PCR vs. In Vivo DNA Replication: A Comprehensive Guide for Biomedical Researchers
This article provides a detailed comparative analysis for researchers, scientists, and drug development professionals on the fundamental differences and applications of in vivo DNA replication and the Polymerase Chain Reaction...
PCR vs. In Vivo DNA Replication: A Comprehensive Guide for Biomedical Researchers
Abstract
This article provides a detailed comparative analysis for researchers, scientists, and drug development professionals on the fundamental differences and applications of in vivo DNA replication and the Polymerase Chain Reaction (PCR). It explores the foundational biological principles of both systems, delves into their methodological applications in research and diagnostics, addresses critical troubleshooting and optimization parameters, and provides a framework for the validation and comparative assessment of these essential processes. The scope covers core mechanisms, key enzymes, technological limitations, and the strategic selection of methods for specific biomedical applications, synthesizing information to guide experimental design and clinical translation.
Core Mechanisms: Contrasting Cellular DNA Replication with Laboratory Amplification
Deoxyribonucleic acid (DNA) synthesis is a fundamental process for life and biotechnology. In living cells, this process occurs in vivo as DNA replication, a complex and highly regulated event essential for cell division and heredity [1]. In the laboratory, the Polymerase Chain Reaction (PCR) facilitates in vitro DNA amplification, a targeted enzymatic process that generates millions of copies of a specific DNA sequence from a minute starting amount [1] [2]. Although both processes produce new DNA strands, they differ profoundly in their mechanism, components, fidelity, and application. This whitepaper provides an in-depth technical comparison of these two systems, framing the analysis within the context of biomedical and pharmaceutical research, where understanding these differences is critical for experimental design and drug development.
Core Principles and Comparative Analysis
At the highest level, in vivo DNA replication is a biological process of producing two identical replicas of an entire genome from one original DNA molecule, occurring inside living cells [3]. In contrast, PCR is a laboratory process used to make millions of copies of a specific, targeted DNA fragment outside of a living organism (in a test tube) [3]. The following table summarizes the fundamental distinctions between these two systems.
Table 1: Comprehensive Comparison of In Vivo DNA Replication and In Vitro PCR
| Basis of Comparison | In Vivo DNA Replication | In Vitro PCR Amplification |
|---|---|---|
| Definition & Purpose | Biological process for copying the entire genome for cell division [3]. | Laboratory technique for amplifying a single, targeted DNA fragment [3]. |
| Cellular Context | Occurs in vivo inside living cells during the S-phase of the cell cycle [1] [3]. | Occurs in vitro in a test tube or well plate, independent of cells [3] [4]. |
| Key Steps | Initiation, Elongation, Termination [5]. | Denaturation, Annealing, Extension [2]. |
| Process Nature | Continuous process proceeding at high speed (~1,000 bases/second) [3]. | Discontinuous process proceeding through 30-40 cycles at a slower speed (1-4 kb/min) [3]. |
| Temperature | Occurs at a constant physiological temperature (e.g., 37°C in humans) [3]. | Requires cyclic temperature changes (denaturation at ~95°C, annealing at ~50-65°C, extension at ~72°C) [3] [2]. |
| Denaturation Mechanism | Enzyme-driven (DNA helicase, e.g., DnaB in E. coli) unwinds the double helix [5]. | Heat-induced (high temperature, ~95°C) separates DNA strands [3]. |
| Primer | Short RNA primers synthesized by primase (e.g., DnaG in E. coli) [3] [5]. | Short, single-stranded DNA primers, synthetically designed and added to the reaction [3] [2]. |
| Polymerizing Enzyme | DNA polymerase (e.g., Pol III in E. coli) with proofreading and repair abilities for high fidelity [3] [6]. | Thermostable DNA polymerase (e.g., Taq polymerase) with no/low proofreading ability in common variants, leading to higher error rates [3] [6]. |
| Error Rate | Approximately 1 in 100,000 bases due to proofreading and repair mechanisms [3]. | Taq polymerase error rate is approximately 1 in 9,000 bases [3]. |
| System Complexity | A complex process dependent on a well-defined set of many enzymes, co-factors, and regulatory proteins [1] [5]. | A simpler process using a minimal set of defined ingredients: template, primers, Taq polymerase, dNTPs, and buffer [1] [2]. |
The quantitative data on fidelity is of paramount importance for research and drug development. A recent 2024 study introduced a novel assay based on loss-of-function mutations in the conditionally toxic sacB gene to directly compare fidelity. The findings were stark: DNA production in E. coli resulted in 80- to 3,000-fold fewer mutations compared to enzymatic DNA replication methods like PCR and rolling circle amplification (RCA) [6]. This underscores that DNA synthesized in vitro can introduce a substantial number of mutation impurities, a critical risk factor for the quality and yield of final pharmaceutical products like gene therapies [6].
Detailed Methodologies and Experimental Protocols
Protocol for Studying Bacterial DNA Replication In Vivo
Studying DNA replication in vivo requires a multifaceted approach to understand its regulation within the complex cellular environment. The following protocol outlines key methodological considerations for in vivo replication research in bacteria, based on current practices [5].
- Step 1: Genetic Background Preparation. The host organism's genetic background must be carefully arranged. This often involves creating gene knockouts, introducing point mutations, or constructing reporter fusions (e.g., GFP) to proteins of interest to monitor their localization and expression levels during the cell cycle.
- Step 2: Cell Synchronization and Culture. To study replication initiation and progression at a population level, cells are often synchronized. This can be achieved through filtration, antibiotic treatment, or temperature shifts for strains with temperature-sensitive mutations in essential replication genes. Synchronized cells are then cultured, and samples are taken at specific time points.
- Step 3: Analysis of Key Replication Parameters. The extracted samples are analyzed using various techniques:
- Origin-to-Terminus Ratio Analysis: Using quantitative PCR (qPCR) to measure copy numbers at the origin (oriC) versus the terminus (ter) of replication. This ratio reveals the replication dynamics and is higher in fast-growing cells undergoing multifork replication [5].
- Flow Cytometry: Used to analyze DNA content per cell, providing information on the timing of replication initiation and completion relative to the cell cycle [5].
- Marker Frequency Analysis (MFA): A genomic technique that maps the number of DNA sequencing reads along the chromosome, creating a profile that shows the location of replication origins and termini and the progression of replication forks.
Protocol for a Standard In Vitro PCR Amplification
The PCR protocol is a highly standardized in vitro procedure for targeted DNA amplification. The following describes a generic endpoint PCR protocol [3] [2].
- Step 1: Reaction Setup. In a sterile, nuclease-free tube, combine the following components on ice:
- Template DNA: 1 pg–1 µg of genomic DNA or 1–100 ng of plasmid DNA.
- Forward and Reverse Primers: 0.1–1 µM each of synthetic oligonucleotide primers complementary to the flanking regions of the target sequence.
- Thermostable DNA Polymerase: 0.5–2.5 units of an enzyme like Taq polymerase.
- Deoxynucleotide Triphosphates (dNTPs): 200 µM of each dNTP (dATP, dCTP, dGTP, dTTP).
- Reaction Buffer: A buffer providing optimal pH, salt conditions (e.g., MgCl₂ is a critical co-factor), and stability.
- Step 2: Thermocycling. Place the reaction tube in a thermal cycler and run a program with the following steps for 25–40 cycles:
- Denaturation: 20–30 seconds at 94–98°C. This step melts the double-stranded DNA into single strands.
- Annealing: 20–40 seconds at 50–65°C. The temperature is set based on the melting temperature (Tm) of the primers, allowing them to bind (anneal) to their complementary sequences on the template DNA.
- Extension: 15–60 seconds per kb at 68–72°C. The DNA polymerase synthesizes a new DNA strand by extending from the primer, adding dNTPs to the growing chain.
- Step 3: Final Hold and Analysis. After the cycles are complete, a final extension step (5–10 minutes at 72°C) ensures all amplicons are fully extended. The reaction is then held at 4–10°C. The PCR products are typically analyzed by agarose gel electrophoresis to confirm the size and yield of the amplified DNA fragment.
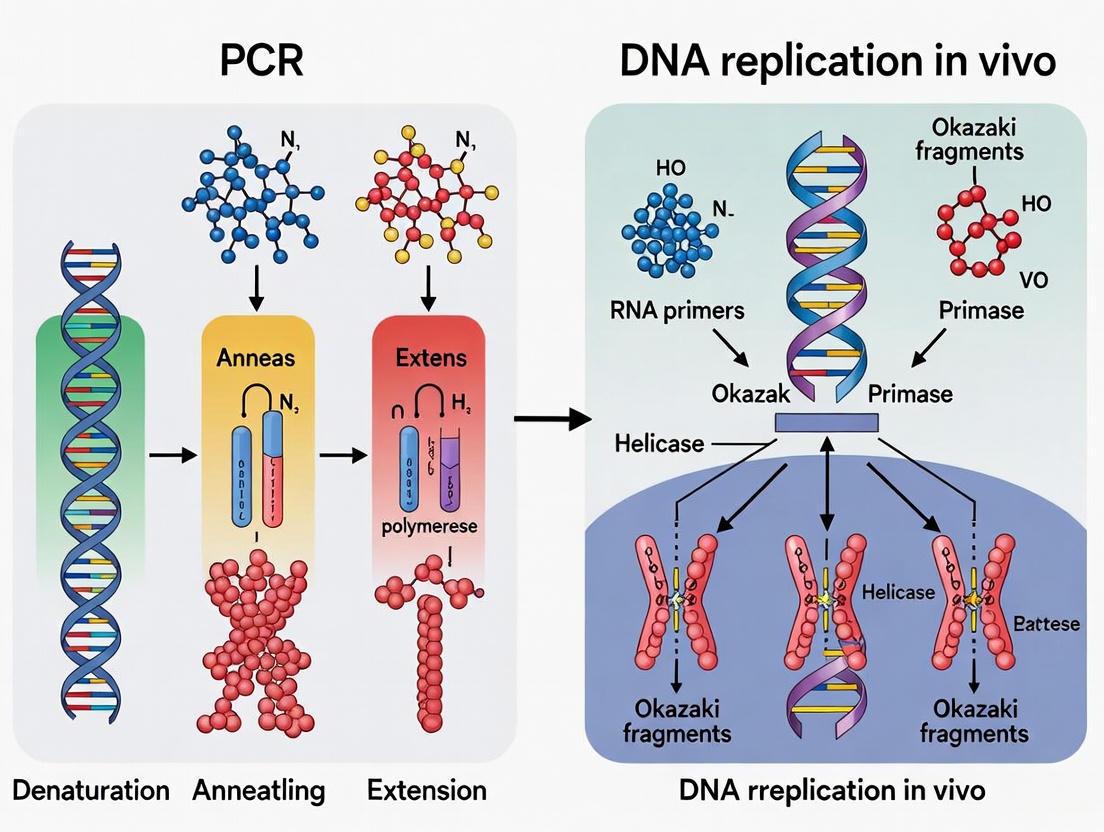
System Visualization and Workflows
The fundamental workflows of in vivo replication and in vitro PCR can be visualized as process maps, highlighting the key stages and their differences.
In Vivo DNA Replication Workflow
In Vitro PCR Amplification Workflow
The Scientist's Toolkit: Essential Research Reagents
Successful experimentation in both in vivo and in vitro DNA synthesis requires a specific set of reagents and tools. The following table details key research reagent solutions for both fields.
Table 2: Essential Research Reagents for DNA Replication and Amplification Studies
| System | Reagent / Tool | Function & Explanation |
|---|---|---|
| In Vivo Replication | Conditional Mutants (e.g., temperature-sensitive) | Allows functional study of essential replication genes (e.g., dnaA, dnaN) by inactivating them under non-permissive conditions [5]. |
| In Vivo Replication | Synchronization Agents (e.g., antibiotics, filtration devices) | Used to arrest a population of bacterial cells at a specific stage of the cell cycle, enabling the study of replication timing and progression [5]. |
| In Vivo Replication | Fluorescent Reporter Fusions (e.g., GFP-tagged proteins) | Enables visualization of replication machinery components (e.g., polymerase, clamp) in live cells using fluorescence microscopy [5]. |
| In Vivo Replication | Origin-Specific Probes | Short DNA sequences complementary to the origin of replication (oriC), used in techniques like qPCR to measure origin firing and copy number [5]. |
| In Vitro PCR | Thermostable DNA Polymerases (e.g., Taq, Q5, Phusion) | Enzymes that withstand high temperatures of PCR. Advanced versions (e.g., Q5) offer high fidelity and processivity, crucial for diagnostic and biomanufacturing applications [3] [6]. |
| In Vitro PCR | Synthetic Oligonucleotide Primers | Short, single-stranded DNA sequences that define the start and end points of the DNA segment to be amplified. They are designed for specificity and optimal annealing temperature [2]. |
| In Vitro PCR | dNTP Mix | A solution containing equimolar concentrations of the four deoxynucleotides (dATP, dCTP, dGTP, dTTP), which serve as the building blocks for the new DNA strands [2]. |
| In Vitro PCR | MgCl₂-Containing Reaction Buffer | Provides the optimal chemical environment (pH, ionic strength) for the PCR. Mg²⁺ is an essential co-factor for DNA polymerase activity [2]. |
Advanced Systems and Future Directions
The boundaries between in vivo and in vitro systems are becoming increasingly blurred with advancements in synthetic biology. A key innovation is the Transcription–Translation coupled DNA Replication (TTcDR) system [7]. This system reconstructs fundamental cellular functions from the bottom-up using a modified PURE (Protein synthesis Using Recombinant Elements) system, which is an in vitro transcription–translation system reconstituted from E. coli proteins, ribosomes, and tRNAs [7]. A central component is the Φ29 DNA polymerase, which can replicate DNA in a stand-alone fashion within this system, for example, via rolling circle amplification (RCA) [7].
Recent work, as highlighted in the search results, has focused on integrating genetic circuitry for external control over these in vitro replication systems. For instance, a TetR-based genetic circuit has been successfully constructed, allowing repression and induction (using anhydrotetracycline, aTc) of DNA replication within the TTcDR system [7]. This demonstrates the potential for controlling in vitro DNA replication with external signals, a critical step toward constructing more complex systems like synthetic cells and for enabling Darwinian evolution in in vitro systems [7]. These hybrid systems represent a powerful platform for studying biological complexity and developing advanced biotechnological tools, bridging the gap between the simplicity of PCR and the regulatory complexity of living cells.
The duplication of genetic material is a fundamental process in biology, achieved with remarkable fidelity in living cells by a sophisticated protein machine known as the replisome. This multi-enzyme complex coordinates numerous catalytic and regulatory functions to copy DNA efficiently and accurately during the synthetic phase (S-phase) of the cell cycle [8]. In contrast, the Polymerase Chain Reaction (PCR) represents a simplified, in vitro mimic of DNA synthesis that amplifies specific target sequences from minute starting material using a minimal set of defined ingredients and controlled thermal cycling [8] [9]. While both processes synthesize new DNA strands, their underlying mechanisms, complexity, and regulatory fidelity differ substantially. This whitepaper examines the intricate architecture and function of the cellular replisome, highlighting the critical distinctions between in vivo replication and its in vitro counterpart, PCR, with implications for basic research and drug development.
The Architecture and Function of the Bacterial Replisome
The replisome is a highly coordinated molecular machine that has evolved to act in a concerted fashion during DNA replication [8]. Its structure is best understood in model organisms like Escherichia coli and Bacillus subtilis.
Core Components and Their Coordination
At its core, the bacterial replisome consists of several essential proteins that work in concert:
- Helicase (DnaB in E. coli): A homohexameric enzyme that unwinds the parental DNA duplex by translocating on single-stranded DNA (ssDNA) in the 5' to 3' direction, using NTP hydrolysis as an energy source [10] [11].
- DNA Polymerase III (Pol III): The primary replicative polymerase in E. coli, consisting of a catalytic subunit (α), a proofreading exonuclease subunit (ε), and an accessory subunit (θ) [10].
- Sliding Clamp (β clamp): A dimeric ring that encircles DNA and tethers Pol III to the template, dramatically increasing its processivity [10].
- Clamp Loader (τ₃δδ'): A pentameric complex that utilizes ATP hydrolysis to assemble the β clamp onto primer-template junctions [10].
- Primase (DnaG): Synthesizes short RNA primers that provide a starting point for DNA polymerases on the lagging strand [10].
- Single-Stranded DNA Binding Protein (SSB): Coats and protects exposed ssDNA generated by helicase activity, preventing secondary structure formation and degradation [10] [12].
caption: The replisome coordinates multiple enzymatic activities through physical interactions between components, with the clamp loader (τ) serving as a central organizer.
The Trombone Model and Replisome Organization
The replisome employs an elegant mechanism known as the trombone model to simultaneously replicate both strands of the DNA duplex despite their antiparallel orientation [10]. While the leading strand is synthesized continuously, the lagging strand is synthesized discontinuously as a series of Okazaki fragments. This process involves the formation and release of transient ssDNA loops on the lagging strand, reminiscent of a trombone slide's movement.
Recent research has revealed unexpected complexity in replisome organization. Contrary to earlier models suggesting two polymerase complexes (one for each strand), in vitro reconstitution and direct visualization in living E. coli cells have demonstrated that the replisome can incorporate three Pol III complexes [10]. This trimeric polymerase architecture is multimerized by the trimeric τ subunit within the clamp loader, with the third polymerase potentially serving as a backup to support efficient lagging strand synthesis [10].
Species-Specific Variations in Replisome Composition
While the core replisome architecture is conserved across bacteria, significant variations exist between species. In E. coli, a single DNA polymerase (Pol III) handles synthesis on both strands [10]. However, in Gram-positive bacteria like B. subtilis, two distinct C-type DNA polymerases—PolC and DnaE—are required for chromosomal replication [10]. Current evidence suggests a division of labor where DnaE extends RNA primers a short distance on the lagging strand before PolC rapidly displaces it to synthesize the majority of DNA on both strands [10].
PCR: A Simplified In Vitro Mimic of DNA Synthesis
In contrast to the complex cellular replisome, PCR employs a radically simplified approach to DNA synthesis, requiring only a DNA polymerase, primers, nucleotides, and a buffer solution in a controlled thermal cycling environment [8] [9]. This simplification enables specific DNA amplification but lacks the sophisticated regulatory mechanisms of in vivo replication.
Key Characteristics of PCR DNA Polymerases
The efficiency and application range of PCR depend critically on the properties of the DNA polymerase used. Key characteristics include:
- Thermostability: Essential for withstanding repeated denaturation temperatures (≥95°C). Enzymes from thermophilic organisms (e.g., Taq from Thermus aquaticus) are preferred, with hyperthermostable variants (e.g., Pfu from Pyrococcus furiosus) offering even greater stability [13].
- Fidelity: The accuracy of DNA sequence replication, determined by the polymerase's proofreading (3'→5' exonuclease) activity. High-fidelity enzymes are crucial for applications like cloning and sequencing [13].
- Processivity: The number of nucleotides incorporated per polymerase binding event. Highly processive enzymes are beneficial for amplifying long templates or GC-rich sequences [13].
- Specificity: The ability to amplify only the intended target, often enhanced through "hot-start" technologies that inhibit polymerase activity at room temperature [13].
Comparative Analysis: Replisome vs. PCR
Table 1: Quantitative Comparison of Replisome and PCR Characteristics
| Parameter | Cellular Replisome | PCR Amplification |
|---|---|---|
| Number of Proteins | Dozens of coordinated proteins [10] [12] | Single DNA polymerase (with potential additives) [8] [9] |
| Initiation Mechanism | Sequence-specific (oriC), regulated by initiator proteins (DnaA) and cell cycle signals [12] | Temperature-mediated DNA denaturation followed by primer annealing [9] |
| Priming Mechanism | De novo RNA primer synthesis by primase (DnaG) [10] [12] | Pre-designed DNA primers added to reaction [9] |
| Replication Rate | ~1,000 nucleotides/second [11] | <100 nucleotides/second (polymerase-dependent) |
| Processivity | Entire chromosome (with clamp) [10] | Typically <10 kb (polymerase-dependent) [13] |
| Error Rate (Fidelity) | ~10⁻⁹–10⁻¹¹ (with proofreading) [13] | ~10⁻⁴–10⁻⁶ (Taq without proofreading); ~10⁻⁷ (with proofreading) [13] |
| Strand Coordination | Coupled leading and lagging strand synthesis [10] | No physical coupling; independent strand synthesis |
Table 2: Key Enzymatic Activities in DNA Replication Systems
| Activity | Cellular Replisome | PCR |
|---|---|---|
| Helicase | Dedicated helicase (DnaB) unwinds DNA [10] [11] | Thermal denaturation (no enzymatic unwinding) [8] |
| 5'→3' Polymerization | DNA Pol III (E. coli) or PolC/DnaE (B. subtilis) [10] | Thermostable DNA polymerase (e.g., Taq, Pfu) [9] [13] |
| 3'→5' Proofreading | Intrinsic to replicative polymerase (ε subunit in E. coli) [10] [13] | Present only in high-fidelity polymerases (e.g., Pfu) [13] |
| Clamp Loading | Dedicated clamp loader complex (τ₃δδ') [10] | Not applicable |
| Primase | Dedicated primase (DnaG) synthesizes RNA primers [10] | Not applicable (pre-synthesized DNA primers used) |
| Strand Repair | Multiple repair pathways associated with replisome | No repair mechanisms |
The fundamental distinction between these systems lies in their complexity and regulation. The replisome represents a highly sophisticated, self-correcting machinery that coordinates numerous enzymatic activities through precise protein-protein interactions and responds to cellular checkpoints [10] [12]. In contrast, PCR employs a minimalist approach that relies on external control (thermal cycling) to drive the reaction, sacrificing the accuracy and coordination of in vivo replication for simplicity and specificity [8].
Experimental Methodologies for Studying Replisome Function
Understanding replisome complexity requires sophisticated experimental approaches that can probe its structure, dynamics, and function. Key methodologies include:
In Vitro Reconstitution and Single-Molecule Studies
Functional replisomes have been reconstituted from purified components in both E. coli and B. subtilis, enabling detailed biochemical characterization [10]. These systems allow researchers to:
- Measure kinetic parameters of DNA unwinding and synthesis
- Determine the processivity of replication
- Analyze the coupling efficiency between helicase and polymerase activities [11]
Advanced biophysical techniques, including single-molecule fluorescence and optical tweezers, have revealed dynamic behaviors such as helicase "slippage" and fork regression that are difficult to observe in bulk assays [11].
Real-Time Fluorescence-Based Assays
A powerful 2-aminopurine (2-AP) fluorescence-based method enables researchers to map the precise positions of helicase and DNA polymerase with respect to the replication fork junction in real time [11]. This approach provides structural information on replisome organization and can quantify individual protein contributions to fork progression.
Radiometric Assays for Activity Coupling
Thin-layer chromatography (TLC)-based radiometric assays simultaneously measure DNA polymerase and exonuclease activities during processive leading strand synthesis [11]. This method provides valuable information on polymerase-exonuclease active-site switching and its dependence on helicase-polymerase coupling.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Replisome and PCR Studies
| Reagent / Tool | Function / Application | Examples / Notes |
|---|---|---|
| Hyperthermostable DNA Polymerases | PCR amplification under demanding conditions | Pfu (Pyrococcus furiosus), KOD (Thermococcus), GBD (Pyrococcus) [13] |
| High-Fidelity Polymerase Blends | Error-sensitive applications like cloning and sequencing | Engineered enzymes with >50–300x fidelity of Taq polymerase [13] |
| Hot-Start Polymerases | Prevention of nonspecific amplification in PCR | Antibody-inhibited, aptamer-blocked, or chemically modified enzymes [13] |
| Processivity-Enhanced Enzymes | Amplification of long templates, GC-rich sequences | Engineered polymerases with DNA-binding domains [13] |
| Defined Origin (oriC) Templates | Study of replication initiation mechanisms | Minimal origin sequences for in vitro replication assays [12] |
| Synthetic Replication Fork Substrates | Analysis of helicase-polymerase coupling | Custom DNA constructs mimicking replication forks [11] |
| Single-Molecule Imaging Systems | Visualization of replisome dynamics in real-time | Fluorescence microscopy, optical tweezers, magnetic tweezers [10] [11] |
| 2-Aminopurine (2-AP) Labeled DNA | Monitoring replication fork junction dynamics | Fluorescent nucleotide analog for structural studies [11] |
Signaling and Workflow Diagrams
caption: Workflow of cellular DNA replication process showing three main phases.
caption: PCR thermal cycling process showing three-step amplification cycle.
Research Implications and Future Directions
The complexity of the replisome presents both challenges and opportunities for drug development. Unlike PCR, which has been successfully adapted for diagnostic applications, the replisome represents a promising but challenging target for therapeutic intervention [14]. Antibiotics that target bacterial replisome components (e.g., novobiocin inhibiting DNA gyrase) demonstrate the potential of this approach, while the complexity of protein-protein interactions within the replisome offers additional targets for drug discovery.
Future research directions include:
- Developing high-throughput screening assays for replisome function [11]
- Engineering DNA polymerases with enhanced properties for both research and diagnostic applications [13] [14]
- Single-molecule analysis of replisome dynamics in live cells [10]
- Structural studies of complete replisome complexes to inform rational drug design
The replisome represents one of the most sophisticated molecular machines in biology, coordinating multiple enzymatic activities through precise temporal and spatial regulation to achieve accurate genome duplication. In contrast, PCR employs a minimalist approach that sacrifices the fidelity and coordination of cellular replication for simplicity and specificity. Understanding the complexity of the replisome not only provides fundamental insights into biology but also informs the development of new research tools and therapeutic strategies that target DNA replication in pathogens and cancer cells. As research methodologies advance, the gap between in vitro simplification and in vivo complexity continues to narrow, offering new opportunities to harness cellular mechanisms for research and clinical applications.
The Polymerase Chain Reaction (PCR) stands as a cornerstone technique in molecular biology, enabling the targeted in vitro amplification of specific DNA sequences. Its remarkable efficiency stems from a fundamentally simple reaction environment, requiring only a minimal set of core components: a thermostable DNA polymerase, primers, deoxynucleoside triphosphates (dNTPs), a magnesium cofactor, and a template DNA. This technical guide delves into the function and optimization of each component within a standard Taq polymerase-based PCR, providing detailed protocols and quantitative data. The composition of this in vitro system is then explicitly contrasted with the vastly more complex, protein-driven machinery of cellular DNA replication, framing the simplicity of the PCR mix as a key differentiator from its in vivo counterpart.
The Polymerase Chain Reaction (PCR), introduced by Kary Mullis in 1985, is a laboratory technique for amplifying a specific region of DNA through repeated cycles of thermal denaturation, primer annealing, and enzymatic extension [9]. Unlike the complex cellular process of DNA replication, which involves a coordinated effort of dozens of proteins to copy an entire genome, PCR is designed for simplicity and specificity, targeting a single fragment for exponential amplification [3]. This process relies on a thermostable DNA polymerase, most famously Taq polymerase isolated from Thermus aquaticus, which can withstand the high temperatures required to denature the DNA double strand [9]. The entire amplification process is accomplished with a surprisingly minimal set of ingredients, mixed in a single tube and cycled through precisely controlled temperatures. This guide explores these core components and their optimization, consistently highlighting the stark contrast between the streamlined in vitro amplification and the intricate in vivo process of genomic duplication.
The Minimal Set of Ingredients: Function and Optimization
A standard PCR reaction contains five essential components that together create the environment for DNA synthesis. The following diagram illustrates the workflow of a single PCR cycle and the role of each component.
DNA Polymerase: The Engine of Amplification
The DNA polymerase is the central enzyme responsible for synthesizing new DNA strands.
- Taq DNA Polymerase: This enzyme, derived from the thermophilic bacterium Thermus aquaticus, is thermostable with a half-life of approximately 40 minutes at 95°C, making it ideal for the repeated high-temperature denaturation steps in PCR [15]. It polymerizes DNA at a rate of about 60 bases per second at 70°C and is typically used at a concentration of 1–2 units per 50 µL reaction [15].
- Key Characteristics: Taq polymerase requires a primer to initiate synthesis and operates exclusively in the 5′ to 3′ direction [16] [9]. Unlike the replicative DNA polymerases in cells, it lacks 3′ to 5′ exonuclease proofreading activity, resulting in a higher error rate (approximately 1 in 9,000 bases) compared to the high-fidelity in vivo replication machinery (error rate of about 1 in 100,000 bases) [3]. This trade-off for robustness and simplicity is a defining feature of standard PCR.
Oligonucleotide Primers: The Guides of Specificity
Primers are short, single-stranded DNA sequences (typically 15–30 nucleotides) that are complementary to the sequences flanking the target region [15]. They are fundamental to PCR's specificity, as they define the start and end points of amplification.
Design Considerations:
- Melting Temperature (Tm): The Tm of both primers should be in the range of 55–70°C and within 5°C of each other [15].
- GC Content: Ideally 40–60%, with a uniform distribution of G and C bases [15].
- 3′ End: Should not contain more than three G or C bases, as this can promote nonspecific priming; having one G or C at the 3′ end can help with anchoring [15].
- Concentration: Typically used at 0.1–1 µM. High concentrations can cause mispriming and nonspecific amplification, while low concentrations yield little to no product (see Table 2) [15].
Deoxynucleoside Triphosphates (dNTPs): The Building Blocks
dNTPs (dATP, dCTP, dGTP, dTTP) are the monomeric substrates from which the DNA polymerase synthesizes the new strand [15]. They are added to the reaction in equimolar concentrations.
- Standard Concentration: The recommended final concentration for each dNTP is 0.2 mM [15].
- Balance is Critical: Excess dNTPs can inhibit PCR and sequester Mg²⁺, while concentrations below the Km of the enzyme (0.01–0.015 mM) can lead to incomplete synthesis [15]. Modifications like using dUTP in place of dTTP can be incorporated for applications like carryover contamination prevention with UDG treatment [15].
Divalent Cations: The Essential Cofactor
Magnesium ions (Mg²⁺) are an absolute requirement for DNA polymerase activity.
- Role: Mg²⁺ acts as a cofactor, facilitating the binding of the dNTP substrate to the enzyme's active site and catalyzing the phosphodiester bond formation [15]. It also helps stabilize the interaction between primers and the template DNA.
- Optimization: The Mg²⁺ concentration must be carefully optimized, as it influences enzyme activity, fidelity, and primer-template specificity. It is typically supplied in the reaction buffer at concentrations between 1.5–5.0 mM.
Template DNA: The Blueprint
The template is the DNA sample containing the target sequence to be amplified.
- Source: Template DNA can be genomic DNA (gDNA), complementary DNA (cDNA), or plasmid DNA [15].
- Amount and Quality: The optimal input amount depends on the complexity of the DNA. For a 50 µL reaction, 5–50 ng of gDNA is commonly used, while only 0.1–1 ng is sufficient for plasmid DNA [15]. The template must be of reasonable purity, as contaminants like phenol, EDTA, or heparin can inhibit the polymerase [9].
The Scientist's Toolkit: Essential PCR Reagents
Table 1: Key Research Reagent Solutions for a Standard Taq PCR
| Reagent | Function | Standard Concentration/Amount |
|---|---|---|
| Taq DNA Polymerase | Thermostable enzyme that synthesizes new DNA strands. | 1–2 units / 50 µL reaction [15] |
| Forward & Reverse Primers | Define the start and end of the target sequence; provide specificity. | 0.1–1 µM each [15] |
| dNTP Mix | The four nucleotides (dATP, dCTP, dGTP, dTTP) used as building blocks for new DNA. | 0.2 mM each [15] |
| MgCl₂ Solution | Essential cofactor for DNA polymerase activity. | 1.5–5.0 mM (optimization required) [15] |
| 10X Reaction Buffer | Provides optimal pH and salt conditions (e.g., Tris-HCl, KCl) for the reaction. | 1X final concentration |
| Nuclease-Free Water | Solvent that brings the reaction to its final volume. | Variable |
Quantitative Comparison of PCR Master Mixes
The performance of a PCR assay is highly dependent on the quality and formulation of the master mix. A 2021 study systematically evaluated seven commercial TaqMan master mixes for detecting porcine DNA, providing quantitative data on their limits of detection (LOD) and PCR efficiency on two different real-time PCR platforms [17]. The results, summarized below, underscore the importance of selecting the right reagents for a specific application.
Table 2: Performance Comparison of Seven Commercial TaqMan Master Mixes [17]
| Manufacturer | Master Mix | Applied Biosystems StepOnePlus LOD (pg/rxn) | Bio-rad CFX Connect LOD (pg/rxn) | PCR Efficiency Range (%) |
|---|---|---|---|---|
| Applied Biosystems | TaqMan Universal PCR Master Mix | 5 | 0.5 | 84.96 – 108.80 |
| CancerROP | MG 2X qPCR MasterMix (TaqMan) with ROX | 0.5 | 5 | 84.96 – 108.80 |
| Invitrogen | Express qPCR Supermix Universal | 0.5 | 0.5 | 84.96 – 108.80 |
| Kogene Biotech | PowerAmp Real-time PCR Master Mix II | 0.5 | 0.5 | 84.96 – 108.80 |
| New England Biolabs | Luna Universal Probe qPCR Master Mix | 0.5 | 0.5 | 84.96 – 108.80 |
| Qiagen | QuantiNova Probe PCR Kit | 0.5 | 0.5 | 84.96 – 108.80 |
| Takara | Premix Ex Taq (Probe qPCR), ROX plus | 5 | 5 | 84.96 – 108.80 |
Key Findings:
- The LOD for the master mixes varied from 0.5 to 5 pg of porcine DNA per reaction, demonstrating that sensitivity is reagent-dependent [17].
- PCR efficiencies across all mixes and platforms ranged from 84.96% to 108.80%, with the best combination achieving 100.49% efficiency [17].
- The study also found that nonspecific amplification of DNA from other species (human, dog, cow, chicken) was observed for four of the seven master mixes, highlighting that specificity is also a variable factor [17].
Experimental Protocol: Setting Up a Standard PCR
This protocol is adapted for a 50 µL reaction using a standard Taq DNA polymerase.
Materials:
- Template DNA (e.g., 10–100 ng gDNA)
- Forward and Reverse Primers (10 µM stock each)
- 10 mM dNTP Mix
- 25 mM MgCl₂
- 10X PCR Buffer
- Taq DNA Polymerase (e.g., 5 U/µL)
- Nuclease-free Water
Method:
- Prepare Reaction Mix on Ice: Combine the following components in a sterile, nuclease-free PCR tube:
- Nuclease-free Water: to a final volume of 50 µL
- 10X PCR Buffer: 5 µL
- 25 mM MgCl₂: 3 µL (1.5 mM final; optimize between 1.5–5.0 mM)
- 10 mM dNTP Mix: 1 µL (0.2 mM each final)
- Forward Primer (10 µM): 1 µL (0.2 µM final)
- Reverse Primer (10 µM): 1 µL (0.2 µM final)
- Template DNA: X µL (e.g., 1 µL of 50 ng/µL gDNA)
- Taq DNA Polymerase: 0.5 µL (2.5 U)
Thermal Cycling: Place the tube in a thermal cycler and run the following program:
- Initial Denaturation: 95°C for 2–5 minutes (1 cycle)
- Amplification (30–40 cycles):
- Denature: 95°C for 15–30 seconds
- Anneal: 55–72°C for 15–60 seconds (temperature is primer-specific)
- Extend: 72°C for 1 minute per 1 kb of amplicon
- Final Extension: 72°C for 5–10 minutes (1 cycle)
- Hold: 4°C ∞
Analysis: Analyze the PCR product by agarose gel electrophoresis.
Contrasting PCR with Cellular DNA Replication
The simplicity of the PCR mix becomes starkly apparent when compared to the complexity of in vivo DNA replication. The following diagram and table delineate the key differences between these two DNA synthesis processes.
Table 3: A Comparative Analysis of PCR and Cellular DNA Replication [16] [3]
| Basis of Comparison | PCR | DNA Replication (In Vivo) |
|---|---|---|
| Definition | A laboratory process to amplify a specific DNA fragment. | A biological process to duplicate the entire genome for cell division. |
| Occurrence | In vitro (in a test tube). | In vivo (inside living cells). |
| Polymerizing Enzyme | A single, thermostable enzyme (e.g., Taq polymerase). | Multiple enzymes (DNA polymerases δ, ε, γ) working in a complex replisome. |
| Primer | DNA oligonucleotides. | Short RNA primers synthesized by primase. |
| Denaturation | Achieved by high heat (~95°C). | Catalyzed by a specific enzyme, helicase, at the replication fork. |
| Strand Synthesis | Discontinuous on both templates after denaturation. | Continuous on the leading strand, discontinuous (via Okazaki fragments) on the lagging strand [16]. |
| Proofreading | Taq polymerase lacks 3′→5′ proofreading, leading to a higher error rate. | DNA polymerases have exonucleolytic proofreading and mismatch repair for extremely high fidelity [16]. |
| Speed | ~1–4 kilobases per minute. | ~1 kilobase per second [3]. |
The power of PCR lies in its elegant simplicity. The technique accomplishes exponential DNA amplification using a minimal set of core ingredients: a thermostable polymerase, two primers, dNTPs, Mg²⁺, and a template. This streamlined in vitro system can be precisely controlled and optimized, as evidenced by the performance variations among commercial master mixes. However, this simplicity comes with inherent trade-offs, such as lower fidelity and a reliance on external thermal cycling, when compared to the native process of DNA replication. The cellular process is a sophisticated, self-correcting, and multi-enzymatic machinery evolved for speed and accuracy across an entire genome. Understanding the composition and limitations of the simple PCR mix is, therefore, fundamental for researchers to effectively harness and optimize this indispensable technique within the broader context of nucleic acid metabolism.
The replication of genetic material is a fundamental process, yet the context in which it occurs—within the living cell versus within the laboratory instrument—dictates entirely different regulatory philosophies. In vivo, DNA replication is a spatially organized and temporally precise event, tightly coupled to the needs of the cell and regulated by a complex network of protein interactions and checkpoints [18]. In contrast, the Polymerase Chain Reaction (PCR) in a thermal cycler is a simplified, automated, and accelerated in vitro process designed for the exponential amplification of specific DNA fragments [19] [9]. This technical guide delves into the core control systems of both processes, framing them within a broader thesis on the fundamental differences between biological fidelity and engineering efficiency. Understanding these distinct regulatory principles is crucial for researchers and drug development professionals who leverage these systems for applications ranging from molecular diagnostics to synthetic biology [20] [21].
The Cell Cycle: A Biologically Integrated Control System
The cell-cycle control system is a sophisticated, autonomous network that ensures the accurate duplication of the entire genome and its faithful distribution to daughter cells. Its operation is deeply integrated with the cell's physiology and environmental cues.
Core Regulatory Machinery
At its heart, the system is driven by cyclin-dependent kinases (Cdks), whose activity oscillates through the synthesis and degradation of regulatory subunits called cyclins [18]. Different cyclin-Cdk complexes trigger specific cell-cycle events:
- G1/S-Cdk commits the cell to DNA replication.
- S-Cdk initiates the actual process of DNA synthesis.
- M-Cdk drives the entry into mitosis [18].
Cdk activity is itself fine-tuned by inhibitory phosphorylation (e.g., by Wee1 kinase) and activating dephosphorylation (e.g., by Cdc25 phosphatase) [18]. This multi-layered regulation creates a robust, binary switch that triggers events in a complete and irreversible manner.
Temporal and Spatial Regulation Mechanisms
The system's sophistication lies in its temporal ordering and spatial organization, which are absent from the thermal cycler.
- Checkpoints as Quality Control: The control system can arrest the cycle at specific checkpoints if previous events are incomplete or if DNA is damaged. These checkpoints operate primarily through negative intracellular signals; for example, unreplicated DNA or an improperly attached chromosome sends a "stop" signal that persists until the problem is resolved [18].
- Adaptability and Robustness: Unlike the fixed programming of a thermal cycler, the cell cycle can be delayed or advanced based on extracellular signals from other cells, which often act at a G1 checkpoint to promote or inhibit proliferation [18]. This provides the system with the adaptability required for complex multicellular life.
Table 1: Key Components of the Mammalian Cell-Cycle Control System
| Component | Function | Key Regulators |
|---|---|---|
| G1-Cdk | Promotes passage through the Start/Restriction point in late G1. | G1-cyclins |
| G1/S-Cdk | Binds Cdks at the end of G1 to commit the cell to DNA replication. | G1/S-cyclins |
| S-Cdk | Required for the initiation of DNA replication during S phase. | S-cyclins |
| M-Cdk | Promotes entry into mitosis and regulates chromosome condensation, nuclear envelope breakdown. | M-cyclins |
| Cdk-Activating Kinase (CAK) | Phosphorylates Cdks for full activation. | - |
| Wee1 Kinase | Inhibitory kinase; phosphorylates Cdks to suppress activity. | - |
| Cdc25 Phosphatase | Activating phosphatase; dephosphorylates Cdks to increase activity. | - |
The Thermal Cycler: An Engineered System for DNA Amplification
The thermal cycler represents a deterministic, externally programmed system designed for a single, powerful function: the exponential amplification of a pre-defined DNA sequence.
Core Operational Principle
PCR in a thermal cycler mimics one aspect of DNA replication—the template-directed synthesis of new strands—but divorces it from all cellular constraints [8]. The process is cyclical and relies on precise, external temperature control:
- Denaturation (90–98°C): The double-stranded DNA is melted into single strands by heating, a step that replaces the enzymatic machinery required for in vivo strand separation [9] [22].
- Annealing (50–65°C): Short, synthetic oligonucleotide primers bind to their complementary sequences on either side of the target DNA. This step provides the specificity that in vivo replication achieves through complex initiation proteins [9] [22].
- Extension (72°C): A thermostable DNA polymerase (e.g., Taq polymerase) synthesizes a new DNA strand from the primers. The enzyme's thermostability is the key engineering breakthrough that allows the process to be automated and repeated [19] [9].
Evolution of Instrument Control
The thermal cycler's control system has evolved for speed, precision, and user convenience, reflecting its engineered nature [19].
- From Manual to Automated: Early PCR required manual transfer between water baths. The first commercial thermal cyclers introduced automated metal blocks with Peltier elements for precise heating and cooling [19].
- Heated Lids and Fast Cycling: The introduction of heated lids prevented evaporation, eliminating the need for mineral oil overlays. Advanced Peltier systems with fast ramp rates (e.g., 6°C/second) enable "fast PCR," reducing run times from hours to under 40 minutes [19].
- Gradient and Verifiable Blocks: Gradient thermal blocks allow for the testing of different annealing temperatures simultaneously. "Better-than-gradient" technology uses separate, insulated blocks for more precise temperature control and faster optimization [19].
Table 2: Core Components of a PCR Reaction Mixture
| Component | Function | In Vivo Analog |
|---|---|---|
| DNA Template | Contains the target sequence to be amplified. | Genomic DNA in nucleus. |
| Thermostable DNA Polymerase (e.g., Taq) | Enzyme that synthesizes new DNA strands; thermostability allows it to survive denaturation temperatures. | DNA polymerases (e.g., Pol δ, Pol ε). |
| Oligonucleotide Primers | Short, single-stranded DNA fragments that define the start and end of the target sequence. | RNA primers synthesized by primase. |
| Deoxynucleotides (dNTPs) | The building blocks (dATP, dCTP, dGTP, dTTP) for new DNA synthesis. | dNTP pools in the nucleus. |
| Buffer System (MgCl₂) | Provides optimal pH and chemical environment. Mg²⁺ is an essential cofactor for DNA polymerase. | Intracellular buffer and ion conditions. |
Comparative Analysis: Biological Fidelity vs. Engineering Efficiency
The fundamental differences between these two systems can be distilled into their approach to regulation, error handling, and overall purpose.
Table 3: Quantitative Comparison of Cell Cycle vs. Thermal Cycler
| Parameter | In Vivo DNA Replication (Cell Cycle) | In Vitro PCR (Thermal Cycler) |
|---|---|---|
| Temporal Regulation | Checkpoint-dependent, variable duration (hours to days). | Fixed, user-defined program (30 mins to 2 hours) [19] [23]. |
| Spatial Regulation | Confined to nucleus during S phase; coordinated with other cellular structures. | Homogeneous reaction in a tube; no spatial organization. |
| Control Mechanism | Autonomous biochemical network (Cdks, cyclins, checkpoints) [18]. | External, pre-programmed instrument (thermal cycler) [19]. |
| Quantifiable Output | Two complete, faithful copies of the entire genome. | Millions to billions of copies of a specific DNA fragment. |
| Error Rate & Fidelity | High fidelity; complex proofreading and mismatch repair systems. | Lower fidelity; error rates of ~10⁻⁵ due to lack of robust proofreading in some polymerases [22]. |
| Sensitivity to Inhibitors | Highly sensitive; arrests in response to internal/external stressors. | Susceptible to inhibitors in sample prep (e.g., heparin, hemoglobin) but can be mitigated (e.g., digital PCR) [9] [24]. |
| Primary Function | Faithful genome duplication for cell division. | Targeted DNA amplification for analysis and detection. |
Key Differentiating Factors
- Purpose and Output: The cell cycle aims for faithful duplication of the entire genome for cell division. PCR aims for mass production of a specific DNA fragment for analytical purposes [18] [8].
- Regulation and Flexibility: The cell cycle is adaptive and responsive, with built-in quality control (checkpoints) that can halt the process. The thermal cycler is deterministic and rigid, executing a fixed program regardless of reaction success, placing the burden of optimization on the user [19] [18].
- Fidelity: In vivo replication is exceptionally accurate due to multiple proofreading and repair mechanisms. PCR is inherently less accurate, as many thermostable polymerases lack robust proofreading, leading to a higher cumulative error rate over multiple cycles [22] [8].
Advanced PCR Technologies and Experimental Protocols
The principles of the thermal cycler have been extended to develop advanced quantification methods that address specific limitations of conventional PCR.
Comparison of Quantitative PCR (qPCR) and Digital PCR (dPCR)
Table 4: Key Differences Between qPCR and dPCR
| Feature | Quantitative PCR (qPCR) | Digital PCR (dPCR) |
|---|---|---|
| Quantification Principle | Relative quantification based on Cycle Threshold (Cq) value; requires a standard curve [20] [25]. | Absolute quantification by counting positive partitions using Poisson statistics; no standard curve needed [20] [24]. |
| Detection Method | Fluorescence measured in real-time during amplification [9] [23]. | Endpoint fluorescence measurement after amplification [20] [24]. |
| Sensitivity & Precision | High sensitivity; precision can be affected by amplification efficiency and standard curve accuracy. | Superior sensitivity and precision, especially for low-abundance targets and small fold-change differences [20] [24]. |
| Tolerance to Inhibitors | Moderately susceptible to PCR inhibitors which can alter amplification efficiency [20]. | Highly tolerant to PCR inhibitors due to partitioning and endpoint detection [24] [23]. |
| Throughput & Cost | High throughput; lower cost per sample [20] [25]. | Lower throughput; higher cost per sample due to specialized consumables [20]. |
| Ideal Application | High-throughput gene expression analysis, pathogen detection where relative quantification is sufficient [20] [25]. | Detection of rare sequences, absolute copy number variation, liquid biopsies, and quantification in complex backgrounds [20] [24]. |
Detailed Experimental Protocol: dPCR for Bacterial Quantification
The following protocol, adapted from a 2025 study comparing dPCR and qPCR for periodontal pathobiont detection, exemplifies a modern application of partitioned PCR [24].
Aim: To absolutely quantify specific bacterial pathogens (Porphyromonas gingivalis, Aggregatibacter actinomycetemcomitans, Fusobacterium nucleatum) in subgingival plaque samples.
Materials & Reagents:
- QIAcuity Four dPCR instrument & QIAcuity Nanoplate 26k 24-well (or equivalent nanoplate-based system).
- QIAcuity Probe PCR Kit, including master mix.
- Primers and double-quenched hydrolysis probes specific for the 16S rRNA genes of each target.
- Restriction enzyme (e.g., Anza 52 PvuII) to reduce background from non-specific amplification.
- QIAamp DNA Mini Kit for DNA extraction from samples.
Methodology:
- Sample Collection and DNA Extraction:
- Collect clinical samples (e.g., using absorbent paper points) and store in appropriate transport medium.
- Extract genomic DNA using a commercial kit (e.g., QIAamp DNA Mini Kit) according to the manufacturer's instructions. Elute DNA in nuclease-free water.
dPCR Reaction Setup:
- Prepare a 40 µL reaction mixture per sample containing:
- 10 µL of sample DNA.
- 10 µL of 4x Probe PCR Master Mix.
- 0.4 µM of each specific forward and reverse primer.
- 0.2 µM of each specific hydrolysis probe (each labeled with a distinguishable fluorophore).
- 0.025 U/µL of the restriction enzyme PvuII.
- Nuclease-free water to volume.
- Mix thoroughly by pipetting.
- Prepare a 40 µL reaction mixture per sample containing:
Partitioning and Amplification:
- Transfer the reaction mixture to a nanoplate well.
- Seal the plate and load it into the dPCR instrument.
- The instrument automatically partitions each sample into ~26,000 nanoscale reactions.
- Run the thermocycling protocol:
- Enzyme activation: 2 min at 95°C.
- 45 cycles of:
- Denaturation: 15 sec at 95°C.
- Annealing/Extension: 1 min at 58°C.
Data Acquisition and Analysis:
- After cycling, the instrument performs endpoint fluorescence imaging for each channel corresponding to the different probes.
- Software (e.g., QIAcuity Software Suite) automatically counts positive and negative partitions for each target.
- The absolute concentration (copies/µL) of each target in the original sample is calculated by the software based on the fraction of positive partitions and the application of Poisson distribution statistics. A reaction is typically considered positive if at least three partitions are positive [24].
The Scientist's Toolkit: Essential Reagents and Materials
Table 5: Key Research Reagent Solutions for Featured Experiments
| Item | Function/Application | Example Product/Catalog |
|---|---|---|
| Thermostable DNA Polymerase | Catalyzes DNA synthesis at high temperatures in PCR. Essential for automation. | Taq DNA Polymerase, Hot Start versions [19] [22]. |
| dNTP Mix | Provides the nucleotide building blocks (dATP, dCTP, dGTP, dTTP) for DNA synthesis in both PCR and in vivo replication studies. | PCR Grade dNTP Set [22]. |
| Fluorescent Hydrolysis Probes (e.g., TaqMan) | Sequence-specific probes for real-time detection and quantification in qPCR and dPCR. Enable multiplexing [24] [23]. | TaqMan Gene Expression Assays [24]. |
| DNA Extraction Kit | Purifies high-quality, inhibitor-free DNA from complex biological samples (tissue, cells, blood), a critical first step for reliable PCR. | QIAamp DNA Mini Kit [24]. |
| Restriction Enzymes | For traditional cloning (REC), Golden Gate Assembly, and can be used in dPCR to reduce non-specific background [24] [21]. | PvuII, EcoRI, etc. [24] [21]. |
| Microfluidic dPCR Plates/Chips | Consumables that partition a PCR reaction into thousands of individual reactions for absolute quantification. | QIAcuity Nanoplate 26k [24]. |
The cell cycle and the thermal cycler represent two diametrically opposed paradigms for controlling DNA synthesis. The cell cycle is an integrated, adaptive biological system where temporal and spatial regulation ensures fidelity and coordination with the organism's needs. In contrast, the thermal cycler is a reductionist, deterministic engineering tool where external programming maximizes speed, output, and specificity for a singular task. For the researcher, this distinction is not merely academic. The choice between using in vivo systems (e.g., for functional genomics) and in vitro tools (e.g., PCR for diagnostics) depends on the question at hand. Furthermore, selecting the appropriate PCR technology—conventional, qPCR, or dPCR—requires a nuanced understanding of their respective strengths, as quantified in this guide. Ultimately, leveraging the power of the thermal cycler while respecting the complexity of the cell cycle allows for a more sophisticated and effective approach in modern molecular research and drug development.
In molecular biology, the accurate duplication of genetic material is a cornerstone of both cellular survival and laboratory science. Two processes are paramount: DNA replication, the natural, in vivo mechanism by which a cell copies its entire genome before division, and the Polymerase Chain Reaction (PCR), an artificial, in vitro technique designed to amplify a specific, targeted DNA fragment. While both are template-dependent processes that synthesize new DNA, their fundamental philosophies diverge dramatically in scope and objective. DNA replication is tasked with the monumental feat of duplicating the entire genome with exceptionally high fidelity to ensure genetic stability across generations of cells. In contrast, PCR is engineered for the targeted amplification of a single, short DNA region from within a vast genome, often sacrificing some degree of accuracy for speed, simplicity, and yield. This technical guide explores the core differences between these two processes, with a focused examination of the fidelity—the accuracy of nucleotide incorporation—that each one achieves. We will delve into the quantitative metrics of error rates, the molecular determinants of fidelity, and the experimental methodologies used to measure them, providing a framework for researchers to select the appropriate process or tool for their specific applications in drug development and basic research.
Core Process Comparison: Entire Genome vs. Targeted Amplification
The fundamental difference between DNA replication and PCR lies in their scope and purpose. A detailed, side-by-side comparison of their characteristics is provided in Table 1.
Table 1: Fundamental Differences Between DNA Replication and PCR
| Characteristic | DNA Replication (In Vivo) | Polymerase Chain Reaction (In Vitro) |
|---|---|---|
| Definition & Objective | Biological process of producing two identical replicas of the entire genome from one original DNA molecule [3] [26]. | Laboratory technique used to make many copies of a specific, targeted DNA fragment [3] [26]. |
| Template | The entire cellular genome. | A single, short DNA region of interest. |
| Key Steps | Initiation, Elongation, Termination [3] [12]. | Denaturation, Annealing, Extension [3]. |
| Primary Enzymes | DNA polymerase with proofreading (3'-5' exonuclease) activity [3]. | Thermostable DNA polymerase (e.g., Taq polymerase), often without proofreading [3]. |
| Primers | Short RNA primers synthesized by primase (RNA polymerase) [3]. | DNA primers supplied in the reaction mixture [3]. |
| Denaturation Method | Enzyme-driven (DNA helicase) [3] [12]. | Heat-induced (high temperature, ~95°C) [3]. |
| Process & Environment | Continuous process occurring at a constant physiological temperature (~37°C) inside living cells (in vivo) [3]. | Discontinuous, cyclic process occurring at three different temperatures in a test tube (in vitro) [3]. |
| Speed | Approximately 1,000 bases per second [3]. | Approximately 1,000 to 4,000 bases per minute [3]. |
DNA Replication: The Cellular Machinery of Whole-Genome Duplication
DNA replication is a complex, enzyme-dependent process that occurs with high fidelity during the cell cycle. In prokaryotes, it initiates at a single origin of replication (oriC) and proceeds bidirectionally to the termination site (ter), faithfully copying the entire chromosome [12]. The process relies on a multi-protein complex known as the replisome, which includes key enzymes such as helicase (unwinds DNA), primase (synthesizes RNA primers), and the DNA polymerase holoenzyme, which is responsible for DNA synthesis and proofreading [12]. The use of RNA primers and the coordination of leading and lagging strand synthesis are hallmarks of this process. Its primary objective is to ensure the accurate and complete duplication of the genome for cell division, making fidelity a non-negotiable requirement.
Polymerase Chain Reaction: The Laboratory Workhorse for Targeted Amplification
PCR, conversely, is an in vitro technique designed to generate millions to billions of copies of a specific DNA sequence. Its simplicity stems from cycling between different temperatures to achieve denaturation, primer annealing, and enzyme-driven extension. The process employs a thermostable DNA polymerase (e.g., Taq polymerase from Thermus aquaticus) that can withstand the high denaturation temperatures [3]. A significant limitation of early PCR enzymes like Taq polymerase is their lack of 3'-5' proofreading exonuclease activity, which is a key fidelity mechanism in cellular DNA polymerases [3]. This inherent difference in the enzymatic machinery is a major contributor to the disparity in error rates between the two processes.
Fidelity and Error Analysis: A Quantitative Deep Dive
The fidelity of a DNA synthesis process is quantitatively defined as the error rate, typically expressed as the number of mutations per base pair per duplication event. The disparity between DNA replication and standard PCR is substantial.
Table 2: Error Rate and Fidelity Comparison
| Process / Enzyme | Reported Error Rate (Errors/bp/duplication) | Relative Fidelity (Compared to Taq) | Key Determinants of Fidelity |
|---|---|---|---|
| In Vivo DNA Replication | ~1 x 10⁻¹⁰ [3] | >10,000x higher | DNA polymerase with proofreading (3'-5' exonuclease), mismatch repair pathways, and other post-replication correction systems. |
| High-Fidelity PCR Enzymes (e.g., Pfu, Phusion) | ~1 x 10⁻⁶ to 5 x 10⁻⁷ [27] | 10x - 100x higher than Taq | Use of DNA polymerases engineered to possess or enhance 3'-5' proofreading exonuclease activity. |
| Standard PCR (Taq Polymerase) | ~1 x 10⁻⁵ to 2 x 10⁻⁵ [3] [27] | 1x (Baseline) | Lacks 3'-5' proofreading activity; primary error type is base substitutions. |
The data in Table 2 highlights a critical concept: the error rate of standard PCR with Taq polymerase is approximately 1 error in every 9,000 to 100,000 bases synthesized, whereas in vivo DNA replication boasts an astonishingly low error rate of about 1 in 100,000,000 bases [3]. This difference of several orders of magnitude underscores the superior accuracy of the cellular machinery. However, it is crucial to note that "high-fidelity" PCR enzymes, such as Pfu and Phusion, have been developed to bridge this gap. These enzymes incorporate proofreading capabilities, reducing error rates to as low as 1 in 1,000,000 to 2,000,000 bases, making them essential for applications like cloning and sequencing where accuracy is paramount [27].
Experimental Methodologies for Assessing Fidelity
Accurately measuring the error rate of DNA polymerases, whether in replication or amplification, is critical for evaluating their performance. Below are detailed protocols for key fidelity assays.
Direct Sequencing of Cloned PCR Products
This method is considered a gold standard for directly quantifying polymerase error rates [27].
Protocol:
- Amplification: Perform PCR on a target plasmid DNA template using a high-fidelity polymerase and a limited template amount (e.g., 25 pg) to maximize the number of template doublings (e.g., 30 cycles).
- Cloning: Ligate the purified PCR product into a sequencing vector and transform into competent E. coli.
- Sequencing: Pick individual bacterial colonies, prepare plasmid DNA, and perform Sanger sequencing of the entire insert for each clone.
- Data Analysis: Align the sequenced clones to the known reference sequence. Identify any base substitutions, insertions, or deletions. The error rate is calculated using the formula:
Error Rate = (Total Number of Mutations Observed) / (Total Number of Base Pairs Sequenced × Number of Template Doublings)[27]. This method allows for the interrogation of a large DNA sequence space and provides a direct, unbiased measurement of errors.
ThelacZForward Mutation Assay
This is a high-throughput screening method that relies on a phenotypic readout.
Protocol:
- Amplification: Amplify a reporter gene (e.g., the lacZ α-complementing fragment) using the polymerase under test.
- Cloning and Transformation: Clone the PCR products and transform them into an appropriate E. coli host strain.
- Phenotypic Screening: Plate the transformed bacteria on media containing a chromogenic substrate (X-gal). Colonies with a functional β-galactosidase enzyme (no mutations in the critical region of lacZ) will turn blue. Mutated, non-functional clones will remain white.
- Validation: Sequence the lacZ gene from a representative number of white (mutant) clones to determine the spectrum and nature of the mutations. The mutation frequency is calculated from the ratio of white to total colonies.
NGS-Based Genome-Wide Off-Target Analysis (for CRISPR Fidelity)
While not a direct measure of polymerase fidelity, the principles of genome-wide fidelity assessment are exemplified by modern CRISPR off-target assays. These unbiased methods are crucial for evaluating the accuracy of genome-editing tools.
Protocol for GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) [28]:
- Transfection: Co-deliver plasmids encoding the CRISPR-Cas9 nuclease and sgRNA along with a proprietary, double-stranded oligonucleotide ("GUIDE-seq tag") into living cells.
- Tag Integration: When a double-strand break (DSB) occurs—either on-target or off-target—the cellular repair machinery incorporates the GUIDE-seq tag into the break site.
- Genomic DNA Preparation & Library Prep: Harvest genomic DNA and shear it. Prepare next-generation sequencing (NGS) libraries. The tag-specific sequences are used to enrich for fragments that contain integrated tags.
- Sequencing & Bioinformatic Analysis: Perform high-throughput sequencing. Bioinformatics pipelines then map the sequenced reads back to the reference genome. Any genomic location with a significant cluster of tag integrations is identified as a nuclease cleavage site, providing a genome-wide profile of off-target activity.
Diagram: Workflow for GUIDE-seq Off-Target Analysis.
The Scientist's Toolkit: Research Reagent Solutions
Selecting the right reagents is critical for controlling fidelity in experimental workflows.
Table 3: Essential Research Reagents for Genetic Amplification and Editing
| Reagent / Tool | Function / Description | Application Notes |
|---|---|---|
| High-Fidelity DNA Polymerases (e.g., Pfu, Phusion) | Engineered PCR enzymes with 3'-5' proofreading exonuclease activity, resulting in lower error rates than Taq polymerase [27]. | Essential for cloning, sequencing, and mutant library generation where sequence accuracy is critical. |
| Taq DNA Polymerase | Thermostable DNA polymerase without proofreading activity; lower fidelity but high processivity and yield [3] [27]. | Suitable for routine PCR, genotyping, and detection where ultimate accuracy is less critical than speed and yield. |
| CRISOT Software Tool | A computational framework that uses RNA-DNA interaction fingerprints from molecular dynamics simulations to predict genome-wide CRISPR off-target effects [29]. | Used in sgRNA design and optimization to improve targeting specificity and reduce experimental validation workload. |
| SpCas9-HF1 (High-Fidelity Variant) | An engineered Cas9 nuclease with point mutations (N497A/R661A/Q695A/Q926A) designed to reduce non-specific DNA contacts, rendering off-target events nearly undetectable [30]. | A key reagent for therapeutic genome editing applications where minimizing off-target effects is a safety imperative. |
| GUIDE-seq Tag | A double-stranded oligonucleotide tag that is incorporated into double-strand breaks by cellular repair pathways, enabling genome-wide identification of nuclease cleavage sites [28]. | An unbiased experimental method for profiling the genome-wide specificity of CRISPR nucleases in a cellular context. |
The dichotomy between replicating the entire genome and amplifying a targeted fragment is fundamentally a trade-off between fidelity and convenience. DNA replication, perfected by evolution, is an intricate, high-fidelity cellular process dedicated to the accurate duplication of the entire genetic blueprint. PCR, a powerful invention of human ingenuity, provides a rapid and simple method for targeted amplification, though often at the cost of absolute accuracy. For the modern researcher, understanding this distinction is not merely academic. It directly informs critical experimental choices: selecting a proofreading polymerase for cloning, opting for a high-fidelity Cas9 variant for gene therapy development, or employing a genome-wide assay like GUIDE-seq to thoroughly assess off-target effects. As molecular techniques continue to evolve and find application in clinical diagnostics and therapeutics, the principles of fidelity—whether in copying a single gene or an entire genome—will remain paramount to ensuring reliable, reproducible, and safe scientific outcomes.
From Bench to Bedside: Research and Clinical Applications of PCR and Native Replication
Polymerase Chain Reaction (PCR) has revolutionized molecular diagnostics by providing a powerful in vitro method for targeted DNA amplification. This technical guide explores PCR's role in detecting pathogens and genetic mutations, framing its capabilities within the fundamental contrast to in vivo DNA replication. While both processes share the core principle of DNA synthesis, their mechanisms, fidelity, and application landscapes differ substantially. We examine the evolution of PCR-based diagnostics from laboratory tools to point-of-care devices, detail experimental protocols adhering to MIQE guidelines, and visualize key workflows. The convergence of enhanced sensitivity, quantification, and portability positions PCR as an indispensable technology in modern clinical research and therapeutic development.
At its core, PCR is an in vitro enzymatic process that amplifies short, predefined regions of DNA. Its power in diagnostics stems from its ability to generate billions of DNA copies from a minute starting amount within hours. Understanding its efficacy requires an appreciation of its relationship with, and divergence from, the biological process of in vivo DNA replication.
Table 1: Key Process Differences between PCR and In Vivo DNA Replication
| Parameter | PCR | In Vivo DNA Replication |
|---|---|---|
| Definition | A laboratory process to amplify a target DNA region [3] | A biological process producing two identical DNA replicas from one original DNA molecule [3] |
| Occurrence | In vitro (test tube) [3] | In vivo (inside living cells) [3] |
| Primary Objective | Copy a single DNA fragment [3] | Copy the entire genome [3] |
| Process Steps | Denaturation, Annealing, Extension [31] | Initiation, Elongation, Termination [3] |
| Operating Temperature | Multiple high temperatures (e.g., 94°C, ~55°C, 72°C) [8] [3] | Physiological temperature (~37°C) [3] |
| Polymerase Enzyme | Thermostable (e.g., Taq); no proofreading in common forms [3] | DNA Pol δ/ε; high-fidelity with proofreading [3] |
| Primers | Short, single-stranded DNA oligonucleotides [31] | Short RNA strands synthesized by primase [3] |
| Strand Separation | Heat-induced denaturation [3] | Enzyme-driven (DNA helicase) [3] |
| Speed | 1-4 kb/min [3] | ~1 kb/second [3] |
| Error Rate | Higher (e.g., ~1 in 9,000 bases for Taq) [3] | Lower (~1 in 100,000 bases) [3] |
While in vivo DNA replication is a complex, concerted action of numerous enzymes and co-factors evolved for high-fidelity genome duplication during the cell cycle, PCR employs a minimalist, automated approach for targeted amplification [8] [3]. This simplicity is its strength, allowing researchers to isolate and exponentially amplify a specific DNA sequence of diagnostic relevance from a complex biological background.
The Evolution of PCR in Molecular Diagnostics
The adoption of PCR in clinical settings was driven by the limitations of conventional, culture-based pathogen identification, which can be slow, insensitive for fastidious organisms, and ineffective for patients who have received antibiotics [31]. PCR answered the need for a rapid, sensitive, and specific diagnostic tool.
Early nucleic acid assays used DNA probe technology, but poor sensitivity due to low target DNA abundance limited their clinical utility [31]. The development of PCR in 1985 enabled direct amplification of target microbial DNA, drastically improving sensitivity and reducing turnaround time [31]. Subsequent modifications vastly expanded its capabilities:
- Multiplex PCR: Allows simultaneous detection of several pathogens or genetic markers in a single reaction by incorporating multiple primer sets [31].
- Reverse Transcriptase PCR (RT-PCR): Enables detection of RNA viruses by first converting RNA into complementary DNA (cDNA) [31].
- Real-Time Quantitative PCR (qPCR): A major breakthrough that couples amplification with fluorescent detection in a closed-tube system. This eliminates post-amplification processing, reduces contamination risk, and provides quantitative data by measuring the cycle at which amplification becomes detectable (Quantification Cycle, Cq) [31].
The subsequent development of digital PCR (dPCR) further advanced quantification by partitioning a sample into thousands of microreactions, allowing absolute nucleic acid quantification without a standard curve and offering enhanced precision for detecting rare mutations [32].
PCR in Practice: Detecting Pathogens and Mutations
Infectious Disease Detection
PCR-based diagnostics have been successfully developed for a wide range of bacteria, viruses, and fungi. Their high sensitivity is critical for identifying organisms that cannot be cultured or have prolonged incubation times [31]. Approved FDA assays exist for pathogens like Chlamydia trachomatis, Group B Streptococcus, and HIV, providing superior sensitivity and specificity over traditional methods [31]. During outbreaks like SARS, the speed of PCR is invaluable for patient triage, infection control, and informed treatment decisions [31].
Genetic Mutation Detection
PCR is fundamental for identifying genetic mutations underlying inherited disorders and cancers. Allele-Specific PCR (ASPCR), a specialized form of PCR, uses primers designed to perfectly match a specific mutant sequence, thereby selectively amplifying it over the wild-type [33]. This principle is being harnessed in novel point-of-care devices. For example, researchers have developed a portable instrument that uses ASPCR combined with electrical impedance measurement on a microchip to detect a point mutation causing hereditary transthyretin amyloidosis from a single blood drop in about 10 minutes [33]. This highlights the ongoing trend to make robust, rapid genetic testing accessible outside central laboratories.
Essential Methodologies and Protocols
Core qPCR Experimental Workflow
Adherence to standardized protocols and reporting guidelines, such as the MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines, is crucial for assay reproducibility and reliability [34].
Diagram 1: Core qPCR workflow.
Key Research Reagent Solutions
Table 2: Essential Materials for PCR-Based Diagnostics
| Reagent / Material | Function | Technical Considerations |
|---|---|---|
| Thermostable DNA Polymerase | Enzymatically synthesizes new DNA strands during extension. | Taq polymerase is common; high-fidelity enzymes with proofreading are preferred for sequencing applications [3]. |
| Primers | Short, single-stranded DNA sequences defining the start and end of the target region. | Must be specific, avoid self-complementarity, and have appropriate melting temperature (Tm) [31]. |
| dNTPs | Deoxynucleotide triphosphates (dATP, dCTP, dGTP, dTTP); the building blocks for new DNA. | Quality and concentration are critical for efficient amplification and fidelity. |
| Buffer Components | Provides optimal chemical environment (pH, ions) for polymerase activity. | Mg²⁺ concentration is a critical co-factor that must be optimized [31]. |
| Fluorescent Probe / Dye | Enables real-time detection of amplified products in qPCR/dPCR. | DNA-binding dyes (SYBR Green) are cost-effective; sequence-specific probes (TaqMan) enhance specificity [31]. |
| Microfluidic Chip | Partitions sample into thousands of nanoliter-scale reactions for dPCR. | Essential for absolute quantification; requires uniform partition volume for statistical accuracy [32]. |
Critical dPCR Workflow and Statistical Foundation
Digital PCR relies on limiting dilution and Poisson statistics to achieve absolute quantification. The sample is partitioned so that each microreaction contains either zero or a few target molecules. After amplification, the fraction of negative reactions is used to calculate the original concentration.
Diagram 2: dPCR workflow and statistics.
For dPCR, the Poisson distribution model is paramount. The key statistical principle states that the accuracy of quantification increases with the number of microreactions, with a significant improvement in relative uncertainty observed up to around 10,000 partitions [32]. Furthermore, consistent volume across all microreactions is critical for maintaining the integrity of the Poisson model and ensuring precise results [32].
Current Trends and Future Directions
The field of PCR diagnostics continues to evolve rapidly. Key trends include:
- Point-of-Care Devices: The miniaturization of PCR into portable, automated systems aims to deliver laboratory-grade accuracy in clinical settings, homes, and community centers [33]. These devices promise results in minutes rather than days, revolutionizing patient management.
- Adherence to Reporting Guidelines: The updated MIQE 2.0 guidelines emphasize transparent reporting of all experimental details to ensure repeatability and reproducibility. This includes providing efficiency-corrected target quantities, detection limits, and dynamic ranges for each assay [34].
- Multiplexing and Comprehensive Panels: The goal is to develop tests that can simultaneously screen for numerous pathogens or genetic mutations. "The Holy Grail would really be to build a test that can do the top 20 different point mutations," as noted by one researcher developing a portable ASPCR device [33].
PCR stands as a diagnostic powerhouse precisely because of its engineered simplicity and efficiency when compared to the complex, biologically constrained process of in vivo DNA replication. Its evolution from a basic amplification tool to a quantitative, digital, and portable technology has solidified its role as a cornerstone of modern clinical research and diagnostic practice. By enabling the rapid, sensitive, and specific detection of pathogens and genetic mutations, PCR directly empowers researchers and clinicians to make informed decisions, paving the way for personalized medicine and advanced therapeutic development.
DNA replication is a fundamental, precision-oriented biological process that ensures the accurate duplication of the genetic material during cell division. This intricate cellular mechanism is fundamental to maintaining genetic continuity across generations of cells and is indispensable for cellular life [35]. The process involves a highly coordinated interplay of numerous enzymes and proteins—including helicase, primase, DNA polymerase, and ligase—working in concert to faithfully replicate the entire genome [36]. Disruptions or errors in this finely tuned process can lead to mutations, genomic instability, and are directly implicated in various human diseases, most notably cancer [35] [36]. Therefore, studying native DNA replication in its physiological context provides critical insights into the fundamental mechanisms preserving genome stability, the origins of disease, and potential therapeutic interventions.
This guide situates the study of native replication within the broader context of molecular biology techniques, contrasting it with in vitro methods like the Polymerase Chain Reaction (PCR). While PCR is a powerful, simplified tool for amplifying specific DNA sequences in a test tube, it lacks the complex regulatory machinery and biological context of in vivo replication [1]. Understanding the differences between these processes is crucial for researchers and drug development professionals aiming to investigate genome dynamics, disease mechanisms, and develop precision medicine approaches that target replication-specific vulnerabilities in conditions like cancer [35] [36].
Fundamental Differences: Native In Vivo Replication vs. PCR
The following table summarizes the core distinctions between the complex, regulated process of native DNA replication in cells and the simplified, enzymatic DNA amplification achieved through PCR.
Table 1: Core Differences Between Native DNA Replication and PCR
| Feature | Native DNA Replication (In Vivo) | Polymerase Chain Reaction (PCR, In Vitro) |
|---|---|---|
| Fundamental Purpose | Accurate duplication of the entire genome for cell division. | Selective amplification of a specific, short DNA sequence for analysis. |
| Biological Context | Occurs within the complex environment of a living cell, with full chromatin context and organization. | Occurs in a simplified test tube reaction mixture. |
| Template | The entire chromosomal DNA within a cell nucleus. | A purified DNA extract containing the target sequence. |
| Key Enzymes & Proteins | A large, coordinated replisome including MCM helicase, primase, multiple DNA polymerases (Polδ, Polε), ligase, topoisomerases, PCNA, RPA, and more [35]. | A single, thermostable DNA polymerase (e.g., Taq polymerase) [37]. |
| Primer Requirements | Requires RNA primers synthesized de novo by primase during replication. | Uses short, single-stranded DNA primers that are chemically synthesized and added to the reaction [37]. |
| Initiation Mechanism | Tightly regulated, occurs at specific genomic origins of replication licensed in the G1 phase [35]. | Chemically and thermally driven; denaturation of DNA strands by heat [37]. |
| Fidelity & Proofreading | High-fidelity polymerases with intrinsic 3'–5' exonuclease proofreading activities and multiple repair pathways [35]. | Varies by polymerase; Taq polymerase has a higher error rate (~10⁻⁴) than cellular replicative polymerases [37]. |
| Regulation | Extremely tight cell-cycle control and checkpoint surveillance (e.g., ATR/CHK1) to ensure replication fidelity and handle stress [35]. | Controlled by manual or automated programming of temperature cycles in a thermal cycler [37]. |
Core Mechanisms and Experimental Analysis of Native Replication
Native DNA replication is a meticulously regulated process that can be separated into three main stages: initiation, elongation, and termination. Advanced research methodologies are essential for dissecting the molecular intricacies of each stage and their connection to genome stability.
The DNA Replication Initiation Phase
The initiation of DNA replication is a highly controlled process that begins at specific genomic locations known as origins of replication. In eukaryotic cells, the assembly of the pre-Replication Complex (pre-RC) during the G1 phase marks the first committed step, a process known as "licensing". This involves the sequential binding of the Origin Recognition Complex (ORC1-6), CDC6, Cdt1, and the core replicative helicase, the MCM2-7 complex, which is loaded as an inactive double hexamer onto the DNA [35]. A critical concept in genome stability is the licensing of many more origins than are typically used, creating "dormant origins" that serve as backups to rescue stalled replication forks under conditions of replication stress [35]. The transition to the S phase and replication initiation involves the activation of the MCM2-7 helicase through its conversion into the active CMG complex (Cdc45-MCM-GINS), which unwinds the DNA to establish bidirectional replication forks [35].
Replication Elongation and the Replisome
Following initiation, the replication fork progresses in a semi-discontinuous manner. The leading strand is synthesized continuously, while the lagging strand is synthesized as short, discontinuous Okazaki fragments. This elongation phase is catalyzed by a multi-protein machine known as the replisome. DNA polymerase α (Polα)/primase initiates synthesis by laying down RNA-DNA primers. The replicative polymerases, polymerase ε (Polε) and polymerase δ (Polδ), then take over, primarily synthesizing the leading and lagging strands, respectively [35]. These polymerases are characterized by their high fidelity and intrinsic 3'–5' exonuclease proofreading activity. The processivity factor PCNA (Proliferating Cell Nuclear Antigen), a homotrimeric ring loaded by RFC (Replication Factor C), forms a sliding clamp that dramatically increases the efficiency of DNA synthesis [35]. Other crucial enzymes include RPA (Replication Protein A), which stabilizes single-stranded DNA; FEN1 (Flap Endonuclease 1) and Dna2 for processing Okazaki fragments; and DNA ligase I, which seals the nicks in the lagging strand [35].
Specialized Replication and Replication Stress
Certain genomic regions, such as centromeres, present unique challenges to the replication machinery. These areas are often "difficult-to-replicate" due to their repetitive DNA sequences and secondary structures. Recent locus-specific proteomics studies have revealed that centromeres harbor a specialized replisome with unique dynamics, making them hotspots for rupture and fragility, particularly under replication stress [38]. Replication stress is a hallmark of pre-cancerous and cancer cells, characterized by the slowing or stalling of replication forks. It can be triggered by various endogenous and exogenous factors, including oncogene activation, nucleotide depletion, and DNA-damaging agents [35] [36]. To cope with this stress, cells activate the DNA Damage Response (DDR), a sophisticated network of signaling pathways that detect lesions, stall the cell cycle, and promote repair. Key mediators of the replication stress response are the kinases ATR and CHK1 [35] [36]. Failure to properly resolve replication stress can lead to the accumulation of DNA damage, mitotic errors, and genome instability—a key driver of tumorigenesis [35] [38].
Table 2: Key Proteins in DNA Replication and Their Research Applications
| Research Reagent / Protein | Primary Function in Native Replication | Utility in Experimental Research |
|---|---|---|
| MCM2-7 Complex | Core of the replicative DNA helicase, essential for unwinding DNA at the fork. | Biomarker for proliferation; studied to understand licensing and origin firing. |
| PCNA (Proliferating Cell Nuclear Antigen) | DNA polymerase processivity factor; sliding clamp that coordinates replication and repair. | Marker for replication forks and S-phase cells via immunofluorescence; used to study protein interactions at the fork. |
| ATR & CHK1 Kinases | Key regulators of the replication stress response; stabilize stalled forks and arrest cell cycle. | Therapeutic targets; inhibitors are in clinical trials for cancers with high replication stress. |
| PARP (Poly ADP-ribose Polymerase) | Enzyme involved in DNA repair, particularly single-strand break repair. | Target for PARP inhibitors, which are clinically approved for BRCA-mutated cancers (synthetic lethality). |
| γ-H2AX | Phosphorylated histone variant (H2AX) that marks sites of DNA double-strand breaks. | Sensitive biomarker for quantifying DNA damage and replication stress in response to genotoxic agents. |
| RPA (Replication Protein A) | Binds and stabilizes single-stranded DNA at the replication fork. | Indicator of replication fork progression and stalling; used in assays to monitor fork integrity. |
Advanced Methodologies for Studying Native Replication
Experimental Workflow for Analyzing Replication Stress
The following diagram and protocol outline a common approach for investigating how replication stress impacts genome stability, particularly at fragile sites like centromeres.
Protocol: Investigating Centromeric Fragility Under Replication Stress [38]
- Cell Culture and Stress Induction: Culture human cell lines (e.g., RPE-1 hTERT or cancer lines). Induce replication stress using chemical agents such as Hydroxyurea (HU, 2mM for 24 hours) or Aphidicolin (0.5-1µM for 24 hours). These agents deplete nucleotide pools or directly inhibit DNA polymerases, respectively, causing fork stalling.
- Immunofluorescence for Damage Markers: Fix cells and perform immunofluorescence staining using antibodies against γ-H2AX (to mark DNA double-strand breaks) and centromere-specific antibodies (e.g., CREST serum). Co-staining allows for the quantification of DNA damage specifically at centromeric regions. Use high-resolution microscopy for imaging.
- Fluorescence In Situ Hybridization (FISH): Employ centromeric DNA probes in a FISH assay to visually detect structural abnormalities such as centromere breaks, gaps, or rearrangements in metaphase spreads prepared from stressed cells.
- Locus-Specific Proteomics: To identify proteins associated with difficult-to-replicate regions, use techniques like Chromatin Immunoprecipitation followed by mass spectrometry (ChIP-MS) with antibodies against centromeric proteins (e.g., CENP-A). This reveals the specialized protein composition at centromeres during unperturbed replication and under stress.
- Analysis of Mitotic Errors and Translocation: Score for mitotic abnormalities like anaphase bridges and micronuclei formation, which are consequences of incomplete replication. Use advanced genomic techniques (e.g., whole-genome sequencing or karyotyping) to identify non-clonal chromosomal rearrangements and whole-arm translocations that arise from prolonged stress.
Key Technological Advances
The field has been revolutionized by several advanced technologies that provide unprecedented insights:
- Single-Molecule Techniques: Methods like optical tweezers and single-molecule fluorescence microscopy allow researchers to observe the real-time behavior of individual replication forks, including kinetics, pausing, and regression [36].
- High-Resolution Structural Biology: Cryo-electron microscopy (cryo-EM) and X-ray crystallography have provided atomic-level structures of key replication complexes, such as the CMG helicase and polymerase holoenzymes, facilitating the structure-based design of inhibitors [36].
- Digital PCR (dPCR): This technology partitions a PCR reaction into thousands of nanoscale reactions, allowing for the absolute quantification of nucleic acids without a standard curve. It is highly precise and is particularly useful for detecting rare genomic alterations and copy number variations that may arise from replication errors [39] [23].
The study of native DNA replication reveals it as a central vulnerability in maintaining genome stability. The inherent difficulties in replicating complex genomic regions like centromeres, combined with frequent challenges from replication stress, create a landscape ripe for chromosomal instability—a key hallmark of cancer [38] [40]. Research has shown that failures in processes like sister chromatid cohesion release or the proper resolution of replication intermediates can lead to anaphase bridges and DNA breaks, generating heritable genome destabilization from a "single-hit" lesion [40].
This deep understanding of replication mechanisms and their failures is now being translated into clinical applications, particularly in precision medicine. The development of ATR and CHK1 inhibitors exploits the heightened replication stress in cancer cells, while PARP inhibitors exemplify the successful application of synthetic lethality in treating BRCA-deficient tumors [36]. Furthermore, insights from replication fork biology are informing the refinement of gene editing technologies like CRISPR-Cas9, improving their safety and efficacy [36]. As research continues to unravel the complexities of the replisome and its regulation, it will undoubtedly yield novel biomarkers, drug targets, and therapeutic strategies to combat cancer and other diseases rooted in genomic instability.
Reverse Transcription PCR (RT-PCR) serves as a critical molecular technique that bridges the world of RNA analysis with the power of PCR amplification. This technical guide provides an in-depth examination of RT-PCR principles, methodologies, and applications relevant to researchers and drug development professionals. By enabling the conversion of RNA into complementary DNA (cDNA), RT-PCR facilitates sensitive detection and quantification of RNA molecules, making it indispensable for gene expression studies, viral load quantification, and biomarker discovery. Framed within the broader context of nucleic acid amplification technologies, this review contrasts the in vitro amplification approach of PCR with the in vivo process of DNA replication, providing a foundation for understanding the technical advantages and limitations of RT-PCR in research and diagnostic applications.
To appreciate the significance of Reverse Transcription PCR, one must first understand its position within the spectrum of nucleic acid amplification technologies. Polymerase Chain Reaction (PCR) and cellular DNA replication represent two fundamentally different approaches to DNA synthesis—one an in vitro laboratory technique, the other a biological process essential to life.
Table 1: Key Differences Between PCR and DNA Replication
| Parameter | PCR | DNA Replication |
|---|---|---|
| Definition | Laboratory process to amplify target DNA regions [3] | Biological process producing two identical DNA replicas [3] |
| Occurrence | In vitro (test tube) [3] | In vivo (inside living cells) [3] |
| Objective | Generate copies of a specific DNA fragment [26] | Copy the entire genome [26] |
| Process Steps | Denaturation, annealing, extension [26] | Initiation, elongation, termination [3] [26] |
| Temperature | Multiple temperatures (thermal cycling) [3] | Constant physiological temperature (~37°C) [3] |
| Enzyme | Thermostable DNA polymerase (e.g., Taq polymerase) [3] | DNA polymerase with proofreading ability [3] |
| Primers | DNA primers [3] | RNA primers [3] |
| Strand Separation | High temperature (denaturation) [3] | Enzymatic (DNA helicase) [3] [26] |
| Speed | 1-4 kb/min [3] | Approximately 1 kb/second [3] |
| Error Rate | Higher (e.g., 1 in 9,000 bases for Taq) [3] | Lower (e.g., 1 in 100,000 bases) [3] |
PCR excels at amplifying specific target sequences through repeated thermal cycles, making it invaluable for research and diagnostics [26]. In contrast, DNA replication is a continuous, organism-controlled process that accurately duplicates the entire genome during cell division [3]. RT-PCR builds upon standard PCR by incorporating an initial reverse transcription step that converts RNA into cDNA, thereby expanding the application of PCR to RNA targets [41].
Fundamental Principles of RT-PCR
RT-PCR revolutionized molecular biology by enabling researchers to detect and quantify RNA molecules with unprecedented sensitivity and specificity [42]. The technique combines the reverse transcription of RNA into complementary DNA (cDNA) with the subsequent amplification of this cDNA using PCR [41]. This two-process system allows for the analysis of RNA transcripts that would otherwise be difficult to detect due to their low abundance and inherent instability [42] [43].
The key advantage of RT-PCR over earlier methods like Northern blot analysis includes its dramatically increased sensitivity—capable of detecting RNA from a single cell—and its ability to provide quantitative data across a broad dynamic range [43]. Furthermore, RT-PCR can tolerate partially degraded RNA samples as long as the region targeted by primers remains intact [41].
Reaction Components and Workflow
A typical RT-PCR reaction requires several key components: RNA template, primers (oligo(dT), random hexamers, or gene-specific), reverse transcriptase enzyme, DNA polymerase, nucleotides (dNTPs), and appropriate reaction buffers [44]. The process follows a defined workflow beginning with RNA extraction and purification, followed by reverse transcription to create cDNA, PCR amplification of the target sequence, and finally detection and analysis of the amplified products [45].
RT-PCR Methodologies and Experimental Design
One-Step vs. Two-Step RT-PCR
Researchers can implement RT-PCR using either one-step or two-step protocols, each with distinct advantages and limitations suited to different experimental needs [42] [45].
Table 2: Comparison of One-Step and Two-Step RT-PCR Methods
| Parameter | One-Step RT-PCR | Two-Step RT-PCR |
|---|---|---|
| Procedure | Reverse transcription and PCR amplification in a single tube [42] | Reverse transcription and PCR performed in separate tubes [42] |
| Primer Usage | Gene-specific primers for both steps [42] | Flexible primer choice (oligo-dT, random hexamers, or gene-specific) for RT step [42] [45] |
| Handling Time | Shorter, less hands-on time [44] | Longer, more pipetting steps [44] |
| Contamination Risk | Lower (fewer transfer steps) [41] [44] | Higher (multiple handling steps) [41] |
| cDNA Storage | Not possible (immediately used in PCR) [45] | Possible (cDNA can be stored for future experiments) [42] [45] |
| Throughput | Ideal for high-throughput applications [45] | Better for analyzing multiple targets from same sample [42] |
| Optimization | Limited individual optimization of RT and PCR [44] | Flexible optimization of each step separately [44] |
| Best Applications | Rapid analysis of single targets, high-throughput screening [42] [45] | Multiple gene analysis from single sample, precious samples [42] [45] |
Detection Chemistries for Real-Time RT-PCR
Real-time RT-PCR (also called quantitative RT-PCR or RT-qPCR) enables monitoring of amplification as it occurs, providing quantitative data throughout the reaction [42]. Multiple detection chemistries are available, each with distinct characteristics.
SYBR Green is a cost-effective fluorescent dye that intercalates nonspecifically into double-stranded DNA [41] [45]. While economical and easy to use, its lack of specificity can lead to overestimation of target concentration due to binding to primer-dimers or non-specific products [41] [45].
TaqMan Probes are target-specific oligonucleotides with a fluorescent reporter at the 5' end and a quencher at the 3' end [43]. During amplification, the 5' nuclease activity of DNA polymerase cleaves the probe, separating reporter from quencher and generating fluorescence proportional to amplicon accumulation [41] [43]. This provides exceptional specificity but requires custom probe synthesis for each target [43].
Molecular Beacons are hairpin-shaped probes with reporter and quencher molecules that remain intact during amplification [41] [43]. When they hybridize to targets, the separation of reporter and quencher generates fluorescence [43]. Like TaqMan probes, they offer high specificity but require careful design [43].
Scorpion Probes integrate primer and probe functions into a single molecule, with a primer linked to a probe sequence that forms a hairpin loop [43] [45]. During amplification, the probe binds to its complement within the extended amplicon, generating fluorescence [45]. This design can improve hybridization efficiency and kinetics [45].
Table 3: Comparison of Detection Chemistries in Real-Time RT-PCR
| Chemistry | Principle | Advantages | Disadvantages |
|---|---|---|---|
| SYBR Green | Binds double-stranded DNA nonspecifically [41] [45] | Economical, easy to use, no probe design needed [41] [45] | Detects nonspecific products and primer-dimers [41] [45] |
| TaqMan Probes | Fluorogenic hydrolysis probes cleaved during amplification [43] | High specificity, minimal optimization, multiplexing capable [43] | Expensive, separate probe for each target [41] [43] |
| Molecular Beacons | Hairpin probes with FRET-based signaling [43] | High specificity, multiplexing capable, reusable probes [43] | Complex design, expensive synthesis [43] |
| Scorpion Probes | Combined primer-probe molecules [45] | Efficient kinetics, single-molecule design [45] | Complex synthesis and design [45] |
Research Reagent Solutions for RT-PCR
Successful RT-PCR experiments depend on appropriate selection of research reagents and materials. The following table outlines essential components and their functions in RT-PCR workflows.
Table 4: Essential Research Reagents for RT-PCR
| Reagent/Material | Function | Examples/Types |
|---|---|---|
| Reverse Transcriptase | Synthesizes cDNA from RNA template [45] | M-MLV, AMV, HIV-1 [45] |
| DNA Polymerase | Amplifies cDNA target [44] | Taq polymerase, hot-start variants [44] |
| Primers | Target sequence recognition and amplification initiation [42] | Oligo-dT, random hexamers, gene-specific [42] [45] |
| Fluorescent Detection Systems | Enable real-time monitoring of amplification [41] [43] | SYBR Green, TaqMan probes, molecular beacons [41] [43] |
| RNA Extraction Kits | Isolate and purify RNA from samples [45] | Column-based, magnetic bead, TRIzol methods [45] |
| dNTPs | Building blocks for DNA synthesis [44] | dATP, dCTP, dGTP, dTTP mixtures [44] |
| RNase Inhibitors | Protect RNA from degradation [46] | Protein-based inhibitors [46] |
| Buffer Systems | Provide optimal enzymatic reaction conditions [44] | Tris-based, magnesium-containing buffers [44] |
Quantitative Analysis in RT-PCR
Quantitative RT-PCR (RT-qPCR) provides precise measurement of RNA abundance through two primary quantification methods: the standard curve method and the comparative Cq (quantification cycle) method (also known as the 2^(-ΔΔCq) method) [43].
The standard curve method involves constructing a dilution series of RNA or DNA standards with known concentrations [43]. This curve serves as a reference for determining target concentration in unknown samples [43]. While this approach can provide absolute quantification, it requires careful preparation and validation of standards [43].
The comparative Cq method calculates relative changes in gene expression between experimental samples and a calibrator (e.g., untreated control) [43]. This method normalizes target gene Cq values to endogenous control genes (housekeeping genes) to account for variations in RNA input and reaction efficiency [43]. The 2^(-ΔΔCq) calculation provides fold-change values that are easily interpretable for gene expression studies [43].
Proper normalization is critical for accurate quantification, typically involving reference genes with stable expression across experimental conditions [42]. Common housekeeping genes include GAPDH, β-actin, and ribosomal RNAs, though ideal reference genes may vary by cell type and experimental treatment [42] [47].
Applications in Research and Diagnostics
Gene Expression Analysis
RT-PCR serves as the gold standard for gene expression analysis, enabling researchers to quantify transcript abundance with exceptional sensitivity and dynamic range [42]. By measuring changes in mRNA levels, researchers can investigate cellular responses to stimuli, compare gene expression across tissues or developmental stages, and validate findings from high-throughput screening methods like microarrays or RNA sequencing [42]. The technique's ability to detect low-abundance transcripts makes it particularly valuable for studying weakly expressed genes or limited sample materials [43].
Viral Detection and Quantification
The COVID-19 pandemic highlighted the critical importance of RT-PCR in viral diagnostics [46] [44]. RT-PCR enables direct detection of RNA viruses like SARS-CoV-2 by targeting conserved regions of the viral genome [46]. Quantitative viral load testing provides clinically relevant information for diagnosing acute infections, guiding treatment decisions, and monitoring therapeutic response [48].
Recent advances include the detection of subgenomic RNAs (sgRNAs) as markers of active viral replication [47]. In SARS-CoV-2 research, E-sgRNA RT-PCR assays have shown strong correlation with viral culture results, providing a reliable indicator of infectivity and replicating virus in rodent models [47].
Methodological Innovations
Recent methodological developments aim to simplify and accelerate RT-PCR workflows. Direct RT-PCR methods eliminate RNA extraction steps through simple thermolysis treatment, disrupting viral particles at 85°C for 5 minutes to release RNA directly into the reaction mixture [46]. This approach reduces processing time and enables rapid diagnosis during outbreaks [46].
Digital RT-PCR (dRT-PCR) provides absolute quantification without standard curves by partitioning samples into thousands of individual reactions [46] [48]. This technology offers unparalleled precision for low-abundance targets and increased resistance to inhibitors, making it valuable for precise viral load quantification and detection of minimal residual disease [46] [48].
Reverse Transcription PCR represents a powerful synthesis of molecular techniques that has transformed RNA analysis in both research and clinical settings. By enabling precise detection and quantification of RNA molecules, RT-PCR provides invaluable insights into gene expression patterns, viral dynamics, and disease mechanisms. The continued evolution of RT-PCR methodologies—including simplified RNA extraction protocols, improved detection chemistries, and advanced digital platforms—ensures this technology will remain essential for biological discovery and diagnostic innovation. As part of the broader landscape of nucleic acid amplification technologies, RT-PCR exemplifies how understanding fundamental biological processes like DNA replication can lead to transformative technical applications that advance both basic science and clinical medicine.
Real-Time Quantitative PCR (qPCR) represents a fundamental advancement in molecular biology, enabling researchers to monitor the amplification of DNA as it occurs and to precisely quantify specific nucleic acid targets. This technical guide details the core principles, methodologies, and analytical frameworks of qPCR, positioning it within the broader context of in vivo research by contrasting its in vitro mechanics with the natural process of DNA replication. Designed for researchers and drug development professionals, this whitepaper provides a comprehensive resource for implementing and interpreting qPCR experiments, complete with structured data, experimental protocols, and essential analytical toolkits.
Real-Time Quantitative PCR (qPCR) is a powerful analytical technique that allows for the monitoring of the Polymerase Chain Reaction (PCR) in real time, as it occurs [49]. Unlike conventional endpoint PCR, data collection happens throughout the entire PCR process, revolutionizing the quantitation of DNA and RNA. In qPCR, reactions are characterized by the point in the cycling process when amplification of a target is first detected, known as the threshold cycle (Ct), rather than the amount of target accumulated after a fixed number of cycles [49]. The fundamental principle is that the higher the starting copy number of the nucleic acid target, the sooner a significant increase in fluorescence is observed.
Framed within the broader thesis of comparing in vitro DNA synthesis (PCR) with in vivo DNA replication, qPCR stands as a sophisticated in vitro application of the core PCR technique. The following table summarizes the key distinctions between the general laboratory technique of PCR and the biological process of DNA replication, highlighting the context in which qPCR is employed [26] [3].
Table 1: Key Differences Between PCR and DNA Replication
| Aspect | PCR (including qPCR) | DNA Replication |
|---|---|---|
| Definition | A laboratory process used to amplify a specific target DNA region [3]. | A biological process of producing two identical DNA replicas from one original DNA molecule [3]. |
| Occurrence | An in vitro process occurring inside a test tube [3]. | An in vivo process occurring inside living cells [26] [3]. |
| Primary Goal | To generate numerous copies of a single DNA fragment [3]. | To copy the entire genome for cell division [3]. |
| Key Steps | Denaturation, Primer Annealing, Strand Extension [26]. | Initiation, Elongation, Termination [26]. |
| Temperature | Occurs at multiple temperatures (denaturation ~95°C, annealing ~50-60°C, extension ~72°C) [26] [3]. | Occurs at a constant physiological temperature (e.g., 37°C in humans) [26] [3]. |
| Polymerase Enzyme | Uses a thermostable DNA polymerase (e.g., Taq polymerase) with no proofreading ability [3]. | Uses DNA polymerase, which operates at body temperature and has proofreading ability [3]. |
| Primers | Uses specifically designed DNA primers [3]. | Uses RNA primers synthesized by primase [3]. |
| Strand Separation | Achieved by applying high heat (denaturation) [26] [3]. | Achieved by the enzyme DNA helicase [26] [3]. |
Core Principles and Detection Chemistries
The ability of qPCR to monitor amplification in real time relies on the detection of a fluorescent signal that increases proportionally to the amount of PCR product. Two primary chemistries are used for this purpose: TaqMan probe-based chemistry and DNA-binding dye chemistry (e.g., SYBR Green I) [49].
TaqMan Fluorogenic Probe Chemistry
TaqMan chemistry uses a sequence-specific probe to enable highly specific detection [49].
- Probe Design: An oligonucleotide probe is constructed with a reporter fluorescent dye on its 5' end and a quencher dye on its 3' end. When the probe is intact, the quencher suppresses the reporter's fluorescence through FRET (Förster Resonance Energy Transfer) [49].
- Probe Cleavage: During PCR, if the target sequence is present, the probe anneals to its complementary sequence. The 5' to 3' nuclease activity of Taq DNA polymerase then cleaves the probe as it extends the primer [49].
- Signal Generation: Cleavage separates the reporter dye from the quencher, leading to a permanent increase in the reporter's fluorescence. This process occurs in every cycle, resulting in an accumulation of fluorescence signal directly proportional to the amount of amplicon produced [49].
SYBR Green I Dye Chemistry
SYBR Green I is a dye that binds indiscriminately to double-stranded DNA (dsDNA) [49].
- Binding: The dye is added to the reaction mixture and binds to any dsDNA present, including both specific and non-specific PCR products.
- Signal Generation: Upon binding to the minor groove of dsDNA, the dye's fluorescence increases dramatically. As the amount of PCR product (dsDNA) increases with each cycle, so does the fluorescence signal [49].
- Considerations: The major advantage is its low cost and simplicity, as no specific probe is required. However, a well-optimized reaction is critical, as the dye will detect any dsDNA, including non-specific reaction products like primer-dimers, which can lead to false positive signals [49].
Table 2: Comparison of qPCR Detection Chemistries
| Characteristic | TaqMan Probes | SYBR Green I Dye |
|---|---|---|
| Specificity | High; requires binding of both primers and a specific probe. | Lower; detects all double-stranded DNA. |
| Cost | Higher due to the need for a fluorescent probe for each target. | Lower; no need for target-specific probes. |
| Multiplexing | Possible by using probes with different, distinguishable reporter dyes. | Not possible in a single reaction. |
| Experimental Setup | More complex; requires probe design and validation. | Simple; works with any pair of primers. |
| Signal Fidelity | Specific signal only from the target amplicon. | Requires post-amplification melt curve analysis to verify specificity. |
Diagram 1: Basic qPCR Workflow
Quantification Methods and Data Analysis
The cornerstone of qPCR data analysis is the Threshold Cycle (Ct), which is the fractional cycle number at which the fluorescence crosses a fixed threshold set above the baseline and within the exponential phase of amplification [49]. A lower Ct value indicates a higher initial amount of the target.
Absolute vs. Relative Quantitation
There are two main approaches to determining the quantity of a target in an unknown sample [49]:
- Absolute Quantitation: Used to determine the exact copy number or concentration of a target by interpolating unknown samples from a standard curve generated using samples of known concentration. This is essential for applications like determining viral load [49].
- Relative Quantitation: Used to determine the relative change in gene expression (or target amount) between different experimental conditions (e.g., treated vs. control). This method does not require a standard curve and is commonly used in gene expression studies [50].
Mathematical Models for Relative Quantitation
Two widely used methods for calculating relative expression or fold change are the Livak (2^–ΔΔCt) method and the Pfaffl method [50].
The Livak Method (2^–ΔΔCt): This method calculates the fold change in expression of a target gene between treatment and control conditions. It assumes that the amplification efficiencies of both the target and reference genes are close to 100% [50].
FC = 2^–[(CT_target – CT_ref)Tr – (CT_target – CT_ref)Co]or simplified,FC = 2^–[ΔCT_Tr – ΔCT_Co]The Pfaffl Method: This method offers a more flexible and accurate approach by incorporating the actual amplification efficiencies (E) of the target and reference genes, which is crucial when efficiencies are not ideal or differ from each other [50].
FC = (E_target)^(CT_Tr – CT_Co) / (E_ref)^(CT_Tr – CT_Co)
Table 3: Key qPCR Data Analysis Terms
| Term | Definition | Significance |
|---|---|---|
| Amplification Plot | The plot of fluorescence signal versus cycle number [49]. | Visual representation of the amplification process. |
| Baseline | The initial cycles of PCR where there is little change in fluorescence signal [49]. | Used to define the background signal level. |
| Threshold | The fluorescence level set above the baseline and within the exponential phase [49]. | Used to determine the Ct value. |
| Ct (Threshold Cycle) | The fractional cycle number at which the fluorescence passes the threshold [49]. | The primary quantitative value in qPCR; inversely proportional to the starting quantity. |
| ΔRn (Delta Rn) | The magnitude of the signal generated by the PCR conditions: (Rn+) – (Rn-), where Rn is the normalized reporter [49]. | Represents the normalized, background-subtracted fluorescence. |
Diagram 2: qPCR Data Analysis Pathways
The Scientist's Toolkit: Essential Reagents and Materials
Successful qPCR experimentation relies on a set of core components, each with a specific function.
Table 4: Essential Research Reagent Solutions for qPCR
| Reagent/Material | Function | Key Considerations |
|---|---|---|
| Thermostable DNA Polymerase | Enzyme that synthesizes new DNA strands by adding nucleotides to the primers. | Must be heat-stable (e.g., Taq polymerase). For probe-based assays, requires 5'→3' nuclease activity [49]. |
| Sequence-Specific Primers | Short DNA oligonucleotides that define the start and end of the target region to be amplified. | Specificity, melting temperature (Tm), and absence of self-complementarity are critical for efficient amplification. |
| Fluorescence Detection System | Chemistry that generates a fluorescent signal proportional to the amplicon produced. | TaqMan Probes: For high specificity and multiplexing [49].SYBR Green I Dye: For cost-effectiveness and simplicity [49]. |
| dNTPs | Deoxynucleoside triphosphates (dATP, dCTP, dGTP, dTTP); the building blocks for new DNA strands. | Quality and concentration are crucial for optimal polymerization and fidelity. |
| Buffer Components | Provides the optimal chemical environment (pH, ions) for polymerase activity and fidelity. | Typically contains MgCl₂, which is a critical co-factor for DNA polymerase. |
| Reference Genes | Genes used for normalization in relative quantitation to control for variations in input RNA/DNA quality and quantity. | Should be stably expressed across all experimental conditions (e.g., GAPDH, β-actin) [50]. |
Experimental Protocol: qPCR for Gene Expression Analysis
This protocol outlines the relative quantitation of a target gene's expression using a two-step RT-qPCR approach with a single reference gene.
Sample Preparation and Reverse Transcription (RT)
- Extract total RNA from treated and control samples using a validated RNA extraction kit, ensuring RNA integrity (e.g., via RIN number).
- Treat RNA with DNase I to remove any contaminating genomic DNA.
- Synthesize cDNA using a Reverse Transcriptase enzyme, oligo(dT) primers, and/or random hexamers, and a pool of the extracted RNA.
qPCR Setup and Run
- Prepare Reaction Mix: For each sample and gene (target and reference), prepare a master mix containing:
- SYBR Green I Master Mix (or TaqMan Universal Master Mix)
- Forward and Reverse Primers (for target or reference gene)
- Nuclease-free water
- Add Template: Aliquot the master mix into a qPCR plate and add a standardized amount of cDNA from each sample as the template.
- Include Controls: Always include a no-template control (NTC) containing water instead of cDNA to check for contamination.
- Run qPCR Program: Load the plate into the real-time PCR instrument and run the following standard program:
- Initial Denaturation: 95°C for 10 minutes (for enzyme activation).
- 40-50 Cycles of:
- Denaturation: 95°C for 15 seconds.
- Annealing/Extension: 60°C for 1 minute (acquire fluorescence at this step).
- (For SYBR Green I) Melt Curve Analysis: 95°C for 15 sec, 60°C for 1 min, then gradually increase to 95°C while continuously acquiring fluorescence.
- Calculate Mean Ct Values: For each sample and gene, calculate the average Ct from technical replicates.
- Determine Amplification Efficiency (E): Calculate efficiency for each primer pair using a dilution series, where E = 10^(–1/slope). Ideal efficiency is 2.0 (100%).
- Calculate Efficiency-Weighted ΔCT (wΔCT): For each sample, calculate
wΔCT = log2(E_target) * CT_target – log2(E_ref) * CT_ref. - Calculate Relative Expression (RE): For each sample,
RE = 2^(–wΔCT). - Calculate Fold Change (FC): Compare the average RE of treated samples to the average RE of control samples.
FC = RE_Tr / RE_Co. Alternatively, use the formula:FC = (E_target)^(CT_Tr – CT_Co) / (E_ref)^(CT_Tr – CT_Co). - Statistical Analysis: Perform appropriate statistical tests (e.g., t-test for two conditions, ANOVA for multiple factors) on the wΔCT or log-transformed FC values to determine significance [50].
The intricate molecular machinery of in vivo DNA replication provides the fundamental blueprint for many modern molecular technologies. This in-depth technical guide explores how the core principles of cellular DNA duplication have been harnessed and adapted to develop two transformative techniques: DNA sequencing and PCR-based molecular cloning. While in vivo DNA replication represents a natural, high-fidelity biological process occurring within living cells, its laboratory derivatives have been optimized for specific research and diagnostic applications [3] [51]. The revolutionary discovery of Taq polymerase from Thermus aquaticus was pivotal in this evolution, providing a thermostable enzyme that could withstand the high temperatures required for PCR automation [51]. This guide examines both the conceptual parallels and technical distinctions between these processes, providing researchers and drug development professionals with a comprehensive framework for leveraging replication principles in experimental design.
The core distinction lies in context and purpose: cellular DNA replication copies the entire genome with exceptional accuracy before cell division, while PCR amplification selectively targets specific DNA fragments for various analytical applications [3]. Understanding how PCR mimics—and deliberately deviates from—natural replication processes enables scientists to optimize these techniques for diverse research needs, from basic gene cloning to diagnostic test development and therapeutic discovery.
Core Principles: PCR Versus In Vivo DNA Replication
Comparative Mechanisms
The following table summarizes the key operational differences between PCR and natural DNA replication, highlighting how laboratory techniques have adapted biological principles for specific applications:
Table 1: Fundamental Differences Between PCR and In Vivo DNA Replication
| Parameter | PCR (In Vitro Amplification) | In Vivo DNA Replication |
|---|---|---|
| Objective | Amplify specific target DNA fragments | Duplicate the entire genome for cell division |
| Cellular Context | Artificial laboratory system | Natural biological process in living cells |
| Process Location | Test tube (in vitro) | Within cell nucleus (in vivo) |
| Temperature Conditions | Cyclical temperature changes (95°C denaturation, 50-72°C annealing, 72°C extension) | Constant physiological temperature (37°C in humans) |
| Enzyme System | Thermostable DNA polymerase (e.g., Taq polymerase) | Multi-enzyme complex including DNA polymerase with proofreading capability |
| Primer Type | Synthetic DNA oligonucleotides | RNA primers synthesized by primase |
| Strand Separation | Heat-induced denaturation | Enzyme-mediated (DNA helicase) |
| Fidelity/Error Rate | Varies by enzyme; Taq polymerase: ~1 error per 9,000 bases | Extremely high fidelity: ~1 error per 100,000 bases |
| Continuity | Discontinuous process through 30-40 cycles | Continuous on leading strand, discontinuous on lagging strand |
| Speed | 1-4 kilobases per minute | Approximately 1 kilobase per second |
Fidelity and Error Correction Mechanisms
A critical distinction between these processes lies in their accuracy and proofreading capabilities. Natural DNA replication incorporates multiple proofreading mechanisms, including exonucleolytic proofreading that immediately removes mispaired nucleotides at the growing chain's 3'-OH terminus [16]. This sophisticated error-correction system, combined with post-replication mismatch repair, achieves remarkable accuracy with approximately one error per 10^9 nucleotides copied [16].
In contrast, conventional PCR utilizing Taq polymerase lacks robust 3'→5' exonuclease activity, resulting in significantly higher error rates [3]. This fidelity difference has direct implications for experimental design—while sufficient for many amplification and detection applications, researchers must employ high-fidelity polymerases with proofreading capability for applications requiring precise sequence replication, such as gene cloning or mutant analysis [52] [53].
Molecular Cloning Leveraging PCR
PCR Cloning Methodologies
PCR-based cloning represents a versatile approach for inserting amplified DNA fragments into plasmid vectors, circumventing many limitations of traditional cloning methods. The fundamental process involves designing PCR primers that incorporate restriction enzyme sites at their 5' ends, enabling seamless fusion of the amplified product with a recipient plasmid [53] [54]. This method is particularly valuable for projects requiring higher throughput than traditional cloning can accommodate and for cloning DNA fragments not available in large quantities [52].
Key advantages of PCR cloning include:
- Elimination of restriction enzyme dependence for the insert fragment
- Rapid cloning of genes with limited starting material
- Flexibility in manipulating sequence ends via primer design
- Amenability to high-throughput applications
Common challenges include potentially higher mutation rates compared to traditional cloning, the need for specialized vectors (adding expense), and limited sequence control at junctions [52]. The following workflow illustrates the standard PCR cloning process:
Figure 1: PCR Cloning Workflow from Primer Design to Verification
Experimental Protocol: PCR Cloning with Restriction Sites
The following detailed protocol enables researchers to efficiently clone PCR-amplified fragments into plasmid vectors:
Primer Design Strategy
- Hybridization Sequence: 18-21 nucleotides complementary to the target sequence, ensuring specificity [54]
- Restriction Site: Incorporate appropriate restriction enzyme sites (6-8 bp) for directional cloning [53]
- Leader Sequence: Add 3-6 extra nucleotides 5' to the restriction site to enhance enzyme cutting efficiency [54]
- Reading Frame Preservation: Ensure maintained ORF when cloning protein-coding sequences
- Restriction Enzyme Selection: Choose enzymes that don't cut within your insert and function in compatible buffers [53]
PCR Amplification and Purification
- Utilize high-fidelity DNA polymerase to minimize mutation introduction during amplification [54]
- Calculate annealing temperature based on the hybridization portion of primers (not full primer sequence)
- Employ 30-35 amplification cycles to balance product yield and potential errors
- Purify PCR products using commercial kits (e.g., QIAquick PCR Purification Kit) to remove enzymes and nucleotides [54]
Restriction Digestion and Ligation
- Digest both purified PCR product and recipient vector with selected restriction enzymes
- Run double digests simultaneously using compatible buffers when possible [53]
- Use phosphatase treatment (CIP or SAP) on linearized vector to prevent self-ligation [54]
- Employ gel purification to isolate properly digested fragments from undigested DNA
- Ligate with insert:vector molar ratio of approximately 3:1 for optimal results [54]
Transformation and Verification
- Transform ligation reactions into appropriate competent E. coli strains (e.g., DH5α, TOP10) [53]
- Include negative controls (vector alone + ligase) to assess background colonies
- Screen multiple colonies by restriction analysis for correct insert incorporation
- Verify final construct by complete sequencing due to PCR error potential [54]
Research Reagent Solutions for PCR Cloning
Table 2: Essential Reagents for PCR-Based Molecular Cloning
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Thermostable DNA Polymerases | Taq polymerase, High-fidelity enzymes (Pfu, Q5) | DNA amplification; selection depends on fidelity requirements and template |
| Cloning Vectors | T-tailed vectors, Linearized suicide vectors | Receive PCR fragments; suicide vectors contain toxic genes for negative selection |
| Restriction Enzymes | EcoRI, NotI, AgeI, SalI | Create compatible ends on insert and vector; should function in same buffer |
| Modifying Enzymes | T4 DNA ligase, Alkaline phosphatase (CIP, SAP) | Join DNA fragments; prevent vector self-ligation |
| Bacterial Strains | DH5α, TOP10, DB3.1 (for toxic genes) | Plasmid propagation; specific strains for different applications |
| Purification Kits | QIAquick PCR Purification, Gel extraction kits | Isolate DNA fragments from reactions and gels |
| Selection Systems | Antibiotic resistance, Blue/white screening | Identify successful recombinants |
DNA Sequencing Technologies
Evolution of Sequencing Methods
DNA sequencing technologies have evolved dramatically, progressing from first-generation methods to today's sophisticated platforms that process millions of fragments in parallel [55]. The journey began with Sanger sequencing ("first-generation"), which served as the workhorse for decades and was used to complete the landmark Human Genome Project [55]. While highly accurate for reading long DNA stretches (500-1000 base pairs), this method was limited by low throughput and high cost [55].
The introduction of next-generation sequencing (NGS) in the mid-2000s represented a paradigm shift, implementing a "massively parallel" approach that sequences millions to billions of DNA fragments simultaneously [55]. This technological leap reduced sequencing costs from billions of dollars per human genome to under $1,000 while dramatically increasing speed from years to hours [55]. More recently, third-generation sequencing technologies, including single-molecule real-time (SMRT) and nanopore sequencing, have further advanced the field by enabling direct reading of much longer DNA fragments, thus simplifying genome assembly and enabling analysis of complex genomic regions [55].
Table 3: Comparative Analysis of DNA Sequencing Technologies
| Sequencing Technology | Read Length | Accuracy | Throughput | Primary Applications |
|---|---|---|---|---|
| Sanger Sequencing | Long (500-1000 bp) | High (>99.9%) | Low | Validation, small-scale targeting, confirmation of variants |
| Next-Generation Sequencing (Illumina) | Short (50-600 bp) | High (>99%) | Very High | Whole genome sequencing, transcriptomics, epigenomics |
| Long-Read Sequencing (PacBio SMRT) | Long (10,000+ bp) | Moderate to High | Moderate | De novo assembly, complex region analysis, structural variants |
| Nanopore Sequencing | Variable (up to millions of bp) | Moderate | Moderate | Real-time analysis, field applications, methylation detection |
NGS Methodology: Sequencing by Synthesis
The most prevalent NGS technology, Illumina's Sequencing by Synthesis, exemplifies how replication principles are harnessed for DNA analysis through these methodical steps:
Figure 2: Next-Generation Sequencing by Synthesis Workflow
Library Preparation: DNA is fragmented into manageable pieces, and specialized adapter sequences are ligated to both ends, enabling attachment to the flow cell and serving as primer binding sites [55]
Cluster Generation: The DNA library is loaded onto a flow cell where fragments bind to the surface and undergo bridge amplification, creating millions of identical clusters—each representing a single template molecule [55]
Sequencing by Synthesis: Fluorescently-labeled nucleotides are added one type at a time. When incorporated, each nucleotide type emits a distinctive color signal detected by a high-resolution camera, building the sequence data cycle by cycle [55]
Data Analysis and Alignment: The massive dataset of short reads is processed through sophisticated algorithms that align sequences to a reference genome or assemble them de novo for further biological interpretation [55]
Applications in Research and Drug Development
PCR Applications in Pharmaceutical Development
PCR technologies have become indispensable throughout the drug development pipeline, providing critical analytical capabilities across multiple therapeutic areas:
Gene Therapy Development: PCR enables precise detection and quantification of viral vector components in adeno-associated virus (AAV)-based therapies, supporting biodistribution studies and monitoring therapeutic response by detecting vector-derived RNA [56]
Small Molecule Drug Support: Quantitative PCR (qPCR) and reverse transcription PCR (RT-PCR) measure gene expression changes in response to drug treatment, identify molecular biomarkers linked to efficacy or toxicity, and track downstream effects of small molecule inhibitors [56]
Personalized Medicine: PCR facilitates pharmacogenetic testing to predict therapeutic response and adverse events, enables cancer diagnostics through detection of mutations in oncogenes like BRCA1 and BRCA2, and supports carrier screening for genetic diseases [56]
CRISPR and Gene Editing: PCR plays a crucial role in confirming successful genetic modifications in CRISPR research, enabling precise mechanism of action studies for emerging gene therapies [56]
Sequencing Applications in Clinical and Research Settings
The implementation of NGS technologies has transformed multiple scientific and clinical domains:
Rare Disease Diagnosis: NGS has dramatically shortened the "diagnostic odyssey" for many families through comprehensive testing approaches like whole-exome or whole-genome sequencing that can screen thousands of genes simultaneously [55]
Oncology and Cancer Research: Comprehensive tumor profiling sequences hundreds of cancer-related genes to identify specific mutations guiding targeted therapies. Liquid biopsies detect circulating tumor DNA for non-invasive monitoring of treatment response and resistance mechanisms [55]
Infectious Disease and Microbiology: NGS enables rapid, unbiased pathogen identification in cases of severe infection and facilitates real-time outbreak tracking through precise strain characterization [55]
Reproductive Health: Non-invasive prenatal testing (NIPT) using sequencing of fetal DNA in maternal blood has transformed prenatal screening for chromosomal abnormalities [55]
The strategic adaptation of natural DNA replication principles has fundamentally advanced modern molecular biology, giving rise to powerful technologies that drive innovation across research and clinical domains. While sharing conceptual foundations with cellular replication processes, PCR amplification, molecular cloning, and DNA sequencing technologies have been deliberately optimized for specific applications where nature's approach requires modification. Understanding these relationships—both the commonalities and the deliberate distinctions—empowers researchers to make informed decisions in experimental design and technology selection.
As these technologies continue to evolve, their implementation across basic research, clinical diagnostics, and therapeutic development highlights the enduring value of fundamental biological principles when creatively applied to address complex scientific challenges. The ongoing refinement of these techniques promises to further accelerate discovery and translation across the life sciences, particularly through enhanced integration with computational approaches and artificial intelligence.
Navigating Technical Challenges and Enhancing Performance in Amplification
In vivo DNA replication is a fundamental, highly regulated cellular process essential for life. It ensures the accurate duplication of the entire genome during cell division through a complex, coordinated machinery involving multiple enzymes, proofreading mechanisms, and precise regulation within the cell cycle [12]. In contrast, the Polymerase Chain Reaction (PCR) is an in vitro technique designed to amplify specific, targeted DNA fragments outside a living organism. While both processes share the core principle of DNA synthesis, their mechanisms, fidelity, and operational contexts differ profoundly [8].
This inherent divergence is the source of PCR's powerful utility but also its significant limitations. The requirement for prior sequence knowledge for primer design makes the amplification of truly "unknown" targets a fundamental challenge. Furthermore, the exponential amplification machinery, so effective for sensitivity, becomes a liability when non-specific products or contaminant DNA are introduced, leading to false positives and erroneous results [57]. This technical guide delves into these two critical limitations—sensitivity to contamination and the amplification of unknown targets—framing them within the conceptual differences between the engineered in vitro process and its natural in vivo counterpart.
Sensitivity to Contamination in PCR
Root Causes and Impact
In vivo DNA replication is protected by cellular compartmentalization and sophisticated regulatory mechanisms that prevent erroneous initiation. PCR lacks these biological safeguards. Its extreme sensitivity, capable of amplifying a single DNA molecule, makes it vulnerable to contamination from various sources, including previously amplified products (amplicons), laboratory environments, and sample cross-contamination. A minute contaminant can be co-amplified, generating false-positive results that compromise diagnostic accuracy, research validity, and clinical decision-making [58] [59].
The core of the problem lies in PCR's exponential amplification. Unlike the strictly controlled, once-per-cycle replication in vivo, every DNA molecule in the PCR mix is a potential template for duplication in every cycle. This means a handful of contaminant molecules present at the start can be amplified into billions of copies, ultimately dominating the reaction products.
Strategies for Contamination Control
Mitigating contamination requires a multi-faceted approach combining procedural, technical, and biochemical strategies.
- Physical and Procedural Controls: These form the first line of defense. Key measures include segregating pre- and post-amplification laboratory areas, using dedicated equipment and supplies, and employing aerosol-barrier pipette tips.
- Biochemical Decontamination: Uracil-DNA Glycosylase (UNG) System: This is one of the most effective technical solutions. The UNG system incorporates dUTP in place of dTTP during PCR amplification. Subsequent reactions are treated with UNG enzyme prior to thermal cycling, which degrades any uracil-containing contaminants from previous runs, preventing their re-amplification. The enzyme is then inactivated during the initial denaturation step, allowing the new amplification to proceed with standard dNTPs [57].
- Digital PCR (dPCR) for Reduced Susceptibility: Partitioning the PCR reaction into thousands of individual droplets or microchambers in dPCR significantly reduces the impact of contamination. The presence of a contaminant in a small subset of partitions has a minimal effect on the final absolute quantification, making dPCR more robust for detecting rare mutations and low-abundance targets in a background of wild-type sequences [58].
Table 1: Key Reagents for Contamination Control
| Research Reagent Solution | Function in Mitigating Contamination |
|---|---|
| Uracil-DNA Glycosylase (UNG) | Enzymatically degrades carryover contaminant amplicons from previous PCR runs that contain dUTP. |
| dUTP | A nucleotide analog used in place of dTTP during PCR to generate amplicons susceptible to UNG digestion. |
| Workflow Segregation | A procedural control involving physical separation of pre-PCR (template preparation) and post-PCR (analysis) areas. |
Amplification of Unknown Targets and Bias
The Fundamental Challenge of Sequence-Unknown Targets
A principal difference between in vivo and in vitro replication is template specificity. Cellular replication machinery duplicates the entire genome without requiring prior knowledge of its sequence. In contrast, PCR requires complementary primer binding to initiate DNA synthesis. This fundamental requirement makes the de novo amplification of sequences flanking a known region—a process known as genome walking—inherently challenging [57].
Traditional genome-walking methods rely on arbitrary or degenerate primers that partially anneal to unknown flanking sequences. However, these methods are plagued by high rates of non-specific amplification, as the primers can bind to multiple non-target sites, resulting in complex background products and failure to isolate the desired target [57]. This is analogous to a loss of replication fidelity in the cell, but arising from a lack of specificity in the initiation mechanism.
PCR Amplification Bias
Even when targets are known, PCR can introduce significant amplification bias, where different DNA templates are amplified with varying efficiencies. This distorts the true representation of template abundances in the final product, a critical problem in quantitative applications and metagenomic sequencing [60] [61].
The primary sources of bias include:
- Primer-Template Mismatches: Variations in the primer-binding site sequence reduce amplification efficiency, leading to the under-representation of specific variants in a mixed template population [62] [60].
- Template GC Content and Secondary Structure: DNA fragments with extremely high or low GC content can be inefficiently denatured or can form stable secondary structures, causing them to be under-represented in the final library [61].
- Amplicon Length: Longer amplicons are generally amplified less efficiently than shorter ones.
- Copy Number Variation (CNV): In metagenomic samples, the varying number of target gene copies across different taxa inherently biases abundance estimates, a factor that affects both amplicon-based and PCR-free methods [60].
Experimental Protocols to Overcome Bias and Access Unknown Targets
Protocol: Uracil Walking Primer PCR (UP-PCR) for Genome Walking
UP-PCR is a novel method designed to specifically suppress non-target amplification during genome walking [57].
- Primer Design: Design a set of three nested, sequence-specific primers (oSP, mSP, iSP) from the known region. Crucially, design an Arbitrary Walking Primer (AWP) with a degenerate sequence but with a uracil base incorporated at the penultimate position of its 3' end.
- Primary UP-PCR: Perform the first PCR using the oSP and the AWP. The cycling program includes low-stringency cycles to allow the AWP to anneal randomly to the unknown flank, generating a mix of target and non-target products, all of which carry the uracil-containing AWP sequence.
- UNG Treatment: After primary PCR, treat the product with Uracil-DNA Glycosylase (UNG). This enzyme excises the uracil base, destroying the binding site for the AWP on all amplicons.
- Secondary (Nested) UP-PCR: Use the UNG-treated product as the template for a second PCR with the mSP and the same AWP. The category of non-target amplicons produced solely by the AWP in the first round cannot be amplified because their AWP binding sites are destroyed. Only the desired target product, which now has binding sites for both the mSP (within the known sequence) and the AWP (on the extended unknown flank), can be exponentially amplified. A tertiary PCR with iSP can further enhance specificity.
This protocol effectively leverages UNG not for decontamination, but as a core component to selectively remove non-target amplicons, dramatically improving the success rate of genome walking [57].
Protocol: Minimizing Amplification Bias in Sequencing Libraries
Optimized protocols are critical for generating representative sequencing libraries [60] [61].
- Primer Design: Use highly degenerate primer pools or target genomic regions with conserved priming sites to maximize the amplification of diverse templates with sequence variations [60].
- PCR Enzyme and Formulation: Select polymerases known for high processivity and robustness. Formulations like AccuPrime Taq HiFi have been shown to outperform others in amplifying across a wide GC spectrum [61].
- Reaction Additives: Include betaine (1-2 M) in the PCR mix. Betaine is a stabilizing osmolyte that equalizes the melting temperatures of DNA templates with different GC contents, promoting more uniform amplification [61].
- Thermal Cycling Optimization:
- Extend Denaturation Time: Increase the denaturation time during cycling (e.g., to 60-80 seconds) to ensure complete melting of GC-rich templates, especially when using thermocyclers with fast ramp rates [61].
- Reduce Cycle Number: Perform the fewest number of PCR cycles necessary to generate sufficient product for sequencing, as bias is compounded with each additional cycle [60].
- Template Concentration: Using higher template concentrations can reduce stochastic amplification effects in early cycles [60].
Table 2: Quantitative Comparison of PCR Bias Mitigation Strategies
| Mitigation Strategy | Experimental Change | Impact on GC-Rich Template Yield (Relative to Mid-GC) | Effective GC Range |
|---|---|---|---|
| Standard Illumina Protocol | Phusion polymerase, 10s denaturation, fast ramping | Severe depletion (>99% loss for >65% GC) | 11% - 56% GC [61] |
| Extended Denaturation | Denaturation time increased to 80s/cycle | Significant improvement | 13% - 84% GC [61] |
| Additive + Extended Denaturation | 2M Betaine + extended denaturation | Optimal for high-GC; slight low-GC depression | 23% - 90% GC [61] |
| Polymerase Blend | AccuPrime Taq HiFi, optimized profile | Balanced performance across spectrum | Broadest and flattest profile [61] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Addressing PCR Limitations
| Reagent / Material | Critical Function |
|---|---|
| Uracil-DNA Glycosylase (UNG) | Core enzyme for both contamination control (degrading dUTP-containing amplicons) and specific genome walking (in UP-PCR) [57]. |
| dUTP | Nucleotide analog used to synthesize amplicons that are susceptible to UNG digestion for contamination control. |
| Betaine | PCR additive that equalizes the melting temperature of DNA templates, mitigating bias against GC-rich and AT-rich sequences [61]. |
| Degenerate Primer Pools | Mixtures of primers with variation at specific positions to bind homologous target sites across a diverse population of organisms, reducing taxonomic bias [62] [60]. |
| High-Fidelity Polymerase Blends | Engineered DNA polymerase mixtures (e.g., AccuPrime Taq HiFi) that offer superior efficiency and fidelity across diverse template sequences, minimizing amplification bias [61]. |
| Non-degenerate Primers with Engineered Protocols | In methods like Thermal-bias PCR, non-degenerate primers are used with specialized cycling to amplify mismatched targets, avoiding the inefficiencies of degenerate pools [62]. |
Visualizing Workflows and Relationships
In Vivo vs. In Vitro DNA Amplification
Uracil Walking Primer (UP-PCR) Workflow
The very factors that make PCR a revolutionary technology—its in vitro nature, specificity, and exponential gain—are also the source of its most critical limitations. Its sensitivity to contamination and inherent difficulty in amplifying unknown targets stand in direct contrast to the robustness and completeness of in vivo DNA replication. Understanding these limitations is not a critique of the technique, but a necessary foundation for its rigorous application.
Fortunately, as detailed in this guide, the scientific community has developed sophisticated methodological and biochemical strategies to mitigate these issues. Techniques like UNG decontamination, digital PCR, and advanced genome-walking methods such as UP-PCR provide powerful tools to enhance specificity and reliability. Furthermore, a deep understanding of the sources of amplification bias and the implementation of optimized protocols ensure that the results generated by this ubiquitous technique are both accurate and meaningful, enabling continued progress in research, diagnostics, and drug development.
In molecular biology, the polymerase chain reaction (PCR) serves as a foundational technique for the in vitro amplification of specific DNA sequences. While its efficiency mimics the core process of cellular DNA replication, the requirements and conditions for PCR are fundamentally different from those of in vivo DNA replication. Understanding these differences is critical for selecting the appropriate DNA polymerase, as the enzyme is the central component that dictates the success, accuracy, and yield of the amplification reaction.
In living cells, DNA replication is a coordinated process involving multiple DNA polymerases (e.g., Pol α, δ, and ɛ in eukaryotes) working in concert with a host of other enzymes and accessory proteins to ensure high-fidelity genome duplication [63]. This process is characterized by sophisticated mechanisms for proofreading and error correction, which are intrinsic properties of the replicative polymerases. In contrast, standard PCR is typically performed with a single, bacterially-derived DNA polymerase that must function under artificial, cyclic temperature changes. This fundamental distinction necessitates a careful balancing act when selecting a polymerase for PCR, weighing the critical parameters of thermostability, fidelity, and processivity to match the specific research or diagnostic application [13] [64].
Core Characteristics of DNA Polymerases for PCR
The performance of a DNA polymerase in PCR is governed by several interdependent biochemical properties. Optimizing these properties through natural discovery and protein engineering has been key to the advancement of PCR technology.
Thermostability
Thermostability refers to an enzyme's ability to retain its structure and function at high temperatures. This is a non-negotiable requirement for PCR, as the reaction involves repeated heating to over 90°C to denature the DNA template.
- Taq Polymerase: Derived from Thermus aquaticus, it has a half-life of approximately 40 minutes at 95°C, making it suitable for standard PCR cycles [65] [66].
- Pfu Polymerase: Sourced from Pyrococcus furiosus, it exhibits superior thermostability with a half-life of over 120 minutes at 95°C. This enhanced stability is beneficial for protocols requiring prolonged high-temperature incubation or for amplifying difficult templates with high secondary structure [13] [66].
Fidelity
Fidelity is a measure of the accuracy of DNA synthesis, defined as the inverse of the error rate (number of misincorporated nucleotides per total nucleotides polymerized). High fidelity is crucial for applications like cloning, sequencing, and functional gene analysis.
The primary mechanism for high fidelity is 3'→5' exonuclease proofreading activity. When a DNA polymerase with this activity incorporates an incorrect nucleotide, it can recognize the mismatch, excise the erroneous base, and replace it with the correct one [13] [63]. Polymerases are broadly categorized based on this capability:
- Polymerases without proofreading: Taq polymerase is the classic example, with an error rate of approximately 1 in 10⁵ nucleotides [66].
- Polymerases with proofreading: Pfu polymerase is a well-known high-fidelity enzyme, with an error rate about 10-fold lower than Taq (1 in 1.3 x 10⁶ nucleotides) [66]. Engineered "next-generation" enzymes can achieve error rates 50- to 300-fold lower than Taq [13].
Processivity
Processivity is defined as the number of nucleotides a polymerase incorporates per single binding event with the template. A highly processive enzyme can synthesize long DNA strands without dissociating, which is essential for efficiently amplifying long targets or templates with complex secondary structures or high GC content.
Early proofreading polymerases like Pfu often exhibited lower processivity, slowing down amplification. Breakthroughs in enzyme engineering have led to polymerases fused with non-specific DNA-binding domains (e.g., Sso7d), enhancing processivity 2- to 5-fold without compromising fidelity [13].
Specificity: The Role of Hot-Start
Specificity in PCR ensures that only the intended target sequence is amplified. A major source of nonspecific amplification is spurious primer binding and extension at low temperatures during reaction setup. Hot-start DNA polymerases address this issue.
These enzymes are rendered inactive at room temperature through antibody-based inhibition or chemical modification. They are only activated after the initial high-temperature denaturation step (e.g., >90°C), preventing undesired primer extension and significantly improving the yield of the specific product [13].
Comparative Analysis of Common DNA Polymerases
The table below summarizes the key properties of widely used DNA polymerases to aid in the selection process.
Table 1: Characteristics of Common DNA Polymerases for PCR
| Polymerase | Source Organism | Proofreading (3'→5' Exo) | Typical Error Rate | Processivity | Thermostability | Primary Application |
|---|---|---|---|---|---|---|
| Taq | Thermus aquaticus | No | 1 x 10⁻⁵ [66] | High | Half-life >40 min @ 95°C [65] | Routine PCR, qPCR |
| Pfu | Pyrococcus furiosus | Yes | ~1.6 x 10⁻⁶ [66] | Lower than Taq [13] | Half-life >120 min @ 95°C [66] | High-fidelity PCR, cloning |
| T4 | Bacteriophage T4 | Yes | 1 x 10⁻⁶ [66] | Low | Inactivated at 75°C [66] | Blunt-end cloning, fill-in |
| Klenow Fragment | E. coli | Yes | 1 x 10⁻⁵ - 10⁻⁷ [66] | Low | Not thermostable | cDNA synthesis, blunting |
Table 2: Advanced and Engineered DNA Polymerase Formulations
| Polymerase Type | Key Features | Mechanism | Best For |
|---|---|---|---|
| Hot-Start | Reduced nonspecific amplification; room-temperature setup [13] | Antibody, aptamer, or chemical inhibition at low temp | High-throughput PCR, low-copy targets |
| High-Fidelity Blends | High accuracy + robust amplification | Engineered enzymes or Taq/Pfu mixtures [64] | Cloning long fragments, NGS library prep |
| Long-Range | High processivity for long amplicons | Engineered with DNA-binding domains [13] | Amplifying large genes (>10 kb) |
| Translesion (e.g., Dpo4) | Bypasses damaged DNA templates [67] | Y-family polymerase with open active site | Forensic, ancient DNA analysis |
A Strategic Guide for Polymerase Selection
Choosing the right polymerase is a strategic decision based on the primary goal of the experiment. The following flowchart provides a decision-making framework for researchers.
Diagram 1: DNA Polymerase Selection Guide
Experimental Protocols for Assessing Polymerase Performance
Protocol: Determining Fidelity by LacZ Assay
This colony-screening assay is a classical method for quantifying polymerase error rates [13].
- Amplification: Use the test polymerase to amplify a segment of the lacZ gene (encoding β-galactosidase) from a plasmid template.
- Cloning: Ligate the PCR product into a suitable vector and transform into an appropriate E. coli host strain.
- Plating & Screening: Plate transformed cells on agar containing X-Gal and IPTG. Cells containing error-free plasmids produce functional β-galactosidase, hydrolyzing X-Gal to form blue colonies. Plasmids with inactivating mutations in the lacZ insert result in white colonies.
- Calculation: The error rate is calculated based on the number of white colonies divided by the total number of colonies, adjusted for the length of the amplified sequence.
Protocol: Evaluating Processivity and Inhibitor Tolerance
This protocol tests a polymerase's ability to perform in suboptimal conditions [13].
- Template & Inhibitors: Prepare reactions with a long genomic DNA template (e.g., >10 kb) and/or common PCR inhibitors (e.g., heparin, humic acid, or blood components).
- Amplification: Run parallel PCRs with the polymerase of interest and a control polymerase.
- Analysis: Analyze the products by agarose gel electrophoresis.
- Processivity: A polymerase with high processivity will produce a strong, clear band of the long amplicon, whereas a low-processivity enzyme will show a weak or absent band.
- Inhibitor Tolerance: Compare the yield of the target amplicon in the presence and absence of inhibitors. A robust polymerase will maintain strong amplification even with inhibitors present.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for PCR
| Reagent/Material | Function/Purpose |
|---|---|
| Hot-Start DNA Polymerase | Minimizes nonspecific amplification during reaction setup; crucial for sensitivity and specificity [13]. |
| High-Fidelity Master Mix | Pre-mixed solution containing a proofreading polymerase, dNTPs, Mg²⁺, and optimized buffer; ensures high accuracy and workflow efficiency. |
| dNTP Mix | The essential building blocks (dATP, dCTP, dGTP, dTTP) for DNA strand synthesis. |
| MgCl₂ Solution | A critical cofactor for DNA polymerase activity; concentration must be optimized for each primer-template system. |
| PCR Optimizer/Buffer | Additives (e.g., DMSO, betaine, glycerol) that help amplify difficult templates like those with high GC content or secondary structure. |
| Nuclease-Free Water | Prevents degradation of primers, templates, and enzymes by contaminating nucleases. |
The selection of a DNA polymerase is a critical determinant of PCR success. The inherent differences between the simplified, in vitro environment of PCR and the complex, regulated process of in vivo DNA replication mean that no single polymerase is perfect for all tasks. By understanding the trade-offs between thermostability, fidelity, and processivity, and by leveraging modern engineered enzymes, researchers can make informed choices. This strategic selection ensures robust, specific, and accurate amplification, thereby underpinning reliable results in genomics, diagnostics, and drug development.
The polymerase chain reaction (PCR) is a cornerstone technique in molecular biology, enabling the in vitro amplification of specific DNA sequences for applications ranging from clinical diagnostics to forensic science [68]. However, its efficiency can be severely compromised by PCR inhibitors—substances that interfere with the amplification process [69]. Successful DNA analysis requires efficient in vitro DNA polymerization and fluorescence detection, both of which are vulnerable to interference from various sample-related substances [69]. The classical approach to mitigating this inhibition involves purifying or diluting DNA extracts, but this often leads to significant DNA loss, which is particularly detrimental when working with low-abundance targets [69]. Understanding the sources and mechanisms of PCR inhibitors, along with developing robust strategies to overcome them, is therefore critical for taking full advantage of cutting-edge DNA analysis techniques like quantitative PCR (qPCR), digital PCR (dPCR), and massively parallel sequencing (MPS) [69].
This guide frames the challenge of PCR inhibition within the broader context of the fundamental differences between in vitro PCR and in vivo DNA replication. Table 1 summarizes these key distinctions, which explain why PCR, a simplified in vitro system, is inherently more vulnerable to inhibition than the complex, highly regulated cellular process of DNA replication [3].
Table 1: Key Differences Between In Vivo DNA Replication and In Vitro PCR
| Aspect | In Vivo DNA Replication | In Vitro PCR |
|---|---|---|
| Process Definition | Biological process of producing two identical DNA replicas from one original DNA molecule [3] | Laboratory process used to make many copies of a target DNA region [3] |
| Environment | Occurs inside living cells (in vivo) [3] | Occurs inside a test tube (in vitro) [3] |
| Polymerase Enzyme | DNA polymerase with proofreading and repair abilities [3] | Thermophilic DNA polymerase (e.g., Taq polymerase) with no proofreading ability [3] |
| Denaturing Mechanism | Enzyme-driven (DNA helicase) [3] | Heat-driven (high temperature) [3] |
| Primers | RNA primers synthesized by primase [3] | DNA primers [3] |
| Temperature | Constant physiological temperature (e.g., 37°C) [3] | Cycled through three different temperatures [3] |
| Scope of Synthesis | Copies the entire genome [3] | Amplifies a single, specific DNA fragment [3] |
| Continuity & Speed | Continuous process proceeding at ~1 kb/s [3] | Discontinuous process (25-40 cycles) proceeding at 1-4 kb/min [3] |
| Complexity & Accuracy | Complex process with a high-fidelity enzyme set; error rate of ~1 in 100,000 bases [3] | Simplified process; error rate of Taq polymerase is ~1 in 9,000 bases [3] |
Common PCR Inhibitors and Their Mechanisms of Action
PCR inhibitors are organic or inorganic molecules that can be introduced from the original sample (e.g., blood, soil, fabrics) or during sample processing and DNA extraction [70]. They exert their effects through several mechanisms, primarily by interacting directly with the DNA polymerase, co-factors like Mg²⁺, or the nucleic acid template itself [71]. Figure 1 illustrates the primary sites of interference within the PCR workflow.
Figure 1: PCR Workflow and Key Inhibition Mechanisms. Inhibitors can enter the process at multiple stages and interfere through various molecular mechanisms.
Table 2 provides a comprehensive overview of common inhibitors, their sources, and their specific mechanisms of action.
Table 2: Common PCR Inhibitors, Their Sources, and Mechanisms of Action
| Inhibitor Category | Specific Examples | Common Sample Sources | Primary Inhibition Mechanism |
|---|---|---|---|
| Blood Components | Hemoglobin, Immunoglobulin G (IgG), Lactoferrin [69] | Blood, blood stains [69] | Binds to DNA polymerase, preventing enzyme activity [71] |
| Soil Components | Humic acid, Fulvic acid [69] | Soil, sediment, compost [69] | Interacts with nucleic acids and DNA polymerase; can also quench fluorescence [69] |
| Anticoagulants | Heparin, EDTA [69] | Clinical blood samples [69] | Heparin co-purifies with DNA; EDTA chelates Mg²⁺ ions essential for polymerase activity [69] [71] |
| Tissue & Cellular Components | Collagen, Melanin [71] | Skin, hair, tissues [71] | Crosslinks with DNA template, preventing strand separation [71] |
| Polysaccharides & Polyphenolics | Complex polysaccharides, Tannins [72] [71] | Feces, plants, wastewater [72] [71] | Binds to polymerase or cofactors; can also sequester nucleic acids [71] |
| Extraction Reagents | Phenol, Ethanol, Isopropanol, Ionic detergents (SDS, Sarkosyl) [70] | Laboratory-prepared samples [70] | Disrupts polymerase activity; salts and solvents interfere with primer annealing [70] |
Quantifying and Detecting PCR Inhibition
Before implementing mitigation strategies, it is crucial to confirm the presence of inhibitors. The simplest method involves sample dilution. In uninhibited qPCR reactions, diluting the sample (e.g., 1:10) results in a higher quantification cycle (Cq) value because the target DNA is less concentrated. If inhibition is present, the diluted sample may show a Cq value equal to or lower than the undiluted sample because the positive effect of diluting the inhibitors outweighs the negative effect of diluting the template [71].
A more robust quantitative method is the use of an internal control. This involves adding a known quantity of a control template to the investigated reaction mixture and comparing its amplification to a control reaction without inhibitors. The difference in amplification efficiency indicates the extent of inhibition [70]. Digital PCR (dPCR) can also be used to detect inhibition. In dPCR, the presence of inhibitors can manifest as a reduction in the expected number of positive droplets or a distortion in the amplitude of the positive droplet cluster [69].
Strategies for Overcoming PCR Inhibition
Strategic Approaches to Mitigate Inhibition
A multi-faceted approach is most effective for handling PCR inhibition. The journey from sample to result offers multiple points for intervention, as outlined in Figure 2.
Figure 2: Strategic Workflow for Overcoming PCR Inhibition. Mitigation strategies can be applied at every stage of the analytical process.
Purification and Clean-up Techniques
DNA purification is a critical step to remove inhibitors co-extracted with nucleic acids.
- Magnetic Bead-Based Purification: This method involves a three-step process of binding, washing, and elution. Magnetic beads are added to the sample, and DNA amplicons bind to them. After magnetic separation, the supernatant containing inhibitors is discarded. A wash step with ethanol removes remaining impurities, and finally, a low-salt buffer elutes the purified DNA [73]. This protocol is easily automated, increasing reproducibility and throughput [73].
- Spin Column Purification: These columns contain a silica membrane with an affinity for nucleic acids. The sample is loaded and centrifuged, whereby DNA binds to the membrane while inhibitors pass through. Wash buffers remove residual contaminants, and DNA is eluted in a low-salt buffer [73]. Specialized columns with matrices designed to bind polyphenolic compounds (e.g., humic acids, tannins) are available for highly inhibitory samples [71].
- Direct PCR Methods: This approach minimizes or completely omits the DNA extraction and purification steps, thereby avoiding associated DNA loss. Its success relies on using highly inhibitor-tolerant DNA polymerase blends and adding a sub-sample of controlled size directly to the PCR [69]. This can reduce profiling time from 10-12 hours to 2-3 hours [69].
PCR Enhancers and Additives
Various compounds can be added to the PCR mix to counteract the effects of inhibitors.
- Proteins: Bovine Serum Albumin (BSA) and T4 gene 32 protein (gp32) are highly effective. They act as "competitive" binding proteins, sequestering inhibitors and preventing them from interacting with the DNA polymerase or nucleic acids [72]. A study on wastewater samples found that adding gp32 at a final concentration of 0.2 μg/μL was the most significant approach for removing inhibition, even outperforming a 10-fold dilution [72].
- Other Chemical Enhancers: Other common additives include Dimethyl Sulfoxide (DMSO) and formamide, which enhance amplification by lowering the melting temperature of DNA, and non-ionic detergents like TWEEN-20, which can counteract inhibitory effects on the DNA polymerase [72].
Table 3 summarizes the most common enhancers and their optimal use cases.
Table 3: Common PCR Enhancers and Their Applications
| Enhancer | Proposed Mechanism of Action | Effective Against | Example Usage |
|---|---|---|---|
| Bovine Serum Albumin (BSA) | Binds to inhibitors, freeing the polymerase and template [72] | Humic substances, polyphenolics, IgG in blood [72] [70] | Commonly added to PCR mix for blood samples [70] |
| T4 Gene 32 Protein (gp32) | Binds single-stranded DNA and inhibitors, stabilizing replication [72] | Humic acids in wastewater and soil [72] | 0.2 μg/μL final concentration in RT-qPCR [72] |
| Dimethyl Sulfoxide (DMSO) | Lowers DNA melting temperature (Tm), destabilizes secondary structure [72] | Inhibitors in complex samples (e.g., wastewater) [72] | Added at varying concentrations to the PCR mix [72] |
| TWEEN-20 | Non-ionic detergent that counteracts inhibitory effects on Taq polymerase [72] | Fecal samples, complex matrices [72] | Added at low concentration to the PCR mix [72] |
Platform and Enzyme Selection
The choice of amplification platform and enzyme can significantly impact inhibitor tolerance.
- Digital PCR (dPCR) vs. Quantitative PCR (qPCR): dPCR has been proven to be less affected by PCR inhibitors than qPCR [69] [74]. The main reason is that dPCR relies on end-point, binary (positive/negative) measurements from thousands of partitioned reactions, rather than on amplification kinetics [69] [74]. This partitioning also reduces the effective concentration of inhibitors in positive droplets [69]. A 2024 study on environmental samples found that dPCR produced precise and statistically significant results even in samples where qPCR was inhibited [74].
- Inhibitor-Tolerant DNA Polymerases: Using DNA polymerases engineered for high resistance to inhibitors is a straightforward and powerful solution [69]. These can be single enzymes or specialized blends that often include a supplementary antibody-based hot-start mechanism. They can often be used with minimal sample purification [69].
The Scientist's Toolkit: Essential Reagents for Inhibition Management
Table 4 lists key reagents and kits cited in recent literature for overcoming PCR inhibition.
Table 4: Research Reagent Solutions for Managing PCR Inhibition
| Reagent / Kit Name | Function / Purpose | Specific Application Context |
|---|---|---|
| OneStep PCR Inhibitor Removal Kit (Zymo Research) [71] | Column-based removal of polyphenolic inhibitors (humic acids, tannins, melanin) | Rapid clean-up of DNA/RNA from soil, fecal, and plant extracts without significant nucleic acid loss [71] |
| T4 Gene 32 Protein (gp32) [72] | PCR enhancer that binds inhibitors and single-stranded DNA | Optimized at 0.2 μg/μL for SARS-CoV-2 detection in inhibited wastewater samples [72] |
| Bovine Serum Albumin (BSA) [72] [70] | PCR enhancer that binds and neutralizes a range of inhibitors | General-purpose additive for reducing inhibition in blood and other complex samples [70] |
| DNeasy PowerSoil Pro Kit (QIAGEN) [74] | DNA extraction kit designed to remove co-extracted inhibitors from complex samples | DNA extraction from activated sludge and environmental samples with high inhibitor content [74] |
| QX200 ddPCR System (Bio-Rad) [74] | Digital PCR platform for absolute quantification | Quantification of ammonia-oxidizing bacteria in inhibited environmental samples where qPCR fails [74] |
| Inhibitor-Tolerant Polymerase Blends (e.g., Phusion Flash) [69] | DNA polymerase enzymes with inherent resistance to common inhibitors | Enables direct PCR from crude samples, minimizing purification and DNA loss [69] |
PCR inhibition remains a significant challenge in molecular biology, but a comprehensive understanding of inhibitor mechanisms and a strategic combination of available tools can effectively overcome it. The core vulnerability of PCR stems from its nature as a simplified in vitro system, lacking the robust, multi-enzyme complexity of in vivo DNA replication. By strategically applying optimized sample collection, advanced purification methods, chemical enhancers, and selecting inhibitor-tolerant enzymes and platforms like dPCR, researchers can ensure the reliability and accuracy of their results, even from the most challenging samples.
In the realm of molecular biology, the polymerase chain reaction (PCR) stands as a cornerstone technique for DNA amplification, yet it faces persistent challenges in specificity that distinguish it from its natural counterpart, cellular DNA replication. While cellular DNA replication is a highly precise in vivo process occurring within living organisms at physiological temperatures with sophisticated proofreading and repair abilities, conventional PCR is an in vitro process susceptible to non-specific amplification artifacts such as primer dimers and mispriming events [3]. This fundamental disparity stems from the enzymatic activity of DNA polymerases at non-permissive temperatures; whereas cellular replication employs DNA polymerase operating at 37°C with intrinsic proofreading capabilities (error rate of 1 in 100,000 bases), standard PCR often utilizes thermophilic polymerases like Taq polymerase that retain residual activity at room temperature, leading to an elevated error rate (approximately 1 in 9,000 bases) [3].
The hot-start technique represents a pivotal methodological advancement designed to bridge this fidelity gap by imposing temporal control over polymerase activation. By inhibiting enzyme activity during reaction setup and initial heating phases, hot-start PCR effectively minimizes off-target amplification, thereby enhancing both the specificity and yield of target amplification [75] [76]. This technical refinement mirrors the exquisite regulation found in biological systems, where enzymatic processes are precisely controlled through compartmentalization and activation cascades rather than through temperature manipulation alone. The development of hot-start methodologies thus represents a convergence of biochemical engineering and biomimicry, addressing a critical limitation of in vitro DNA synthesis by incorporating principles of temporal control observed in natural DNA replication machinery.
Mechanisms of Hot-Start PCR: Principles and Molecular Implementation
The fundamental principle underlying hot-start PCR technology is the reversible inhibition of DNA polymerase activity during reaction setup until the optimal temperature for specific primer annealing is reached. This approach prevents the polymerase from extending primers that have bound non-specifically at lower temperatures, a common occurrence during conventional PCR reaction preparation [75]. Several molecular strategies have been developed to implement this temporal control, each with distinct mechanisms and biochemical characteristics.
Antibody-Mediated Inhibition
Antibody-based hot-start methods employ specialized antibodies that bind specifically to the active site of DNA polymerases, forming complexes that remain inactive during reaction setup at room temperature [75] [77]. The activation of the polymerase occurs during the initial denaturation step of the PCR cycle (typically 95°C for 2-5 minutes), where the elevated temperature causes antibody denaturation and subsequent dissociation from the enzyme, thereby restoring full polymerase activity [77]. This method offers rapid activation kinetics (30 seconds to 5 minutes) and maintains the inherent enzymatic characteristics of the unmodified polymerase [77]. Examples include DreamTaq Hot Start DNA Polymerase and Platinum II Taq Hot Start DNA Polymerase [75]. A potential consideration with this approach is the presence of animal-derived antibodies and higher levels of exogenous proteins in the reaction mixture [75].
Chemical Modification
Chemical hot-start methods utilize small chemical molecules (such as anhydrides) that form covalent bonds with amino acid side chains (e.g., lysine residues) in the polymerase's active site, effectively blocking enzymatic activity at lower temperatures [75] [77] [78]. The activation of chemically modified polymerases requires prolonged exposure to high temperatures (typically >95°C for 10-15 minutes) to cleave the chemical bonds and restore enzyme function [77]. This method provides strict inhibition at room temperature and is free from animal-derived components, making it suitable for diagnostic applications [75]. However, the extended activation time required and potential incomplete enzyme reactivation can be limiting factors for some applications, particularly when amplifying longer fragments (>3kb) [75]. AmpliTaq Gold DNA Polymerase is a prominent example of this technology [75].
Aptamer-Based Inhibition
Aptamer-mediated hot-start approaches utilize short, single-stranded DNA or RNA molecules (typically 25-60 nucleotides) that fold into specific three-dimensional structures capable of binding to and inhibiting DNA polymerases through non-covalent interactions [75] [77]. These nucleic acid aptamers dissociate from the polymerase at elevated temperatures (above 60-70°C), restoring enzymatic activity [77]. This technology offers short activation times (1-5 minutes) and lacks animal-derived components [75] [77]. However, aptamer-based inhibitors may demonstrate reduced inhibition stringency compared to other methods, particularly at intermediate temperatures (around 40°C), and may not provide prolonged stability at room temperature after reaction setup [75] [77]. The AptaTaq Fast DNA Polymerase system employs this technology [76].
Affibody-Based Technology
A less common but effective approach utilizes Affibody molecules (small α-helical peptide domains) that bind to the polymerase active site, providing inhibition similar to antibodies but with a smaller protein footprint [75]. Examples include Phire Hot Start II DNA Polymerase and Phusion Hot Start II DNA Polymerase [75]. This method combines short activation times with reduced exogenous protein compared to antibody-based methods and contains no animal-derived components [75]. However, it may offer slightly less stringent inhibition than antibody-based approaches [75].
Table 1: Comparison of Major Hot-Start Modification Methods
| Feature | Antibody Modification | Chemical Modification | Aptamer Modification |
|---|---|---|---|
| Inhibition Mechanism | Antibody binding to active site | Covalent modification of active site | Oligonucleotide binding to enzyme |
| Activation Conditions | >70°C for 30s-5min [77] | >95°C for 10-15min [77] | >60°C for 1-5min [77] |
| Key Advantages | Rapid activation; maintains native enzyme properties [75] [77] | Strict inhibition; animal-component-free [75] [77] | Short activation; animal-component-free [75] [77] |
| Key Limitations | Animal-derived components; exogenous protein [75] | Longer activation required; may affect long amplicons [75] | Less stringent inhibition; temperature-sensitive [75] [77] |
| Representative Enzymes | DreamTaq Hot Start, Platinum II Taq [75] | AmpliTaq Gold [75] | AptaTaq Fast [76] |
Comparative Analysis: Hot-Start PCR Versus Conventional PCR
The implementation of hot-start technology confers substantial advantages over conventional PCR methods, primarily through the suppression of non-specific amplification events that compromise reaction efficiency and analytical sensitivity.
Specificity and Yield Enhancements
The principal benefit of hot-start PCR is the marked reduction in non-specific amplification, including both mispriming events and primer-dimer formation [75] [76]. In conventional PCR, the residual activity of DNA polymerases at room temperature allows for extension of misannealed primers during reaction setup, generating non-target products that compete with the desired amplicon for reaction components [75]. This competition leads to reduced target amplicon yield and diminished detection sensitivity [75]. Hot-start methods eliminate this pre-activation amplification by maintaining polymerase inhibition until the reaction reaches denaturation temperatures, thereby ensuring that primer extension only occurs under stringent temperature-controlled conditions [75] [78]. Studies demonstrate that hot-start protocols can significantly improve amplification efficiency, particularly for challenging templates such as those with low copy numbers, high GC content, or complex secondary structures [76] [77].
Practical Workflow Advantages
Beyond specificity improvements, hot-start PCR offers practical benefits for laboratory workflow. The technology enables room temperature reaction assembly without compromising reaction specificity, facilitating its use in high-throughput screening platforms and automated liquid handling systems [75]. This characteristic enhances experimental reproducibility across multiple users and platforms. Furthermore, the implementation of hot-start methods can reduce the need for extensive reaction optimization, particularly when working with suboptimal primer pairs or complex template mixtures [76]. The improved specificity also translates to cleaner amplification products for downstream applications, including cloning, sequencing, and diagnostic assays [77] [78].
Table 2: Impact of Hot-Start Technology on PCR Performance Parameters
| Performance Parameter | Conventional PCR | Hot-Start PCR |
|---|---|---|
| Non-specific amplification | Common occurrence [75] | Significantly reduced [75] [76] |
| Primer-dimer formation | Frequently observed [75] | Minimized [75] [76] |
| Low-copy target sensitivity | Often compromised [75] | Enhanced [75] [77] |
| Room temperature setup | Not recommended [75] | Suitable [75] [76] |
| Multiplexing capability | Limited | Improved [76] |
| Downstream application success | Variable | More reliable [75] [78] |
Experimental Design and Protocol Implementation
The successful implementation of hot-start PCR requires consideration of several methodological factors, including enzyme selection, activation parameters, and cycling conditions tailored to specific research applications.
Standardized Hot-Start PCR Protocol
The following protocol provides a generalized framework for hot-start PCR implementation, with specific modifications required based on the polymerase system employed:
Reaction Assembly:
- Combine PCR components on ice or at room temperature, including:
- Template DNA (1-100 ng genomic DNA or 0.1-10 ng plasmid DNA)
- Forward and reverse primers (0.1-1.0 μM each)
- Hot-start DNA polymerase (0.5-2.5 units per reaction)
- dNTP mixture (200 μM each)
- Reaction buffer with MgCl₂ (1.5-2.5 mM Mg²⁺ final concentration)
- Brief centrifugation to collect reaction mixture at tube bottom
- Combine PCR components on ice or at room temperature, including:
Initial Denaturation/Activation:
Amplification Cycling (25-40 cycles):
- Denaturation: 95°C for 15-30 seconds
- Annealing: Primer-specific temperature (typically 55-65°C) for 15-60 seconds
- Extension: 72°C for 15-60 seconds per kilobase of amplicon
Final Extension:
- 72°C for 5-10 minutes to ensure complete extension of all amplified products
Hold:
- 4°C for short-term storage or -20°C for long-term preservation
Application-Specific Methodologies
High-GC Content Amplification
For templates with high GC content (>70%), enhance protocol with:
- Supplementation with PCR additives such as DMSO, betaine, or glycerol (5-10%)
- Implementation of a temperature gradient during annealing to determine optimal specificity
- Use of specialized polymerase blends such as KOD Xtreme hot-start DNA polymerase, designed for challenging templates [76]
Long-Range Amplification
For amplification of long fragments (>5 kb):
- Select polymerases with strong proofreading activity (e.g., Phusion Hot Start II)
- Extend extension time to 2-4 minutes per kilobase
- Implement a two-step cycling protocol (combining annealing and extension at 68°C)
- Use of KOD hot-start DNA polymerase capable of amplifying up to 12 kb from genomic DNA and 21 kb from lambda DNA templates [76]
Multiplex PCR Applications
For multiplex PCR with multiple primer pairs:
- Carefully balance primer concentrations (0.05-0.3 μM each)
- Employ antibody-based hot-start enzymes for rapid activation and reduced non-specific amplification
- Optimize Mg²⁺ concentration (typically 2.5-4.0 mM for multiplexing)
- Consider the use of PCR additives like ThermaStop and ThermaGo which work synergistically with hot-start polymerases to further enhance specificity in multiplex reactions [79]
Visualization of Hot-Start PCR Mechanisms and Workflow
The following diagrams illustrate the fundamental mechanisms of hot-start PCR inhibition and activation, along with a standardized experimental workflow.
Hot-Start PCR Inhibition and Activation Mechanism
Hot-Start PCR Experimental Workflow
Research Reagent Solutions for Hot-Start PCR
The successful implementation of hot-start PCR relies on appropriate selection of specialized reagents and enzyme systems tailored to specific research requirements.
Table 3: Essential Research Reagents for Hot-Start PCR Implementation
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Antibody-Based Hot-Start Enzymes | DreamTaq Hot Start DNA Polymerase [75], Platinum II Taq Hot Start DNA Polymerase [75], KOD Hot-Start DNA Polymerase [76] | Rapid activation (30s-5min at >70°C); ideal for standard PCR, qPCR, and multiplex applications; maintains native enzyme characteristics [75] [76] |
| Chemically Modified Hot-Start Enzymes | AmpliTaq Gold DNA Polymerase [75], FastStart Taq DNA Polymerase [76] | Strict inhibition at room temperature; requires longer activation (10-15min at >95°C); animal-component-free; suitable for diagnostic applications [75] [76] |
| Aptamer-Modified Enzymes | AptaTaq Fast DNA Polymerase [76] | Short activation time (1-5min at >60°C); animal-component-free; suitable for fast cycling protocols [76] |
| Affibody-Modified Enzymes | Phire Hot Start II DNA Polymerase [75], Phusion Hot Start II DNA Polymerase [75] | Short activation with reduced exogenous protein; no animal-derived components; balance of stringency and rapid activation [75] |
| Specialized Polymerase Blends | FastStart High Fidelity PCR System [76], KOD Xtreme Hot-Start DNA Polymerase [76] | Combination of hot-start Taq polymerase with proofreading enzymes; enhanced accuracy and processivity; ideal for long amplicons (>10kb) and high-GC templates [76] |
| PCR Additives | ThermaStop, ThermaGo [79] | Chemical additives that provide additional thermal control; inhibit non-specific amplification during setup and cooling phases; compatible with hot-start polymerases [79] |
| Pre-mixed Formulations | JumpStart Taq ReadyMix [76], AptaTaq Fast PCR Master Mix [76] | Ready-to-use mixtures containing hot-start polymerase, dNTPs, buffer, and Mg²⁺; optimized for convenience and reproducibility; reduce pipetting steps [76] |
The development and refinement of hot-start PCR technologies represent a significant advancement in molecular biology, effectively addressing the specificity limitations inherent in conventional PCR. By imposing temporal control over polymerase activity through diverse biochemical mechanisms—including antibody-mediated inhibition, chemical modification, and aptamer-based strategies—hot-start methods have substantially improved the reliability, sensitivity, and reproducibility of DNA amplification across diverse research and diagnostic applications [75] [76] [77].
The continuing evolution of hot-start methodologies reflects a broader trend toward biomimicry in molecular techniques, where artificial systems increasingly incorporate the precision and regulation observed in biological processes. Future developments will likely focus on enhancing the stringency of inhibition while minimizing activation requirements, potentially through novel inhibitor classes or engineered polymerases with intrinsic thermal responsiveness. Furthermore, the integration of hot-start technology with emerging amplification platforms and point-of-care diagnostic devices will continue to expand its applications in clinical medicine, environmental monitoring, and basic research [77] [78].
As PCR methodologies evolve toward greater multiplexing capabilities, digital quantification, and single-cell analysis, the fundamental principles of hot-start technology will remain essential for maintaining specificity amidst increasing reaction complexity. The ongoing optimization of these techniques thus represents a critical frontier in the continued integration of molecular amplification technologies into both basic research and translational applications.
The accurate transfer of genetic information is fundamental to life, a process dependent on the fidelity of DNA polymerases. These enzymes catalyze template-directed DNA synthesis during both cellular DNA replication and in vitro polymerase chain reaction (PCR) amplification, with error rates having profound consequences for genomic stability, disease pathogenesis, and the reliability of molecular biology techniques [80] [12]. While the underlying chemistry of phosphodiester bond formation is identical in both contexts, the cellular environment and PCR conditions create fundamentally different landscapes for error introduction, detection, and correction.
This guide examines the mechanisms of DNA polymerase fidelity and error rates within the framework of a core thesis: that DNA replication in vivo occurs within a sophisticated cellular machinery designed to maximize accuracy, while PCR amplification in vitro presents a simplified system where biochemical and thermal constraints dominate error accumulation. We explore this dichotomy through quantitative error rate comparisons, detailed experimental methodologies for fidelity measurement, structural bases of polymerase inaccuracy, and the specialized reagent solutions enabling this research.
Quantitative Landscape of DNA Synthesis Errors
The fidelity of DNA polymerases varies by several orders of magnitude depending on enzyme family, proofreading capability, and reaction conditions. The following tables summarize key error rate measurements across biological and experimental contexts.
Table 1: DNA Polymerase Fidelity in Replication and PCR
| Polymerase / System | Error Rate (per base pair) | Primary Error Types | Context |
|---|---|---|---|
| In Vivo Replication | 10⁻⁹ to 10⁻¹¹ [81] | Not specified | Cellular replication with proofreading and mismatch repair |
| Family A Replicative DNAPs | Varies by family [80] | Family-specific error profiles | In vitro replication assay |
| KOD DNA Polymerase | ~1.1 × 10⁻⁶ [82] [83] | Not specified | High-speed PCR |
| Taq DNA Polymerase | ~1 × 10⁻⁴ [84] | Multiple substitution patterns | Standard PCR (20 cycles) |
| Pfu DNA Polymerase | "Outstanding fidelity" [82] | Not specified | PCR with proofreading activity |
| Pol θ (Human) | 2.4 × 10⁻³ [85] | T:G and T:T mismatches | Translesion synthesis/DSB repair |
Table 2: Error Type Distribution Across Polymerases in PCR
| Polymerase | Dominant Substitution Pattern | Less Frequent Substitutions | Reference |
|---|---|---|---|
| Kapa HF | C>T / G>A | A>C / T>G | [84] |
| Encyclo | A>G / T>C | C>A / G>T | [84] |
| SD-HS | A>G / T>C | A>T / T>A | [84] |
| TruSeq | C>T / G>A | C>G / G>C | [84] |
Biochemical Basis of Polymerase Fidelity
Replicative DNA polymerases achieve remarkable accuracy through a dual-step mechanism: selective nucleotide incorporation at the polymerase active site followed by proofreading exonuclease activity that removes misincorporated nucleotides [80]. The polymerase active site enforces geometric complementarity between incoming nucleotides and template bases, with precise alignment of catalytic residues and metal ions essential for correct incorporation [86]. High-fidelity enzymes exhibit remarkably precise steric exclusion of incorrectly paired nucleotides and altered transition state stabilization for non-complementary base pairs.
The polymerase active site architecture varies significantly across enzyme families. Family A and B DNA polymerases possess a Klenow-like DNA polymerase active site, Family C features a β-like active site, while Family D contains a double-Ψ-β-barrel (DPBB) polymerase active site [80]. These structural differences contribute to family-specific error profiles observed in fidelity measurements [80].
The fundamental difference between in vivo replication and PCR amplification lies in their error management systems:
In Vivo Replication:
- Multi-layered fidelity: Base selection, proofreading, and post-replication mismatch repair
- Optimal physiological conditions (temperature, dNTP concentrations, pH)
- Replisome coordination with processivity factors and accessory proteins
- Error rate: 10⁻⁹ to 10⁻¹¹ with all correction systems [81]
PCR Amplification:
- Limited to intrinsic polymerase fidelity and proofreading (if present)
- Non-physiological thermal cycling causing DNA damage
- Variable dNTP ratios and buffer conditions
- Error rate: 10⁻³ to 10⁻⁶ depending on enzyme [82] [84] [83]
PCR errors accumulate from two primary sources: polymerase misincorporation during extension and DNA thermal damage during denaturation steps. Thermal damage includes A+G depurination (creating abasic sites), cytosine deamination to uracil, and oxidative guanine damage to 8-oxoG [82] [83]. These lesions become fixed as mutations in subsequent amplification cycles, with thermal damage contributing significantly to total error counts in conventional PCR protocols.
Experimental Methodologies for Fidelity Measurement
Advanced Sequencing-Based Fidelity Assays
Modern polymerase fidelity measurement employs high-throughput sequencing to comprehensively characterize error rates and profiles. The PMC study (2025) utilized Pacific Biosciences SMRT sequencing with circular consensus sequencing (CCS) to achieve extremely high accuracy without PCR amplification bias [80]. Their workflow encompasses:
- DNA Polymerase Primer Extension: Enzymatic DNA synthesis under controlled conditions
- PacBio Library Preparation: Ligation of SMRTbell adapters without PCR amplification
- SMRT Sequencing: Long-read, single-molecule real-time sequencing
- Error Analysis: Computational identification of polymerase errors from CCS reads
This approach directly measures both error rates and family-specific error profiles across A, B, C, and D DNA polymerase families, revealing remarkable diversity in error signatures despite similar biological functions [80].
UMI-Based High-Throughput Error Quantification
An advanced methodology combining unique molecular identifier (UMI) tagging with high-throughput sequencing provides exceptional resolution for PCR error measurement [84]. This protocol involves:
This method enables discrimination between errors introduced during the first PCR versus subsequent steps by analyzing consensus sequences within UMI groups. The approach revealed that linear amplification errors occur at 5±1 times higher frequency than per-cycle PCR errors, attributed to higher dNTP concentrations and polymerase efficiency differences [84].
Mathematical Modeling of Error Accumulation
Quantitative models of PCR error accumulation incorporate both polymerase misincorporation kinetics and thermal damage rates [82] [83]. These models segment each PCR cycle into small time intervals, calculating:
- Polymerase errors based on kinetic parameters (nucleotide insertion time, fidelity)
- Thermal damage from temperature-dependent rate constants for depurination and deamination
- Error propagation through amplification cycles
The models demonstrate that thermal management significantly impacts total errors, with rapid cyclers that minimize high-temperature exposure substantially reducing error frequencies compared to conventional protocols [83].
Structural Basis of Error-Prone Synthesis
Recent structural studies illuminate how DNA polymerases accommodate errors. Cryo-EM structures of human Pol θ (an A-family polymerase specialized in error-prone synthesis) reveal a unique ability to accommodate mismatched base pairs with a fully closed finger domain, contrasting with high-fidelity homologs that adopt ajar conformations with incorrect nucleotides [85].
Key structural features enabling error-prone synthesis include:
- Well-closed finger domain even with T:G and T:T mismatches
- Unique active site residues that stabilize mismatched base pairs
- Alternative metal ion coordination in mismatch contexts
- Efficient mismatch extension capability through template/primer looping-out mechanisms
These structural insights explain how Pol θ achieves error rates 10-100 times higher than replicative A-family polymerases while maintaining similar catalytic efficiency [85].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for DNA Polymerase Fidelity Research
| Reagent / Material | Function / Application | Examples / Specifications |
|---|---|---|
| High-Fidelity Polymerases | PCR amplification with minimal errors | Pfu, KOD, Phusion, Q5 [82] [84] |
| Proofreading-Deficient Mutants | Studying polymerase activity without exonuclease correction | exo- variants (e.g., Klenow Fragment exo-) [80] |
| PacBio SMRT Sequencing | Long-read, PCR-free fidelity measurement | Circular Consensus Sequencing [80] |
| UMI Oligonucleotides | Molecular barcoding for error source discrimination | 14-mer random tags [84] |
| dNTP Pools | Controlling nucleotide concentration ratios | Variable dNTP ratios to modulate fidelity [80] |
| Processivity Factors | Studying replisome components in fidelity | PCNA, sliding clamps [80] [12] |
| Site-Directed Mutagenesis Kits | Creating polymerase active site mutants | Studying residue-specific fidelity roles [80] [86] |
The study of DNA polymerase error rates reveals fundamental biological constraints on genetic information transfer and practical limitations for molecular biology applications. The dichotomy between in vivo replication with its multi-layered fidelity systems and PCR amplification with its inherent error vulnerabilities underscores the remarkable efficiency of cellular machinery. As research advances, particularly through structural biology and high-throughput fidelity assays, our understanding of polymerase fidelity mechanisms continues to deepen, enabling both improved biotechnological tools and enhanced comprehension of disease-associated mutagenesis.
Future directions include engineering novel polymerases with customized fidelity profiles [86], developing increasingly accurate error measurement technologies, and translating mechanistic insights into therapeutic strategies targeting error-prone synthesis in diseases such as cancer. The continuing dialogue between in vivo and in vitro studies of DNA polymerase fidelity promises to yield both fundamental biological insights and practical advancements for the life sciences.
Assessing Accuracy, Reliability, and Strategic Method Selection
Polymersse Chain Reaction (PCR) stands as a foundational technology in modern molecular biology and clinical diagnostics, earning its status as the "gold standard" for nucleic acid detection. Its development mirrors, in a simplified in vitro context, the essential process of in vivo DNA synthesis that occurs within living cells. In living cells, DNA replication depends on a "well defined but complex set of enzymes and co-factors, which have evolved to act in a concerted fashion during the synthetic phase (S-phase) of the cell cycle" [1]. This in vivo process is regulated by intricate mechanisms to ensure genomic stability and high fidelity, involving numerous enzymes, co-factors, and checkpoint controls.
In contrast, PCR facilitates in vitro DNA synthesis in a much simpler fashion, making use of a smaller set of defined ingredients and reaction conditions involving relatively high temperatures [1] [8]. This simplification is key to its utility; by distilling the core principle of DNA amplification—thermal denaturation, primer annealing, and enzyme-mediated extension—into a automated thermal cycling process, PCR achieves exponential amplification of specific target sequences from minimal starting material. Understanding this fundamental relationship between the complex biological process of cellular DNA replication and its streamlined in vitro counterpart is crucial for appreciating both the power and the limitations of PCR technology in clinical and research settings.
The Fundamental Divide: In Vitro Amplification vs. In Vivo Replication
While both PCR and cellular DNA replication share the common goal of synthesizing new DNA strands, their mechanisms, components, and biological contexts differ significantly. Table 1 summarizes the core distinctions between these two processes.
Table 1: Fundamental Differences Between In Vivo DNA Replication and In Vitro PCR
| Characteristic | In Vivo DNA Replication | In Vitro PCR |
|---|---|---|
| Purpose | Faithful duplication of the entire genome for cell division | Targeted amplification of specific DNA sequences for analysis |
| Template | Entire chromosomal DNA | Short, specific DNA regions or genes |
| Enzymes | Complex replisome: multiple DNA polymerases (δ, ε), primase, helicase, ligase, etc. | Single thermostable DNA polymerase (e.g., Taq polymerase) |
| Primers | Short RNA primers synthesized by primase | Specific, synthetic DNA oligonucleotides |
| Temperature | Isothermal (physiological temperature) | Cyclical, high temperatures (denaturation ~95°C) |
| Fidelity & Proofreading | High-fidelity polymerases with exonuclease proofreading | Varies; Taq has no innate proofreading, others (e.g., Pfu) do |
| Regulation | Tight cell-cycle control, checkpoint mechanisms, licensed origins | Controlled by thermal cycler programming and reaction components |
| Specificity | Genome-wide, bidirectional replication from thousands of origins | Determined by primer design to target a unique sequence |
| Initiation | Defined origins of replication; can be "focused" or "dispersed" [87] | Defined by primer binding sites |
A key distinction lies in initiation. In vivo replication in human cells initiates from tens of thousands of sites, with recent single-molecule sequencing revealing that only a minority (~20%) occur within previously identified "focused" initiation zones, while the majority are "dispersed throughout the genome" [87]. PCR initiation, conversely, is entirely determined by the researcher's choice of primers.
Furthermore, in vivo replication must contend with endogenous replication barriers, such as "repetitive DNA, non-B DNA structures, and protein barriers" that can cause replication fork stalling [88]. Cells employ specialized mechanisms, including "helicase-mediated unwinding" and "fork restart," to overcome these challenges [88]. The in vitro environment of PCR is designed to minimize such obstructions, using high temperatures to denature secondary structures, thus ensuring efficient amplification.
The Evolution of a Gold Standard: PCR Technology Advancements
Since its inception, PCR has evolved into a family of sophisticated techniques that have solidified its role as a gold standard. The initial method of conventional PCR, which provides end-point detection, was transformative. However, the advent of quantitative real-time PCR (qPCR) marked a major advancement by enabling "real-time monitoring of DNA amplification through fluorescent signals proportional to the initial DNA template quantity" [89]. This innovation allowed for precise quantification of target nucleic acids, dramatically improving the method's utility in diagnostics and research.
Subsequent developments have further expanded PCR's capabilities and applications. Digital PCR (dPCR) represents a paradigm shift by enabling absolute quantification without standard curves. It achieves this by "partitioning the sample into droplet emulsions (digital droplet PCR) or physically isolated chambers (microchamber PCR)," which allows for the precise absolute quantification and detection of "single DNA molecules" [90]. Other significant advancements include multiplex PCR, which allows for the simultaneous amplification of multiple targets in a single reaction, and isothermal nucleic acid amplification (INAA) methods, which offer a "cost-effective and practical alternative to qPCR" by operating at a constant temperature, thereby eliminating the need for thermocyclers [91]. Photonic PCR is an emerging technology that utilizes "photothermal effects to accelerate thermal cycling, significantly reducing thermal inertia and enabling ultrafast amplification with lower energy consumption" [90].
The trajectory of PCR development is characterized by enhancements in speed, sensitivity, specificity, and quantitation, alongside a push towards miniaturization and point-of-care applicability. These advances have cemented PCR's status as an indispensable tool.
PCR in Clinical and Laboratory Practice: Applications and Protocols
The "gold standard" status of PCR is derived from its unparalleled performance across a vast spectrum of applications. Its reliability, sensitivity, and specificity make it the benchmark against which other diagnostic methods are measured.
Key Application Domains
- Infectious Disease Detection: PCR is the cornerstone of modern pathogen identification. It allows for the accurate detection of pathogens at the genus or species level, and in some cases, allows for the identification of single-nucleotide polymorphisms [91]. During the COVID-19 pandemic, its role in confirming cases was essential. Its high analytical sensitivity, specificity, and reproducibility, coupled with a "10–100-fold speed advantage over traditional culture methods," positions qPCR as the gold standard [91].
- Oncology Diagnostics: In cancer care, PCR is crucial for detecting genetic mutations associated with specific cancers (e.g., BRCA in breast cancer) and for monitoring minimal residual disease (MRD) after treatment to assess relapse risk [92].
- Genetic Disorder Screening: PCR enables the identification of inherited conditions like cystic fibrosis and sickle cell anemia, facilitating early intervention and personalized treatment plans [92].
- Food Safety and Cosmetic Quality Control: PCR has been successfully implemented for the rapid and reliable detection of contaminants. In cosmetic quality control, real-time PCR (rt-PCR) "consistently demonstrated superior sensitivity and reliability" compared to traditional plate counts, achieving a 100% detection rate for pathogens like E. coli, S. aureus, P. aeruginosa, and C. albicans [89].
Detailed Experimental Protocol: rt-PCR for Pathogen Detection
The following protocol, adapted from a 2025 study on cosmetic quality control, exemplifies a gold-standard rt-PCR workflow for pathogen detection [89].
1. Sample Preparation and Enrichment:
- Inoculate 1 g of the sample (e.g., cosmetic product, tissue) into 9 mL of Eugon broth.
- Spike with a low inoculum (3–5 colony forming units (CFU)) of the target pathogen(s).
- Incubate the enriched culture at 32.5°C for 20–24 hours. For complex matrices with antimicrobial properties, a longer incubation (36 h) and/or sample dilution (1:100) may be required.
2. Automated DNA Extraction:
- Use 250 μL of the enrichment culture for DNA extraction.
- Employ a commercial kit (e.g., PowerSoil Pro Kit) and an automated extractor (e.g., QIAcube Connect) per manufacturer's instructions.
- Critical steps include: vortexing with a lysis solution for 10 minutes, centrifugation, and transfer of supernatant for automated nucleic acid purification and elution.
- Include extraction controls: a medium control, a zero control, and an extraction control.
3. Real-Time PCR (rt-PCR) Setup:
- Use commercial, validated rt-PCR kits that include an internal reaction control.
- Prepare a rt-PCR plate and analyze each DNA extract in duplicate.
- Include necessary controls on each plate: a no-template control (NTC) and a positive control provided in the kit.
- For bacteria like E. coli, S. aureus, and P. aeruginosa, the R-Biopharm SureFast PLUS real-time PCR kit can be used. For fungi like C. albicans, the Biopremier Candida albicans dtec-rt-PCR kit is suitable.
4. Thermal Cycling and Data Analysis:
- Configure the thermal cycler according to the kit manufacturer's instructions. A typical protocol may involve: initial denaturation (95°C for 2 min), followed by 40 cycles of denaturation (95°C for 5 sec) and combined annealing/extension (60°C for 30 sec) with fluorescence acquisition.
- Analyze the amplification curves and set the fluorescence threshold to determine the cycle threshold (Ct) value for each sample.
- A sample is considered positive if it produces an amplification curve that crosses the threshold within the defined cycle number, and the result is concordant between replicates.
Essential Research Reagent Solutions
The robustness of PCR is built upon well-characterized reagents and consumables. The following table details key components essential for successful PCR experiments, particularly in a diagnostic context.
Table 2: Key Research Reagent Solutions for PCR-Based Diagnostics
| Reagent / Material | Function | Example & Notes |
|---|---|---|
| Thermostable DNA Polymerase | Enzyme that synthesizes new DNA strands at high temperatures. | Taq polymerase (from Thermus aquaticus). High-fidelity enzymes (e.g., Pfu) offer proofreading. |
| Primers | Synthetic oligonucleotides that define the start and end of the target amplification region. | Designed to target conserved regions of pathogens (e.g., HBV S gene, HCV 5'-UTR) [91]. |
| dNTPs | Deoxynucleotide triphosphates (dATP, dCTP, dGTP, dTTP); the building blocks for new DNA. | Quality is critical for efficient amplification and low error rates. |
| Buffer Systems | Provide optimal chemical environment (pH, salts, co-factors) for polymerase activity. | Often includes MgCl₂, which is a critical co-factor. |
| Fluorescent Probes/Dyes | Enable real-time detection of amplified DNA in qPCR/dPCR. | SYBR Green, TaqMan probes, molecular beacons. |
| Commercial rt-PCR Kits | Integrated solutions containing optimized reagents for specific targets. | e.g., R-Biopharm SureFast PLUS kit for bacteria; Biopremier kit for C. albicans [89]. |
| Nucleic Acid Extraction Kits | For purifying high-quality DNA/RNA from complex sample matrices. | e.g., PowerSoil Pro Kit (Qiagen), used with automated extractors like QIAcube Connect [89]. |
| Internal Controls | Non-target nucleic acids added to the reaction to monitor for PCR inhibition and validate the process. | Included in commercial kits to distinguish true negatives from failed reactions. |
The Future Trajectory: Point-of-Care and Isothermal Alternatives
The landscape of nucleic acid testing is evolving, with a clear trend toward decentralization. The integration of PCR with microfluidic chips is a key driver for point-of-care testing (POCT), making "rapid on-site diagnosis outside central laboratories" a reality [90]. This shift is supported by the development of portable, automated platforms that reduce detection time and operational complexity.
A significant challenge to traditional qPCR comes from isothermal nucleic acid amplification (INAA) methods. Techniques like Multiple Cross Displacement Amplification (MCDA) can achieve "diagnostic accuracy comparable to conventional qPCR, but with significantly reduced turnaround time and cost" [91]. MCDA uses "a set of 10 primers targeting distinct regions within a specific gene," enhancing assay specificity and sensitivity to that of qPCR, but under isothermal conditions (e.g., 64°C) without the need for expensive thermocyclers [91]. When coupled with simple visual readouts like gold nanoparticle-based lateral flow biosensors (AuNPs-LFB), these platforms create highly suitable tests for decentralized POC diagnostic applications [91].
The question then arises: what is the continued necessity for qPCR? While INAA technologies are "poised to become the next gold-standard technique" for specific decentralized applications [91], qPCR will likely remain the reference method in centralized, high-throughput laboratories due to its well-established protocols, extensive validation, and robust quantitative capabilities. The future diagnostic ecosystem will therefore be characterized by a complementary coexistence of laboratory-based qPCR and rapid, simple POC isothermal tests, each serving distinct needs within the healthcare continuum.
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology, serving as an in vitro tool for amplifying specific DNA sequences. To fully appreciate its application in drug development and basic research, it is essential to frame its principles—specifically, the quantification cycle (Cq) and amplification efficiency—within the context of its natural counterpart: in vivo DNA replication. While both processes synthesize new DNA strands, their mechanisms, components, and purposes differ significantly. PCR is a simplified, enzymatic method that uses thermal cycling to exponentially amplify a targeted DNA fragment from a complex mixture outside a living organism [9]. In contrast, in vivo DNA replication is a complex, coordinated cellular process that accurately duplicates the entire genome during the S phase of the cell cycle, ensuring genetic inheritance [93].
This technical guide will detail how precise quantification in qPCR hinges on understanding Cq values and PCR efficiency. The core principle is that the Cq value—the cycle number at which amplification is first detected—is inversely related to the starting concentration of the target nucleic acid [94]. However, this relationship is profoundly influenced by the reaction's amplification efficiency. A thorough grasp of these concepts is crucial for researchers applying qPCR to gene expression analysis, pathogen detection, and biomarker validation in drug development, enabling robust, interpretable, and reproducible data.
Core Concepts: Cq and PCR Efficiency
The Quantification Cycle (Cq)
In quantitative PCR (qPCR), the amplification of DNA is monitored in real time by measuring fluorescence. The Quantification Cycle (Cq), also known as the threshold cycle (Ct), is a critical parameter defined as the fractional cycle number at which the fluorescent signal exceeds a predefined threshold, indicating confident detection of amplification [95] [94]. This threshold is set within the exponential phase of the amplification curve, where the reaction is most efficient and reproducible.
The fundamental relationship between Cq and the target concentration is described by the equation: [ Cq = \frac{\log(Nq) - \log(N0)}{\log(E)} ] Where:
- ( N_0 ) is the initial number of target copies.
- ( N_q ) is the number of amplicons at the threshold.
- ( E ) is the amplification efficiency (fold-increase per cycle) [94].
This equation shows that Cq is inversely proportional to the logarithm of the initial target amount. A lower Cq value indicates a higher starting concentration, while a higher Cq suggests a lower starting concentration. It also highlights that Cq is dependent on both the chosen threshold (( N_q )) and the PCR efficiency (( E )), meaning that these factors must be carefully controlled for valid comparisons [94].
The Principle of PCR Efficiency
PCR efficiency (E) quantifies the effectiveness of the amplification reaction in each cycle. It is defined as the fold-increase in the number of amplicon molecules per cycle. An ideal, or "100% efficient," reaction has an efficiency (E) of 2, meaning the number of DNA molecules doubles with every cycle [96] [97]. The kinetics of the PCR reaction are described by: [ NC = N0 \times E^C ] Where ( N_C ) is the number of amplicons after cycle ( C ) [97].
In practice, however, efficiencies are often below 2 due to factors like suboptimal primer design, reagent limitations, or the presence of inhibitors [9]. Efficiencies can also appear to exceed 100%, which is typically an artifact caused by the presence of polymerase inhibitors in more concentrated samples. These inhibitors cause a delay in the Cq, flattening the standard curve and leading to a calculated efficiency above 100% [98].
Table 1: Interpretation of PCR Efficiency Values.
| Efficiency Value | Efficiency (%) | Interpretation | Common Causes |
|---|---|---|---|
| 2.0 | 100% | Ideal reaction | Optimal primer design and reaction conditions. |
| 1.8 - 2.0 | 90% - 100% | Acceptable range | Slight sub-optimal conditions; generally usable with efficiency correction. |
| < 1.8 | < 90% | Low efficiency; requires investigation | Poor primer design, inhibitor presence, or degraded reagents. |
| > 2.0 | > 100% | Artifact, not biologically possible | Piperror or inhibition in concentrated samples [98]. |
Comparative Framework: PCR vs. In Vivo DNA Replication
Understanding PCR efficiency is aided by contrasting it with the flawless efficiency of in vivo DNA replication. The cellular machinery is engineered for high-fidelity, processive synthesis of the entire genome.
Table 2: Key Differences Between In Vivo DNA Replication and In Vitro PCR.
| Feature | In Vivo DNA Replication | In Vitro PCR Amplification |
|---|---|---|
| Purpose | Duplicate entire genome for cell division [93]. | Amplify a specific, short DNA target from a complex sample [9]. |
| Initiation | Specific genomic origins of replication; regulated by cell cycle [93]. | Temperature-mediated denaturation; not sequence-specific outside the target [9]. |
| Enzymes | Complex replisome: multiple DNA polymerases (δ, ε, α), primase, helicase, ligase, topoisomerase [93]. | Single, thermostable DNA polymerase (e.g., Taq polymerase) [9]. |
| Fidelity | High; multiple proofreading and error-correction mechanisms [93]. | Lower; relies on fidelity of the polymerase; no cellular repair mechanisms [9]. |
| Strand Synthesis | Simultaneous continuous (leading) and discontinuous (lagging) synthesis with Okazaki fragments [93]. | Both strands synthesized continuously during each cycle. |
| Efficiency & Yield | Exactly one copy of the entire genome per cell cycle. | Billions of copies of a specific target after 30-40 cycles [9]. |
| Priming | Requires RNA primers synthesized by primase for each Okazaki fragment [93]. | Requires a single pair of specific DNA oligonucleotide primers [9]. |
Experimental Protocols for Determining PCR Efficiency
Accurate quantification in qPCR is impossible without knowing the amplification efficiency of the assay. The following section provides detailed methodologies for determining this critical parameter.
Standard Curve Method for Efficiency Determination
This is the most common and recommended method for determining PCR efficiency.
Protocol:
- Template Preparation: A sample with a known, high concentration of the target sequence (e.g., a plasmid containing the amplicon or a cDNA sample) is serially diluted. A 10-fold dilution series is standard, typically spanning 5-6 orders of magnitude (e.g., from 10 ng/µL to 0.001 ng/µL) [96].
- qPCR Run: The entire dilution series is amplified in the same qPCR run, using the same master mix and cycling conditions as the experimental samples. Each dilution should be run in replicate (at least triplicate) to account for pipetting error.
- Cq Determination: For each dilution, the mean Cq value is calculated from the replicates.
- Standard Curve Plotting: A plot is generated with the logarithm of the starting template concentration (or relative dilution factor) on the X-axis and the observed mean Cq value on the Y-axis. The data points should form a straight line [96] [95].
- Efficiency Calculation: The slope of the standard curve is determined by linear regression. The PCR efficiency (E) is then calculated using the formula: [ E = 10^{(-1/slope)} ] The corresponding efficiency percentage is given by ( (E-1) \times 100\% ) [96].
Table 3: Interpretation of Standard Curve Slope and Efficiency.
| Slope | Efficiency (E) | Efficiency (%) | Interpretation |
|---|---|---|---|
| -3.32 | 2.00 | 100% | Ideal, perfect doubling every cycle. |
| -3.58 | 1.90 | 90% | Acceptable; efficiency correction required for precise quantification. |
| -3.00 | 2.16 | 116% | Unacceptable; indicates potential artifact or inhibition. |
Data Analysis and the Critical Role of Baseline and Threshold
Accurate Cq assignment depends on two crucial preprocessing steps: baseline correction and threshold setting. Errors here directly impact Cq values and, consequently, efficiency calculations [95] [97].
- Baseline Correction: The baseline is the fluorescent background signal present during the initial cycles of PCR before exponential amplification. Software algorithms model this baseline (often from cycles 5-15) and subtract it from the entire amplification curve. Incorrect baseline settings can distort the curve's shape and lead to inaccurate Cq values [95].
- Threshold Setting: The quantification threshold must be set manually or by the instrument within the exponential phase of all amplification curves. The exponential phase is best identified by plotting fluorescence on a logarithmic (log) scale, where it appears as a straight line. The threshold should be set at a level where all curves of interest are parallel, ensuring consistent Cq determination across samples with different starting concentrations [95].
The Scientist's Toolkit: Essential Reagents and Materials
Successful and reproducible qPCR requires a set of high-quality, specific reagents.
Table 4: Key Research Reagent Solutions for qPCR.
| Reagent/Material | Function | Technical Considerations |
|---|---|---|
| Thermostable DNA Polymerase | Enzyme that synthesizes new DNA strands; thermostability allows it to survive repeated denaturation steps. | Taq polymerase is most common. High-fidelity enzyme blends are available for applications requiring lower error rates [9]. |
| Primers (Oligonucleotides) | Short, single-stranded DNA sequences that are complementary to the flanking regions of the target. They define the amplicon and provide a starting point for synthesis. | Must be specifically designed for the target. Optimal length is 18-25 bases with balanced GC content and minimal self-complementarity to avoid dimers [9]. |
| dNTPs | Deoxynucleoside triphosphates (dATP, dCTP, dGTP, dTTP); the building blocks for new DNA strands. | Quality is critical; impurities can inhibit the polymerase and reduce efficiency [9]. |
| Fluorescent Detection System | Reports amplicon accumulation. Intercalating dyes (e.g., SYBR Green) bind dsDNA. Hydrolysis probes (e.g., TaqMan) provide sequence specificity. | Dyes are cost-effective but less specific. Probes are specific and enable multiplexing but are more expensive [9] [94]. |
| Reaction Buffer | Provides optimal chemical environment (pH, salt concentration) for polymerase activity. | Often contains MgCl₂, which is a critical cofactor for polymerase. Its concentration can be optimized for each assay [9]. |
| Nuclease-Free Water | Solvent for the reaction. | Must be free of nucleases and contaminants to prevent degradation of reagents or inhibition of the reaction. |
| Calibrator Sample | A reference sample used in relative quantification to which all other samples are compared. | Often a control or untreated sample. Its use allows for fold-change calculations across multiple experiments [96]. |
Advanced Analysis: From Cq to Biological Interpretation
Relative Quantification: The ΔΔCq Method
The most common application of qPCR is determining the relative change in gene expression between samples. The ΔΔCq method is a widely used approach, but it is strictly valid only when the amplification efficiencies of the target and reference genes are approximately equal and close to 100% [96] [94].
Protocol for the ΔΔCq Method:
- Efficiency Validation: Confirm that the amplification efficiencies for both the target gene and the endogenous reference gene (e.g., GAPDH, β-actin) are similar and close to 2.0 (90-105%).
- Calculate ΔCq: For each sample, subtract the Cq of the reference gene from the Cq of the target gene. ( \Delta Cq = C{q,\ target} - C_{q,\ reference} )
- Calculate ΔΔCq: Subtract the ΔCq of the calibrator sample (e.g., untreated control) from the ΔCq of each test sample. ( \Delta\Delta Cq = \Delta C{q,\ sample} - \Delta C_{q,\ calibrator} )
- Calculate Fold Change: The normalized relative expression level is calculated as: ( \text{Fold Change} = 2^{-\Delta\Delta C_q} ) [96]
Efficiency-Corrected Quantification
When the efficiencies of the target and reference gene ((E{target}) and (E{reference})) differ, the ΔΔCq method introduces significant errors. In this case, an efficiency-corrected model, such as the Pfaffl method, must be used [95] [94]. The formula for the expression ratio is: [ \text{Ratio} = \frac{(E{target})^{\Delta C{q,\ target}}}{(E{reference})^{\Delta C{q,\ reference}}} ] This calculation requires knowing the precise efficiency for each assay, underscoring the necessity of the standard curve protocol described in Section 3.1 [95].
Troubleshooting and Clinical Implications
Inconsistent Cq values or anomalous efficiencies can derail an experiment. Common issues include:
- Inhibitors: Compounds like phenol, heparin, or hemoglobin carried over from sample preparation can inhibit the polymerase, leading to reduced efficiency and higher Cq values. This can be mitigated by diluting the template or re-purifying the nucleic acids [9] [98].
- Poor Primer Design: Primers with secondary structures (dimers, hairpins) or non-optimal melting temperatures are a leading cause of low efficiency and non-specific amplification [9].
In clinical diagnostics, such as SARS-CoV-2 testing via RT-qPCR, the Cq value is not just a quantitative measure but also a potential indicator of viral load and transmissibility. Lower Cq values generally correlate with higher viral loads [9]. However, direct translation of a single Cq value into a viral titer is unreliable without a standardized, efficiency-corrected calibration curve. Reporting and interpreting Cq values without context on PCR efficiency can lead to dramatically incorrect conclusions, with assumed expression ratios being orders of magnitude off from the true value [94]. Therefore, adhering to the MIQE guidelines (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) by reporting efficiency values and analysis methods is paramount for scientific rigor and clinical accuracy [94].
The replication of DNA is a fundamental process in molecular biology, serving as the cornerstone for genetic inheritance, cellular function, and numerous biotechnological applications. This whitepaper presents a side-by-side technical analysis of two principal methods for DNA synthesis: in vivo DNA replication, the natural, biological process occurring within living cells, and in vitro Polymerase Chain Reaction (PCR), the foundational laboratory technique for amplifying specific DNA sequences. Within the context of research and drug development, understanding the distinct operational parameters, capabilities, and limitations of each system is critical for selecting the appropriate tool for applications ranging from basic genetic research to the development of advanced cell and gene therapies. The purpose of this document is to provide researchers, scientists, and drug development professionals with an in-depth, comparative framework to inform experimental design and technological application.
Fundamental Principles and Mechanisms
In Vivo DNA Replication
In vivo DNA replication is a complex, enzyme-driven process that accurately duplicates the entire genome of a cell during the S-phase of the cell cycle. This biological mechanism is dependent on a well-defined and intricate set of enzymes and co-factors that function in a highly coordinated manner [8]. The replisome, the multiprotein machinery responsible for replication, works with high precision to unwind the DNA double helix and simultaneously replicate both strands [99]. This process is continuous and occurs at the physiological temperature of the organism, approximately 37°C for humans [3]. Key characteristics include its reliance on RNA primers synthesized by RNA primase to initiate synthesis, and the use of DNA polymerase enzymes that possess proofreading and repair abilities, contributing to an exceptionally high fidelity with an error rate of approximately 1 in 100,000 bases [3]. Its primary biological object is to copy the entire genome accurately for cell division.
In Vitro Polymerase Chain Reaction (PCR)
The Polymerase Chain Reaction (PCR), introduced by Kary Mullis in 1985, is a laboratory technique that facilitates in vitro DNA amplification of a specific, targeted fragment [9]. In principle, PCR mimics aspects of DNA replication but in a vastly simplified format, using a minimal set of defined ingredients and reaction conditions that involve rapid cycling through three elevated temperatures [8]. A single cycle consists of denaturation (separation of DNA strands at ~95°C), annealing (binding of DNA primers to their complementary sequences at 55-72°C), and extension (synthesis of new DNA strands by a thermostable polymerase at 72°C) [9]. This process is repeated for 30-40 cycles in a thermal cycler, resulting in the exponential amplification of the target sequence. PCR typically uses thermostable DNA polymerases (e.g., Taq polymerase) which lack proofreading ability, leading to a higher error rate than in vivo replication, approximately 1 in 9,000 bases [3]. It is a discontinuous process, optimized for speed over a short duration but for a limited fragment length.
Comparative Analysis: Operational Parameters
The following tables provide a detailed, side-by-side comparison of the core characteristics, components, and performance metrics of in vivo DNA replication and in vitro PCR.
Table 1: Core Characteristics and Key Differentiators
| Parameter | In Vivo DNA Replication | In Vitro PCR |
|---|---|---|
| Definition | Biological process of producing two identical DNA replicas from one original DNA molecule [3] | Laboratory process used to make many copies of a target DNA region [3] |
| Occurrence | In vivo (inside living cells) [3] | In vitro (inside a test tube) [3] |
| Primary Objective | Copy the entire genome for cell division [3] | Generate millions of copies of a single, specific DNA fragment [3] |
| Process Complexity | Complex, dependent on a well-defined but intricate set of enzymes and co-factors [8] | Simple process for in vitro DNA synthesis [3] |
| Key Applications in Research | Study of cell cycle, genomic stability, and disease models (e.g., cohesinopathies) [100] | Pathogen detection, genetic disorder screening, gene expression analysis, forensics, mutagenesis [9] |
Table 2: Molecular Components and Mechanisms
| Component/Mechanism | In Vivo DNA Replication | In Vitro PCR |
|---|---|---|
| Polymerizing Enzyme | DNA polymerase (e.g., Pol δ, ε). Operates at ~37°C, has proofreading and repair abilities [3] | Thermostable DNA polymerase (e.g., Taq polymerase). Operates at ~72°C, typically no proofreading ability [9] [3] |
| Primers | RNA primers synthesized by RNA primase [3] | Short, synthetically produced DNA primers [9] |
| Denaturation Method | Enzyme-driven (DNA helicase) [3] | Thermal (high temperature of ~95°C) [9] |
| Initiation | Specific genomic origins of replication | Requires prior knowledge of flanking sequences for primer design [101] |
| Fidelity/Error Rate | High fidelity; error rate of ~1 in 100,000 bases [3] | Lower fidelity; Taq polymerase error rate of ~1 in 9,000 bases [3] |
Table 3: Performance and Practical Metrics
| Metric | In Vivo DNA Replication | In Vitro PCR |
|---|---|---|
| Speed of Synthesis | Continuous process at ~1 kb/s [3] | Discontinuous process through cycles at ~1-4 kb/min [3] |
| Amplification Length | Entire genome (billions of base pairs) | Limited length; standard PCR effective for up to 5 kb, specialized long-range PCR up to ~40 kb [101] |
| Throughput & Scalability | Limited by cell culture and in vivo model systems | High throughput and easily scalable with automation (thermal cyclers) [9] |
| Sensitivity | N/A (occurs in every cell) | Extremely high; can detect a single copy of a DNA template [102] |
| Quantification Capability | N/A | Yes, particularly with quantitative real-time PCR (qPCR) and digital PCR (dPCR) [9] [58] |
Experimental Protocols
Analysis of In Vivo DNA Replication Machinery
Studying the in vivo replication machinery often involves sophisticated genetic and cellular techniques to understand its function and regulation.
- In Vivo Micronucleus (MN) Screen Protocol: This protocol is used to identify genes regulating genomic instability, a key readout of replication fidelity in a living organism [100].
- 1. Model System Generation: Create loss-of-function mutant lines (e.g., in mice) for genes of interest.
- 2. Sample Collection & Staining: Collect peripheral blood cells from mutant and wild-type control animals. Use a nucleic acid stain (e.g., propidium iodide) to label DNA.
- 3. Flow Cytometry Analysis: Analyze red blood cells using flow cytometry to enumerate the frequency of micronuclei, which are extranuclear structures containing mis-segregated or damaged chromatin.
- 4. Data Analysis & Validation: Compare MN frequency in mutants versus controls to identify genes whose loss significantly increases (+MN) or decreases (-MN) MN formation. Validate hits in human cell lines (e.g., CHP-212) using CRISPR-Cas9 knockout and microscopy, potentially with exposure to a replication stressor like hydroxyurea [100].
- In Vitro Single-Molecule Analysis of Replisome Components: This methodology provides novel, quantitative insights into the dynamics and mechanics of individual replication proteins and reconstituted replisomes, information not obtainable through classical ensemble-averaging methods [99]. Techniques include force spectroscopy (e.g., optical tweezers) and single-molecule fluorescence to observe the real-time activity of replication proteins.
Standard Quantitative PCR (qPCR) Protocol
This protocol allows for the amplification and quantification of a specific DNA target in real-time [9].
- 1. Reaction Mixture Assembly: In a sterile, nuclease-free tube, combine the following components on ice:
- Template DNA: 1-100 ng of genomic DNA or cDNA.
- Primers: Forward and reverse primers (20-25 nucleotides each), specific to the target sequence.
- PCR Master Mix: Contains Thermostable DNA Polymerase (e.g., Taq), dNTPs, MgCl₂, and reaction buffer.
- Fluorescent Probe: A sequence-specific probe (e.g., TaqMan) or a DNA-intercalating dye (e.g., SYBR Green).
- 2. Thermal Cycling: Place the reaction tube in a real-time PCR thermal cycler and run the following program:
- Initial Denaturation: 95°C for 2-10 minutes.
- Amplification Cycles (Repeated 40-50 times):
- Denaturation: 95°C for 15-30 seconds.
- Annealing: 55-65°C (primer-specific) for 30-60 seconds.
- Extension: 72°C for 30-60 seconds (time depends on amplicon length).
- Fluorescence Detection: The fluorescence is measured at the end of each annealing/extension step.
- 3. Data Analysis: The instrument software plots fluorescence versus cycle number. The quantification cycle (Cq), the cycle at which the fluorescence crosses a predetermined threshold, is determined. The Cq value is inversely proportional to the starting quantity of the target, enabling relative quantification when compared to a standard curve or reference genes [9].
Advantages and Limitations of PCR Technology
PCR offers a powerful and versatile tool for molecular biology but comes with specific constraints that must be managed.
Key Advantages:
- High Sensitivity and Specificity: PCR can detect a single copy of a DNA template and can distinguish between sequences that differ by only a single nucleotide [102] [101]. This makes it ideal for diagnosing low-level infections or detecting rare genetic variants.
- Speed and Efficiency: The entire process, from sample to result, can be completed within a few hours, generating billions of copies from a minimal amount of starting material [9] [101].
- Versatility and Automation: PCR can be applied to a vast range of applications, including genetic testing, forensic analysis, and pathogen detection. The process is easily automated using thermal cyclers, ensuring consistency and reproducibility [9] [101].
Key Limitations:
- Requirement for Sequence Knowledge: Designing primers requires prior knowledge of the nucleotide sequences flanking the target region. While primers can be designed based on conserved regions of related genes, this remains a fundamental constraint for discovering entirely novel sequences [102] [101].
- Amplicon Length Constraints: Standard PCR efficiently amplifies fragments up to 5 kilobases. While long-range PCR protocols can extend this to ~40 kb, they require specialized enzyme mixes and optimized conditions [101].
- Error Rate: The use of thermostable polymerases like Taq, which lack proofreading ability, can introduce errors during amplification, potentially leading to false results in sequencing or cloning applications [3] [102].
- Sensitivity to Contamination: The technique's extreme sensitivity makes it highly susceptible to contamination from exogenous DNA or amplicons from previous reactions, which can cause false-positive results [102] [101]. Rigorous laboratory practices and dedicated workspaces are essential.
Advantages and Limitations of In Vivo DNA Replication as a Research Model
Using in vivo systems to study DNA replication provides biological context but presents practical challenges for research.
- Advantages: Provides a complete physiological context, including all native enzymes, co-factors, and regulatory mechanisms (e.g., cell cycle checkpoints). Allows for the study of replication fidelity and its impact on organismal health and disease, as demonstrated in models studying genomic instability [100].
- Limitations: Complex and difficult to manipulate or observe directly in a living organism. Studies are often low-throughput, time-consuming, and expensive, requiring sophisticated model systems like genetically engineered mice [100].
Technology Evolution: Digital PCR
Digital PCR (dPCR) represents the third generation of PCR technology, offering a paradigm shift in quantification [58]. The core principle involves partitioning a single PCR reaction mixture into thousands to millions of individual nanoliter-scale reactions (either droplets or microchambers). Following end-point PCR amplification, each partition is analyzed for fluorescence. The fraction of positive partitions is then used to compute the absolute concentration of the target nucleic acid in the original sample using Poisson statistics, without the need for a standard curve [58].
Key Advantages over conventional qPCR:
- Absolute Quantification: Removes the reliance on external standards, improving accuracy and reproducibility.
- Superior Sensitivity and Precision: Enables detection of rare genetic events (e.g., somatic mutations) and small fold-changes in gene expression, which is crucial for applications like liquid biopsy in oncology and prenatal diagnosis [58].
- Increased Resilience to Inhibitors: The partitioning effect can dilute out PCR inhibitors present in the sample, making dPCR more robust for analyzing complex biological samples [58].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagent Solutions for DNA Synthesis Studies
| Research Reagent | Function/Application |
|---|---|
| Taq DNA Polymerase | Thermostable enzyme for catalyzing DNA synthesis during PCR amplification [9] |
| DNA Primers | Short, single-stranded DNA oligonucleotides that define the start and end of the target sequence to be amplified in PCR [9] |
| dNTPs (deoxynucleotide triphosphates) | The building blocks (dATP, dCTP, dGTP, dTTP) used by DNA polymerases to synthesize new DNA strands [9] |
| Fluorescent Probes/Dyes (e.g., SYBR Green, TaqMan) | Enable real-time detection and quantification of amplified DNA during qPCR [9] |
| Restriction Enzymes | Proteins that cleave DNA at specific recognition sequences; foundational for traditional molecular cloning and analysis of replication products [21] |
| DNA Ligase | Enzyme that catalyzes the joining of DNA strands; essential for in vivo replication (Okazaki fragment ligation) and in vitro cloning [21] |
| CRISPR-Cas9 System | A genome editing tool used to create knock-out mutant lines (e.g., in human cells) for validating the function of replication-related genes identified in screens [100] |
Workflow and Pathway Visualizations
PCR Amplification Thermal Cycling Workflow
Digital PCR (dPCR) Partitioning and Analysis
Quality Control and Contamination Prevention in the PCR Laboratory
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology that harnesses the core principles of cellular DNA replication in a controlled, in vitro setting. In vivo, DNA replication is a complex, enzyme-driven process that ensures the faithful duplication of the entire genome before cell division. This cellular process involves a suite of enzymes, including helicases to unwind the DNA double helix and DNA polymerases to synthesize new strands, and occurs at a constant physiological temperature [103] [2].
PCR, in contrast, is a targeted amplification technique designed to replicate only a specific segment of DNA, bounded by two short, synthetic primers. It simplifies the in vivo process by using a single, thermostable DNA polymerase (e.g., Taq polymerase) to carry out synthesis and employs repeated temperature cycles—denaturation, annealing, and extension—to achieve exponential amplification of the target sequence [2]. This very power of amplification, however, is also the source of its greatest vulnerability: contamination from previously amplified products (amplicons), which can lead to devastating false-positive results [104]. A robust framework of quality control and contamination prevention is, therefore, not merely good practice but a critical necessity for any laboratory performing PCR.
Foundational Concepts: PCR vs. In Vivo DNA Replication
Understanding the key differences between PCR and cellular DNA replication is essential for appreciating the unique contamination risks and control strategies in the PCR laboratory. The following table outlines these core distinctions.
Table 1: Key differences between in vivo DNA replication and in vitro PCR amplification.
| Feature | In Vivo DNA Replication | In Vitro PCR Amplification |
|---|---|---|
| Scope | Replication of the entire genome [2] | Targeted amplification of a specific DNA segment [2] |
| Primers | Short RNA primers synthesized by primase [2] | Short, single-stranded DNA primers; synthetically designed [2] |
| Polymerase | Multiple, complex DNA polymerases (e.g., δ, ε) [103] | A single, thermostable DNA polymerase (e.g., Taq) [2] |
| Temperature | Constant physiological temperature [2] | Cyclical temperature changes (denaturation, annealing, extension) [2] |
| Template | Chromosomal DNA | Any DNA containing the target sequence |
| Primary Contaminant | Not applicable | Previously generated amplification products (amplicons) [104] |
Preventing Amplification Product Carryover Contamination
A typical PCR reaction can generate up to 10^9 copies of the target sequence. If aerosolized, these amplicons can contaminate laboratory reagents, equipment, and ventilation systems, leading to false-positive results in subsequent experiments [104]. Prevention requires a multi-layered strategy involving both physical barriers and chemical sterilization.
Mechanical and Chemical Barriers
- Spatial Separation: The most critical mechanical barrier is the strict, unidirectional separation of laboratory areas. Laboratories should have physically separated, dedicated spaces for reagent preparation, sample preparation, amplification, and post-amplification analysis. Movement of personnel and materials must flow from "clean" to "dirty" areas without backtracking [104].
- Decontamination Protocols: Work surfaces must be routinely decontaminated. A 10% sodium hypochlorite (bleach) solution is highly effective, as it causes oxidative damage to nucleic acids, rendering them unamplifiable. The bleach should be followed with ethanol to remove residue. Any item moved from a contaminated area to a clean area must be soaked in bleach overnight and washed extensively [104].
Sterilization Techniques
Both pre- and post-amplification sterilization methods should be employed to control contamination.
Pre-Amplification Sterilization
- Ultraviolet (UV) Irradiation: UV light (254-300 nm) induces thymidine dimers in DNA, rendering contaminating amplicons inactive as templates. While simple and inexpensive, its efficacy is suboptimal for short or G+C-rich templates and can damage PCR reagents like primers and polymerase if not used judiciously [104].
- Enzymatic Inactivation with Uracil-N-Glycosylase (UNG): This is the most widely used contamination control method. The technique involves substituting dUTP for dTTP in the PCR master mix. In subsequent reactions, the enzyme UNG is added, which recognizes and hydrolyzes any uracil-containing contaminating amplicons from previous runs before the new PCR cycle begins. The UNG is then inactivated during the first high-temperature denaturation step, allowing the new amplification to proceed with dUTP incorporation [104].
Post-Amplification Sterilization
- Psoralen Inactivation: Psoralen compounds can be added to the completed PCR reaction. When activated by UV light, they intercalate into the double-stranded amplicons and form covalent cross-links, blocking them from being used as templates in future reactions [104].
Implementing a Comprehensive QC Strategy
A proactive quality control (QC) system is vital for ensuring the ongoing accuracy and reliability of PCR results.
Experimental Controls
Including the correct controls in every PCR run is non-negotiable. Their function is to monitor for contamination, inhibition, and reagent failure.
Table 2: Essential experimental controls for a PCR workflow.
| Control Type | Function | Interpretation of Results |
|---|---|---|
| No-Template Control (NTC) | Contains all PCR reagents except the template DNA. Monitors for amplicon contamination [105]. | A positive signal in the NTC indicates contamination in the reagents or environment. |
| Positive Control | Contains a known, low-copy number of the target sequence. Verifies that the PCR reaction is working efficiently [105]. | A negative result indicates a problem with the PCR reagents or conditions. |
| Internal Positive Control (IPC) | A non-target control sequence spiked into each reaction. Distinguishes between true target-negative results and PCR inhibition [105]. | Failure of the IPC signal indicates the sample contains PCR inhibitors. |
| External Quality Control (EQC) | Known reference samples included in each run to assess the entire process, from extraction to amplification. Detects reagent drift and instrument variation [105]. | EQC values outside established acceptance limits (e.g., mean Ct ± 1) indicate a systemic issue requiring investigation. |
Quantitative vs. Conventional PCR
The choice of detection method itself influences QC sensitivity. Quantitative PCR (qPCR) offers significant advantages over conventional PCR for contamination monitoring. A 2015 study comparing the two methods for detecting Human Adenoviruses in environmental samples found that qPCR detected nearly twice as many positive samples (87.3% in water samples) compared to conventional PCR (47.3%) [106]. This is because qPCR measures the target during the exponential phase of amplification and is less susceptible to the effects of partial inhibition that can cause false negatives in end-point conventional PCR analysis [106].
A Practical Workflow for Contamination Prevention
The following diagram synthesizes the key concepts and procedures into a single, streamlined workflow for a contamination-aware PCR laboratory.
Diagram 1: PCR contamination prevention workflow.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key research reagent solutions for quality control and contamination prevention.
| Reagent/Material | Function in QC/Contamination Prevention |
|---|---|
| Uracil-N-Glycosylase (UNG) | Enzymatically degrades carryover contaminant amplicons from previous PCRs that contain dUTP [104]. |
| dUTP | A nucleotide substitute for dTTP; allows newly synthesized amplicons to be distinguishable from native DNA and susceptible to UNG degradation [104]. |
| Sodium Hypochlorite (Bleach) | A chemical decontaminant that causes oxidative damage to nucleic acids on laboratory surfaces and equipment [104]. |
| Validated EQC Materials | Standardized reference samples (e.g., synthetic DNA, inactivated virus) used to monitor the accuracy and consistency of the entire PCR process over time [105]. |
| Aerosol-Barrier Pipette Tips | Prevent the carryover of aerosolized amplicons or sample material into pipette shafts, a common source of cross-contamination [104]. |
| Psoralen Compounds | Post-amplification sterilization agents that intercalate into amplicons and cross-link them upon UV activation, preventing re-amplification [104]. |
Maintaining the integrity of PCR-based assays demands constant vigilance against contamination. By understanding PCR as an in vitro derivation of in vivo replication, laboratories can better appreciate its unique vulnerabilities. A successful strategy integrates physical segregation, rigorous decontamination protocols, enzymatic sterilization methods like UNG, and a comprehensive QC system with appropriate controls. Adhering to these principles allows researchers to harness the full, powerful potential of PCR while ensuring the generation of reliable and meaningful data.
DNA replication is a fundamental biological process, and its study can be approached through two fundamentally different paradigms: investigating the native, in vivo replication within living cells or utilizing the in vitro polymerase chain reaction (PCR) to amplify specific DNA sequences. The choice between these approaches is not merely technical but fundamentally shapes the biological questions a researcher can address. Native replication research seeks to understand the complex, regulated cellular process of entire genome duplication, while PCR provides a powerful tool for targeted amplification and detection of specific nucleic acid sequences. This guide provides a structured framework to help researchers select the appropriate methodology based on their specific research goals, whether for basic science, diagnostic development, or therapeutic discovery.
Core Conceptual Differences: In Vivo Replication vs. PCR
Understanding the fundamental distinctions between native DNA replication and PCR is critical for selecting the right experimental path.
- In Vivo DNA Replication is the natural, biological process by which a cell duplicates its entire genome before cell division. It is a complex, coordinated process dependent on a well-defined but intricate set of enzymes and co-factors that act in a concerted fashion during the synthetic phase (S-phase) of the cell cycle [1]. Its primary purpose is to ensure the faithful inheritance of genetic material.
- Polymerase Chain Reaction (PCR) is an in vitro enzymatic synthesis that mimics, in a much simpler fashion, the core principle of DNA replication to amplify a specific, targeted DNA sequence exponentially [1] [107] [2]. Its purpose is not genome maintenance but the generation of millions to billions of copies of a particular DNA fragment from a minute starting amount.
The table below summarizes the key operational differences between these two processes.
Table 1: Fundamental Differences Between In Vivo DNA Replication and PCR
| Feature | In Vivo DNA Replication | PCR |
|---|---|---|
| Purpose & Scope | Copies the entire genome for cell division [3] | Amplifies a specific, targeted DNA fragment [3] |
| Process Location | Inside living cells (in vivo) [3] | In a test tube (in vitro) [3] |
| Key Enzymes | DNA polymerase (with proofreading), DNA helicase, primase (synthesizes RNA primers), ligase, and other replisome components [5] [3] | Thermostable DNA polymerase (e.g., Taq polymerase, often without proofreading) [3] |
| Denaturation | Achieved by helicase enzymes [5] [3] | Achieved by high-temperature incubation (~95°C) [3] |
| Primers | RNA primers synthesized by primase [3] | DNA primers (synthetically produced) [3] |
| Temperature Regimen | Occurs at a constant physiological temperature (e.g., 37°C) [3] | Requires cyclic temperature changes (denaturation, annealing, extension) [3] |
| Process Nature | Continuous and coordinated with the cell cycle [5] | Discontinuous, involving 25-40 repeated cycles [3] |
| Fidelity | High, with proofreading; error rate ~1 in 100,000 bases [3] | Lower fidelity (for Taq polymerase); error rate ~1 in 9,000 bases [3] |
Methodological Guide: Experimental Approaches and Workflows
The experimental protocols for studying native replication versus performing PCR are vastly different, from the required reagents to the procedural steps.
Research Reagent Solutions
The core materials needed for each approach reflect their underlying complexity.
Table 2: Essential Research Reagents and Their Functions
| Category | Reagent/Material | Function in the Experiment |
|---|---|---|
| Studying Native Replication | Genetic Tools (e.g., mutant strains, plasmids) | To manipulate and study the function of specific replication genes and proteins in vivo [5] |
| Synchronized Cell Cultures | To obtain a population of cells that are at the same stage of the cell cycle, allowing replication analysis [5] | |
| Pulse-Labelling Compounds (e.g., radioactive or heavy isotopes) | To track newly synthesized DNA strands and measure replication dynamics [5] | |
| PCR Amplification | Thermostable DNA Polymerase (e.g., Taq) | Enzymatically synthesizes new DNA strands during the high-temperature extension step [107] [2] |
| Synthetic Oligonucleotide Primers | Short, single-stranded DNA sequences that define the 5' and 3' boundaries of the target DNA region to be amplified [107] [2] | |
| dNTP Mix (dATP, dCTP, dGTP, dTTP) | The four deoxynucleotide triphosphates that serve as the building blocks for the new DNA strands [107] | |
| Target DNA Template | The source DNA containing the sequence of interest to be copied [2] | |
| Buffer with Mg²⁺ | Provides the optimal chemical environment (pH, ions) for polymerase activity and primer binding [107] |
Experimental Workflow Visualization
The following diagrams illustrate the high-level workflows for key methodologies in both domains.
PCR Amplification Workflow: This process involves repeated thermal cycling to achieve exponential amplification of a specific DNA target [107] [2].
Studying Bacterial Replication Initiation: This workflow outlines a genetic and molecular approach to probe the complex initiation of replication in live bacterial cells [5].
Decision Framework: Selecting the Right Tool for Your Research Goal
The choice between PCR and native replication studies is dictated by the core research question. The following table provides guidance on which approach is best suited for specific applications.
Table 3: Application-Based Selection Guide: PCR vs. Native Replication Studies
| Research Goal | Recommended Approach | Rationale and Technical Considerations |
|---|---|---|
| Pathogen Detection / Diagnostic Assay | PCR (especially qPCR/dPCR) | Unmatched sensitivity and speed for detecting low copy numbers of a specific pathogen sequence (e.g., viral RNA/DNA) [107] [20] [106]. |
| Generate Probes/Sequencing Templates | PCR | The most efficient method to produce large quantities of a specific DNA fragment for use in downstream molecular applications [107]. |
| Gene Expression Analysis | Reverse Transcription qPCR (RT-qPCR) | The gold standard for quantifying transcript levels from specific genes by first converting RNA to cDNA and then monitoring amplification in real-time [107] [20]. |
| Absolute Quantification of DNA | Digital PCR (dPCR) | Provides absolute quantification without a standard curve and is more tolerant to inhibitors, making it ideal for detecting rare mutations or copy number variations [20]. |
| Understand Genome Duplication Mechanics | Native Replication Studies | Essential for elucidating the roles of replication machinery (replisome), regulation, and coordination with the cell cycle [5]. |
| Study Replication Fork Dynamics & Stability | Native Replication Studies | Requires in vivo systems to investigate how replication forks respond to stress, DNA damage, and other intracellular factors [5]. |
| Investigate Chromosome Segregation | Native Replication Studies | The process of segregating newly replicated chromosomes is intrinsically linked to in vivo replication and must be studied in a cellular context [1] [5]. |
Advanced PCR Technologies: qPCR vs. dPCR
Within the PCR paradigm, choosing between quantitative PCR (qPCR) and digital PCR (dPCR) is a critical modern decision.
Quantitative PCR (qPCR): Also known as real-time PCR, it allows for the quantification of DNA (or cDNA) as it is amplified. The key operational characteristics for diagnostic tests based on MIQE guidelines include [20]:
- Reliability (Precision): High, suitable for high-throughput applications.
- Accuracy: High for relative quantification when using standard curves.
- Analytical Sensitivity (LoD): Can detect low target copies, but can be influenced by PCR inhibitors.
- Specificity: High, confirmed via melting curve analysis.
Digital PCR (dPCR): A newer variant that partitions a sample into thousands of nanoreactions, allowing for absolute quantification of nucleic acids by counting positive and negative partitions [20].
When to choose qPCR: For high-throughput routine diagnostics (e.g., SARS-CoV-2 testing), relative gene expression quantification, and when cost-effectiveness and operational ease are priorities [20]. When to choose dPCR: For applications requiring absolute quantification without a standard curve, detection of rare genetic variants (e.g., in liquid biopsies), and when analyzing samples with potential PCR inhibitors [20].
The decision to use PCR or to study native DNA replication is foundational to experimental design in molecular biology. PCR is an unparalleled tool for targeted amplification, detection, and quantification of specific nucleic acid sequences, forming the backbone of modern diagnostics, genotyping, and recombinant DNA work. In contrast, studying native replication is indispensable for basic science aimed at understanding the fundamental mechanisms of genome duplication and its regulation within the complex environment of the living cell. By aligning your research question with the strengths and limitations of each approach, as outlined in this guide, you can ensure the selection of the most appropriate and powerful tool for your scientific inquiry.
Conclusion
In vivo DNA replication and PCR, while sharing a fundamental principle of DNA synthesis, serve distinct and complementary roles in biomedical science. In vivo replication is a complex, highly regulated cellular process essential for life, whereas PCR is a simplified, powerful in vitro tool for targeted DNA amplification. For researchers and clinicians, PCR's unparalleled speed, sensitivity, and specificity make it indispensable for diagnostics, pathogen detection, and genetic analysis. However, its limitations, including primer dependency, contamination susceptibility, and polymerase error rates, necessitate rigorous optimization and validation. The future of this field lies in the continued engineering of more advanced DNA polymerases and the development of even more robust, automated platforms. Understanding the core differences between these processes is not just an academic exercise; it is critical for making informed decisions in experimental design, diagnostic development, and the advancement of personalized medicine.
References
- 1. A Brief Comparison Between In Vivo DNA Replication and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7120002/]
- 2. 4.2: In vitro applications of DNA replication [https://bio.libretexts.org/Courses/University_of_Massachusetts_Boston/Bio_252_254%3A_Genetics/04%3A__SPOC_IV_-_DNA_Replication/4.02%3A_In_vitro_applications_of_DNA_replication]
- 3. What are the differences between PCR and DNA replication? [https://www.aatbio.com/resources/faq-frequently-asked-questions/what-are-the-differences-between-pcr-and-dna-replication]
- 4. In vivo vs. in vitro: What is the difference? [https://www.medicalnewstoday.com/articles/in-vivo-vs-in-vitro]
- 5. Concise Overview of Methodologies Employed in the Study ... [https://www.mdpi.com/1422-0067/26/2/446]
- 6. Manufacturing DNA in E. coli yields higher-fidelity ... [https://www.sciencedirect.com/science/article/pii/S2329050124000433]
- 7. Genetically Encoded Control of In Vitro Transcription ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12538582/]
- 8. A Brief Comparison Between In Vivo DNA Replication and ... [https://link.springer.com/chapter/10.1007/978-1-4020-6241-4_2]
- 9. Polymerase Chain Reaction (PCR) - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK589663/]
- 10. Frontiers | A Replisome ’s journey through the bacterial chromosome [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2015.00562/full]
- 11. methods to study helicase, DNA polymerase, and... Quantitative [https://pmc.ncbi.nlm.nih.gov/articles/PMC9933136/]
- 12. Concise Overview of Methodologies Employed in the Study of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11764726/]
- 13. DNA Polymerase—Four Key Characteristics for PCR [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/dna-polymerase-characteristics.html]
- 14. Integrating biotechnology into diagnostic labs: Innovations ... [https://www.sciencedirect.com/science/article/pii/S2590262825000565]
- 15. PCR Setup—Six Critical Components to Consider [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-component-considerations.html]
- 16. DNA Replication Mechanisms - Molecular Biology of the Cell [https://www.ncbi.nlm.nih.gov/books/NBK26850/]
- 17. Comparison of Seven Commercial TaqMan Master Mixes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7810389/]
- 18. Components of the Cell-Cycle Control System - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK26824/]
- 19. PCR Thermal Cyclers Education [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-thermal-cyclers.html]
- 20. Quantitative or digital PCR? A comparative analysis for ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993624001584]
- 21. A comparative review of DNA assembly strategies [https://www.sciencedirect.com/science/article/pii/S2452014425002493]
- 22. PCR Machine: Principle, Parts, Steps, Types, Uses ... [https://microbenotes.com/pcr-machine-principle-parts-steps-types-uses-examples/]
- 23. PCR vs qPCR vs RT-PCR vs RT-qPCR: What's the Difference [https://molsentech.com/post/pcr-vs-qpcr-vs-rt-pcr-vs-rt-qpcr-whats-the-difference/]
- 24. Digital PCR outperforms quantitative real-time PCR for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288185/]
- 25. PCR and Real-Time PCR: Differences in Results, Cost ... [https://www.labconsulting.at/pcr-and-real-time-pcr-differences/]
- 26. DNA Replication [https://byjus.com/biology/difference-between-pcr-and-dna-replication/]
- 27. Error Rate Comparison during Polymerase Chain Reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4150459/]
- 28. BLOG: Selecting the Right Gene Editing Off-Target Assay [https://seqwell.com/guide-to-selecting-right-gene-editing-off-target-assay/]
- 29. Genome-wide CRISPR off-target prediction and ... [https://www.nature.com/articles/s41467-023-42695-4]
- 30. High-fidelity CRISPR-Cas9 variants with undetectable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4851738/]
- 31. PCR-based diagnostics for infectious diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC7106425/]
- 32. Mastering dMIQE Guidelines for the Publication of dPCR ... [https://www.thermofisher.com/us/en/home/life-science/pcr/digital-pcr/education/basic-research/mastering-dmiqe-guidelines.html]
- 33. Engineers Develop Genetic Testing Device to Detect Rare ... [https://www.rutgers.edu/news/engineers-develop-genetic-testing-device-detect-rare-mutations]
- 34. MIQE 2.0: Revision of the Minimum Information for ... [https://pubmed.ncbi.nlm.nih.gov/40272429/]
- 35. DNA replication: Mechanisms and therapeutic interventions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9899494/]
- 36. Advances In DNA Replication Research For Precision ... [https://www.genesispcl.com/articles/vol-2-issue-4/jber-25-24-0001/]
- 37. Polymerase Chain Reaction (PCR): Principle and ... [https://www.intechopen.com/chapters/67558]
- 38. Specialized replication mechanisms maintain genome ... [https://www.sciencedirect.com/science/article/pii/S1097276524000546]
- 39. Comparing the precision of two digital PCR applications for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12297429/]
- 40. One-hit wonders of genomic instability - Cell Division [https://celldiv.biomedcentral.com/articles/10.1186/1747-1028-5-15]
- 41. Reverse transcription polymerase chain reaction [https://en.wikipedia.org/wiki/Reverse_transcription_polymerase_chain_reaction]
- 42. Introduction to Quantitative PCR (qPCR) Gene Expression ... [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/gene-expression-analysis-real-time-pcr-information/introduction-gene-expression.html]
- 43. The Basics: RT-PCR [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rtpcr-analysis/general-articles/rt--pcr-the-basics.html]
- 44. The Basics of Reverse Transcription PCR (RT-PCR) [https://www.excedr.com/resources/the-basics-of-reverse-transcription-pcr-rt-pcr]
- 45. From RNA to results: the essential guide to RT-PCR [https://www.integra-biosciences.com/united-states/en/blog/article/rna-results-essential-guide-rt-pcr]
- 46. One-Step RT-qPCR for Viral RNA Detection Using Digital ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8948435/]
- 47. Reverse transcription-quantitative PCR assays for ... [https://www.sciencedirect.com/science/article/pii/S2405844025008837]
- 48. Viral load quantification [https://www.qiagen.com/de/applications/digital-pcr/applications/viral-load-quantification]
- 49. Essentials of Real-Time PCR [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/essentials-real-time-pcr.html]
- 50. rtpcr: a package for statistical analysis and graphical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530193/]
- 51. 3.2: Replication of DNA [https://bio.libretexts.org/Courses/West_Los_Angeles_College/Biotechnology/03%3A_Molecular_Biology_Fundamentals/3.02%3A_Replication_of_DNA]
- 52. PCR Cloning Method [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/pcr-cloning?srsltid=AfmBOopmn9HSekFWaSsXLcsnu1OgiBpNpEwSy-ZajRK_L3vtrizdSgjq]
- 53. Molecular cloning using polymerase chain reaction, an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4350901/]
- 54. Plasmid Cloning by PCR (with Protocols) [https://www.addgene.org/protocols/pcr-cloning/]
- 55. Next-generation sequencing: 2025 Revolution [https://lifebit.ai/blog/next-generation-sequencing/]
- 56. Applications of PCR in Drug Development [https://kcasbio.com/blogs/pcr-applications-drug-development/]
- 57. Uracil walking primer PCR : An accurate and efficient genome-walking... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11957515/]
- 58. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 59. Towards practical point-of-care quick, ubiquitous ... [https://www.nature.com/articles/s41378-025-01057-4]
- 60. Estimating and mitigating amplification bias in qualitative ... [https://www.nature.com/articles/s41598-017-17333-x]
- 61. Analyzing and minimizing PCR amplification bias in Illumina ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2011-12-2-r18]
- 62. Thermal-bias PCR: generation of amplicon libraries without ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12558157/]
- 63. Fidelity of DNA replication—a matter of proofreading - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6153641/]
- 64. Engineering polymerases for new functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6745271/]
- 65. Taq polymerase [https://en.wikipedia.org/wiki/Taq_polymerase]
- 66. So Many DNA Polymerases, So Little Time [https://www.goldbio.com/blogs/articles/somanypolymerasessolittletime?srsltid=AfmBOoqw7BRxHXH1pmolExNIO6vP2F_kqLxEJow8R0xUyjAeWf2_kosv]
- 67. Novel thermostable Y-family polymerases - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC1373694/]
- 68. Current PCR Methods [https://www.labome.com/method/Current-PCR-Methods.html]
- 69. PCR inhibition in qPCR, dPCR and MPS—mechanisms and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7072044/]
- 70. Polymerase chain reaction inhibitors [https://en.wikipedia.org/wiki/Polymerase_chain_reaction_inhibitors]
- 71. What Are PCR Inhibitors? [https://www.zymoresearch.de/blogs/blog/what-are-pcr-inhibitors]
- 72. Evaluation of PCR-enhancing approaches to reduce ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969724069250]
- 73. PCR purification | Magnetic beads and spin columns | INTEGRA [https://www.integra-biosciences.com/united-states/en/stories/introduction-pcr-purification]
- 74. Comparative Study Between Droplet Digital PCR and Real ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12654130/]
- 75. 热启动PCR-特异性扩增-赛默飞| Thermo Fisher Scientific - CN [https://www.thermofisher.cn/cn/zh/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/hot-start-technology-benefits-PCR.html]
- 76. 热启动PCR [https://www.sigmaaldrich.com/HK/zh/technical-documents/technical-article/genomics/pcr/hot-start-pcr?srsltid=AfmBOopAJMIos2_35oNyi4RtOWcJwjGNvWPP304PrvYmvrR8FxSzvqS1]
- 77. 提高PCR特异性的方法:热启动型PCR全面解析 [https://www.yeasen.com/h5/solutiondetail/322]
- 78. 热启动Taq酶的原理与应用 [https://www.detaibio.com/topics/hotstart-theory.html]
- 79. ThermaGenix PCR 添加剂可降低背景并提高产量 [https://www.sigmaaldrich.com/HK/zh/technical-documents/technical-article/genomics/pcr/thermagenix?srsltid=AfmBOorXlpTYCdORePuj132zOxUBxQDyrF6g27xESI5RNikrCbeRSYI4]
- 80. The A, B, C, D's of replicative DNA polymerase fidelity [https://pmc.ncbi.nlm.nih.gov/articles/PMC12634111/]
- 81. 24.2: DNA Mutations, Damage, and Repair [https://bio.libretexts.org/Bookshelves/Biochemistry/Fundamentals_of_Biochemistry_(Jakubowski_and_Flatt)/03%3A_Unit_III-_Information_Pathway/24%3A_DNA_Metabolism/24.02%3A_DNA_Mutations_Damage_and_Repair]
- 82. A QUANTITATIVE MODEL OF ERROR ACCUMULATION ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1544370/]
- 83. A quantitative model of error accumulation during PCR ... [https://www.sciencedirect.com/science/article/abs/pii/S1476927105001258]
- 84. A high-throughput assay for quantitative measurement of ... [https://www.nature.com/articles/s41598-017-02727-8]
- 85. Structural basis of error-prone DNA synthesis by DNA ... [https://www.nature.com/articles/s41467-025-57269-9]
- 86. Implications for the creation of mutant DNA polymerases [https://www.sciencedirect.com/science/article/abs/pii/S1568786405002570]
- 87. Most human DNA replication initiation is dispersed throughout ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03591-w]
- 88. Overcoming natural replication barriers formed by DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582535/]
- 89. Development of real-time PCR methods for quality control ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12058689/]
- 90. A comprehensive review of methodological and ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002058]
- 91. The new gold rush in clinical diagnostics - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12584703/]
- 92. Diagnostics Pcr in the Real World: 5 Uses You'll Actually ... [https://www.linkedin.com/pulse/diagnostics-pcr-real-world-5-uses-youll-2yzje/]
- 93. Biochemistry, DNA Replication - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK482125/]
- 94. Use and Misuse of Cq in qPCR Data Analysis and Reporting [https://pmc.ncbi.nlm.nih.gov/articles/PMC8229287/]
- 95. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOoo9yIGUqzXk70Xfx1Hsp7krvFL8vOzQtRlBUs5ZdjKAoqRtOzET]
- 96. PCR efficiency | Determining amplification efficiencies [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/pcr-quantification/determining-amplification-efficiencies]
- 97. Analysis of qPCR Data: From PCR Efficiency to Absolute ... [https://www.preprints.org/manuscript/202510.2485]
- 98. Understanding qPCR Efficiency and Why It Exceeds 100% [https://biosistemika.com/blog/qpcr-efficiency-over-100/]
- 99. DNA replication machinery: Insights from in vitro single- ... [https://www.sciencedirect.com/science/article/pii/S2001037021001240]
- 100. Genetic determinants of micronucleus formation in vivo [https://www.nature.com/articles/s41586-023-07009-0]
- 101. The Limitations and Advantages of Polymerase Chain ... [https://www.letstalkacademy.com/limitations-and-advantages-of-pcr/]
- 102. Advantages and disadvantages of PCR technology [https://facellitate.com/advantages-and-disadvantages-of-pcr-technology/]
- 103. A Brief Comparison Between In Vivo DNA Replication and ... [https://www.semanticscholar.org/paper/A-Brief-Comparison-Between-In-Vivo-DNA-Replication/0c62f5cee6b8762767ca61558549458e3e8303a6]
- 104. Preventing PCR Amplification Carryover Contamination in ... [https://www.annclinlabsci.org/content/34/4/389.long]
- 105. Implementing External Quality Control in PCR Workflows [https://www.mapi-research.fr/implementing-external-quality-control-in-pcr-workflows-a-step-by-step-laboratory-guide/]
- 106. QUANTITATIVE VS. CONVENTIONAL PCR FOR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4616914/]
- 107. Polymerase chain reaction and its applications [https://www.sciencedirect.com/science/article/abs/pii/S0968605302001023]