3'-complementarity" to prevent hairpins and primer-dimers.
Troubleshooting Guides
Guide 1: Addressing Primer-Dimer and Non-Specific Amplification
| Symptom | Possible Cause | Recommended Solution |
|---|---|---|
| Faint, fast-migrating bands (primer-dimers) on gel [3] | Primers annealing to each other; Low annealing temperature; Polymerase active at room temp [3] | Optimize primer design [11]; Increase annealing temperature [38]; Use a Hot-Start DNA polymerase [39] [15] [40] |
| Multiple non-specific bands on gel [38] | Mispriming; Low annealing/extension specificity [38] | Employ Touchdown PCR [15] [38]; Optimize Mg²⁺ concentration [37]; Use a gradient thermal cycler to find optimal annealing temperature [37] |
| No product or weak target band | Primer-dimers consuming reagents; Overly stringent conditions [3] [41] | Use Nested PCR for difficult templates [15] [41]; Verify primer and template quality/quantity [37] |
Guide 2: Optimizing Specialized PCR Protocols
| Method | Core Principle | Best for Troubleshooting |
|---|---|---|
| Hot-Start PCR [39] [15] [40] | DNA polymerase is chemically inactivated or blocked (e.g., by antibody, aptamer) until initial denaturation at high temp. | Room-temperature setup; Reactions with high primer concentration; Multiplex PCR; Routine prevention of pre-amplification artifacts. |
| Touchdown PCR [15] [38] | PCR starts with high annealing temp (above primer Tm), then temp decreases incrementally over cycles to the optimal Tm. | When primer Tm is difficult to calculate; When non-specific bands persist after standard optimization. |
| Nested PCR [15] [41] | Two consecutive PCR rounds: 1st with outer primers, 2nd with nested primers binding inside the 1st amplicon. | Low template samples; Highly complex templates (e.g., genomic DNA); Reactions with significant non-specific amplification in the first round. |
Research Reagent Solutions
| Reagent / Tool | Function in Preventing Primer-Dimer | Examples & Notes |
|---|---|---|
| Hot-Start DNA Polymerase [15] [40] [42] | Prevents enzymatic activity during setup, reducing primer-dimer and non-specific synthesis. | Antibody-based (Platinum Taq, DreamTaq HS); Chemical modification (AmpliTaq Gold); Aptamer-based (AptaTaq). Choose based on stringency, activation time, and component origin needs [40]. |
| Primer Design Software [3] [11] [43] | Automates checks for self-complementarity, hairpins, and optimal Tm/GC content. | Primer3, Primer-BLAST, Eurofins Genomics tools. Use to enforce design rules and assess specificity [11] [43]. |
| PCR Additives [15] [41] | Can help destabilize non-specific primer-template interactions and secondary structures. | DMSO is common for GC-rich templates. Use judiciously as it can lower primer Tm [15]. |
| Optimized Buffer Systems [15] [37] | Provides optimal pH, salt, and Mg²⁺ concentrations for specific polymerase fidelity and processivity. | Often supplied with enzyme. Mg²⁺ concentration is critical and may require titration [37]. |
Experimental Protocols & Workflows
Protocol 1: Hot-Start PCR
Detailed Methodology:
- Reaction Setup: Assemble all PCR components (template, primers, dNTPs, buffer, Mg²⁺, and Hot-Start polymerase) at room temperature [39] [40].
- Initial Activation/Denaturation: Place tubes in thermocycler and run first step at 95°C for 2-10 minutes. This heat step simultaneously activates the polymerase by dissociating the antibody or chemical modifier and denatures the template DNA [15] [40].
- Cycling:
- Denature: 95°C for 15-30 seconds.
- Anneal: 45-65°C for 15-30 seconds (optimize for primer set).
- Extend: 72°C for 1 minute per kb.
- Repeat for 25-35 cycles.
- Final Extension: 72°C for 5-10 minutes [39].
Protocol 2: Touchdown PCR
Detailed Methodology [38]:
- Reaction Setup: Keep reagents on ice and use a Hot-Start polymerase for maximum specificity.
- Initial Denaturation: 95°C for 2-3 minutes.
- Touchdown Phase (Stage 1): 10-15 cycles of:
- Denature: 95°C for 30 seconds.
- Anneal: Start at 10°C above the calculated Tm (e.g., 67°C for a 57°C Tm), then decrease by 1°C per cycle.
- Extend: 72°C for 30-45 seconds.
- Standard Phase (Stage 2): 20-25 cycles of:
- Denature: 95°C for 30 seconds.
- Anneal: Use the final, optimal temperature from the touchdown phase (e.g., 57°C).
- Extend: 72°C for 30-45 seconds.
- Final Extension: 72°C for 5 minutes.
Protocol 3: Nested PCR
Detailed Methodology [15] [41]:
- First Round PCR:
- Use the outer primer pair that flanks the target region.
- Perform 15-20 cycles of standard PCR.
- This amplifies the target but may also produce non-specific products.
- Second Round PCR:
- Dilute the product from the first reaction (e.g., 1:50).
- Use a small aliquot (1-2 µl) as the template for a new PCR.
- Use the nested primer pair that binds within the first amplicon.
- Perform 20-25 cycles of standard PCR.
- The nested primers will only amplify the correct product from the first round, dramatically increasing specificity.
In polymerase chain reaction (PCR) research, the unintended formation of primer dimers is a significant obstacle to experimental success. These artifacts are small, spurious DNA fragments that form when PCR primers anneal to each other instead of to the intended target DNA template [2]. This nonspecific amplification consumes reaction resources—including primers, polymerase, and nucleotides—thereby reducing the yield and sensitivity of the desired amplification product [10]. The challenge intensifies with advanced applications requiring high sensitivity, excellent single-nucleotide polymorphism (SNP) discrimination, or high levels of multiplexing, where multiple targets are amplified simultaneously [10] [44].
Self-Avoiding Molecular Recognition Systems (SAMRS) offer a sophisticated chemical solution to this problem. SAMRS are synthetically modified DNA nucleobases designed to bind complementarily with natural DNA but not with other SAMRS analogs [45]. By incorporating these bases into PCR primers, researchers can create primers that efficiently amplify natural target DNA while strategically avoiding the primer-primer interactions that lead to dimer formation [10] [46].
Mechanism: How SAMRS Nucleotides Work
The core principle of SAMRS technology lies in the strategic re-engineering of the hydrogen-bonding patterns of nucleobases. The standard SAMRS components include 2-Aminopurine (A*), N4-Ethyl-2'-deoxycytidine (C*), 2'-Deoxyinosine (G*), and 2'-Deoxy-2-thiothymidine (T*) [45] [46].
A SAMRS-modified primer maintains the critical ability for SAMRS:Natural base pairing. For example, SAMRS base A* pairs with natural T, and T* pairs with natural A. These interactions are comparable in strength to a natural A:T pair, allowing the primer to bind stably to its natural DNA template and initiate polymerization [10] [45].
The key innovation is the suppression of SAMRS:SAMRS base pairing. When two SAMRS-containing primers encounter each other, the potential for hydrogen bonding between their modified bases is dramatically weakened or eliminated. For instance, while natural adenine and thymine form two hydrogen bonds, the A:T pair is thermodynamically disfavored, often forming only one hydrogen bond or experiencing steric hindrance [45] [46]. This effectively prevents the primers from initiating extension off one another, thereby suppressing primer-dimer formation.
The following diagram visualizes this selective binding logic:
Designing Effective SAMRS-Modified Primers
Successful implementation of SAMRS requires careful primer design. The goal is to incorporate enough SAMRS components to disrupt primer-primer interactions without significantly impairing the primer's ability to bind its target.
Strategic Placement and Number of Modifications
Incorporating SAMRS modifications is a balancing act. The following table summarizes the key design rules derived from experimental studies [10] [46]:
| Design Parameter | Recommendation | Rationale |
|---|---|---|
| Primer Length | At least 20 nucleotides | Ensures sufficient contact area with the target despite the slightly weaker SAMRS:natural binding [46]. |
| Number of SAMRS Bases | 1 to 3 modifications per primer | Provides sufficient "self-avoidance" without critically destabilizing primer-template binding [46]. |
| 3'-End Position | Should be a natural DNA base | The polymerase's extension efficiency is highest when the 3'-terminal base is natural [46]. |
| Modification Strategy | Place SAMRS bases in regions of primer-primer complementarity | Strategically disrupts intermolecular interactions between forward and reverse primers [10]. |
Relative Destabilization of SAMRS Components
Not all SAMRS bases impact duplex stability equally. When designing primers, consider their relative destabilizing effects, from least to most impactful [46]:
- T* (2-Thio-dT): Least destabilizing.
- A* (2-Aminopurine).
- C* (N4-Ethyl-dC).
- G* (2'-Deoxyinosine): Most destabilizing. G-rich primers generally have lower amplification efficiency.
Experimental Data and Protocol
Impact on Melting Temperature and SNP Discrimination
The incorporation of SAMRS bases affects the melting temperature of the primer-template duplex. Research has quantified this effect to guide experimental design. The table below summarizes the change in melting temperature (ΔTm) for different SAMRS:natural base pairs compared to their natural:natural counterparts, as measured in PCR buffer [10]:
| Base Pair | Experimentally Observed ΔTm (°C) |
|---|---|
| A*:T | -1.5 to -2.5 |
| T*:A | -2.0 to -3.0 |
| G*:C | -3.5 to -4.5 |
| C*:G | -4.0 to -5.0 |
This predictable lowering of Tm is a critical factor for calculating the appropriate annealing temperature in a PCR protocol.
The value of SAMRS extends beyond preventing primer dimers. Because SAMRS primers are less tolerant of mismatches, they can provide superior discrimination of single-nucleotide polymorphisms (SNPs) compared to conventional allele-specific PCR. Studies have shown that with appropriately chosen polymerases, SAMRS-based PCR can achieve exceptional specificity, distinguishing between alleles that differ by only a single base [10] [44].
Step-by-Step Experimental Protocol
This protocol outlines the process for testing and implementing SAMRS-modified primers in a PCR experiment, based on methodologies from the literature [10].
Step 1: Primer Design and Synthesis
- Design primers according to the rules in Section 3.1.
- Synthesize SAMRS-containing oligonucleotides using standard phosphoramidite chemistry. SAMRS phosphoramidites are commercially available and couple with the same efficiency as standard bases [10].
- Purify primers via ion-exchange HPLC to a high purity standard (>85-90%) to ensure optimal performance [10].
Step 2: Resuspension and Storage
- Resuspend purified primers in molecular-grade water or TE buffer (pH 8.0).
- Aliquot and store properly at recommended temperatures to prevent degradation [8].
Step 3: PCR Setup with Optimized Components
- Use a hot-start DNA polymerase. This is highly recommended to further suppress nonspecific amplification and primer-dimer formation that can occur during reaction setup [8] [2].
- Optimize primer concentration. A typical starting range is 0.1–1 μM. Using a concentration gradient (e.g., 0.1, 0.5, 1.0 μM) can help identify the lowest concentration that yields robust amplification without dimers [8] [7].
- Include a No-Template Control (NTC). This is essential for diagnosing primer-dimer formation, as dimers will amplify in the absence of any target DNA [2].
Step 4: Thermal Cycling
- Calculate annealing temperature. Use the predicted Tm of your SAMRS-modified primer (accounting for the ΔTm values in the table above) and a standard formula (e.g., Tm - 3°C to 5°C). Using a thermal cycler with a gradient function to test a range of annealing temperatures is ideal [8].
- Cycling profile: A typical three-step protocol is recommended:
- Initial Denaturation/Activation: 95°C for 2-3 minutes (activates hot-start polymerase).
- Amplification (35-40 cycles):
- Denature: 95°C for 10-30 seconds.
- Anneal: At optimized temperature (from gradient) for 15-45 seconds.
- Extend: 72°C for 15-60 seconds per kilobase of amplicon.
- Final Extension: 72°C for 5-10 minutes.
Step 5: Analysis
- Analyze PCR products by gel electrophoresis. Primer dimers typically appear as a fuzzy smear or band below 100 bp [2].
- Compare your test reactions with the NTC to confirm that the desired amplicon band is specific to the presence of the template.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in SAMRS Experiment |
|---|---|
| SAMRS Phosphoramidites (A, C, G, T) | Building blocks for the solid-phase synthesis of SAMRS-modified oligonucleotides [10] [46]. |
| Ion-Exchange HPLC Columns | For purification of synthesized SAMRS primers to achieve the required >85% purity for reliable diagnostics [10]. |
| Hot-Start DNA Polymerase | A proofreading or high-fidelity enzyme that remains inactive until a high-temperature step, minimizing nonspecific amplification prior to PCR cycling [8]. |
| dNTPs | Standard deoxynucleotide triphosphates (dATP, dCTP, dGTP, dTTP) serve as the building blocks for the nascent DNA strand during amplification [8]. |
| Thermal Cycler with Gradient Function | Essential for empirically optimizing the annealing temperature for SAMRS-modified primers, which have altered melting temperatures [8]. |
| Buffer Additives (e.g., DMSO, GC Enhancer) | Can be used to help denature complex templates (e.g., GC-rich sequences) and improve amplification efficiency, though concentrations must be optimized [8]. |
FAQs and Troubleshooting
Q1: Can I create a primer made entirely of SAMRS bases? No. Primers should be at least 20 nucleotides long and contain only 1-3 SAMRS modifications. Using too many SAMRS bases can critically impair the primer's ability to bind stably to its natural DNA target [46].
Q2: My SAMRS-PCR has low yield. What should I check?
- Annealing Temperature: This is the most common issue. Re-calculate the Tm using the ΔTm penalties for your specific SAMRS bases and run a gradient PCR to find the optimal temperature.
- Primer Concentration: Test a range of concentrations. High primer concentrations promote dimer formation, while very low concentrations can reduce yield.
- 3'-Terminus: Verify that the base at the 3'-end of your primer is a natural DNA base, as this is critical for efficient polymerase extension [46].
Q3: Are SAMRS compatible with other PCR techniques beyond conventional PCR? Yes. SAMRS technology has been successfully applied in various isothermal amplification techniques, including Recombinase Polymerase Amplification (RPA) and Loop-Mediated Isothermal Amplification (LAMP), where it similarly helps to prevent spurious amplification products [46].
Q4: I see a band in my No-Template Control (NTC). Does this mean SAMRS failed? Not necessarily. A band in the NTC confirms the presence of primer-dimers. However, the intensity of this band should be compared to that of reactions with template. A successful SAMRS implementation will show a strong specific band in the test reaction and a very faint or absent band in the NTC, indicating that primer-dimer formation has been significantly reduced, allowing resources to be channeled into specific amplification [2].
Systematic Troubleshooting and Protocol Optimization for Pristine Results
FAQs
What does a primer-dimer look like on a gel?
Primer-dimers are short, unintended DNA fragments that appear as a fuzzy smear or a distinct band at the bottom of an agarose gel, typically below 100 base pairs (bp) and often in the 30-50 bp range [2] [21] [1]. In contrast, your target amplicon is usually a sharp, well-defined band higher up on the gel, corresponding to the expected size of your specific PCR product [2].
Why are primer-dimers a problem?
Primer-dimers compete with your target DNA for essential PCR reagents, such as primers, nucleotides, and DNA polymerase. This competition can reduce the efficiency and yield of your desired amplification product [3] [1]. In quantitative PCR (qPCR), they can also interfere with accurate fluorescence quantification, leading to false positives or inaccurate data [1].
How can I confirm that a band is a primer-dimer and not my target?
The most reliable method is to run a No-Template Control (NTC). This reaction contains all PCR components except the DNA template. If the band in question appears in the NTC lane, it confirms the band is a primer-dimer, as these can form and amplify without a template [2].
My gel shows a smear instead of distinct bands. Is this related to primer-dimers?
A smear can have several causes, but it is often a sign of non-specific amplification [21]. While a primer-dimer is one type of non-specific product, a general smear can also be caused by degraded DNA template, primers binding to non-target sequences, or an annealing temperature that is too low [21]. A primer-dimer-specific smear is usually located very low on the gel [2].
Troubleshooting Guide: Strategies to Minimize Primer-Dimer Formation
The following table summarizes the key strategies for preventing primer-dimer formation, from primer design to reaction setup and cycling conditions.
| Strategy | Implementation | Rationale |
|---|---|---|
| Primer Design [2] [11] | Use software to design primers with low self-complementarity and 3'-end complementarity. Avoid 3' GC-rich ends. | Prevents primers from annealing to themselves or each other, which is the primary cause of dimer formation. |
| Primer Concentration [2] [47] | Lower primer concentration (typical range 0.1-1 µM); optimize for your reaction. | Reduces the chance of primer-primer interactions by lowering primer-to-template ratio. |
| Hot-Start Polymerase [2] [8] | Use a hot-start DNA polymerase. | Inactivates the enzyme until the high-temperature denaturation step, preventing enzymatic activity during reaction setup at low temperatures. |
| Annealing Temperature [2] [8] | Increase the annealing temperature incrementally (1-2°C at a time). | Promotes stringent binding of primers only to their perfect target sequences, reducing non-specific annealing. |
| Reaction Setup [8] [21] | Assemble reactions on ice and use pre-heated thermal cyclers. | Minimizes non-specific primer interactions before the PCR cycle begins. |
Experimental Protocol: Using a No-Template Control (NTC)
Purpose: To definitively identify primer-dimer artifacts in your PCR results [2].
Materials:
- Same PCR master mix used for your experimental samples.
- Nuclease-free water.
- PCR tubes.
Method:
- When preparing your PCR reactions, allocate enough master mix for at least one extra tube.
- For the NTC tube, add all reaction components—except the DNA template.
- Replace the volume of the missing template with nuclease-free water.
- Run the NTC alongside your experimental samples under identical PCR cycling conditions.
- Analyze all reactions together on an agarose gel.
Interpretation: Any amplification product visible in the NTC lane is the result of primer-artifact amplification (e.g., primer-dimer) and is not derived from your template. Bands of the same size in your sample lanes can therefore be identified as non-target products [2].
Research Reagent Solutions
The following reagents are essential for troubleshooting and preventing primer-dimer formation.
| Reagent | Function in Prevention |
|---|---|
| Hot-Start DNA Polymerase [2] [8] | A modified enzyme that is inactive at room temperature, preventing primer-dimer extension during reaction setup. It is activated only at high temperatures (e.g., 94-95°C). |
| PCR Additives (e.g., DMSO, Betaine) [14] | Can help improve specificity and reduce non-specific annealing, especially for complex templates, which can indirectly suppress competing reactions like primer-dimer formation. |
| Mg²⁺ Ions [8] [47] | A critical cofactor for DNA polymerase. Its concentration can be optimized (typically 0.5-5.0 mM); lowering it can sometimes increase reaction specificity and reduce primer-dimer artifacts. |
Workflow for Troubleshooting Primer-Dimers
The diagram below outlines a logical, step-by-step workflow for diagnosing and addressing primer-dimer issues in your experiments.
NTC Troubleshooting Guide
Why is my No-Template Control (NTC) showing amplification?
If you observe amplification in your NTC, it typically indicates one of two main issues: contamination of your reaction components or the formation of primer-dimers. Correctly identifying the cause is the first step toward resolving the problem [48] [49].
The table below summarizes the key characteristics and solutions for each scenario.
| Cause | Characteristics of Amplification | How to Investigate | Primary Solutions |
|---|---|---|---|
| DNA Contamination [48] [49] | - Amplicon size matches your target product- CT values may be random or consistent across replicates | - Run a melt curve analysis; the peak should match the target.- Check if the band in gel electrophoresis is the same size as the expected product. | - Use separate pre- and post-PCR work areas [49].- Decontaminate workspaces with 10% bleach or UV light [50] [49].- Use aliquoted reagents and filter tips [50] [49].- Incorporate UNG/UDG to prevent carryover contamination [48]. |
| Primer-Dimer Formation [48] [2] [49] | - Low molecular weight product (often <100 bp)- In a melt curve, shows a low Tm peak distinct from the target- In a gel, appears as a fuzzy smear near the bottom [2] | - Perform melt curve analysis to identify a low-temperature peak.- Run gel electrophoresis; primer-dimers appear as a faint, fast-moving band/smear [2]. | - Optimize primer design to avoid 3' complementarity [7] [2].- Increase annealing temperature [2].- Use a hot-start DNA polymerase [2].- Lower primer concentration [7]. |
My NTC is clean, but my sample reactions are inefficient. Could primer-dimers still be a problem?
Yes. Even if primer-dimers are not visible in the NTC, they can form in sample reactions and compete for reagents, reducing the efficiency and yield of your target amplification [48] [3]. This is a common challenge in PCR research that necessitates careful optimization.
Frequently Asked Questions (FAQs)
Q1: What is the fundamental purpose of a No-Template Control (NTC)? The NTC is a critical quality control used to detect contamination in your PCR reagents. It contains all reaction components—master mix, primers, water—except for the template DNA. Any amplification in the NTC indicates that one or more of your reagents are contaminated with nucleic acid, casting doubt on all results from that run [51] [49].
Q2: How can I distinguish between contamination and primer-dimer formation in my NTC? The most reliable method is to analyze the amplification product. If you are using SYBR Green chemistry, perform a melt curve analysis. A single, sharp peak at the same melting temperature (Tm) as your positive sample indicates target contamination. A peak at a lower, different Tm suggests primer-dimer formation [48]. In gel electrophoresis, a band identical in size to your target points to contamination, while a small, faint band or smear at the bottom of the gel (<100 bp) is characteristic of primer-dimers [2] [49].
Q3: I work with bacterial 16S rRNA genes and consistently get false positives in my NTC. What should I do? This is a common issue because 16S rRNA sequences are ubiquitous and can be present in reagents, including some polymerase enzymes [50]. Solutions include:
- Choose a hypervariable region: Design your primers to target a unique hypervariable region of the 16S rRNA gene rather than a highly conserved one [50].
- Use a different master mix: Test different master mixes, as some are certified to be free of bacterial DNA contamination [50].
- Employ blocking oligos: Use peptide nucleic acid (PNA) clamps or other blocking oligos to suppress the amplification of contaminating bacterial DNA [50].
Q4: What are the best laboratory practices to prevent NTC contamination?
- Physical Separation: Maintain separate, dedicated areas for pre-PCR (reaction setup) and post-PCR (product analysis) work. Never bring amplified PCR products into the clean pre-PCR area [48] [49].
- Dedicated Equipment: Use a dedicated set of pipettes, tips (always use filter tips), and lab coats for the pre-PCR area [49].
- Reagent Management: Aliquot all reagents (polymerase, water, primers, dNTPs) into single-use volumes to minimize the risk of contaminating your entire stock [50] [49].
- Rigorous Decontamination: Regularly clean workspaces and equipment with a 10% bleach solution or commercial DNA decontaminants. Use UV irradiation in PCR hoods when available [50] [49].
Experimental Protocol: Diagnosing and Resolving NTC Amplification
Follow this step-by-step workflow to systematically address amplification in your No-Template Control.
Research Reagent Solutions
The following table lists key reagents and materials essential for implementing effective NTCs and preventing related issues like primer-dimer formation.
| Reagent / Material | Function in Control and Troubleshooting |
|---|---|
| Hot-Start DNA Polymerase | Remains inactive until a high-temperature step, preventing non-specific primer extension and primer-dimer formation during reaction setup [2]. |
| AmpErase UNG / UDG | Enzyme that degrades PCR products from previous reactions (carryover contamination) by breaking down uracil-containing DNA, preventing false positives in the NTC [48]. |
| Nuclease-Free Water | Sterile, DNA/RNA-free water used to prepare master mixes and NTCs, ensuring it is not a source of contamination [49]. |
| Filter Pipette Tips | Contain a barrier to prevent aerosols and liquids from contaminating the pipette shaft, a major vector for DNA cross-contamination [49]. |
| Optimized Primer Pairs | Primers designed with software to lack self-complementarity and 3' end complementarity, minimizing the potential for primer-dimer formation [7] [2]. |
Primer Optimization Table
If primer-dimer formation is the suspected or confirmed cause, optimizing your primer concentrations is a critical step. The table below, derived from experimental optimization strategies, provides a template for testing different primer combinations [48].
| Reverse Primer (nM) | Forward Primer: 100 nM | Forward Primer: 200 nM | Forward Primer: 400 nM |
|---|---|---|---|
| 100 nM | 100/100 | 200/100 | 400/100 |
| 200 nM | 100/200 | 200/200 | 400/200 |
| 400 nM | 100/400 | 200/400 | 400/400 |
The combination that yields the strongest target signal with the least primer-dimer should be selected for future experiments [48].
FAQs on Temperature Optimization for PCR
1. Why is the annealing temperature critical for preventing primer-dimer formation? The annealing temperature is critical because primer-dimers form when primers anneal to each other instead of the target DNA, which occurs more readily at low temperatures [2]. A sufficiently high annealing temperature ensures stable and specific binding between the primer and its intended target sequence, while discouraging these nonspecific interactions [24]. If the temperature is too low, spurious priming and primer-dimer formation are likely; if it is too high, primer binding may be inefficient, leading to reduced or no amplification [52].
2. How do I determine the starting point for my annealing temperature? A good starting point for your annealing temperature is 3–5°C below the calculated melting temperature (Tm) of your primers [8] [53]. For optimal results, the forward and reverse primers should have Tms within 2–5°C of each other, allowing you to select a single, effective annealing temperature for both [11] [54] [14].
3. What is the relationship between denaturation time and complex DNA templates? Complex templates, such as those with high GC content, can form strong secondary structures that are difficult to denature [8]. For such templates, increasing the denaturation time and/or temperature can help to fully separate the DNA strands, making them more accessible for primer binding [8]. Standard denaturation times are typically 15-30 seconds at 95°C, but GC-rich sequences may require longer durations [54].
4. When should I consider using a specialized polymerase? Hot-start DNA polymerases are highly recommended to prevent primer-dimer formation. These enzymes remain inactive until a high-temperature activation step (e.g., 95°C), thereby preventing any enzymatic activity during reaction setup on the bench where primer-dimers are most likely to form [2] [8]. For templates with complex secondary structures or high GC content, polymerases with high processivity are also beneficial [8].
5. How can I systematically optimize the annealing temperature? The most robust method is to perform a gradient PCR [52]. This involves running identical reactions across a range of annealing temperatures (e.g., from 55°C to 65°C) in a single thermocycler run [52]. The optimal temperature is the one that produces the highest yield of the specific product and the lowest level of nonspecific products or primer-dimers, as verified by gel electrophoresis [8] [52].
Key Temperature Parameters for PCR Optimization
The following table summarizes the standard and optimization ranges for critical temperature-related parameters in PCR.
| Parameter | Standard / Starting Range | Optimization Considerations |
|---|---|---|
| Primer Melting Temp (Tm) | 52–65°C [11] [54] [14] | Ensure both primers have Tms within 2–5°C of each other [54] [14]. |
| Annealing Temperature (Ta) | 3–5°C below the primer Tm [8] [53] | Optimize using a gradient PCR; increase temperature to enhance specificity [8] [52]. |
| Initial Denaturation | 95°C for 2 minutes [54] | Increase time or temperature for GC-rich templates (>60% GC) [8]. |
| Cycle Denaturation | 95°C for 15–30 seconds [54] | Increase to 10–30 seconds for GC-rich templates or templates with strong secondary structures [8]. |
| Final Extension | 68–72°C for 5–15 minutes [54] [8] | Ensures all amplicons are fully replicated. |
Experimental Protocol: Optimizing Annealing Temperature via Gradient PCR
This protocol provides a detailed methodology for determining the optimal annealing temperature for a primer set.
1. Prepare the Master Mix
- Calculate the number of reactions (e.g., 8 for a temperature gradient) plus one for a negative control.
- In a nuclease-free tube, combine the following components on ice for a single 50 µl reaction [54] [14]:
- Mix the master mix thoroughly by pipetting up and down. Gently vortex and briefly centrifuge to collect the contents at the bottom of the tube [14].
2. Aliquot and Set Up Controls
- Aliquot an equal volume of the master mix into each PCR tube.
- For the No-Template Control (NTC), add water instead of DNA template to one tube. This is crucial for detecting primer-dimer formation or contamination [2] [52].
3. Program the Thermocycler
- Use the following three-step cycling program, setting the annealing step to a gradient across the desired range (e.g., 55°C to 65°C) [52]:
| Step | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 95°C | 2 minutes | 1 |
| Denaturation | 95°C | 15–30 seconds | 25–35 |
| Annealing | Gradient (e.g., 55°C–65°C) | 15–30 seconds | 25–35 |
| Extension | 68–72°C | 1 minute per kb | 25–35 |
| Final Extension | 68–72°C | 5–15 minutes | 1 |
| Hold | 4–10°C | ∞ | 1 |
4. Analyze Results
- Run the PCR products on an agarose gel [14].
- The optimal annealing temperature is identified by the lane that produces the brightest, correct-sized band and the absence of a smear or primer-dimer band (which typically appears as a fuzzy smear below 100 bp) [2].
- Confirm the results by comparing the NTC to the sample lanes; the NTC should show no amplification [2].
Troubleshooting Guide for Temperature-Related PCR Issues
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Primer-dimer formation | Annealing temperature too low [2] [3] | Increase annealing temperature in 1–2°C increments [2] [8]. |
| High primer concentration [8] | Lower primer concentration (e.g., to 0.1–0.5 µM) [54] [8]. | |
| Enzyme activity during setup [2] | Use a hot-start DNA polymerase [2] [8]. | |
| No PCR product | Annealing temperature too high [52] | Lower the annealing temperature stepwise [8] [52]. |
| Inefficient denaturation [8] | Increase denaturation time or temperature, especially for GC-rich templates [8]. | |
| Non-specific bands/smearing | Low annealing temperature [8] | Increase annealing temperature [8] [52]. |
| Excessive cycle number [8] | Reduce the number of PCR cycles [8]. | |
| Low yield | Suboptimal denaturation [8] | Increase denaturation time for complex templates [8]. |
| Insufficient extension time | Use extension time of 1 minute per 1 kb [54] [53]. |
Research Reagent Solutions for PCR Optimization
| Reagent / Material | Function / Role in Optimization |
|---|---|
| Hot-Start DNA Polymerase | Prevents enzymatic activity before the initial denaturation step, drastically reducing primer-dimer formation during reaction setup [2] [8]. |
| Gradient Thermocycler | Allows a single PCR run to test a range of annealing temperatures, enabling rapid and systematic optimization [8] [52]. |
| dNTPs (Deoxynucleotides) | Building blocks for DNA synthesis. Unbalanced concentrations can increase error rates; typical final concentration is 200 µM of each dNTP [54] [8]. |
| Magnesium Chloride (MgCl₂) | Cofactor for DNA polymerase. Concentration (typically 1.5-2.0 mM) must be optimized, as it profoundly affects primer annealing, specificity, and yield [54] [8]. |
| PCR Additives (e.g., DMSO) | Can help denature templates with high GC content or strong secondary structures, improving amplification efficiency [8] [53]. |
For researchers, scientists, and drug development professionals, achieving specific and efficient amplification in the polymerase chain reaction (PCR) is critical. A common and persistent challenge in this process is the formation of primer-dimers and other non-specific products, which can drastically reduce assay sensitivity, compromise data accuracy, and hinder downstream applications. Within the broader context of preventing primer-dimer formation, the selection of the appropriate DNA polymerase is a fundamental decision. This guide details the advantages of Hot-Start DNA polymerases, providing a technical resource to troubleshoot issues and enhance experimental outcomes by suppressing the mechanisms that lead to off-target amplification [55] [56].
The Core Problem: Primer-Dimer and Mis-Priming
What are primer-dimers and how do they form? A primer-dimer is a small, unintended DNA fragment that forms when PCR primers anneal to each other instead of to the specific target DNA template. This can occur through self-dimerization of a single primer or, more commonly, cross-dimerization between forward and reverse primers [2]. When these primers bind to each other, their 3' ends provide a free starting point for DNA polymerase to extend, creating short, spurious products [2] [14].
Why are they problematic? Primer-dimers and other mis-primed products compete with the desired amplification target for essential reaction components, including primers, dNTPs, and DNA polymerase [57]. This competition leads to:
- Reduced yield of the specific target amplicon.
- Lower sensitivity, making detection of low-copy-number targets difficult or impossible.
- Unreliable results that are challenging to interpret and quantify, especially in quantitative PCR (qPCR) where primer-dimers can generate false positive signals [2] [57].
The greatest amount of primer-dimer formation often occurs before the thermal cycling even begins, during reaction setup at room temperature, where DNA polymerase can be active enough to extend these mistakenly paired primers [55] [2].
FAQs on Hot-Start DNA Polymerases
1. What is Hot-Start PCR and how does it prevent primer-dimer formation? Hot-Start PCR is a modified technique that inhibits DNA polymerase activity during the reaction setup at lower temperatures (e.g., room temperature or on ice) [56]. By keeping the polymerase inactive, it prevents the enzyme from extending primers that have bound to each other (forming primer-dimers) or to non-specific, partially homologous regions on the DNA template (mis-priming) [55] [58]. The polymerase is only activated at high temperatures (typically during the initial denaturation step at 95°C), ensuring that primer extension begins under the stringent conditions of the thermal cycler [56].
2. What are the key benefits of using a Hot-Start DNA polymerase?
- Prevents Primer-Dimer Formation: Blocks extension of primers that have bound to each other during reaction setup [55].
- Reduces Mis-Priming: Inhibits extension of primers bound to template sequences with low homology [55].
- Increases Target Yield and Sensitivity: By eliminating competing off-target amplification, more reagents are available for the desired product, leading to higher yields and better detection of low-abundance targets [55] [57].
- Enables Robust Workflows: Reactions are stable at room temperature, making them suitable for high-throughput or automated liquid-handling platforms without compromising specificity [55] [58].
- Provides Cleaner Results: Results in gels or qPCR plots are easier to interpret, with minimal background noise [2].
3. I am setting up a multiplex PCR assay with significant primer-dimer issues. Can Hot-Start help? Yes, Hot-Start technology is particularly beneficial for multiplex PCR. These assays contain multiple primer pairs, dramatically increasing the probability of cross-talk and primer-dimer formation between different primers [57]. Hot-Start polymerases, or the use of modified Hot-Start primers, have been shown to significantly reduce this off-target amplification, allowing for efficient co-amplification of multiple targets without extensive re-optimization and improving the detection limit for each target in the reaction [57].
4. Are there any limitations or special considerations when using Hot-Start polymerases? While highly beneficial, consider the following:
- Activation Time: Some methods, particularly chemical modification, require a longer initial denaturation step (e.g., 10 minutes) to fully activate the enzyme, which can be a drawback for fast-cycling protocols or could potentially damage the template DNA with extended heat [55] [56].
- Compatibility: Certain Hot-Start methods may not be compatible with procedures requiring low-temperature steps before amplification, such as some one-tube reverse transcription-PCR protocols [56].
- Cost: Hot-Start polymerases can be more expensive than their standard counterparts.
Hot-Start Mechanism Comparison and Selection Guide
Hot-Start technology employs various strategies to inhibit polymerase activity at low temperatures. The table below summarizes the common methods, their benefits, and key considerations to guide your selection.
Table 1: Comparison of Common Hot-Start Technologies
| Hot-Start Technology | Mechanism of Inhibition | Benefits | Considerations |
|---|---|---|---|
| Antibody-Based [55] | An antibody binds the polymerase's active site, blocking it. | Short activation time (released during initial denaturation); full enzyme activity restored; features similar to native polymerase. | Antibodies may be of animal origin; higher level of exogenous proteins in reaction. |
| Chemical Modification [55] | Polymerase is covalently modified with chemical groups to block activity. | Generally more stringent inhibition; free of animal-origin components. | Longer activation time required; full enzyme activity may not always be restored; can affect long target (>3kb) amplification. |
| Affibody-Based [55] | A small, engineered protein (Affibody) binds the active site. | Less exogenous protein than antibody method; short activation time; animal-origin free. | May be less stringent than antibody-based method; bench-top stability may be limited. |
| Aptamer-Based [55] | A specific oligonucleotide (aptamer) binds to the polymerase. | Short activation time; free of animal-origin components. | May be less stringent, potentially allowing some nonspecific amplification; bench-top stability may be limited. |
| Primer-Based [59] [57] | Primers are synthesized with thermolabile groups (e.g., OXP, CleanAmp) at the 3' end. | Does not require a specialized enzyme; offers flexible activation kinetics ("Turbo" vs "Precision"); compatible with many standard polymerases. | Requires synthesis of modified primers; thermolabile group must be compatible with your experimental design. |
| Physical Barrier [56] | A wax bead creates a physical barrier between polymerase and other components. | A simple, effective method to separate components. | Requires an extra heating step to melt the wax; less convenient for high-throughput setups. |
The following diagram illustrates the general mechanism of how Hot-Start polymerases remain inactive during setup and become active only at high temperatures, preventing early missteps.
Troubleshooting Guide: Addressing Primer-Dimer Formation
Even with Hot-Start polymerases, primer-dimers can occasionally occur. The following table outlines common issues and solutions.
Table 2: Troubleshooting Primer-Dimer and Non-Specific Amplification
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Pronced primer-dimer band in gel | Non-Hot-Start polymerase used; enzyme activated too early. | Switch to a Hot-Start DNA polymerase [55] [2]. Set up reactions on ice if using a standard polymerase [8]. |
| Primer concentration is too high. | Lower the primer concentration (typical range 0.1–1.0 μM) and optimize [2] [8] [9]. | |
| Primer design has complementarity at 3' ends. | Re-design primers using software tools to minimize self-complementarity and 3'-end complementarity between forward and reverse primers [2] [14] [9]. | |
| Non-specific bands and primer-dimer | Annealing temperature is too low. | Increase the annealing temperature stepwise by 1-2°C increments. Use a gradient thermal cycler if available [2] [8] [60]. |
| Excessive template or enzyme amount. | Reduce the amount of template DNA and/or decrease the units of DNA polymerase used per reaction [8] [9]. | |
| Mg²⁺ concentration is too high. | Optimize Mg²⁺ concentration; high Mg²⁺ can reduce stringency and promote non-specific binding [8] [60]. | |
| Low yield of target with Hot-Start polymerase | Polymerase not fully activated. | Ensure a long enough initial denaturation step (e.g., 2-10 minutes as manufacturer recommends), especially for chemically modified enzymes [55]. |
| The Hot-Start method is too stringent for the target. | If using primer-based methods, consider a "Turbo" (faster-activating) version over a "Precision" (slower-activating) version [57]. |
Experimental Protocol: Evaluating Hot-Start Polymerase Performance
This protocol provides a methodology to compare the performance of a standard DNA polymerase versus a Hot-Start DNA polymerase in the context of primer-dimer suppression.
Objective: To demonstrate the efficacy of a Hot-Start DNA polymerase in reducing primer-dimer formation and improving amplicon specificity.
Materials (The Scientist's Toolkit):
- DNA Template: High-quality genomic or plasmid DNA containing your target.
- Primers: A set specific to your target. For a convincing demonstration, include a primer set known to be prone to dimerization.
- DNA Polymerases: Standard Taq DNA polymerase and a Hot-Start version (e.g., antibody-mediated or chemically modified).
- dNTP Mix: 10 mM solution containing equimolar dATP, dCTP, dGTP, dTTP.
- PCR Buffer: 10X buffer, typically supplied with the enzyme.
- MgCl₂: 25 mM solution (if not included in the buffer).
- Sterile Water: Nuclease-free.
- Equipment: Thermal cycler, agarose gel electrophoresis system, gel documentation system.
Procedure:
- Prepare Two Master Mixes:
- Tube A (Standard): Combine all components for multiple reactions, including standard Taq polymerase.
- Tube B (Hot-Start): Combine identical components, but substitute with the Hot-Start DNA polymerase. Note: For the Hot-Start enzyme, follow the manufacturer's instructions regarding the initial activation step. [14]
Aliquot and Add Template:
- Aliquot the master mixes into individual PCR tubes.
- Add template DNA to the sample tubes. Include a No-Template Control (NTC) for each master mix by adding sterile water instead of DNA. The NTC is critical for visualizing primer-dimer, as it will be the only amplification product if present [2].
Thermal Cycling:
- Use the following standard cycling conditions, modifying the initial denaturation according to the Hot-Start enzyme's requirements:
- Initial Denaturation: 95°C for 2-10 minutes (see manufacturer's guidelines).
- 35 Cycles of:
- Denaturation: 95°C for 30 seconds
- Annealing: (Tm -5°C) for 30 seconds
- Extension: 72°C for 1 minute per kb
- Final Extension: 72°C for 5-10 minutes.
- Use the following standard cycling conditions, modifying the initial denaturation according to the Hot-Start enzyme's requirements:
Analysis:
- Run the PCR products, including the NTCs, on an agarose gel.
- Interpretation: In the NTC for the standard polymerase, you will likely see a bright, smeary band below 100 bp, indicating primer-dimer [2]. The specific target band in the sample lane may be weak. In contrast, the NTC for the Hot-Start polymerase should be clear, and the sample lane should show a strong, specific band with little to no background.
The workflow for this experiment is summarized below:
The strategic selection of Hot-Start DNA polymerases is a highly effective approach within a comprehensive strategy to prevent primer-dimer formation in PCR. By understanding the different inhibition mechanisms and their respective benefits, researchers can make an informed choice that best suits their specific application, whether it's routine genotyping, sensitive diagnostics, or complex multiplex assays. Integrating a well-chosen Hot-Start polymerase with optimized primer design and reaction conditions ensures higher specificity, greater sensitivity, and more reliable results, ultimately accelerating the pace of research and drug development.
FAQs: Troubleshooting GC-Rich Template Amplification
Q1: Why is amplifying GC-rich templates so challenging, and what are the primary symptoms of failure?
GC-rich templates (typically >65% GC content) are difficult to amplify because the strong triple hydrogen bonding between guanine (G) and cytosine (C) nucleotides promotes stable secondary structures and incomplete denaturation [61]. This results in premature termination of polymerase extension and truncated amplicons [61]. Common symptoms include complete amplification failure, smeared bands on a gel, or lower yield of the desired product [8].
Q2: Which specialized PCR additives can improve amplification of GC-rich regions, and at what concentrations should they be used?
Specific additives help denature stable GC-rich templates by disrupting base pairing. The table below summarizes common reagents and their optimal concentrations [62] [20] [61].
Table: Additives for Optimizing GC-Rich PCR
| Additive | Recommended Final Concentration | Mechanism of Action | Important Considerations |
|---|---|---|---|
| DMSO | 2–10% (2.5–5% is common) [62] [61] | Lowers template melting temperature (Tm), prevents secondary structures [20] | Concentrations >5% can reduce polymerase activity; 10% is often inhibitory [62] |
| Betaine | 0.5–2 M [62] | Equalizes the thermal stability of AT and GC base pairs [62] | Can be used in combination with DMSO [62] |
| Formamide | 1.25–10% [20] | Weakens base pairing, increases primer specificity [20] | - |
| Glycerol | 5–25% [62] | Stabilizes enzymes and lowers DNA denaturation temperature [62] | - |
| GC-RICH Resolution Solution | 0.5–2.5 M (titrate in 0.25 M steps) [62] | A proprietary solution designed to resolve complex templates [62] | Part of specialized commercial systems [62] |
Q3: How should thermal cycling conditions be modified for GC-rich targets?
Optimizing the thermal profile is critical for success [61]:
- Denaturation: Use a higher denaturation temperature (e.g., 98°C instead of 94–95°C) and/or increase the denaturation time to ensure complete strand separation [61] [8].
- Annealing: Use primers with a higher Tm (>68°C) and keep annealing times as short as possible to enhance specificity [61].
- Polymerase Selection: Employ polymerases specifically engineered for high GC content, which often have higher processivity and affinity for difficult templates [61] [8].
FAQs: Troubleshooting Multiplex PCR and Primer-Dimer Formation
Q4: What is primer dimer, and why is it a particularly severe problem in multiplex PCR?
A primer dimer is a small, unintended DNA fragment that forms when primers anneal to each other via complementary regions instead of binding to the target template [2]. This consumes reagents and reduces amplification efficiency [3]. In multiplex PCR, the problem grows quadratically; for a set of 2N primers, there are (2N choose 2) potential primer-dimer interactions [63]. A 96-plex reaction (192 primers) has over 18,000 potential pairwise interactions, making efficient amplification of all targets exceptionally challenging [63].
Q5: What are the fundamental strategies to minimize primer-dimer formation?
The core strategy is to reduce opportunities for primers to interact nonspecifically [2]. Key methods include:
- Primer Design: Design primers with minimal self-complementarity and 3'-end complementarity using specialized software [3] [8].
- Lower Primer Concentration: Optimize and reduce primer concentrations (typically 0.1–1 µM) to lower the primer-to-template ratio [2] [8] [64].
- Hot-Start Polymerases: Use these enzymes to prevent polymerase activity during reaction setup at low temperatures, a period when primer dimer formation is most likely [2] [8].
- Higher Annealing Temperature: Increase the temperature to promote strict primer-template binding [2] [64].
- Computational Optimization: For highly multiplexed assays, use advanced algorithms like SADDLE (Simulated Annealing Design using Dimer Likelihood Estimation) to design primer sets that minimize dimer potential from the outset [63].
Q6: How can I identify primer dimer in my results?
In gel electrophoresis, primer dimers typically [2]:
- Appear as a smear or fuzzy band at a low molecular weight.
- Run below 100 base pairs, often below the smallest band of your DNA ladder. To confirm, always run a no-template control (NTC); the presence of a band in the NTC is a classic indicator of primer dimer [2].
Experimental Protocols
Protocol 1: Systematic Optimization of a GC-Rich PCR
This protocol provides a step-by-step method for establishing a robust GC-rich PCR.
Materials:
- Template DNA: 10–100 ng of high-quality, intact genomic DNA or equivalent [61].
- Primers: Designed with a Tm >68°C [61].
- Specialized Polymerase: A polymerase blend formulated for GC-rich templates (e.g., GC-RICH PCR System, PrimeSTAR GXL DNA Polymerase) [62] [61].
- Additives: DMSO, Betaine, or a proprietary GC Enhancer solution [62] [61].
- MgCl₂ Solution: For separate titration (if required by your system) [61].
- Nuclease-free Water and Standard PCR Buffers.
Method:
- Set Up a Master Mix: Combine all common reagents (water, buffer, dNTPs, polymerase) on ice. If your buffer does not contain Mg²⁺, add it separately [20].
- Titrate Additives: Aliquot the master mix into several tubes. Add your chosen additive (e.g., DMSO) at varying concentrations across the tubes (e.g., 0%, 2.5%, 5%, 7.5%) [62].
- Add Template and Primers: Add a fixed, optimal amount of template and primers to each tube.
- Run a Thermal Cycler Program:
- Analyze Results: Use agarose gel electrophoresis to assess yield and specificity. The optimal additive concentration will show a strong, specific band with minimal smearing.
Protocol 2: Computational Workflow for Minimizing Primer-Dimer in Multiplex PCR
This protocol outlines the SADDLE algorithm for designing large, multiplex primer sets with minimal dimer formation [63].
Materials:
- Target Genomic Sequences in FASTA format.
- Computational Resources: A workstation capable of running the design algorithm.
- SADDLE Algorithm or similar multiplex primer design software [63].
Method:
- Primer Candidate Generation:
- For each target (e.g., a gene exon), define a "pivot" nucleotide that must be covered.
- Systematically generate multiple "proto-primers" of varying lengths flanking the pivot.
- Trim the 3' ends of these proto-primers to achieve a target binding free energy (ΔG°) of approximately -11.5 kcal/mol, which balances efficiency and specificity. Apply filters for GC content (e.g., 25–75%) [63].
Initial Primer Set Selection:
- Randomly select one primer pair candidate for each target amplicon to form the initial primer set S₀ [63].
Iterative Optimization via Simulated Annealing:
- Evaluate the "Loss Function": Calculate a score L(S) for the current primer set that sums the "Badness" (likelihood of dimer formation) for every possible pair of primers [63].
- Generate a New Candidate Set: Create a temporary set T by randomly swapping one or more primers in the set with other candidates from the pool [63].
- Evaluate and Select: Calculate L(T). The new set T is always accepted if it is better (lower Loss), and accepted with a certain probability if it is worse. This probabilistic acceptance helps the algorithm escape local minima to find a globally optimal solution [63].
- Repeat this process for thousands of generations until a primer set with a satisfactorily low Loss value is obtained [63].
Research Reagent Solutions
Table: Essential Reagents for Complex PCR Applications
| Reagent Category | Specific Examples | Function in Resolving Complex Cases |
|---|---|---|
| Specialized Polymerases | GC-RICH PCR System [62], PrimeSTAR GXL [61], Q5 High-Fidelity [64] | Engineered for high processivity and affinity to denature difficult templates (GC-rich, long amplicons) and/or provide high fidelity. |
| Hot-Start Enzymes | OneTaq Hot Start [64], various Hot-Start Taq formulations [8] | Remains inactive until a high-temperature activation step, dramatically reducing primer-dimer formation during reaction setup [2] [8]. |
| PCR Additives | DMSO, Betaine, Proprietary GC Enhancers [62] [61] [8] | Disrupts secondary structures and stabilizes DNA, facilitating the amplification of GC-rich templates. |
| Primer Design Technologies | SAMRS (Self-Avoiding Molecular Recognition Systems) [46] | Incorporates modified bases that prefer binding to natural DNA over other SAMRS bases, reducing primer-primer interactions in multiplex assays [46]. |
| Buffer Components | MgCl₂/MgSO₄ solutions [20] [61], optimized salt buffers (KCl) [61] | Mg²⁺ is an essential cofactor for polymerases; its concentration must be optimized. Salt concentration influences denaturation efficiency of long vs. short amplicons [61]. |
Step-by-Step Troubleshooting Flowchart for Common Scenarios
This technical support guide provides targeted solutions for researchers addressing common PCR complications, with a particular focus on preventing primer-dimer formation, a key factor in ensuring the accuracy and efficiency of your experiments.
Troubleshooting Common PCR Problems
1. I am observing low or no yield of my desired PCR product. What should I check?
A lack of sufficient amplified product can stem from issues with the template, primers, or reaction conditions.
- Template DNA: Verify the quality, quantity, and integrity of your template DNA. Ensure it is free from common PCR inhibitors like phenol or EDTA. For a 50 µL reaction, typical amounts are 1 pg–10 ng for plasmid DNA or 1 ng–1 µg for genomic DNA [65]. If the template is of low purity, consider re-purifying it or using a DNA polymerase with high tolerance to inhibitors [8].
- Primers: Confirm that your primers are designed with optimal length (18–24 nucleotides) and GC content (40–60%) [11]. Check that they have been reconstituted to the correct concentration and are not degraded. Optimize the primer concentration in the reaction, typically between 0.1–1 µM [8].
- Reaction Components and Cycling: Ensure all reagents, especially the DNA polymerase, are active and have not undergone multiple freeze-thaw cycles. Increase the number of cycles (up to 40 cycles for low-copy-number templates) or extend the extension time to allow for full-length product synthesis [8]. Use a thermal cycler that has been properly calibrated [65].
2. My gel shows multiple bands or a single band of the wrong size, indicating non-specific amplification. How can I improve specificity?
Non-specific products, including primer-dimers, occur when primers bind to incorrect sites on the template or to each other.
- Primer Design and Concentration: Re-analyze your primer sequences for specificity to the target. Avoid regions with direct repeats and ensure the 3' ends do not have long stretches of G or C nucleotides, which can promote mis-priming [8] [11]. High primer concentration can exacerbate this; titrate to find the lowest effective concentration [8].
- "Hot-Start" Polymerase: Use a hot-start DNA polymerase. These enzymes are inactive until a high-temperature activation step, preventing enzymatic activity during reaction setup at lower temperatures and thereby reducing primer-dimer formation and non-specific amplification [8] [3].
- Optimize Thermal Cycling Conditions: The most common fix is to increase the annealing temperature. Use a gradient thermal cycler to determine the highest possible annealing temperature that still provides robust yield of your specific product [8] [65]. You can also reduce the number of cycles and ensure the denaturation temperature and time are sufficient to fully melt complex templates [8].
3. My sequencing results reveal mutations not present in the original sample. What causes these fidelity errors?
Errors incorporated during amplification can compromise downstream applications like cloning.
- DNA Polymerase Fidelity: Use a high-fidelity DNA polymerase with proofreading (3'→5' exonuclease) activity, especially for cloning applications [8] [66].
- Reaction Chemistry: Excess Mg²⁺ concentration and unbalanced dNTP concentrations can both increase the misincorporation rate of nucleotides. Optimize the Mg²⁺ concentration and always use fresh, equimolar dNTP stocks [8] [65].
- Cycle Number: Reduce the number of PCR cycles. Each cycle presents another opportunity for polymerase error; fewer cycles with more input template can reduce the accumulation of errors [8].
The Scientist's Toolkit: Essential Research Reagent Solutions
The table below lists key reagents and their specific functions in optimizing PCR and preventing common issues like primer-dimer formation.
| Item | Function & Rationale |
|---|---|
| Hot-Start DNA Polymerase | Remains inactive until a high-temperature activation step, drastically reducing primer-dimer formation and non-specific amplification during reaction setup [8] [3]. |
| High-Fidelity DNA Polymerase | Contains proofreading activity (3'→5' exonuclease) to correct misincorporated nucleotides, essential for generating accurate sequences for cloning [8] [66]. |
| PCR Additives (e.g., GC Enhancer, DMSO) | Aids in denaturing GC-rich templates and sequences with secondary structures, improving yield and specificity for difficult targets [8]. |
| Magnesium Salts (MgCl₂, MgSO₄) | Cofactor essential for DNA polymerase activity. Its concentration must be optimized, as excess can cause non-specific products and errors, while insufficient amounts lead to low yield [8] [65]. |
| Nuclease-Free Water | Ensures the reaction is not compromised by external nucleases that could degrade primers, templates, or products. |
| dNTPs | The building blocks for DNA synthesis. Must be fresh, undegraded, and provided in equimolar concentrations to prevent misincorporation errors [8] [65]. |
Experimental Protocol: Systematic Optimization of Annealing Temperature
A key method for enhancing specificity and preventing primer-dimer formation is to empirically determine the optimal annealing temperature (Ta).
1. Principle The calculated melting temperature (Tm) of a primer is an estimate. An empirical test determines the actual highest Ta that allows specific primer binding while disabling non-specific binding and primer-dimer formation.
2. Materials
- Prepared PCR master mix (containing template, primers, polymerase, dNTPs, and buffer)
- Gradient thermal cycler
- Gel electrophoresis equipment
3. Procedure
- Prepare the Reaction: Aliquot your PCR master mix into a single tube and dispense equal volumes into the wells of the gradient thermal cycler.
- Set the Gradient: In the cycler's program, set the annealing temperature to a gradient range that spans at least 5°C above and below the calculated Tm of your primer pair (e.g., if Tm is 60°C, set a gradient from 55°C to 65°C) [8] [11].
- Run PCR: Execute the full PCR cycling program.
- Analyze Results: Separate the PCR products via gel electrophoresis. Identify the well(s) with the strongest band of the correct size and the absence of non-specific bands or smearing. The annealing temperature used in that well is your optimal Ta.
Primer Design and Problem Flowchart
The following diagram outlines a systematic approach to diagnosing and resolving common PCR issues, with a special emphasis on pathways leading to primer-dimer formation.
Frequently Asked Questions (FAQs)
Q1: What are the most critical factors in primer design to prevent primer-dimer formation? The most critical factors are minimizing self-complementarity (within a single primer) and cross-complementarity (between the forward and reverse primer), particularly at the 3' ends. Furthermore, avoid consecutive G or C nucleotides (a "GC clamp") at the 3' end, as this stabilizes non-specific binding. Using a reliable primer design tool is essential to check these parameters [11].
Q2: My primer design looks good, but I still get primer-dimer. What is a common laboratory practice I might be missing? A common oversight is setting up PCR reactions at room temperature. Even with well-designed primers, the DNA polymerase can have low-level activity that facilitates primer-dimer formation before thermal cycling begins. Always prepare your reactions on ice and use a hot-start DNA polymerase, which is inactive until the initial high-temperature denaturation step [8] [65].
Q3: How does increasing the annealing temperature help with primer-dimer? Primer-dimers form because short regions of complementarity between primers are stable at lower temperatures. By increasing the annealing temperature, you create a more stringent environment where only the perfectly matched primer-template hybrids are stable. The weaker bonds holding the primer-dimers together will not form, thereby suppressing their amplification [8] [3].
Validation, Comparative Analysis, and Ensuring Assay Specificity
FAQs: Troubleshooting Primer-Dimer Formation
FAQ 1: What is a primer dimer and how does it form? A primer dimer is a small, unintended DNA fragment that forms when PCR primers anneal to each other instead of binding to their intended target in the template DNA [2]. This occurs primarily through two mechanisms: self-dimerization, where a single primer contains regions complementary to itself, and cross-dimerization, where two different primers have complementary regions that bind to each other [2]. Once bound, they create free 3' ends that DNA polymerase can extend, consuming reaction resources [10].
FAQ 2: Why is it critical to prevent primer-dimer formation in sensitive applications like qPCR and sequencing? Preventing primer dimers is crucial because they compete with the target DNA for reaction components (polymerase, dNTPs, primers), thereby reducing the efficiency and yield of your desired amplicon [3]. In SYBR Green qPCR, the dye binds non-specifically to all double-stranded DNA, including primer dimers, leading to inaccurate fluorescence quantification and false positives [67]. In sequencing, adapter dimers (which contain full-length adapter sequences) can cluster efficiently on the flow cell and generate meaningless sequences, wasting a significant portion of your sequencing reads and potentially causing runs to fail prematurely [68].
FAQ 3: How can Melt-Curve Analysis help identify primer dimers in qPCR? Melt-curve analysis is an essential quality control step performed after a SYBR Green qPCR run to verify amplification specificity [67]. After amplification, the temperature is gradually increased from about 60°C to 95°C. As the DNA denatures, the fluorescence decreases. A single, specific PCR product will typically produce a single, sharp peak in the derivative melt curve. The presence of primer dimers, which have a different melting temperature (Tm) due to their shorter length and sequence, results in additional, distinct peaks—often at lower temperatures—or causes broad, asymmetrical peaks, alerting you to potential issues [67].
FAQ 4: How does Capillary Electrophoresis aid in detecting primer dimers? Capillary electrophoresis (CE), including lab-on-chip systems, offers superior resolution and sensitivity compared to conventional agarose gel electrophoresis [69]. It automates the separation and detection of DNA fragments, providing a detailed electropherogram. Primer dimers, typically under 100 base pairs (bp), will appear as a sharp peak at around 100-120 bp [2] [68]. This high-resolution separation allows you to clearly distinguish the smeary, low molecular weight primer-dimer peak from your specific, larger amplicon, and is particularly useful for optimizing multiplex PCR assays [69].
Troubleshooting Guide: Resolving Primer-Dimer Issues
Prevention Strategies: Experimental Design and Setup
| Strategy | Description | Key Parameters |
|---|---|---|
| Primer Design [7] [2] | Design primers with minimal self-complementarity or 3'-end complementarity to avoid intra- and inter-primer binding. | Use primer design software; avoid complements at 3' ends. |
| Primer Concentration [7] [2] | Lowering primer concentration reduces chances of primer-primer interactions. | Use a concentration gradient to find the lowest effective amount. |
| Hot-Start Polymerase [2] | Inactive at room temperature, preventing polymerase activity during reaction setup. | Activates only at high temperatures (e.g., 94-95°C). |
| Annealing Temperature [2] | Higher temperatures promote specific primer-template binding. | Increase temperature incrementally by 1-2°C. |
| Touchdown PCR [10] | Starts with high annealing temperature, incrementally lowering it in later cycles. | Favors specific amplification in early cycles. |
Analysis and Validation Techniques
1. Melt-Curve Analysis for qPCR Validation
- Protocol: After the final qPCR cycle, run a melt-curve protocol on your instrument. A standard method involves heating from 60°C to 95°C with a continuous fluorescence measurement (e.g., 0.1°C per second) [67].
- Interpretation: Inspect the derivative melt curve plot. A single, sharp peak indicates specific amplification. Multiple peaks, shoulders on the main peak, or broad peaks suggest the presence of primer dimers or non-specific products [67].
- Troubleshooting: If anomalous peaks are detected, consider purifying your PCR product and re-running the melt curve, or proceed to capillary electrophoresis for further analysis.
2. Capillary Electrophoresis for End-Point PCR and Library QC
- Protocol: Use a lab-on-chip or similar CE system according to the manufacturer's instructions. This typically involves loading the PCR product onto a chip alongside a DNA ladder and running the analysis [69].
- Interpretation: Analyze the resulting electropherogram. The primer dimer will appear as a small, sharp peak around 100-120 bp (for conventional PCR) or 120-170 bp (for sequencing adapter dimers) [2] [68]. Compare its size and peak area to your target amplicon.
- Troubleshooting: If a significant primer-dimer peak is present, perform a bead-based clean-up (e.g., with AMPure XP beads) using a 0.8x to 1x bead-to-sample ratio to selectively remove the short fragments before proceeding with sequencing or other downstream applications [68].
Diagram 1: Pathways of primer-dimer formation and subsequent detection methods. Self- or cross-dimerization of primers can be extended by DNA polymerase, leading to primer-dimer artifacts. These can be detected via melt curve analysis in qPCR or as a characteristic low molecular weight peak in capillary electrophoresis.
Quantitative Data for Experimental Thresholds
Table 1: Quantitative Specifications for Primer-Dimer Analysis
| Parameter | Typical Value for Primer Dimer | Detection Method | Acceptance Threshold for Downstream Applications |
|---|---|---|---|
| Size | < 100 bp [2] | Agarose Gel CE | N/A |
| Adapter Dimer Size | 120-170 bp [68] | Capillary Electrophoresis | N/A |
| Adapter Dimer in Sequencing Library | N/A | Capillary Electrophoresis | ≤ 0.5% (patterned flow cells) [68] |
| Melting Temperature (Tm) | Lower than specific amplicon [67] | Melt-Curve Analysis | N/A |
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Preventing and Analyzing Primer-Dimers
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Hot-Start DNA Polymerase | Remains inactive until high temperature is reached, preventing enzymatic activity during reaction setup [2]. | Essential for all PCR setups to minimize primer-dimer formation before thermal cycling begins. |
| SYBR Green Master Mix | Fluorescent dye that binds indiscriminately to double-stranded DNA [67]. | Used in qPCR for real-time detection of amplification, requiring subsequent melt-curve analysis to confirm specificity. |
| AMPure/SPRI Beads | Magnetic beads used for size-selective purification of DNA fragments [68]. | Cleaning up sequencing libraries by removing adapter dimers (e.g., using a 0.8x bead ratio). |
| Lab-on-Chip Kits | Microfluidic chips for automated capillary electrophoresis [69]. | Providing high-sensitivity, automated sizing and quantification of PCR products and primer dimers. |
| Primer Design Software | In-silico tools to check for self-complementarity and secondary structures [2] [3]. | The first line of defense; used to design primers with minimal potential for dimerization. |
Troubleshooting Guides
Issue 1: Non-specific Amplification and Primer-Dimer Formation
Problem Description Researchers observe multiple bands or a smear on an agarose gel, or a dominant short product (~50-100 bp) indicative of primer-dimer, which competes with the desired amplicon for reaction resources [14] [2].
Possible Causes and Solutions
| Cause | Solution |
|---|---|
| Polymerase activity during setup | Use hot-start DNA polymerases (antibody-mediated or chemically modified) to inhibit activity until initial denaturation [8] [70]. |
| Low annealing temperature | Increase annealing temperature incrementally by 1-2°C; use a gradient thermal cycler. Employ touchdown PCR, starting 5-10°C above calculated Tm [8] [15]. |
| Suboptimal primer design | Redesign primers using tools (NCBI Primer-BLAST). Ensure 3' ends lack complementarity; optimal length 18-25 bases, GC content 40-60% [14] [8]. |
| Excess primers or enzyme | Lower primer concentration (0.1-0.5 µM). Use recommended amount of DNA polymerase; excess can increase mispriming [8] [2]. |
| Insufficient denaturation | Increase denaturation temperature or time (e.g., 98°C) to separate complex templates, disrupting primer interactions [8] [2]. |
Issue 2: Amplification Failure with Complex Templates
Problem Description The reaction fails to yield any product, or yield is very low, when targeting GC-rich sequences, long amplicons, or templates from inhibitor-containing samples [8] [71].
Possible Causes and Solutions
| Cause | Solution |
|---|---|
| Polymerase with low processivity | Switch to a high-processivity blend (e.g., MyFi, KOD-Sto7d). These enzymes remain bound longer, navigating secondary structures and inhibitors [72] [73]. |
| GC-rich secondary structures | Use PCR additives: DMSO (1-10%), formamide (1.25-10%), or Betaine (0.5-2.5 M). Combine with a high denaturation temperature (98°C) [14] [8] [15]. |
| PCR inhibitors in the sample | Re-purify template DNA. Use high-processivity polymerases known for inhibitor tolerance (e.g., MyFi). Add BSA (10-100 µg/ml) to bind contaminants [8] [73] [71]. |
| Insufficient extension time | Increase extension time for long targets (>5 kb). For fast, high-processivity enzymes, calculate time based on synthesis speed (e.g., KOD-Sto7d: 10 s/kb) [8] [72]. |
Issue 3: Low Fidelity and Incorporation of Errors
Problem Description The amplified product sequence contains unintended mutations, compromising downstream applications like cloning or sequencing [8] [71].
Possible Causes and Solutions
| Cause | Solution |
|---|---|
| Use of non-proofreading polymerase | Use high-fidelity polymerases with 3'→5' exonuclease (proofreading) activity (e.g., Pfu, KOD). Fidelity can be >50x higher than Taq [70] [71]. |
| Unbalanced dNTP or excess Mg²⁺ | Use equimolar dNTP concentrations. Optimize Mg²⁺ concentration (e.g., 1.5-2.5 mM); excess Mg²⁺ reduces fidelity [8]. |
| Excessive cycle number | Reduce the number of PCR cycles (25-35 cycles) to minimize cumulative errors, especially when template input is sufficient [8]. |
Frequently Asked Questions (FAQs)
Q1: What is the primary mechanism by which high-processivity blends reduce primer-dimer formation? High-processivity blends often incorporate a hot-start mechanism, preventing enzymatic activity during reaction setup at room temperature when primer-dimer initiation primarily occurs [70] [73]. Furthermore, their engineered DNA-binding domains (e.g., Sso7d, Sto7d) enhance affinity for the correct, longer DNA template, favoring its amplification over short, nonspecific primer-dimers once cycling begins [72] [70].
Q2: Can I use the same thermal cycling protocol for a high-processivity blend as I do for my standard Taq polymerase? Not optimally. High-processivity enzymes are significantly faster. You can often reduce extension time by half or more. For example, the KOD-Sto7d variant can amplify a 2 kb target in 20 seconds and a 7 kb target in 70 seconds [72]. You may also combine annealing/extension into a two-step PCR protocol. Always consult the manufacturer's recommendations.
Q3: My high-fidelity, proofreading polymerase gives low yield. Is this normal, and how can I improve it? Yes, this is a common trade-off. Native proofreading enzymes like Pfu often have lower processivity and extension rates than Taq [70] [71]. To improve yield:
- Use polymerase blends that mix a high-fidelity enzyme with a processive one.
- Ensure adequate template quantity and quality.
- Optimize Mg²⁺ concentration and increase the number of cycles slightly.
- Select engineered high-fidelity enzymes that have been optimized for both speed and accuracy [70] [73].
Q4: How do I choose between a standard and a high-processivity polymerase for my experiment? Use the following decision workflow:
Quantitative Data Comparison of Polymerase Enzymes
Table 1: Performance Characteristics of Common Polymerase Types
| Polymerase Type | Example Enzymes | Processivity | Fidelity (Relative to Taq) | Extension Speed (seconds/kb) | Suitable Amplicon Length | Primary Best Use Case |
|---|---|---|---|---|---|---|
| Standard | Taq | Low | 1x | ~60 | < 5 kb | Routine amplification, genotyping |
| High-Fidelity | Pfu, KOD-WT | Medium | ~10x | >30 | < 10 kb | Cloning, sequencing, mutagenesis |
| High-Processivity Blend | MyFi, KOD-Sto7d, KOD-Sso7d | High | ~3.5x (MyFi) | ~10 | Up to 10 kb | Complex templates (GC-rich, long), inhibitor-rich samples |
| Engineered Reverse Transcriptase | Novel Taq variants [74] | High | Varies | Varies | Varies | Single-enzyme quantitative multiplex RT-PCR |
Table 2: Resistance Profiles and Buffer Additives
| Polymerase Type | Tolerance to Common Inhibitors | Often Requires Additives for GC-Rich Templates | Hot-Start Availability |
|---|---|---|---|
| Standard | Low | Yes | Common |
| High-Fidelity | Low to Medium | Yes | Common |
| High-Processivity Blend | High [73] | No (Often has built-in enhancers) [73] | Standard feature |
Experimental Protocols
Protocol 1: Evaluating Primer-Dimer Reduction with Hot-Start Technology
Objective: To visually demonstrate the reduction of primer-dimer formation using a hot-start high-processivity polymerase versus a standard non-hot-start enzyme.
Materials:
- Test primers (known to form dimers)
- Standard Taq DNA polymerase
- High-processivity hot-start DNA polymerase (e.g., MyFi)
- dNTPs, reaction buffers, DNA template
- Thermal cycler, agarose gel electrophoresis system
Methodology:
- Reaction Setup: Prepare two identical 25 µL master mixes containing all reagents (1X buffer, 200 µM dNTPs, 0.4 µM each primer, 1 ng template DNA).
- Enzyme Addition: Aliquot the master mix into two tubes. Add standard Taq to one tube and hot-start high-processivity blend to the other. Include a no-template control (NTC) for each.
- Incubation Challenge: Hold the completed reactions at room temperature for 30 minutes before placing them in the thermal cycler. This challenges the hot-start capability.
- Thermal Cycling: Run a standard PCR protocol (e.g., 30 cycles of 98°C/10s, 55°C/15s, 72°C/30s/kb).
- Analysis: Analyze 10 µL of each reaction and its NTC on a 2% agarose gel.
Expected Outcome: The NTC for the standard Taq will show a strong primer-dimer smear (~50-100 bp), while the NTC for the hot-start enzyme should be clean. The test reaction with the hot-start enzyme will show a single, strong band of the correct size [70] [2].
Protocol 2: Amplification of a GC-Rich Target
Objective: To successfully amplify a difficult GC-rich (>70%) target using a high-processivity polymerase blend, with and without additives.
Materials:
- GC-rich DNA template (e.g., human genomic DNA)
- Primers for a GC-rich target (e.g., EGFR gene)
- High-processivity polymerase (e.g., KOD-Sto7d, MyFi) and standard Taq
- 5M Betaine, 100% DMSO
Methodology:
- Master Mix Prep: Create several master mixes for different conditions:
- Condition A: Standard Taq + standard buffer
- Condition B: Standard Taq + buffer + 1M Betaine
- Condition C: High-processivity blend + standard buffer
- Condition D: High-processivity blend + buffer + 1M Betaine
- Cycling: Use a protocol with a higher denaturation temperature (98°C). Extension time can be short (e.g., 20-30 seconds/kb) for the high-processivity enzyme.
- Analysis: Run products on an agarose gel.
Expected Outcome: Condition A will likely fail. Condition B may show weak product. Conditions C and D should yield strong, specific bands, demonstrating the inherent capability of the high-processivity enzyme to handle difficult templates [73] [15].
Research Reagent Solutions
Table 3: Essential Reagents for PCR Optimization and Troubleshooting
| Reagent | Function/Benefit | Example Use Case |
|---|---|---|
| Hot-Start High-Processivity Polymerase Blends (e.g., MyFi, KOD-Sso7d/Sto7d) | Robust amplification of complex targets; reduced primer-dimer; inhibitor tolerance. | Primary solution for challenging PCRs (GC-rich, long amplicons, crude samples) [72] [73]. |
| Betaine | Reduces secondary structure formation; equalizes Tm of GC- and AT-rich regions. | Added to 0.5-2.5 M final concentration for GC-rich templates [14] [15]. |
| DMSO | Disrupts base pairing, aiding DNA denaturation. | Added to 1-10% final concentration for GC-rich templates and long amplicons [8] [15]. |
| BSA (Bovine Serum Albumin) | Binds to and neutralizes common PCR inhibitors. | Added to 10-100 µg/ml final concentration when using samples like blood or plant tissue [14] [71]. |
| MgCl₂/MgSO₄ | Essential cofactor for DNA polymerase activity. Concentration critically affects specificity and yield. | Optimized in 0.25-0.5 mM increments, typically from 1.5 to 5.0 mM [14] [8]. |
FAQ: Primer Performance and Troubleshooting
What are the key criteria for evaluating primer set performance?
Primer sets are evaluated based on several key performance metrics in a comparative study. The most critical criteria are sensitivity and specificity, which measure a primer set's ability to correctly identify true positive cases and avoid false positives, respectively [75].
Additional important factors include:
- Consistency of amplification: The ability to produce single, distinct amplicons without non-specific bands [75]
- Diagnostic reliability: Overall accuracy in detecting the target under field conditions [75]
- Amplification efficiency: How effectively the primers generate the desired product [15]
How can I systematically compare multiple primer sets in my research?
A structured experimental approach ensures reliable comparison of primer sets. Below is a standardized protocol adapted from validated methodology [75]:
Table: Experimental Protocol for Primer Set Comparison
| Step | Parameter | Specification |
|---|---|---|
| 1. Sample Preparation | Sample Type | 100 wild rat blood samples |
| Storage | Preserved in Crookes tubes at -20°C | |
| 2. DNA Extraction | Method | Genomic DNA Mini Kit |
| Sample Volume | 300 μL whole blood | |
| Storage | -20°C until PCR amplification | |
| 3. PCR Amplification | Primer Sets | TC121/TC122, CATLew F/CATLew R, LEW1S/LEW1R |
| Conditions | Optimized thermal cycling for each set | |
| 4. Product Analysis | Method | Agarose gel electrophoresis |
| Evaluation | Visualize amplification products |
Experimental Workflow for Primer Comparison
What specific performance differences might I observe between primer sets?
Substantial variation in performance metrics can occur between different primer sets targeting the same sequence. The following table summarizes quantitative findings from a comparative evaluation study [75]:
Table: Comparative Performance of Three PCR Primer Sets
| Primer Set | Positives Detected | Sensitivity (%) | Specificity (%) | Key Observations |
|---|---|---|---|---|
| LEW1S/LEW1R | 30 | 100 | 97.22 | Consistently produced single, distinct amplicons with no non-specific bands |
| CATLew F/CATLew R | 29 | 96.43 | 97.22 | High performance with minor reduction in detection |
| TC121/TC122 | 21 | 67.86 | 97.22 | Significantly lower sensitivity despite good specificity |
How does primer design influence performance and primer-dimer formation?
Proper primer design is fundamental to preventing primer-dimer formation and ensuring optimal performance. Consider these evidence-based design principles [76] [11]:
Table: Optimal Primer Design Parameters to Minimize Artifacts
| Parameter | Optimal Range | Rationale | Consequences of Deviation |
|---|---|---|---|
| Length | 18-30 nucleotides [76] [11] | Balances specificity and hybridization efficiency | Short primers: reduced specificity; Long primers: slower hybridization |
| GC Content | 40-60% [11] | Stable binding without excessive strength | High GC: mismatches, primer-dimers; Low GC: weak binding |
| Melting Temperature (Tₘ) | 60-64°C [76] | Ideal for enzyme function | Low Tₘ: nonspecific amplification; High Tₘ: reduced efficiency |
| 3'-End Stability | Avoid >3 G/C residues [11] | Prevents non-specific binding | Promotes mispriming and primer-dimer formation |
| Self-Complementarity | ΔG > -9.0 kcal/mol [76] | Minimizes secondary structures | Primer-dimers, hairpins, reduced product yield |
What troubleshooting strategies address poor primer performance?
When encountering suboptimal primer performance, systematic troubleshooting is essential:
For No or Weak Amplification:
- Verify primer design quality and check for secondary structures [77] [76]
- Optimize annealing temperature using a gradient PCR cycler [8]
- Increase primer concentration (typically 0.1-1 μM) [8]
- Check template quality and concentration [77]
- Ensure all reaction components are present and properly mixed [77]
For Non-Specific Bands or Primer-Dimers:
- Use hot-start DNA polymerases to prevent room-temperature activity [15]
- Increase annealing temperature incrementally (1-2°C steps) [8]
- Implement touchdown PCR (starting 5°C above Tₘ) [15]
- Optimize Mg²⁺ concentration in 0.2-1 mM increments [77]
- Reduce primer concentration if excessively high [8]
Troubleshooting Primer Performance Issues
What advanced techniques improve primer specificity?
Several advanced PCR methods can significantly enhance specificity and reduce artifacts:
Hot-Start PCR: Employs modified DNA polymerases inactive at room temperature, preventing nonspecific amplification during reaction setup. Activation occurs only during initial high-temperature denaturation step [15].
Touchdown PCR: Begins with annealing temperature 5°C above primer Tₘ, gradually decreasing by 1°C per cycle until optimal temperature is reached. This enriches specific products early in amplification process [15].
Nested PCR: Uses two sequential primer sets where the second set (nested primers) amplifies a region within the first product. This dramatically improves specificity but requires additional time and reagents [15].
Research Reagent Solutions
Table: Essential Materials for Primer Performance Evaluation
| Reagent/Equipment | Function | Application Notes |
|---|---|---|
| Hot-Start DNA Polymerase | Reduces nonspecific amplification & primer-dimers | Essential for multiplex PCR; prevents enzyme activity during setup [15] |
| Genomic DNA Extraction Kit | Provides high-quality template | Critical for sensitivity; ensures removal of PCR inhibitors [75] |
| Gradient Thermal Cycler | Optimizes annealing temperatures | Enables simultaneous testing of multiple temperatures [8] |
| Agarose Gel Electrophoresis System | Visualizes amplification products | Standard method for analyzing product size and specificity [75] |
| PCR Primers | Target-specific amplification | Designed with 18-30 bp length, 40-60% GC content [76] [11] |
Assessing Specificity and Sensitivity in SNP Detection Assays
What are the core metrics for evaluating a SNP detection assay?
The performance of a Single Nucleotide Polymorphism (SNP) detection assay is primarily evaluated using two core metrics: sensitivity and specificity [78].
- Sensitivity (also known as the true positive rate) measures the assay's ability to correctly identify the presence of a SNP. It is calculated as the proportion of true positive samples correctly identified among all samples that actually contain the SNP [78].
- Specificity (the true negative rate) measures the assay's ability to correctly identify the absence of a SNP. It is calculated as the proportion of true negative samples correctly identified among all samples that do not contain the SNP [78].
These metrics are crucial because they directly impact the reliability of your research findings. High sensitivity minimizes false negatives, ensuring you don't miss real SNP signals. High specificity minimizes false positives, preventing misidentification of non-specific amplification or artifacts as genuine SNPs [78].
Troubleshooting Guides
Low Sensitivity
Issue: My SNP assay is failing to detect known positives (Low Sensitivity).
Low sensitivity can result in false negatives, compromising your data's completeness.
| Possible Cause | Recommended Solution |
|---|---|
| Suboptimal Primer/Probe Design | Design primers with minimal self-complementarity; use tools to ensure low 3’ complementarity to avoid primer-dimer formation that competes with target amplification [2]. |
| Insufficient Template Quality/Quantity | Verify template DNA concentration and purity (260/280 ratio of 1.8-2.0); increase template amount if necessary; use polymerases with high sensitivity [8]. |
| Inefficient PCR Amplification | Optimize Mg2+ concentration; use robust, high-fidelity DNA polymerases; ensure equimolar dNTP concentrations to avoid misincorporation [8]. |
| Suboptimal Thermal Cycling | Increase the number of PCR cycles (e.g., to 40 cycles for low-copy templates); optimize annealing temperature; ensure adequate extension times [8]. |
Low Specificity
Issue: My assay is producing false positives or non-specific amplification (Low Specificity).
Low specificity can lead to false positives, often from non-specific binding or primer-dimer artifacts.
| Possible Cause | Recommended Solution |
|---|---|
| Primer-Dimer Formation | Increase annealing temperature; use hot-start DNA polymerases to prevent activity during setup; lower primer concentration [2] [8]. |
| Non-specific Primer Binding | Increase annealing temperature stepwise; use a gradient cycler for optimization; design longer primers; consider touchdown PCR [8]. |
| Excess Reaction Components | Lower primer concentration (typically 0.1–1 μM); optimize and potentially reduce Mg2+ concentration; avoid excess DNA polymerase [8]. |
| Contamination | Always include a No-Template Control (NTC); use uracil N-glycosylase (UNG) and dUTP in qPCR to prevent amplicon carryover; physically separate pre- and post-PCR workspaces [79]. |
Frequently Asked Questions (FAQs)
Q1: How can I visually identify primer-dimer in my results, and why does it affect my assay's specificity?
In gel electrophoresis, primer dimers typically appear as a smeary band or a fuzzy smear at a low molecular weight, usually below 100 base pairs (bp) [2]. In qPCR melt curve analysis, they produce a broad peak that melts at a lower temperature than your specific PCR product [79]. Primer-dimer affects specificity because it competes for reaction reagents (primers, enzymes, dNTPs), thereby reducing the efficiency and yield of your desired SNP-specific amplification product [2] [79].
Q2: What are the best practices for primer design to maximize both sensitivity and specificity from the start?
To maximize assay performance, adhere to the following primer design principles [8]:
- Ensure primers are specific to the target sequence with minimal homology to other regions.
- Avoid regions of self-complementarity or consecutive G/C nucleotides at the 3' end to prevent primer-dimer formation.
- Use reputable online primer design tools that automatically check for potential secondary structures and complementarity.
- Verify the primer sequence and its specificity using tools like NCBI Primer-BLAST [80].
Q3: Beyond sensitivity and specificity, what other performance metrics should I consider when validating a SNP assay?
A robust validation includes multiple performance metrics that provide a comprehensive view of assay quality [78]:
- Youden Index: Combines sensitivity and specificity into a single metric (Youden Index = Sensitivity + Specificity - 1). A value closer to 1 indicates better overall performance.
- Diagnostic Odds Ratio (DOR): The ratio of the odds of positivity in true positive samples versus false positive samples. Higher values indicate better discriminatory power.
- Area Under the Curve (AUC): Measures the overall ability of the test to discriminate between positive and negative samples. An AUC of 1 represents a perfect test.
- Positive Predictive Value (PPV) & Negative Predictive Value (NPV): These are influenced by disease prevalence and indicate the probability that a positive/negative test result is correct.
The following table summarizes performance data from key studies and technologies to provide benchmark expectations.
Table 1: Performance Metrics of SNP Detection Methods
| Method / Technology | Reported Sensitivity | Reported Specificity | Key Factors Influencing Performance |
|---|---|---|---|
| High-Resolution Melting Analysis [81] | 100% (for products ≤300 bp); 96.1% (400-1000 bp) | 100% (for products ≤300 bp); 99.4% (400-1000 bp) | PCR product size; SNP type (false negatives more common with A/T wild type). |
| TaqMan SNP Genotyping Assays [82] | Implied "gold standard" level; high due to MGB probe technology. | Implied "gold standard" level; superior allele discrimination. | Robustness of design pipeline; quality of input DNA. |
| MassARRAY-based Assay [83] | 100% for species ID; 99.15% for serotyping | 100% for species ID and serotyping | Specificity of extension primers; optimized multiplex PCR conditions. |
| Cooperative Primer qPCR [80] | At least 10-fold lower LOD vs. conventional primers. | Implied high specificity due to reduced primer-dimer propagation. | Effective limitation of primer-dimer formation and propagation. |
Detailed Experimental Protocols
Protocol: High-Resolution Melting Analysis for SNP Scanning
This protocol is adapted from Reed et al. (2004) for identifying heterozygous single-base changes in PCR products [81].
1. Reagents and Equipment:
- PCR reagents (primers, dNTPs, buffer)
- Saturating DNA dye (e.g., LCGreen I)
- High-resolution real-time PCR instrument capable of precise temperature control and melting curve acquisition.
2. Procedure:
- PCR Amplification: Set up PCR reactions in the presence of the LCGreen I dye. For optimal results, design amplicons to be 300 bp or less, as this size demonstrated 100% sensitivity and specificity in validation studies [81].
- Post-PCR Melting: After amplification, subject the products to a high-resolution melting analysis.
- Fluorescence Normalization: Normalize the fluorescence data for all samples.
- Temperature Overlay: Align the melting curves by temperature.
- Curve Shape Analysis: Analyze the shape of the melting curves. Heterozygous samples will produce distinct curve shapes compared to homozygous wild-type or mutant samples due to the presence of heteroduplexes.
3. Critical Notes:
- The technique is homogeneous and closed-tube, reducing contamination risk.
- Sensitivity decreases slightly for larger amplicons (400-1000 bp) and for SNPs where the wild-type base is an A or T [81].
Protocol: Implementing Cooperative Primers to Suppress Primer-Dimer
This protocol, based on Ofori et al. (2021), uses modified primer structures to drastically reduce primer-dimer formation in SYBR Green-based qPCR [80].
1. Reagent Preparation:
- Cooperative Primers: Synthesize primers where each consists of a low melting temperature short primer sequence and a capture sequence, connected by two units of a spacer (e.g., hexaethylene glycol, Spacer 18) [80].
- Conventional Primer: The paired primer is a standard sequence.
- Master Mix: Use a SYBR Green-based qPCR master mix (e.g., Luna Universal qPCR Master Mix).
2. Experimental Workflow:
3. Procedure Steps:
- Primer Design: Design cooperative primers targeting conserved genomic regions. A cooperative primer is structured as:
5'-[Short Primer Sequence][Spacer18][Spacer18][Capture Sequence]-3'[80]. - Reaction Setup: Prepare a 15 μL reaction containing:
- 1x qPCR Master Mix
- 250 nM of each cooperative and conventional primer
- 3 μL of template DNA.
- Thermal Cycling: Run the qPCR with the following conditions:
- Initial Denaturation: 95°C for 3 minutes.
- 45 Cycles of:
- Denaturation: 95°C for 15 seconds.
- Annealing/Extension: 50°C for 40 seconds, then 60°C for 40 seconds.
- Analysis: Confirm specificity by analyzing the melting curves and running the products on an agarose gel. The cooperative primer system should yield a single, specific product with a clean melt peak and no smearing on the gel [80].
The Scientist's Toolkit
Table 2: Essential Reagents and Kits for Robust SNP Genotyping
| Reagent / Kit | Primary Function | Key Feature / Consideration |
|---|---|---|
| Hot-Start DNA Polymerase [2] [8] | Amplifies target DNA; hot-start version reduces nonspecific amplification and primer-dimer formation at room temperature. | Inactive until activated by high temperature, improving specificity. Essential for assays prone to primer-dimer. |
| TaqMan SNP Genotyping Assays [82] | Specifically detects and discriminates SNPs in qPCR. | Uses Minor Groove Binder (MGB) probes for superior allele discrimination against highly homologous sequences. |
| Saturating DNA Dyes (e.g., LCGreen I) [81] | Binds dsDNA for detection in high-resolution melting analysis. | Suitable for homogeneous, closed-tube SNP scanning without needing probes. |
| Uracil N-glycosylase (UNG) [79] | Prevents carryover contamination from previous PCR products. | Incorporated into master mixes; degrades uracil-containing amplicons from prior runs. |
| Cooperative Primers [80] | Drastically limits primer-dimer formation and propagation in qPCR. | Unique structure with a spacer and capture sequence reduces dimerization by up to 2.5 million-fold. |
Establishing Robust Standard Operating Procedures (SOPs) for Reproducibility
Troubleshooting Guides and FAQs
This technical support center provides targeted guidance to address common issues in PCR experiments, with a special focus on preventing primer-dimer formation to ensure research reproducibility.
FAQ: Understanding and Preventing Primer-Dimers
Q1: What are primer-dimers and why are they a problem for my PCR results? Primer-dimers are short, unintended amplification products formed when PCR primers anneal to each other instead of the target DNA template. They consume reaction components, reduce the yield of your desired product, and can lead to inaccurate quantification, especially in qPCR, potentially causing false-positive or false-negative results [3] [5].
Q2: What are the most common causes of primer-dimer formation? The most frequent causes include poorly designed primers with self-complementary regions, excess primer concentration, low annealing temperatures, and prolonged PCR cycles [5]. Preparation of reactions at room temperature can also allow primers to anneal randomly before thermal cycling begins [5].
Q3: How can I design primers to minimize the risk of primer-dimers? Follow these key design principles [11]:
- Length: Keep primers between 18-24 nucleotides.
- GC Content: Maintain GC content between 40-60%.
- 3' End Stability: Avoid more than three G or C nucleotides at the 3' end (a "GC clamp") to prevent non-specific binding [11].
- Complementation: Ensure primers have low self-complementarity and low complementarity to each other to prevent self-dimers and cross-dimers [8] [11]. Use reliable primer design software to check these parameters.
Q4: My primers are well-designed, but I still see primer-dimers. What laboratory practices can help? Adopt the following SOPs in your workflow [8] [5]:
- Use Hot-Start DNA Polymerases: These enzymes remain inactive until a high-temperature activation step, preventing spurious amplification during reaction setup [8] [15].
- Prepare Reactions on Ice: Keep all components and the reaction tube on ice during setup to minimize enzyme activity at low temperatures [8].
- Add Polymerase Last: Introduce the DNA polymerase to the reaction mix just before transferring tubes to the pre-heated thermal cycler [5].
- Optimize Primer Concentration: Use the minimum necessary primer concentration, typically between 0.1–1 µM, to reduce the amount of unused primer available for dimer formation [8] [5].
Troubleshooting Guide: Resolving Common PCR Issues
The table below outlines specific problems, their causes, and recommended solutions.
| Observation | Possible Cause | Recommended Solution |
|---|---|---|
| No Amplification [84] | Poor template quality or quantity | Re-purify template DNA; assess integrity by gel electrophoresis; increase amount of input DNA if necessary [8] [84]. |
| Incorrect annealing temperature | Calculate primer Tm using a reliable tool; perform a gradient PCR to optimize Ta [8] [84]. | |
| Insufficient Mg2+ concentration | Optimize Mg2+ concentration; remember EDTA or high dNTPs can chelate Mg2+, requiring higher concentration [8]. | |
| Multiple or Non-Specific Bands [84] | Low annealing temperature | Increase annealing temperature incrementally (1-2°C steps) to improve specificity [8]. |
| Excess primer or Mg2+ | Lower primer concentration within the 0.1–1 µM range; reduce Mg2+ concentration [8] [84]. | |
| Enzyme activity at setup | Use a hot-start DNA polymerase and set up reactions on ice [8] [84]. | |
| Primer-Dimer Formation [3] [5] | Primer complementarity | Redesign primers to avoid 3'-end complementarity; check for homology with software [11]. |
| Low annealing temperature | Increase annealing temperature; use a gradient to find the optimal temperature [5]. | |
| High primer concentration | Titrate primer concentration downwards (e.g., from 1 µM to 0.1 µM) to find the lowest efficient concentration [8] [5]. | |
| Low Fidelity/Sequence Errors [8] | Low-fidelity polymerase | Switch to a high-fidelity polymerase for applications like cloning and sequencing [8] [84]. |
| Unbalanced dNTP concentrations | Use fresh, equimolar dNTP mixes to prevent misincorporation [8] [84]. | |
| Excess Mg2+ | Lower Mg2+ concentration, as high levels can reduce fidelity [8]. |
The Scientist's Toolkit: Research Reagent Solutions
The following reagents are essential for robust and reproducible PCR.
| Reagent / Solution | Function & Importance in PCR |
|---|---|
| Hot-Start DNA Polymerase | Critical for specificity. Remains inactive until a high-temperature step, preventing primer-dimer formation and non-specific amplification during reaction setup [8] [15]. |
| MgCl2 or MgSO4 | Essential co-factor for DNA polymerase activity. Concentration must be optimized, as it significantly impacts yield, specificity, and fidelity [8] [84]. |
| PCR Additives (e.g., DMSO) | Aids in denaturing complex templates, such as GC-rich sequences, by disrupting DNA secondary structures. Note: it can lower the effective primer Tm [8] [15]. |
| GC Enhancer / Co-solvents | Specially formulated additives that help amplify difficult targets like GC-rich sequences without adversely affecting the enzyme, unlike common additives like DMSO [8] [15]. |
| dNTP Mix | The building blocks for new DNA strands. Requires fresh, equimolar concentrations of dATP, dCTP, dGTP, and dTTP to maintain high fidelity and prevent misincorporation [8]. |
Experimental Protocols for PCR Optimization
Protocol 1: Optimizing Annealing Temperature Using Gradient PCR
This protocol is fundamental for establishing the specific annealing conditions for any new primer set [5].
- Primer and Template Preparation: Dilute primers to a working concentration (e.g., 10 µM) and prepare a master mix with all standard PCR components.
- Thermal Cycler Programming: Set up your thermal cycler's gradient function across a temperature range. A good starting point is from 5°C below to 5°C above the calculated average Tm of your primer pair [84].
- Reaction Setup: Aliquot the master mix into PCR tubes and place them in the gradient block.
- PCR Amplification: Run the PCR with the gradient annealing step.
- Analysis: Analyze the results by gel electrophoresis. The optimal annealing temperature yields the highest amount of the desired specific product with the least amount of non-specific products or primer-dimers.
Protocol 2: Implementing Hot-Start PCR for Enhanced Specificity
This SOP minimizes non-specific amplification and is crucial for sensitive applications like multiplex PCR [15].
- Reagent Selection: Use a DNA polymerase formulated with a hot-start mechanism (e.g., antibody-based, chemical modification) [15].
- Reaction Assembly: Prepare the master mix on ice, according to the manufacturer's instructions.
- Initial Denaturation/Activation: Program the thermal cycler with an extended initial denaturation step (often 2-5 minutes at 95°C or higher). This step simultaneously activates the hot-start enzyme and denatures the template DNA [15].
- Cycling: Proceed with the standard cycling steps for your protocol.
SOP Development and Implementation Workflows
Workflow for SOP Lifecycle Management
Primer Design and Validation Pathway
Conclusion
Primer-dimer formation is a manageable challenge, not an inevitable flaw in PCR. A proactive, integrated approach—combining meticulous in-silico primer design with optimized reaction conditions and rigorous validation—is paramount for success. The strategies outlined, from foundational design principles to advanced troubleshooting, provide a clear path to highly specific and efficient amplification. For the future of biomedical and clinical research, mastering these techniques is crucial. It directly enhances the reliability of diagnostic assays, improves the accuracy of SNP detection, and enables more robust multiplexing for complex applications like next-generation sequencing and personalized medicine. Continued innovation in polymerase engineering, primer chemistry, and computational prediction will further empower scientists to eliminate these artifacts, paving the way for more sensitive and reproducible molecular assays.
References
- 1. Primer dimer [https://en.wikipedia.org/wiki/Primer_dimer]
- 2. Troubleshooting primer dimer in PCR [https://www.minipcr.com/primer-dimer-pcr/]
- 3. PCR Optimization: Strategies To Minimize Primer Dimer [https://genemod.net/blog/pcr-optimization]
- 4. What are Primer Dimers? - A Beginner's Guide [https://geneticeducation.co.in/what-are-primer-dimers-a-beginners-guide/]
- 5. PCR Troubleshooting 103: How to Address Primer-Dimers [https://geneticeducation.co.in/pcr-troubleshooting-103-how-to-address-primer-dimers/]
- 6. The influence of quality of primers on the formation ... [https://pubmed.ncbi.nlm.nih.gov/32799617/]
- 7. How can I avoid primer-dimer formation during PCR ... [https://www.qiagen.com/us/resources/faq/544]
- 8. PCR Troubleshooting Guide | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-troubleshooting.html]
- 9. PCR Troubleshooting Guide [https://www.genscript.com/pcr-troubleshooting-guide.html]
- 10. Eliminating primer dimers and improving SNP detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7200914/]
- 11. Primer design guide - 5 tips for best PCR results [https://the-dna-universe.com/2022/09/05/primer-design-guide-the-top-5-factors-to-consider-for-optimum-performance/]
- 12. DNA Amplification | No Complementarily to other primers [https://nij.ojp.gov/nij-hosted-online-training-courses/dna-amplification/overview/primer-design/no-complementarily-other-primers]
- 13. Guide to Primer Design for PCR [https://www.benchling.com/primer-design-for-pcr]
- 14. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 15. PCR Methods - Top Ten Strategies [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-methods.html]
- 16. Primer Dimer [https://mugenomicscore.missouri.edu/primerdimer.html]
- 17. Troubleshooting your PCR [https://www.takarabio.com/learning-centers/pcr/faq/troubleshooting?srsltid=AfmBOoosQ8XJWC4eNI7yXToNRAha_3MHd9lgLTMBanL9bPOlKA-Jh74V]
- 18. Overcoming the challenge of primer-dimers in multiplexed ... [https://blog.biosearchtech.com/overcoming-the-challenge-of-primer-dimers-in-multiplexed-pcr-assays]
- 19. Quantitative Experimental Determination of Primer-Dimer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3416024/]
- 20. Optimizing PCR: Proven Tips and Troubleshooting Tricks [https://www.the-scientist.com/optimizing-pcr-proven-tips-and-troubleshooting-tricks-71660]
- 21. Troubleshooting Non-Specific Amplification [https://bento.bio/resources/bento-lab-advice/troubleshooting-non-specific-amplification-with-bento-lab/]
- 22. How to Interpret DNA Gel Electrophoresis Results [https://www.goldbio.com/blogs/articles/interpreting-gel-electrophoresis-results?srsltid=AfmBOoq3HJj_DtUulUEUy5FBrEIXZlh7f8_f6R1xn8oIrmWXsvKZUJDH]
- 23. Primer Dimer - an overview [https://www.sciencedirect.com/topics/neuroscience/primer-dimer]
- 24. The Pain of Primer Dimer [https://kilobaser.com/post/the-pain-of-primer-dimer]
- 25. and homodimers? Selecting primers to avoid problems [https://eu.idtdna.com/pages/support/faqs/what-are-self--and-homo-dimers-and-how-do-i-select-primers-to-avoid-these-problems-]
- 26. PCR Primer Design Tips - Behind the Bench [https://www.thermofisher.com/blog/behindthebench/pcr-primer-design-tips/]
- 27. PCR Troubleshooting Guide [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/pcr-troubleshooting-guide?srsltid=AfmBOop5ebyftDx4eTqig1ZdqNNueStobsLfj4f9C8qNDnM9sBDhU3nS]
- 28. What Is A GC Clamp In PCR Primers? [https://toptipbio.com/gc-clamp-pcr/]
- 29. Effect of single mismatches at 3′–end of primers on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3174695/]
- 30. PrimerROC: accurate condition-independent dimer using... prediction [https://www.nature.com/articles/s41598-018-36612-9?error=cookies_not_supported&code=68f7a666-8c70-4007-89d5-626142341782]
- 31. Primer designing tool - NCBI - NIH [https://www.ncbi.nlm.nih.gov/tools/primer-blast/]
- 32. PrimerQuest PCR & qPCR Primer Design Tool | IDT [https://www.idtdna.com/pages/tools/primerquest]
- 33. PCR Primer Design Tool [https://eurofinsgenomics.eu/en/ecom/tools/pcr-primer-design/]
- 34. PCR Assay Optimization and Validation [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/assay-optimization-and-validation?srsltid=AfmBOorlkHQlFBo3RJq6CxGGvsmdfQB-vJ3InTe2ZtVoYktHo7yhnu3z]
- 35. Optimizing PCR Reaction Conditions for High Fidelity and ... [https://www.labx.com/resources/optimizing-pcr-reaction-conditions-for-high-fidelity-and-yield/5579]
- 36. PCR Assay Development: Common Pitfalls and How to ... [https://synapse.patsnap.com/article/pcr-assay-development-common-pitfalls-and-how-to-avoid-them]
- 37. 5 Steps to a Better PCR: A Troubleshooting and ... [https://blog.abclonal.com/blog/5-steps-to-a-better-pcr]
- 38. Touchdown PCR: Easy Intro Plus 5 Expert Tips [https://bitesizebio.com/2203/touchdown-pcr-a-primer-and-some-tips/]
- 39. What is Hot Start PCR? [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/what-is-hot-start-pcr?srsltid=AfmBOorrzB-EWmYyZOChL4ZLS4N9gFW4wKkLqH8iXw51qYSZZdlknF6w]
- 40. How is Hot-Start Technology Beneficial For Your PCR [https://www.thermofisher.com/tr/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/hot-start-technology-benefits-PCR.html]
- 41. Time to Instigate Nested PCR [https://bitesizebio.com/24592/time-to-instigate-nested-pcr/]
- 42. Hot Start PCR [https://www.merckmillipore.com/HN/es/technical-documents/technical-article/genomics/pcr/hot-start-pcr]
- 43. Prediction of PCR amplification from primer and template ... [https://www.nature.com/articles/s41598-021-86357-1]
- 44. Eliminating primer dimers and improving SNP detection using ... [https://pubmed.ncbi.nlm.nih.gov/32395633/]
- 45. Self-avoiding molecular recognition systems (SAMRS ... [https://www.biosyn.com/tew/Members-of-the-%E2%80%9CSelf-Avoiding-Molecular-Recognition-Systems%E2%80%9D-or-SAMRS-bind-to-natural-DNA-but-not-to-other-SAMRS-analogs.aspx]
- 46. Application Note Polymerase Chain Reaction (PCR) ... [https://www.glenresearch.com/reports/gr36-23]
- 47. PCR Troubleshooting Guide [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/pcr-troubleshooting-guide?srsltid=AfmBOopXCDadqOVySsuDVmx6n41HUnAD3OtHRqTyBcD3wfbSjsLkvYJU]
- 48. Amplification of the No Template Control (NTC) [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/real-time-pcr-troubleshooting-tool/gene-expression-quantitation-troubleshooting/amplification-no-template-control.html]
- 49. Why Your Negative Control Has a Band (And How to Fix It) [https://www.clyte.tech/post/troubleshooting-pcr-why-your-negative-control-has-a-band-and-how-to-fix-it?srsltid=AfmBOooDB22C-cr4hbtBOnTzKuTz7E3TzJgUltrz1C-BFufk3wh6tdX8]
- 50. Avoiding false positives with PCR | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-avoid-false-positives-in-pcr-and-what-to-do-if-you-get-them/]
- 51. PCR controls [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/reference-genes-and-controls/pcr-controls]
- 52. PCR Assay Optimization and Validation [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/assay-optimization-and-validation?srsltid=AfmBOorURx9aqGyt_23phrQ0_SI4HdTdkhCZyk5Uxm5xc18wi7OpVMEr]
- 53. Four Tips for Optimizing Your PCR Amplification [https://www.genscript.com/synthetic-biology-news/four-tips-for-optimizing-your-pcr-amplification.html]
- 54. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOoo682EcFIBqv72hDUTxaveBfDfukksEgYhG-KL0YmMqORIUfwLp]
- 55. How is Hot-Start Technology Beneficial For Your PCR [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/hot-start-technology-benefits-PCR.html]
- 56. Hot start PCR [https://en.wikipedia.org/wiki/Hot_start_PCR]
- 57. Hot Start PCR update: CleanAmp™ Primers [https://www.glenresearch.com/reports/gr21-11]
- 58. Why hot start is so hot? [https://solisbiodyne.com/EN/why-hot-start-is-so-hot/]
- 59. Hot Start PCR with heat-activatable primers [https://pmc.ncbi.nlm.nih.gov/articles/PMC2582603/]
- 60. PCR Troubleshooting: Common Problems and Solutions [https://www.mybiosource.com/learn/pcr-troubleshooting-common-problems-and-solutions/]
- 61. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOorGe0wkWx4_jkF2oTphAa1hXzz93mFmQsjc0unYkPJB2-wpTz1d]
- 62. GC-RICH PCR System Troubleshooting [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/gc-rich-pcr-system?srsltid=AfmBOoofp7h7vUuC_D4FUS5gIC-6m2yxty08jHl8PerxXeGmeN8Jh8jF]
- 63. Designing highly multiplex PCR primer sets with Simulated ... [https://www.nature.com/articles/s41467-022-29500-4]
- 64. PCR Troubleshooting Guide [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/pcr-troubleshooting-guide?srsltid=AfmBOornKUCYxyS8nkRoDyu9nzhHzEr1Pmh9gZrtOyB02s6MCW9IgiYD]
- 65. PCR Basic Troubleshooting Guide [https://www.creative-biogene.com/blog/pcr-basic-troubleshooting-guide]
- 66. PCR Basics | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-basics.html]
- 67. Trust your SYBR Green qPCR Data [https://www.thermofisher.com/blog/behindthebench/trust-your-sybr-green-qpcr-data/]
- 68. Adapter dimers causes, effects, and how to remove them [https://knowledge.illumina.com/library-preparation/general/library-preparation-general-troubleshooting-list/000001911]
- 69. Microchip capillary electrophoresis (Lab-on-chip ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996910000864]
- 70. DNA Polymerase—Four Key Characteristics for PCR [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/dna-polymerase-characteristics.html]
- 71. Modified DNA polymerases for PCR troubleshooting [https://link.springer.com/article/10.1007/s13353-016-0371-4]
- 72. Enhanced performance of Thermococcus kodakarensis ... [https://www.sciencedirect.com/science/article/abs/pii/S1046592825001512]
- 73. MyFi™ DNA Polymerase [https://www.bioline.com/myfi-dna-polymerase.html]
- 74. Engineering of novel DNA polymerase variants for single ... [https://www.nature.com/articles/s41598-025-10211-x]
- 75. Comparative evaluation of three polymerase chain reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12501571/]
- 76. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 77. PCR Troubleshooting Guide [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/pcr-troubleshooting-guide?srsltid=AfmBOoqIS3LN6jt3efZeqZNbsBh_HfuCcHZd5pe71w5Y158RM2tSWrcJ]
- 78. Performance Metrics for Selecting Single Nucleotide ... [https://www.nature.com/articles/srep36155]
- 79. Tips for successful PCR and Troubleshooting PCR-qPCR [https://info.abmgood.com/polymerase-chain-reaction-pcr-qpcr-troubleshooting]
- 80. Development of Cooperative Primer-Based Real-Time PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8591562/]
- 81. Sensitivity and specificity of single-nucleotide polymorphism ... [https://pubmed.ncbi.nlm.nih.gov/15308590/]
- 82. SNP Genotyping Analysis Using TaqMan Assays [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/snp-genotyping-taqman-assays.html]
- 83. Development of a high-throughput MassARRAY-based ... [https://www.nature.com/articles/s41598-025-92524-5]
- 84. PCR Troubleshooting Guide [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/pcr-troubleshooting-guide?srsltid=AfmBOoqa4jG5QmANCZCl3Nop8WFRuXKSaPqhH70FecZBPtqaSx3-4B7Y]