PCR Optimization Guide 2025: Proven Strategies for High-Yield, High-Fidelity Results
This guide provides new researchers and drug development professionals with a comprehensive framework for mastering Polymerase Chain Reaction (PCR) optimization.
PCR Optimization Guide 2025: Proven Strategies for High-Yield, High-Fidelity Results
Abstract
This guide provides new researchers and drug development professionals with a comprehensive framework for mastering Polymerase Chain Reaction (PCR) optimization. It covers foundational principles, from reagent function and primer design to advanced methodological strategies like hot-start and quantitative PCR. The article delivers a systematic troubleshooting guide to resolve common amplification issues and outlines rigorous validation protocols to ensure results are reproducible, specific, and suitable for sensitive downstream applications like cloning and sequencing. By integrating foundational knowledge with practical optimization techniques and validation standards, this resource aims to accelerate experimental success in both research and clinical diagnostics.
PCR Fundamentals: Core Principles and Reaction Components
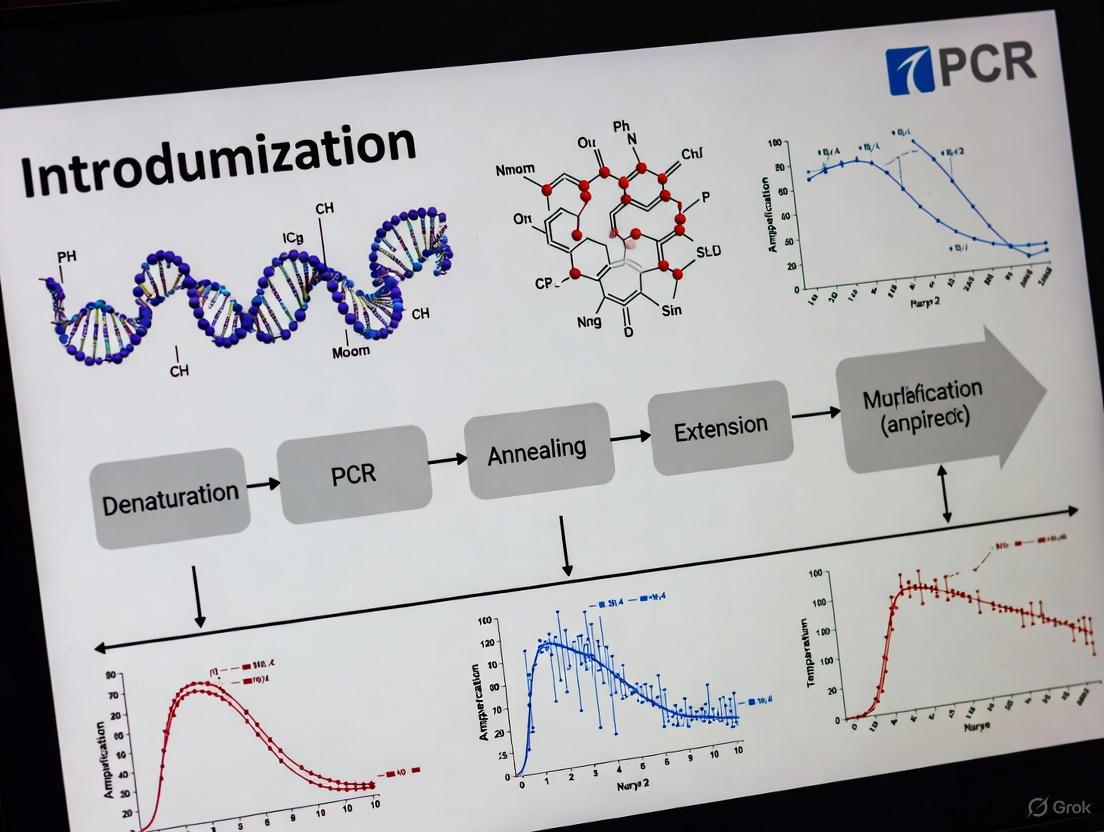
The Polymerase Chain Reaction (PCR) is a cornerstone technique in molecular biology, enabling the amplification of trace amounts of DNA or RNA into millions of copies for a wide array of applications, from gene cloning and diagnostic testing to forensic analysis and biomedical research [1] [2]. For new researchers, a deep understanding of the core PCR process is the essential first step toward effective experimentation and optimization. This guide provides an in-depth technical examination of the four fundamental steps of a standard PCR—denaturation, annealing, extension, and analysis—framed within the critical context of PCR optimization for robust and reliable results.
The Core Principles of PCR
At its heart, PCR is an enzymatic process that amplifies a specific DNA segment through repeated temperature cycles. Each cycle theoretically doubles the amount of the target DNA fragment, leading to exponential amplification [1]. The process relies on a thermostable DNA polymerase, primers designed to flank the target sequence, and nucleotides to build the new DNA strands [3]. The standard PCR formula for yield is PCR product yield = 2N copies, where N is the number of cycles. However, as cycles progress, reagents are consumed and byproducts accumulate, eventually leading to a plateau phase where amplification efficiency drops [1]. Understanding and optimizing the reaction components and cycling conditions is key to maximizing yield and specificity before this plateau occurs.
Detailed Breakdown of the Four PCR Steps
The following diagram illustrates the three temperature-dependent steps of a PCR cycle—Denaturation, Annealing, and Extension—which are repeated 25-40 times to exponentially amplify the target DNA sequence. Analysis is performed after cycling is complete to evaluate the results.
Step 1: Denaturation
The denaturation step is crucial for initiating the reaction by separating the double-stranded DNA template into single strands, providing the necessary template for primer binding.
Process and Purpose: During denaturation, the reaction mixture is heated to a high temperature (typically 94–98°C), which breaks the hydrogen bonds between complementary base pairs of the double-stranded DNA molecule [4] [2]. This results in single-stranded DNA molecules that are accessible to primers in the subsequent annealing step. The initial denaturation at the beginning of the PCR program is often longer (* 1–3 minutes* ) to ensure complete separation of complex DNA, such as genomic DNA, and to activate hot-start DNA polymerases [4]. Subsequent denaturation steps in each cycle are shorter, often 10–60 seconds [5].
Optimization Considerations:
- Template Complexity: Mammalian genomic DNA or DNA with high GC content (>65%) may require longer denaturation times or higher temperatures for complete separation [4].
- Enzyme Stability: While Taq polymerase can withstand brief denaturation temperatures, prolonged incubation above 95°C can lead to enzyme denaturation. Highly thermostable enzymes from Archaea are more suitable for challenging templates requiring harsh denaturation conditions [4].
- Additives: Reagents like glycerol, DMSO, or formamide can help denature DNA with strong secondary structures, potentially reducing the need for extreme temperatures or extended times [4].
Step 2: Annealing
In the annealing step, the reaction temperature is lowered to allow primers to bind specifically to their complementary sequences on the single-stranded DNA template.
Process and Purpose: The temperature is rapidly cooled to a range typically between 45°C and 72°C for 30 seconds to 2 minutes [4] [5]. This enables the forward and reverse primers to hybridize (anneal) to the target DNA flanking the region to be amplified. The annealing temperature is a critical parameter determined by the primer's melting temperature (Tm), which is the temperature at which 50% of the primer-duplex dissociates [4].
Optimization Considerations:
- Tm Calculation: The simplest formula for estimating Tm is Tm = 4(G + C) + 2(A + T), where G, C, A, and T represent the number of each nucleotide in the primer [4] [6]. A more accurate method is the Nearest Neighbor method, which accounts for salt concentrations [4].
- Annealing Temperature: A common starting point is to set the annealing temperature 3–5°C below the calculated Tm of the primers [4]. If nonspecific amplification occurs, increase the temperature in 2–3°C increments. If yield is low, decrease the temperature similarly [4] [5].
- Primer Design: Primers should be 15–30 nucleotides long with a GC content of 40–60% [5]. The 3' end should be rich in G or C bases to enhance binding stability and reduce mismatches [1] [5]. The Tm for both primers should be similar (within 5°C), and their sequences should be checked to avoid primer-dimer formation [5].
- Universal Annealing: Some advanced reaction buffers are formulated to allow for a universal annealing temperature (e.g., 60°C), simplifying protocol setup and reducing optimization time [4].
Step 3: Extension
During extension, the DNA polymerase synthesizes a new DNA strand complementary to the template, starting from the bound primers.
Process and Purpose: The temperature is raised to the optimal range for the DNA polymerase, typically 68–72°C [4] [6]. The enzyme adds nucleotides (dNTPs) to the 3' end of the primer, synthesizing a new DNA strand in the 5' to 3' direction. The extension time depends on the length of the amplicon and the speed of the polymerase. A common guideline is 1 minute per 1000 base pairs for Taq polymerase, though faster enzymes are available [4] [5].
Optimization Considerations:
- Polymerase Selection: Different DNA polymerases have varying extension rates, fidelity (accuracy), and processivity (number of nucleotides added per binding event). For example, Pfu polymerase has proofreading activity (high fidelity) but a slower extension rate than Taq [5].
- Two-Step PCR: If the annealing temperature is within 3°C of the extension temperature, the annealing and extension steps can be combined into a single two-step PCR, shortening the total run time [4].
- Final Extension: A final extension step of 5–15 minutes after the last cycle is often added to ensure all amplicons are fully synthesized and to facilitate proper adenylation (A-tailing) for TA cloning [4].
Step 4: Analysis
After thermal cycling, the amplified PCR products must be analyzed to confirm the success of the reaction, including the presence, size, and specificity of the amplicon.
Process and Purpose: The most common method for analyzing conventional PCR products is agarose gel electrophoresis [3] [2]. The DNA fragments are separated by size in an electric field and visualized using fluorescent dyes like ethidium bromide or safer alternatives. The resulting band pattern is compared to a DNA ladder of known sizes to verify the amplicon size.
Optimization and Advanced Techniques:
- Troubleshooting: The absence of a band, smearing, or multiple bands on the gel indicates issues with amplification specificity or efficiency, requiring optimization of primers, Mg2+ concentration, or cycling conditions [1] [5].
- Quantitative Analysis: In quantitative real-time PCR (qPCR), amplification is monitored in real-time using fluorescent dyes or probes. The quantification cycle (Cq) is determined, which is the cycle number at which the fluorescence crosses a defined threshold, allowing for precise quantification of the initial DNA template [7].
- High-Resolution Melting (HRM): Following qPCR, HRM analysis can be used to distinguish between PCR products based on their melting temperature (Tm), which is sensitive to sequence, length, and GC content. This is useful for species identification, genotyping, and mutation detection without the need for gel electrophoresis [8].
Critical PCR Components and Optimization
A successful PCR requires careful optimization of its core components. The following table summarizes the key reagents, their functions, and critical optimization parameters.
Table 1: Key Research Reagent Solutions for PCR Optimization
| Reagent | Function | Standard Concentration/Range | Optimization Tips |
|---|---|---|---|
| DNA Polymerase | Enzyme that synthesizes new DNA strands [2]. | Varies by enzyme; e.g., 1.25-2.5 U/50µL reaction [5]. | Use hot-start versions to reduce nonspecific amplification [5]. Select high-fidelity enzymes for cloning [5]. |
| Primers | Short oligonucleotides that define the start and end of the target sequence [3]. | 0.1-1.0 µM each; often optimal at 0.4-0.5 µM [1] [5]. | Avoid primer-dimer formation; ensure Tm values are similar and 3' ends are G/C-rich [1] [5]. |
| Template DNA | The DNA sequence to be amplified. | 10 ng - 1 µg genomic DNA; 1e4 - 1e6 copies [5] [6]. | Ensure high quality and purity. Re-quantify DNA stored for long periods [1]. |
| dNTPs | Nucleotide building blocks (dATP, dCTP, dGTP, dTTP) for new DNA strands [3]. | 20-200 µM of each dNTP [5]. | Use balanced equimolar concentrations. Degraded dNTPs are a common cause of PCR failure. |
| MgCl₂ | Essential cofactor for DNA polymerase activity [5]. | 1.5-2.5 mM (often included in buffer) [5] [6]. | Critical optimization parameter; titrate in 0.5-1 mM increments between 1-8 mM [6]. |
| Buffer | Provides optimal chemical environment (pH, salts) for the reaction [5]. | Usually supplied as 10X concentrate, used at 1X [5]. | May contain additives like (NH4)2SO4 or KCl to enhance specificity and yield. |
| Additives | Enhance amplification of difficult templates (e.g., high GC%) [5]. | DMSO: 1-10%; BSA: ~400 ng/µL [5]. | Use DMSO or formamide for GC-rich templates; BSA can alleviate inhibition from sample contaminants [5]. |
Experimental Protocol: Standard Endpoint PCR
This detailed protocol is adapted from methodologies used in recent research and commercial kits, providing a robust starting point for new researchers [1] [8] [6].
Table 2: PCR Master Mix Setup for a 50 µL Reaction
| Component | Stock Concentration | Final Concentration | Volume per 50 µL Reaction |
|---|---|---|---|
| PCR Buffer | 10X | 1X | 5 µL |
| dNTP Mix | 10 mM each | 200 µM each | 1 µL |
| Forward Primer | 10 µM | 0.4 µM | 2 µL |
| Reverse Primer | 10 µM | 0.4 µM | 2 µL |
| Template DNA | Variable (e.g., 50 ng/µL) | ~100 ng | 2 µL |
| MgCl₂ | 25 mM | 1.5-2.5 mM* | 3-5 µL |
| DNA Polymerase | 5 U/µL | 1.25-2.5 U | 0.5 µL |
| Nuclease-Free Water | - | - | To 50 µL |
*Concentration may require optimization; if already present in the buffer, do not add extra.
Procedure:
- Prepare Master Mix: In a sterile, nuclease-free tube, combine all components except the template DNA on ice. This minimizes pipetting errors and contamination. A common order is water → primers → template → PCR Mix enzymes [1].
- Aliquot and Add Template: Dispense the master mix into individual PCR tubes or a multi-well plate, then add the template DNA to each reaction.
- Thermal Cycling: Place the tubes in a thermal cycler and run the program with the parameters outlined in the following table.
Table 3: Standard Three-Step PCR Thermal Cycling Conditions
| Step | Temperature | Time | Cycles | Purpose |
|---|---|---|---|---|
| Initial Denaturation | 94-98°C | 2-5 minutes | 1 | Complete denaturation of complex DNA and enzyme activation [4] [6]. |
| Denaturation | 94-98°C | 20-60 seconds | 25-35 | Separate newly synthesized DNA strands for the next cycle. |
| Annealing | 45-72°C* | 20-60 seconds | 25-35 | Primer binding to the specific target sequence. |
| Extension | 68-72°C | 20-60 sec/kb | 25-35 | Synthesis of new DNA strands by the polymerase. |
| Final Extension | 68-72°C | 5-10 minutes | 1 | Complete synthesis of all PCR fragments and A-tailing for cloning [4]. |
| Hold | 4-10°C | ∞ | 1 | Short-term storage of samples. |
*Temperature is primer-specific and must be optimized. _*Time is dependent on amplicon length and polymerase speed.
- Post-PCR Analysis: Analyze 5-10 µL of the PCR product using agarose gel electrophoresis alongside an appropriate DNA size marker to confirm amplification success.
Mastering the four steps of standard PCR—denaturation, annealing, extension, and analysis—provides the foundation upon which all successful molecular biology experiments are built. For the new researcher, a meticulous approach to optimizing each component, from primer design and reagent concentrations to thermal cycling parameters, is not merely a procedural requirement but a critical investment in research efficacy. As PCR technologies continue to evolve with innovations like digital PCR, faster enzymes, and microfluidic platforms, this foundational knowledge will remain essential for adapting to new methods and driving scientific discovery in diagnostics, drug development, and basic research [9] [10].
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology, enabling the exponential amplification of specific DNA sequences from minimal starting material [11]. Its development revolutionized fields from medical diagnostics to biomedical research [12]. The power of PCR hinges on the precise interplay of core reaction components, each fulfilling a critical biochemical role. For researchers embarking on PCR optimization, a deep understanding of these components—template DNA, primers, deoxynucleoside triphosphates (dNTPs), and the reaction buffer—is not merely beneficial but essential for experimental success. This guide provides an in-depth technical examination of these core elements, framing their functions within the context of robust, reproducible PCR setup and optimization for drug development and scientific research.
Core Component 1: Template DNA
The template DNA is the target sequence that will be amplified. It can originate from various sources, including genomic DNA (gDNA), complementary DNA (cDNA), or plasmid DNA [13]. The composition and complexity of the DNA directly influence the optimal input amount for efficient amplification.
Quantity and Quality Considerations
The optimal amount of template DNA varies significantly based on its type and complexity. For instance, 0.1–1 ng of pure plasmid DNA is often sufficient, whereas 5–50 ng may be required for more complex genomic DNA in a standard 50 µL reaction [13]. Using too much DNA can increase the risk of nonspecific amplification, while too little can reduce the final product yield. The sensitivity of the DNA polymerase used also influences the required template input; enzymes engineered for higher sensitivity require less starting material [13]. In some protocols, especially those involving gDNA, the template input may be defined by copy number, which can be calculated using Avogadro's constant and the molar mass of the DNA [13]. In theory, PCR can amplify a target from a single DNA copy, but in practice, efficiency is highly dependent on reaction components and parameters [13].
Technical Guidelines for Use
Purification: For best results, template DNA should be of high purity. While unpurified PCR products can sometimes be re-amplified, carryover salts, dNTPs, and primers from the previous reaction can inhibit amplification [13]. Diluting the initial reaction in water or, preferably, purifying the amplicons beforehand is recommended.
Handling: The DNA template should be quantified accurately, typically using spectrophotometric or fluorometric methods. It is crucial to avoid introducing nucleases or contaminants that could degrade the template or inhibit the DNA polymerase.
Core Component 2: Primers
PCR primers are short, synthetic single-stranded DNA oligonucleotides, typically 15–30 bases in length, that are designed to be complementary to sequences flanking the target region [13] [14]. They are the key ingredient that defines the specific DNA sequence to be amplified, serving as the starting point for DNA synthesis by the polymerase [14].
Design Principles for Specificity and Efficiency
Careful primer design is paramount for successful PCR amplification. The following principles should be adhered to:
- Length and Melting Temperature (Tm): Primers should be 15-30 nucleotides long, with a calculated Tm between 55–70°C. The Tms of the forward and reverse primers should be within 5°C of each other to ensure both bind with similar efficiency during the annealing step [13] [15].
- GC Content: The GC content should ideally be between 40–60%, with a uniform distribution of G and C bases to prevent mispriming [13] [16].
- 3' End Specificity: The 3' end of the primer is critical for initiation. It should end in a G or C base (a "GC clamp") to promote stable binding due to stronger hydrogen bonding, but should not contain more than three consecutive G or C bases, as this can promote nonspecific priming [13] [15].
- Secondary Structures: Primers must be checked for self-complementarity (which can form hairpin loops) and complementarity to each other (which can form primer-dimers), particularly at their 3' ends [13] [15].
Table 1: Primer Design Guidelines Summary
| Parameter | Recommended Value | Rationale |
|---|---|---|
| Length | 15–30 nucleotides | Balances specificity and binding efficiency. |
| Melting Temperature (Tm) | 55–70°C (within 5°C for a pair) | Ensures simultaneous primer annealing. |
| GC Content | 40–60% | Provides optimal stability; too high increases nonspecific binding. |
| 3' End | One G or C; avoid >3 consecutive G/C | Promotes specific anchoring and extension; reduces mispriming. |
| Secondary Structures | Avoid self-complementarity and primer-dimer formation | Prevents amplification failure and spurious products. |
Optimization in Reaction Setup
In a standard PCR, primers are typically used at a final concentration of 0.1–1 µM [13]. Higher concentrations can lead to mispriming and nonspecific amplification, while lower concentrations may result in little or no amplification of the desired target. For challenging applications like long PCR or when using degenerate primers, concentrations of 0.3–1 µM are often favorable [13].
Core Component 3: Deoxynucleoside Triphosphates (dNTPs)
Deoxynucleoside triphosphates (dNTPs) are the essential building blocks from which DNA polymerase synthesizes a new DNA strand. The four dNTPs—dATP, dCTP, dGTP, and dTTP—provide the adenine, cytosine, guanine, and thymine nucleotides required for DNA replication [16] [17].
Biochemical Function and Reaction Energetics
DNA polymerase catalyzes the formation of a phosphodiester bond between the 3'-hydroxyl group of the last nucleotide in the growing DNA chain and the 5'-phosphate group of the incoming dNTP [17]. The hydrolysis of the dNTP's triphosphate group into pyrophosphate releases energy, which drives the polymerization reaction forward [17]. The four dNTPs are typically added to the PCR in equimolar amounts to ensure balanced and accurate base incorporation [13] [16].
Concentration and Purity in Optimization
The recommended final concentration for each dNTP in a standard PCR is typically 200 µM [15]. However, concentrations can be adjusted within a 20–200 µM range depending on the application [18]. Lower dNTP concentrations (e.g., 20–40 µM) can increase specificity and, when used with non-proofreading polymerases, improve fidelity [13] [18]. Conversely, higher concentrations may be needed for long PCR fragments but can be inhibitory if they exceed optimal levels [13] [18]. The purity of dNTPs is critical; degraded or impure dNTPs can introduce mutations and reduce amplification efficiency [16].
Table 2: dNTP Concentration Guidelines
| Application | Typical Final Concentration (per dNTP) | Notes |
|---|---|---|
| Standard PCR | 150–200 µM | Standard starting point for most applications. |
| High-Fidelity PCR | 20–50 µM | Reduces misincorporation by non-proofreading enzymes. |
| Long-range PCR | Up to 200 µM | Ensures sufficient building blocks for long fragments. |
| General Range | 20–200 µM | Adjustments may be needed based on other components like Mg²⁺. |
Specialized dNTPs for Advanced Applications
Modified dNTPs expand the utility of PCR in research and diagnostics. For example, dTTP can be partially or fully replaced with deoxyuridine triphosphate (dUTP). Subsequent treatment with Uracil DNA Glycosylase (UDG) prior to PCR degrades any carryover amplicons from previous reactions, preventing false positives [13]. Other modified dNTPs (e.g., fluorescently labeled or biotinylated) are used for labeling amplicons for downstream applications like sequencing or detection, though the DNA polymerase must be compatible with these modifications [13] [17].
Core Component 4: Reaction Buffer
The PCR buffer provides a stable chemical environment that optimizes the activity and stability of the DNA polymerase. It typically contains a pH buffer, salts, and magnesium ions [18].
Key Constituents and Their Roles
- Magnesium Ions (Mg²⁺): This is a critical cofactor for all DNA polymerases. Mg²⁺ facilitates the binding of the polymerase to the primer-template complex and catalyzes the incorporation of dNTPs during phosphodiester bond formation [13] [18]. The concentration of Mg²⁺ (usually supplied as MgCl₂) is one of the most important variables to optimize, typically ranging from 0.5–5.0 mM [18] [15]. Excessive Mg²⁺ reduces specificity and can promote non-specific amplification, while insufficient Mg²⁺ renders the polymerase inactive [18].
- pH Buffer: Tris-HCl is commonly used to maintain a stable pH, usually around 8.3, which is optimal for Taq polymerase activity [18].
- Potassium Salt (KCl): Potassium chloride, typically at 35–100 mM, helps promote primer annealing by stabilizing the primer-template duplex [18] [15].
Additives and Enhancers
Various additives can be incorporated into the buffer to overcome amplification challenges:
- Dimethyl Sulfoxide (DMSO): Added at 1–10% (though often <5%), DMSO disrupts base pairing, which helps reduce secondary structures in GC-rich templates and effectively lowers the Tm [18] [15]. High concentrations can inhibit Taq polymerase.
- Betaine: Used at 0.5–2.5 M, betaine can enhance the amplification of GC-rich templates and reduce the DNA Tm's dependence on dNTP concentration [18] [15].
- Bovine Serum Albumin (BSA): BSA (up to 0.8 mg/mL) can bind to inhibitors present in the DNA sample, such as polyphenols or salts, thereby neutralizing their effects [18].
Integrated PCR Workflow and Component Interaction
The PCR process is a cyclic sequence of temperature changes designed to exploit the functions of each core component. The following diagram illustrates the workflow and highlights where each key component acts.
The Scientist's Toolkit: Essential Research Reagents
Successful PCR optimization requires not only the core components but also a suite of reliable reagents and tools. The following table details essential materials for setting up and analyzing PCR experiments.
Table 3: Essential Research Reagents for PCR
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| Thermostable DNA Polymerase | Enzyme that synthesizes new DNA strands. | Choice depends on application (e.g., standard, high-fidelity, long-range). Taq is common; Pfu offers proofreading [11] [12]. |
| dNTP Mix | Pre-mixed equimolar solution of dATP, dCTP, dGTP, dTTP. | Sourced as purified, high-quality solutions to ensure fidelity and efficiency. Verify concentration and avoid freeze-thaw cycles [19] [17]. |
| Oligonucleotide Primers | Custom-designed sequences defining the amplification target. | Must be designed with specificity in mind. Require proper resuspension and storage to prevent degradation [14] [15]. |
| Nuclease-Free Water | Diluent for the reaction mixture. | Essential for preventing degradation of reaction components by environmental nucleases. |
| Thermal Cycler | Instrument that automates temperature cycling. | Must be calibrated for accurate and reproducible temperature control across all wells. |
| Agarose & Electrophoresis Equipment | For post-PCR analysis to separate and visualize amplicons by size. | Requires DNA ladder (size standard) for product verification [12]. |
| PCR Additives (e.g., DMSO, BSA) | Enhancers to overcome challenges like GC-richness or sample inhibitors. | Concentration must be optimized, as high levels can be inhibitory [18] [15]. |
Mastering the core components of PCR—template DNA, primers, dNTPs, and buffer—is a fundamental requirement for any researcher seeking to harness the full power of this technique. The journey from a failed reaction to a specific, high-yield amplification often lies in the systematic optimization of these elements. By applying the detailed principles and guidelines outlined in this whitepaper, from precise primer design and dNTP balancing to strategic buffer formulation, scientists can transform PCR from a black-box procedure into a reliably controlled and powerfully adaptable tool. This deep understanding is the bedrock upon which robust, reproducible, and innovative research in drug development and molecular biology is built.
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology, enabling the specific amplification of target DNA sequences from minimal starting material [2]. At the heart of every PCR reaction lies the DNA polymerase, an enzyme responsible for synthesizing new DNA strands by adding nucleotides to a growing DNA chain during the extension phase of thermal cycling [20]. Since the introduction of Taq DNA polymerase from Thermus aquaticus in the 1980s, significant advancements have been made in developing specialized DNA polymerases with enhanced properties tailored for specific applications [20]. These developments have transformed PCR from a relatively simple amplification method to a sophisticated tool capable of meeting the rigorous demands of modern research, clinical diagnostics, and therapeutic development.
The selection of an appropriate DNA polymerase represents one of the most critical factors in PCR optimization, directly impacting amplification success, yield, specificity, and sequence accuracy [20]. Researchers now face a choice among conventional polymerases like Taq, high-fidelity enzymes with proofreading capabilities, and specialized hot-start formulations designed to prevent nonspecific amplification. Each polymerase category offers distinct advantages and limitations that must be carefully considered in the context of experimental goals, template characteristics, and downstream applications. This technical guide provides an in-depth comparison of these polymerase classes, equipping researchers with the knowledge needed to make informed decisions for their specific PCR applications within the broader context of PCR optimization.
Key Characteristics of DNA Polymerases
Thermostability
Thermostability refers to a DNA polymerase's ability to withstand the high temperatures required for DNA denaturation during PCR cycling without significant loss of activity. Taq DNA polymerase, derived from the thermophilic bacterium Thermus aquaticus, has a half-life of approximately 40 minutes at 95°C, making it suitable for standard PCR applications [20] [13]. However, polymerases from hyperthermophilic archaea such as Pyrococcus furiosus (Pfu polymerase) exhibit dramatically enhanced thermostability—approximately 20 times more stable than Taq at 95°C [20]. This property is particularly valuable for protocols requiring prolonged high-temperature incubations or for amplifying templates with extensive secondary structure that may require longer denaturation times.
Processivity
Processivity defines the number of nucleotides a DNA polymerase can incorporate per single binding event with the template DNA [20]. Highly processive enzymes remain bound to the DNA template for extended periods, incorporating more nucleotides before dissociating. Taq polymerase has moderate processivity, incorporating approximately 60 bases per second at 70°C [13]. Early proofreading polymerases like Pfu historically exhibited lower processivity due to competition between polymerization and exonuclease activities, but modern engineered polymerases have addressed this limitation through the incorporation of DNA-binding domains that enhance template affinity without compromising enzymatic activity [20]. Enhanced processivity is particularly beneficial for amplifying long templates (>5 kb), GC-rich sequences that form stable secondary structures, and when PCR inhibitors are present in the sample [20].
Fidelity and Proofreading Activity
Fidelity refers to the accuracy with which a DNA polymerase replicates the template sequence, minimizing misincorporation of incorrect nucleotides [21]. This characteristic is quantified as the error rate, typically expressed as the number of errors per base per duplication event [22]. Fidelity is primarily determined by two mechanisms: selective nucleotide incorporation at the polymerase active site and 3'→5' exonuclease (proofreading) activity that removes misincorporated nucleotides [20] [21].
The geometric constraints of the polymerase active site promote selective incorporation of correct nucleotides through optimal alignment of catalytic groups. When an incorrect nucleotide is incorporated, the resulting suboptimal architecture causes a synthetic delay, increasing the opportunity for the incorrect nucleotide to dissociate before chain elongation continues [21]. Proofreading enzymes contain a separate exonuclease domain that detects and excises misincorporated nucleotides from the 3' end of the growing DNA strand before further extension occurs [21]. Polymerases with robust proofreading capabilities can achieve error rates up to 280 times lower than Taq polymerase [21].
Specificity and Hot-Start Activation
Specificity refers to a polymerase's ability to amplify only the intended target sequence without producing nonspecific byproducts such as primer-dimers or misprimed amplification artifacts [20]. Conventional DNA polymerases exhibit residual activity at room temperature, allowing them to extend primers that have bound to non-target sequences with partial complementarity during reaction setup [23]. These nonspecific products are then amplified throughout subsequent PCR cycles, reducing target yield and compromising downstream applications.
Hot-start technology addresses this limitation by inhibiting polymerase activity during reaction setup until elevated temperatures are reached in the thermal cycler [23]. This inhibition is achieved through various mechanisms including antibody-mediated blocking, chemical modification, Affibody molecules, or aptamers that bind to the enzyme's active site at lower temperatures [23] [24]. When the reaction mixture reaches the initial denaturation temperature (typically >90°C), the inhibitory molecule is denatured or released, restoring full polymerase activity under conditions where primer binding is highly specific [20]. This approach significantly reduces nonspecific amplification and increases target yield, particularly for complex templates or low-copy-number targets [23].
Table 1: DNA Polymerase Characteristics and Their Impact on PCR Performance
| Characteristic | Definition | Impact on PCR | Representative Enzymes |
|---|---|---|---|
| Thermostability | Ability to withstand high temperatures without denaturation | Determines suitability for high-temperature protocols and template denaturation requirements | Pfu (high), Taq (moderate) |
| Processivity | Number of nucleotides incorporated per binding event | Affects amplification efficiency for long templates, GC-rich regions, and in presence of inhibitors | Engineered polymerases (high), Taq (moderate), Pfu (lower) |
| Fidelity | Accuracy of DNA sequence replication | Critical for cloning, sequencing, and mutagenesis; reduces downstream sequencing burden | Q5 (very high), Pfu/Phusion (high), Taq (standard) |
| Specificity | Ability to amplify only intended targets | Reduces nonspecific products and primer-dimers; improves yield and downstream application success | Hot-start formulations (high), Standard polymerases (variable) |
Comparative Analysis of DNA Polymerase Types
Taq DNA Polymerase
Taq DNA polymerase, isolated from Thermus aquaticus, represents the foundational enzyme that enabled the automation of PCR [2]. This 94 kDa enzyme exhibits robust DNA-synthesizing capability with a typical incorporation rate of 60 nucleotides per second at 70°C [13]. Its moderate thermostability (half-life of approximately 40 minutes at 95°C) makes it suitable for standard PCR applications with denaturation temperatures of 94-95°C [13]. A distinctive feature of Taq polymerase is its terminal transferase activity, which adds a single deoxyadenosine (A) to the 3' ends of PCR products. This property is exploited in TA cloning strategies, facilitating direct ligation of PCR products into vectors with complementary 3'-T overhangs.
The primary limitation of Taq polymerase is its relatively low fidelity, with error rates typically ranging from 1.0-20.0×10⁻⁵ errors per base per duplication [22] [21]. This accuracy limitation stems from the absence of 3'→5' proofreading exonuclease activity, leaving the enzyme unable to correct misincorporated nucleotides. Consequently, Taq polymerase is generally unsuitable for applications requiring high sequence accuracy, such as cloning without subsequent sequencing verification, site-directed mutagenesis, or long-amplicon generation where errors accumulate across the sequence.
Table 2: Taq DNA Polymerase Characteristics and Applications
| Property | Specification | Optimal Reaction Conditions | Primary Applications |
|---|---|---|---|
| Origin | Thermus aquaticus | 1.5-2.0 mM Mg²⁺, pH 8.3-9.0 | Routine PCR, genotyping, allele-specific PCR |
| Error Rate | 1.0-20.0×10⁻⁵ errors/bp/duplication [22] | 200 µM each dNTP | Diagnostic applications not requiring sequence perfection |
| Thermal Stability | Half-life ~40 min at 95°C [13] | Denaturation: 94-95°C | Standard-length amplifications (≤5 kb) |
| Processivity | ~60 nt/second at 70°C [13] | Extension: 68-72°C | TA cloning strategies |
| Special Features | 5'→3' polymerase activity, A-tailing | Annealing: 5°C below primer Tm | Educational demonstrations, routine amplification |
High-Fidelity DNA Polymerases
High-fidelity DNA polymerases represent a significant advancement over Taq through the incorporation of 3'→5' exonuclease (proofreading) activity that dramatically reduces error rates during DNA synthesis [20]. These enzymes typically fall into two categories: naturally occurring polymerases from hyperthermophilic archaea and engineered enzymes optimized through directed evolution. The proofreading mechanism involves enzymatic removal of misincorporated nucleotides from the 3' end of the growing DNA strand before continuing synthesis, improving accuracy by 10-300-fold compared to Taq polymerase [21].
Naturally occurring proofreading enzymes include Pfu (from Pyrococcus furiosus), Pwo (from Pyrococcus woesii), and KOD (from Thermococcus kodakarensis). These polymerases typically achieve error rates in the range of 1.0×10⁻⁶ to 1.2×10⁻⁵ errors per base per duplication [22] [21]. Engineered high-fidelity enzymes such as Phusion, Q5, and Platinum SuperFi II represent further refinements, combining proofreading activity with enhancements to processivity, thermostability, and amplification efficiency. These engineered enzymes can achieve error rates as low as 5.3×10⁻⁷ errors per base per duplication, approaching 300-fold greater accuracy than Taq polymerase [21].
A notable consideration when working with archaeal proofreading polymerases is their inability to amplify uracil-containing templates due to the presence of a uracil-binding pocket that functions as part of a DNA repair mechanism [20]. This property prevents their use in applications requiring dUTP incorporation for carryover prevention or bisulfite-treated DNA analysis. Additionally, some proofreading enzymes exhibit slower synthesis rates compared to Taq polymerase, potentially requiring longer extension times, particularly for long amplicons.
Table 3: High-Fidelity DNA Polymerase Error Rates and Fidelity
| Enzyme | Error Rate (errors/bp/duplication) | Relative Fidelity (Compared to Taq) | Proofreading Activity |
|---|---|---|---|
| Taq | 1.5×10⁻⁴ [21] | 1X | No |
| KOD | 1.2×10⁻⁵ [21] | 12X | Yes |
| Pfu | 5.1×10⁻⁶ [21] | 30X | Yes |
| Deep Vent | 4.0×10⁻⁶ [21] | 44X | Yes |
| Phusion | 3.9×10⁻⁶ [21] | 39X | Yes |
| Q5 | 5.3×10⁻⁷ [21] | 280X | Yes |
Hot-Start DNA Polymerases
Hot-start DNA polymerases employ inhibition strategies to prevent enzymatic activity during reaction setup until elevated temperatures are reached in the thermal cycler [23]. This technology addresses a fundamental limitation of conventional PCR: the extension of misprimed sequences and primer-dimers that occur when reactions are assembled at room temperature [24]. By blocking polymerase activity until the first denaturation step, hot-start methods ensure that DNA synthesis initiates only under conditions of high stringency, dramatically improving amplification specificity and yield [23].
Multiple hot-start technologies have been developed, each with distinct mechanisms and performance characteristics. Antibody-based inhibition utilizes monoclonal antibodies that bind to the polymerase's active site, with dissociation occurring during the initial denaturation step (typically 2-3 minutes at 95°C) [23] [24]. This approach offers rapid activation and restoration of full enzymatic activity but introduces exogenous protein into the reaction. Chemical modification methods employ covalently attached inhibitory groups that block enzyme activity, with gradual reactivation occurring over multiple thermal cycles [23]. While offering stringent inhibition and animal-component-free formulation, chemical activation may require longer initialization times (up to 10-15 minutes) and is less suitable for long amplicons (>3 kb) [24].
Alternative approaches include Affibody molecules (engineered binding proteins) and aptamers (inhibitory oligonucleotides) that block polymerase activity until thermal denaturation [23]. These methods offer shorter activation times and animal-component-free formulations but may provide less stringent inhibition compared to antibody-based methods. Recent innovations include heat-activatable primers containing thermolabile phosphotriester modifications at 3'-terminal positions, which block primer extension until converted to natural phosphodiester linkages at elevated temperatures [25].
Diagram 1: Hot-Start PCR activation mechanism and inhibition methods. Polymerase activity is blocked during reaction setup until heat activation enables specific amplification.
Experimental Protocols for Fidelity Assessment
Direct Sequencing Method
The direct sequencing approach provides the most comprehensive assessment of polymerase fidelity by identifying all types of errors (substitutions, insertions, and deletions) across the entire amplified sequence [22]. This method involves amplifying a target sequence, cloning the products, and sequencing individual clones to identify mutations introduced during amplification.
Protocol:
- Template Preparation: Select a well-characterized template (e.g., plasmid containing a target gene of approximately 1-2 kb). Use 25 pg of plasmid DNA per reaction to ensure sufficient amplification cycles for error detection [22].
- PCR Amplification: Perform amplification using standardized conditions: 30 cycles with denaturation at 95°C for 15-30 seconds, annealing at appropriate temperature for 15-30 seconds, and extension at enzyme-specific optimal temperature (1-2 minutes per kb) [22] [26].
- Product Cloning: Purify PCR products using agarose gel extraction or PCR purification kits. Clone into sequencing vector using restriction digestion/ligation or recombination-based cloning systems such as Gateway technology [22].
- Sequencing and Analysis: Sequence 50-100 individual clones using Sanger sequencing. Align sequences to the reference template and identify mutations. Calculate error rate using the formula: Error rate = total mutations / (total bp sequenced × number of doublings) [22].
The number of template doublings can be calculated from the fold-amplification using the equation: Doublings = log₂(fold-amplification) [22]. This method provides a direct measurement of all error types but requires significant sequencing effort to achieve statistical significance for high-fidelity enzymes, particularly those with error rates below 1×10⁻⁶ [21].
LacZ-Based Screening Assay
The LacZ fidelity assay provides a higher-throughput alternative for initial fidelity assessment through phenotypic screening of mutations in a reporter gene [21]. This method amplifies the lacZα gene, which produces the α-peptide of β-galactosidase, and clones the products into an appropriate vector.
Protocol:
- Template Amplification: Amplify the lacZα gene (approximately 500 bp) using the polymerase under test conditions. Standardize cycle number (typically 16-25 cycles) and template input across comparisons [21].
- Ligation and Transformation: Ligate PCR products into lacZ-deficient vector and transform into an appropriate Escherichia coli host strain.
- Phenotypic Screening: Plate transformed bacteria on media containing X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) and IPTG (isopropyl β-D-1-thiogalactopyranoside). Colonies containing error-free inserts produce functional β-galactosidase and develop blue color, while those with mutations in critical regions remain white [21].
- Calculation: Determine the mutation frequency as the ratio of white colonies to total colonies. Sequence white colonies to characterize the specific mutations introduced.
While higher-throughput than direct sequencing, this method only detects mutations within the critical 349-base region of the lacZ gene that affect β-galactosidase function, potentially underestimating total error rates [21]. Additionally, not all sequence alterations necessarily disrupt enzyme function, creating potential false negatives.
Next-Generation Sequencing Approaches
Next-generation sequencing (NGS) platforms enable comprehensive fidelity assessment by sequencing millions of PCR products simultaneously, providing statistically robust error rate measurements even for high-fidelity enzymes [21]. Single-molecule real-time (SMRT) sequencing by PacBio is particularly valuable as it sequences individual PCR molecules without an intermediate amplification step that could introduce additional errors.
Protocol:
- Library Preparation: Amplify target sequence (typically 1-2 kb) using standardized conditions. Purify products and prepare sequencing library without bacterial cloning.
- Sequencing: Perform SMRT sequencing to generate circular consensus sequences with high accuracy (background error rate of approximately 9.6×10⁻⁸ errors/base) [21].
- Bioinformatic Analysis: Align sequences to reference template using appropriate algorithms. Identify substitutions, insertions, and deletions. Filter potential sequencing errors using quality scores and consensus requirements.
- Error Rate Calculation: Calculate error rates using the formula: Error rate = total confirmed errors / (total bases sequenced × number of doublings). Include standard deviations from multiple replicates.
This approach provides the most statistically rigorous fidelity measurements for high-fidelity enzymes but requires specialized instrumentation and bioinformatic expertise [21].
Selection Guidelines for Specific Applications
Application-Based Polymerase Selection
Different experimental applications impose distinct requirements on DNA polymerase characteristics, necessitating careful selection to achieve optimal results. The following guidelines outline polymerase recommendations for common research scenarios:
Cloning and Expression Studies: Applications involving gene cloning, protein expression, or functional analysis require maximum sequence accuracy to ensure encoded proteins maintain proper amino acid sequences and biological activity. High-fidelity polymerases with proofreading capability such as Q5, Phusion, or Pfu are strongly recommended [20] [21]. These enzymes provide error rates 10-280 times lower than Taq polymerase, minimizing the need for extensive clone sequencing to identify error-free constructs [21]. For TA cloning specifically, non-proofreading enzymes like Taq may be necessary to generate the required 3'A overhangs, but should be followed by thorough sequence verification.
Diagnostic PCR and Genotyping: Applications focused on target detection, such as pathogen identification, genetic screening, or genotyping, prioritize specificity and robustness over ultimate sequence accuracy. Hot-start Taq formulations (antibody-mediated or chemically modified) provide excellent specificity while maintaining cost-effectiveness for high-throughput applications [23] [2]. The hot-start mechanism prevents false positives from mispriming while offering rapid activation for shorter protocol times.
Long-Range PCR: Amplification of targets >5 kb requires polymerases with high processivity and strong strand displacement activity. Engineered polymerases such as PrimeSTAR GXL or specialty long-range mixes combine proofreading activity with enhanced processivity [20]. These formulations often include accessory proteins that improve polymerase binding and progression through complex template regions.
Quantitative PCR (qPCR): qPCR applications require robust amplification efficiency and minimal primer-dimer formation to ensure accurate quantification. Hot-start polymerase formulations, particularly antibody-mediated systems, provide excellent specificity and consistent CT values across replicates [23] [2]. For high-resolution melting curve analysis, polymerases producing uniform amplicons without spurious products are essential.
High-Throughput and Automated Applications: Robotic liquid handling systems benefit from polymerases with room temperature stability during setup. Modern hot-start formulations, particularly antibody-based and chemical modification methods, maintain inhibition during extended room temperature incubation, enabling reliable high-throughput processing without specialized chilled equipment [23].
Table 4: Polymerase Selection Guide for Specific Applications
| Application | Recommended Polymerase Type | Key Considerations | Alternative Options |
|---|---|---|---|
| Cloning & Mutagenesis | High-fidelity proofreading (Q5, Phusion, Pfu) | Lowest error rate critical for sequence integrity | Standard Taq with extensive sequencing |
| Diagnostic PCR | Hot-start Taq (antibody or chemical) | Specificity, cost-effectiveness, rapid results | Standard Taq with optimized conditions |
| Long Amplicons (>5 kb) | Engineered high-processivity enzymes | Enhanced processivity, strand displacement capability | Polymerase mixtures with processivity factors |
| Quantitative PCR | Hot-start (antibody-mediated) | Minimal primer-dimer, consistent efficiency | Chemically modified hot-start |
| Multiplex PCR | Stringent hot-start (antibody or chemical) | Reduced mispriming with multiple primer pairs | Standard hot-start with optimized Mg²⁺ |
| TA Cloning | Standard Taq | A-tailing activity required | Proofreading enzymes with A-tailing protocol |
Template-Specific Considerations
Template characteristics significantly influence polymerase selection and performance. The following template-specific guidelines ensure optimal amplification across diverse scenarios:
High-GC Content Templates: GC-rich sequences (>65% GC) form stable secondary structures that impede polymerase progression. Polymerases with high processivity and enhanced strand displacement capability are essential [20]. Buffer additives such as DMSO, betaine, or GC enhancers can improve melting of secondary structures. Engineered polymerases with DNA-binding domains often outperform natural enzymes for these challenging templates.
Low-Copy-Number Targets: Amplification of rare targets requires maximal specificity to prevent amplification of nonspecific products that can overwhelm the desired signal. Stringent hot-start formulations (antibody-based or chemical modification) provide the strongest inhibition during reaction setup [23] [24]. Additionally, polymerases with high affinity for template (low Km) improve efficiency with limited starting material.
Complex Templates: Templates with extensive secondary structure, hairpins, or repetitive elements benefit from polymerases with both high processivity and proofreading activity [20]. The combination of efficient strand displacement and high fidelity ensures complete and accurate amplification of challenging regions. Elevated denaturation temperatures possible with hyperthermostable enzymes like Pfu can improve melting of stable structures.
Uracil-Containing Templates: Applications involving dUTP incorporation (for carryover prevention) or bisulfite-treated DNA (for methylation analysis) require polymerases capable of amplifying uracil-containing templates [20]. Most archaeal proofreading enzymes cannot amplify these templates due to uracil-binding pockets, making engineered versions or Taq-based systems necessary.
The Scientist's Toolkit: Essential Research Reagents
Table 5: Essential Reagents for PCR Optimization and Fidelity Assessment
| Reagent/Category | Function/Purpose | Application Notes |
|---|---|---|
| High-Fidelity Polymerases | Accurate DNA synthesis for cloning and sequencing | Q5, Phusion, Pfu for low-error amplification; require dNTP optimization [21] |
| Hot-Start Polymerases | Specificity enhancement through temperature activation | Antibody-based: rapid activation; Chemical: stringent inhibition [23] |
| dNTP Mixtures | Nucleotide substrates for DNA synthesis | 200 µM each dNTP standard; lower concentrations (50-100 µM) may enhance fidelity [26] |
| MgCl₂ Solutions | Cofactor for polymerase activity | 1.5-2.0 mM optimal for Taq; concentration affects specificity and yield [26] [13] |
| Fidelity Assessment Systems | Error rate measurement | lacZ assay for screening; Sanger sequencing for validation; NGS for comprehensive analysis [21] |
| Buffer Additives | Enhancement of specific amplification | DMSO, betaine, formamide for GC-rich templates; BSA for inhibitor resistance [20] |
| Cloning Kits | Downstream application of PCR products | TA cloning for Taq products; blunt-end cloning for proofreading enzymes [22] |
The selection of an appropriate DNA polymerase represents a critical decision point in PCR experimental design, with significant implications for amplification success, data quality, and downstream application performance. Taq DNA polymerase remains suitable for routine applications where ultimate sequence accuracy is not paramount, while high-fidelity enzymes with proofreading capability are essential for cloning, protein expression, and any application requiring precise DNA replication. Hot-start formulations, available through multiple inhibition technologies, provide enhanced specificity across all polymerase classes by preventing nonspecific amplification during reaction setup.
The continuing evolution of DNA polymerase technology, including engineered enzymes with combined advantages of high fidelity, processivity, and specificity, continues to expand PCR capabilities. By understanding the fundamental characteristics of each polymerase class and their performance in specific experimental contexts, researchers can make informed selections that optimize results while conserving resources. As PCR maintains its position as a cornerstone technique in molecular biology, appropriate polymerase selection remains fundamental to experimental success across diverse research domains.
Polymersse chain reaction (PCR) is a foundational technique in molecular biology, and its success critically depends on the design of the oligonucleotide primers used to initiate DNA synthesis. Effective primers must specifically bind to the target DNA sequence with high efficiency while avoiding structures that compromise amplification. This guide details the core principles of PCR primer design, providing researchers with a structured framework to create robust assays for applications ranging from basic gene amplification to diagnostic test development.
Core Parameters for Effective Primer Design
The performance of a PCR assay is governed by several key physicochemical properties of the primers. Adhering to the following quantitative ranges ensures optimal binding, specificity, and yield.
Table 1: Core Parameter Guidelines for PCR Primer Design
| Parameter | Recommended Range | Rationale & Impact |
|---|---|---|
| Primer Length | 18–30 nucleotides (bp) [27] [28]; 18–24 bp is often optimal [29] | Balances specificity (longer) with efficient binding and annealing (shorter) [27] [28]. |
| Melting Temperature (Tm) | 55–65°C [30]; 60–64°C is ideal [31]; Primer pairs should be within 2–5°C of each other [27] [31] [30] | Ensures both primers anneal to the template simultaneously under a single reaction temperature [27] [31]. |
| GC Content | 40–60% [27] [28] [5] | Provides stable primer-template binding without promoting non-specific interactions [27] [32]. |
| GC Clamp | Presence of G or C bases at the 3' end | Strengthens local binding via stronger hydrogen bonding, providing a stable start point for polymerase [27] [30] [32]. |
Designing to Avoid Secondary Structures and Mispriming
A well-designed primer must not only bind to its intended target but also avoid unintended interactions with itself, its partner primer, or off-target sequences.
Avoid Secondary Structures: Primers should be screened for hairpins (intra-primer homology), which occur when a region of three or more bases is complementary to another region within the same primer [29]. The stability of these structures is measured by Gibbs free energy (ΔG); any self-dimers or hairpins should have a ΔG weaker (more positive) than –9.0 kcal/mol [31].
Prevent Primer-Dimer Formation: Self-dimers (two same-sense primers annealing) and cross-dimers (forward and reverse primers annealing to each other) consume reagents and reduce yield [29]. This is often caused by inter-primer homology, particularly at the 3' ends [27].
Eliminate Repeated Sequences: Avoid runs of the same nucleotide (e.g., ACCCC) or dinucleotide repeats (e.g., ATATAT), as these can cause mispriming [27] [29].
Ensure Specificity: Always verify primer specificity by performing a sequence similarity search (e.g., NCBI BLAST) against the genome of your organism to ensure they are unique to the intended target [31] [29].
The Primer Design and Validation Workflow
A systematic approach to primer design, from in silico planning to bench-side validation, significantly increases the chance of a successful PCR experiment. The following diagram and protocol outline this process.
Experimental Protocol: Primer Validation and Annealing Temperature Optimization
After designing and obtaining primers, empirical validation is crucial. This protocol uses a gradient PCR to determine the optimal annealing temperature (Ta) [29].
- Primer Resuspension and Dilution: Resuspend the lyophilized primers in sterile TE buffer or nuclease-free water to create a high-concentration stock (e.g., 100 µM). Prepare a working dilution (e.g., 10 µM) for use in PCR reactions [28].
- PCR Master Mix Setup: Assemble reactions using a standard master mix. A typical 50 µL reaction may contain [5]:
- Gradient PCR Cycling:
- Initial Denaturation: 94–98°C for 1–5 minutes [5].
- Amplification Cycles (25–35 cycles):
- Denaturation: 94–98°C for 10–60 seconds.
- Annealing: Set a temperature gradient across the thermal cycler block, typically from 5–10°C below the calculated average Tm of the primers to a few degrees above it [29]. Maintain for 30 seconds.
- Extension: 70–80°C (depending on the polymerase) for 1 minute per 1000 bp [5].
- Final Extension: 70–80°C for 5 minutes [5].
- Hold: 4°C ∞.
- Analysis: Separate the PCR products by agarose gel electrophoresis. The optimal annealing temperature is the one that produces a single, intense band of the expected size with minimal to no non-specific products or primer-dimer [29].
The Scientist's Toolkit: Essential Reagents for PCR
A successful PCR experiment relies on a suite of carefully selected reagents, each serving a critical function in the reaction.
Table 2: Essential Research Reagents for PCR
| Reagent / Solution | Function in the Reaction |
|---|---|
| Thermostable DNA Polymerase (e.g., Taq, Pfu) | Enzyme that synthesizes new DNA strands by adding nucleotides to the 3' end of the primers [2] [5]. |
| dNTP Mix (dATP, dTTP, dCTP, dGTP) | The essential building blocks (nucleotides) for the synthesis of new DNA strands [5]. |
| Oligonucleotide Primers (Forward & Reverse) | Short, single-stranded DNA sequences that define the start and end of the target amplicon by binding complementarily to the template [2] [29]. |
| PCR Reaction Buffer (with Mg²⁺) | Provides the optimal chemical environment (pH, salts) for polymerase activity. Mg²⁺ is a critical cofactor for the enzyme [5]. |
| Template DNA | The sample DNA containing the target sequence to be amplified [5]. |
| Nuclease-Free Water | Solvent used to bring the reaction to its final volume, ensuring no enzymatic degradation of reagents [5]. |
Advanced Considerations and Troubleshooting
- GC-Rich Targets: For templates with high GC content (>60%), additives like DMSO (1-10%) or formamide (1.25-10%) can be included in the reaction mix to help disrupt stable secondary structures and improve amplification [5].
- qPCR Probes: When designing hydrolysis (TaqMan) probes for quantitative PCR, the probe should have a Tm that is 5–10°C higher than the primers, be 20–30 bases long, and avoid a guanine (G) base at the 5' end [31].
- Cloning and Modification: For cloning applications, primers can include 5' extensions containing restriction enzyme sites. Typically, 3-4 extra nucleotides are added 5' of the restriction site to allow for efficient enzyme cutting [27].
Within the framework of polymerase chain reaction (PCR) optimization, the precise control of reaction components is paramount for achieving specific and efficient DNA amplification. Among these components, divalent cations, particularly magnesium ions (Mg²⁺), stand out as a critical cofactor that is indispensable for enzymatic activity and reaction fidelity. For new researchers, understanding the role of Mg²⁺ extends beyond recognizing it as a simple buffer ingredient; it is a fundamental regulator of PCR thermodynamics and kinetics. Magnesium ions directly influence DNA polymerase function, primer-template binding stability, and the overall specificity of the amplification process [33] [34]. Consequently, the optimization of Mg²⁺ concentration is not an optional step but a necessary one for successful PCR, especially when dealing with challenging templates such as genomic DNA or GC-rich sequences [35]. This guide provides an in-depth examination of the role of magnesium in PCR, offering detailed methodologies for its optimization to enhance the reproducibility and reliability of experimental results.
Biochemical Mechanisms of Magnesium in PCR
Magnesium ions (Mg²⁺) serve as an essential cofactor for thermostable DNA polymerases, with their role rooted in well-defined biochemical mechanisms. The primary function of Mg²⁺ is to facilitate the catalytic activity of the DNA polymerase enzyme. It does this by forming a coordinate bond between the enzyme's active site and the phosphate groups of the incoming deoxynucleoside triphosphates (dNTPs) [34]. Specifically, the Mg²+ ion binds to the alpha phosphate group of a dNTP, enabling the nucleophilic attack by the 3'-hydroxyl group of the primer terminus and the subsequent release of pyrophosphate. This interaction is crucial for the formation of the phosphodiester bond that elongates the DNA chain [13]. Without adequate free Mg²⁺, DNA polymerases remain enzymatically inactive, leading to PCR failure [33] [36].
A secondary, but equally critical, function of Mg²⁺ is to stabilize the interaction between the primer and the template DNA. The phosphate backbone of DNA is negatively charged, creating electrostatic repulsion between two complementary nucleic acid strands. Mg²⁺ ions, being divalent cations, effectively shield these negative charges, reducing repulsion and facilitating the annealing of primers to their target sequences [34] [37]. This charge stabilization increases the melting temperature (Tm) of the DNA duplex. Quantitative analyses have demonstrated a logarithmic relationship between MgCl₂ concentration and DNA melting temperature, with every 0.5 mM increase in MgCl₂ within the 1.5–3.0 mM range raising the Tm by approximately 1.2°C [35]. This dual role in both enzyme catalysis and nucleic acid stabilization makes Mg²⁺ a central player in the PCR process.
Table 1: Core Biochemical Functions of Magnesium Ions in PCR
| Function | Molecular Mechanism | Effect on PCR |
|---|---|---|
| Enzyme Cofactor | Binds to dNTPs at the polymerase active site, catalyzing phosphodiester bond formation [34] [13]. | Essential for DNA polymerase activity; without it, DNA elongation cannot proceed. |
| Charge Shielding | Neutralizes the negative charge on the DNA phosphate backbone, reducing electrostatic repulsion [34] [37]. | Stabilizes the primer-template duplex, facilitating proper annealing and increasing melting temperature. |
| Fidelity Regulation | Influences the accuracy of nucleotide incorporation by the DNA polymerase [33] [37]. | Optimal concentrations maximize fidelity; excess Mg²⁺ can reduce enzyme specificity and increase error rates. |
Quantitative Effects and Optimal Concentration Ranges
The concentration of Mg²⁺ is a critical variable that requires precise optimization, as both deficiency and excess can be detrimental to PCR success. A comprehensive meta-analysis of PCR studies established an optimal MgCl₂ concentration range of 1.5 to 3.0 mM for efficient PCR performance [35]. Within this range, a consistent logarithmic relationship with DNA melting temperature is observed, which is fundamental for calculating accurate annealing temperatures [35]. For many standard applications with Taq DNA polymerase, a concentration of 1.5-2.0 mM is often optimal [38].
The requirement for Mg²⁺ is not absolute but is dynamically influenced by the concentrations of other reaction components that can chelate or bind the ion. dNTPs, primers, and the DNA template itself all compete for the available free Mg²⁺ [33] [36]. Notably, dNTPs are strong chelators; therefore, an increase in dNTP concentration must be balanced with an increase in Mg²⁺ concentration to ensure an adequate pool of free ions remains available for the DNA polymerase [13]. Furthermore, the complexity and type of DNA template influence the optimal Mg²⁺ level. Genomic DNA, with its high complexity, often requires higher Mg²⁺ concentrations compared to more straightforward templates like plasmid DNA [35]. The presence of chelating agents from sample preparation, such as EDTA or citrate, can also sequester Mg²⁺, necessitating higher starting concentrations to compensate [33] [34].
Table 2: Factors Influencing Optimal Magnesium Concentration in PCR
| Factor | Influence on Mg²⁺ Requirement | Practical Consideration |
|---|---|---|
| dNTP Concentration | dNTPs chelate Mg²⁺. Higher [dNTP] requires higher [Mg²⁺] to maintain free ion levels [33] [13]. | Standard dNTP concentration is 200 µM each. Adjust Mg²⁺ if dNTP concentration is altered. |
| Template Type & Complexity | Complex templates (e.g., genomic DNA) require more Mg²⁺ than simple ones (e.g., plasmid DNA) [35]. | Start with 2.0 mM for genomic DNA and 1.5 mM for plasmid DNA, then optimize. |
| Presence of Chelators | Agents like EDTA (from DNA storage buffers) bind Mg²⁺, making it unavailable [33] [37]. | Ensure Mg²⁺ is in excess of EDTA concentration. Use minimal EDTA in template storage buffers. |
| Primer Sequence & Tm | Higher Mg²⁺ stabilizes duplexes, effectively lowering the optimal annealing temperature [35]. | If increasing annealing temperature is not an option, slightly reducing Mg²⁺ can increase stringency. |
Deviations from the optimal Mg²⁺ window have clear and predictable consequences. Insufficient Mg²⁺ results in low enzyme activity, yielding little to no PCR product due to inefficient primer extension and poor polymerase function [34] [38] [37]. Conversely, excess Mg²⁺ reduces the fidelity of the DNA polymerase and decreases reaction specificity. This can lead to non-specific amplification, as seen by multiple bands or smears on an agarose gel, because the stabilized primer-template complexes allow primers to bind to incorrect, partially complementary sites on the template DNA [33] [37] [36].
Experimental Optimization Protocols
Magnesium Titration Methodology
A systematic approach to Mg²⁺ optimization is crucial for developing robust PCR assays, particularly for novel targets or challenging templates. The following protocol provides a detailed methodology for determining the optimal MgCl₂ concentration.
- Reaction Setup: Prepare a master mix containing all PCR components except MgCl₂ and the DNA template. Aliquot this master mix into a series of thin-walled PCR tubes. A typical 50 µL reaction will include 1X PCR buffer (without Mg²⁺), 200 µM of each dNTP, 0.1–0.5 µM of each primer, 0.5–2.5 units of DNA polymerase, and a defined amount of template DNA (e.g., 10–100 ng genomic DNA) [38] [15]. The final volume of the master mix should account for the variable MgCl₂ and water to be added.
- MgCl₂ Titration: Supplement each reaction tube with MgCl₂ from a stock solution (e.g., 25 mM) to create a concentration gradient. A recommended starting range is 1.0 mM to 4.0 mM in increments of 0.5 mM [38] [37]. For example, to achieve a final concentration of 1.5 mM in a 50 µL reaction, add 3 µL of 25 mM MgCl₂ stock. Include a negative control (no template DNA) for each Mg²⁺ level to detect contamination or primer-dimer formation.
- Thermal Cycling and Analysis: Run the reactions under standard cycling conditions appropriate for the primer set and amplicon size. Analyze the resulting PCR products using agarose gel electrophoresis. The optimal Mg²⁺ concentration is identified as the one that produces a single, intense band of the expected amplicon size with a clean background and minimal to no non-specific products or primer dimers [15] [39].
Integrated Optimization with Annealing Temperature
Mg²⁺ concentration and annealing temperature (Ta) are interdependent parameters. A higher Mg²⁺ concentration stabilizes the primer-template duplex, which is analogous to lowering the effective Ta. Therefore, optimization should consider both variables simultaneously. The most efficient method is to perform a gradient PCR combined with Mg²⁺ titration [37]. This involves setting up the Mg²⁺ titration series as described and then running the thermal cycler with an annealing temperature gradient across the block. This two-dimensional approach allows for the identification of the best combination of [Mg²⁺] and Ta that yields specific amplification with high yield. Computational models have been developed that achieve excellent predictive capabilities (R² = 0.9942 for MgCl₂) by integrating Tm, GC content, amplicon length, and dNTP concentration, providing a theoretical starting point for empirical optimization [40].
Diagram 1: PCR Magnesium and Temperature Optimization Workflow.
The Scientist's Toolkit: Research Reagent Solutions
A successful PCR experiment relies on high-quality reagents. The following table details essential materials and their specific functions, with a focus on magnesium-related components.
Table 3: Essential Reagents for PCR Optimization with Magnesium
| Reagent | Function/Description | Optimization Consideration |
|---|---|---|
| Thermostable DNA Polymerase (e.g., Taq) | Enzyme that synthesizes new DNA strands. Requires Mg²⁺ as a cofactor for activity [33] [38]. | Use 0.5–2.5 units/50 µL reaction. Higher amounts may tolerate inhibitors but can increase non-specific bands. |
| MgCl₂ Stock Solution (e.g., 25 mM) | Source of divalent magnesium cations (Mg²⁺) for the reaction. | The most common variable for optimization. Supplied separately for fine-tuning [33] [36]. |
| PCR Buffer (Mg²⁺-free) | Provides optimal pH and ionic strength (e.g., Tris-HCl, KCl). | Using a Mg²⁺-free buffer allows for precise, independent control over Mg²⁺ concentration [33]. |
| dNTP Mix | Building blocks (dATP, dCTP, dGTP, dTTP) for new DNA synthesis. | Standard final concentration is 200 µM of each dNTP. They chelate Mg²⁺, so [Mg²⁺] must be in excess [38] [13]. |
| Template DNA | The DNA sample containing the target sequence to be amplified. | Quality is critical. Common amounts: 10–100 ng genomic DNA, 1–10 ng plasmid DNA. Excess template can increase background [38] [13]. |
| Oligonucleotide Primers | Short, single-stranded DNA sequences that define the start and end of the amplicon. | Typical final concentration is 0.1–0.5 µM each. Higher concentrations can promote mispriming [38] [13]. |
Troubleshooting Common Magnesium-Related PCR Issues
Even with a standardized protocol, researchers may encounter suboptimal results. The table below outlines common PCR problems linked to Mg²⁺, their potential causes, and corrective actions.
Table 4: Troubleshooting Guide for Magnesium-Related PCR Issues
| Observed Problem | Potential Cause | Recommended Solution |
|---|---|---|
| No PCR Product | Mg²⁺ concentration is too low; DNA polymerase is inactive [38] [37]. | Increase MgCl₂ concentration in 0.5 mM steps, ensuring it exceeds total [dNTP]. Check for EDTA in template sample. |
| Multiple Bands or Smear | Mg²⁺ concentration is too high, leading to non-specific priming and reduced fidelity [33] [37]. | Decrease MgCl₂ concentration in 0.5 mM steps. Simultaneously, increase the annealing temperature. |
| Primer-Dimer Formation | Excess Mg²⁺ and low annealing temperature facilitate primer-to-primer annealing [34] [39]. | Optimize Mg²⁺ concentration and increase annealing temperature. Ensure 3' ends of primers are not complementary. |
| Low Yield | Suboptimal Mg²⁺ concentration, either too high or too low, reducing efficiency [34]. | Titrate MgCl₂ to find the concentration that maximizes product yield without introducing non-specific products. |
| GC-Rich Template Failure | Stable secondary structures prevent primer binding or polymerase elongation. | Increase Mg²⁺ (e.g., up to 4 mM) to enhance duplex stability. Consider adding 2–10% DMSO or 1–2 M betaine as enhancers [37] [36]. |
Diagram 2: Effect of Magnesium Concentration on PCR Outcome.
Magnesium is far more than a passive component in a PCR buffer; it is a critical cofactor that sits at the crossroads of enzymatic catalysis, nucleic acid thermodynamics, and reaction specificity. Its concentration directly dictates the success or failure of amplification. For researchers embarking on PCR optimization, a methodical titration of MgCl₂ represents one of the most impactful and accessible steps toward achieving robust and reproducible results. By understanding its biochemical roles, recognizing the factors that influence its availability, and applying systematic optimization and troubleshooting protocols, scientists can effectively harness the power of this essential divalent cation to advance their molecular biology research.
Advanced PCR Methods and Their Specific Applications
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology, enabling the exponential amplification of specific DNA sequences. However, conventional PCR is often plagued by issues of non-specific amplification and primer-dimer formation, which drastically reduce yield, sensitivity, and reliability [23]. These artifacts typically occur during reaction setup at room temperature, where DNA polymerase retains partial enzymatic activity and can extend primers that are misprimed or bound to each other [41].
Hot-Start PCR represents a critical advancement in PCR optimization by employing specialized mechanisms to inhibit DNA polymerase activity at lower temperatures, preventing unwanted amplification events before thermal cycling begins [23]. This technical guide explores the principles, methodologies, and applications of Hot-Start PCR, providing researchers with comprehensive protocols to enhance assay specificity and efficiency, particularly for demanding applications in diagnostics, genetic testing, and drug development.
The Problem: Non-Specific Amplification in Conventional PCR
In conventional PCR, the reaction mixture is assembled at room temperature, creating conditions where non-specific amplification can occur through two primary mechanisms:
- Mispriming: DNA polymerase can extend primers that bind to template sequences with low homology at lower temperatures [23]. These misprimed sequences compete with the target amplicon for reaction components, significantly reducing the efficiency of specific amplification [25].
- Primer-Dimer Formation: Primers can bind to each other through complementary sequences, creating short artificial templates that DNA polymerase efficiently amplifies [23]. Primer-dimers consume reaction reagents and can dominate the reaction, especially when target template is limited [25].
Impact on Experimental Results
The consequences of non-specific amplification are particularly pronounced in applications requiring high sensitivity:
- Reduced Target Yield: Non-specific products compete with the desired amplicon for enzymes, nucleotides, and primers [23]
- Compromised Sensitivity: Detection of low-copy number targets becomes unreliable amid background amplification [25]
- Downstream Application Failure: Non-specific products can interfere with cloning, sequencing, and other molecular biology applications [23]
Hot-Start PCR: Fundamental Principles and Mechanisms
Core Concept
Hot-Start PCR employs biochemical modifications to prevent DNA polymerase extension at room temperature, with activation occurring only after an initial high-temperature incubation step [23]. This simple yet powerful principle ensures that the polymerase becomes active only when the reaction temperature provides sufficient stringency for specific primer-template hybridization [41].
Molecular Mechanisms of Popular Hot-Start Technologies
Several biochemical approaches have been developed to implement the Hot-Start principle, each with distinct advantages and considerations:
Table 1: Comparison of Major Hot-Start Technologies
| Technology | Mechanism of Action | Activation Requirements | Advantages | Limitations |
|---|---|---|---|---|
| Antibody-Based | Anti-Taq antibody binds polymerase active site [23] | Initial denaturation (95°C, 2-10 min) [42] | Rapid activation; full enzyme activity restored [23] | Animal-origin components; exogenous proteins in reaction [23] |
| Chemical Modification | Covalent linkage of inhibitory chemical groups [23] | Extended pre-incubation (95°C, 10-15 min) [23] | High stringency; animal-component free [23] | Longer activation time; may affect long amplicons [23] |
| Aptamer-Based | Oligonucleotide aptamers bind and inhibit polymerase [41] | Elevated temperature (>70°C) [43] | Short activation time; animal-component free [23] | Potential reversibility; lower stringency [23] |
| Affibody-Based | Engineered protein domains block active site [23] | Initial denaturation [23] | Low protein content; animal-component free [23] | Potential lower stringency [23] |
| Primer-Based | Thermolabile groups (OXP) block 3' extension [25] | Temperature-dependent deprotection [25] | Targeted inhibition; no polymerase modification needed [25] | Requires specialized primer synthesis [25] |
| Physical Separation | Wax barriers or separate compartments [41] | Wax melting (~70°C) [41] | Simple principle; no specialized reagents [41] | Manual intensive; potential contamination risk [41] |
Diagram: Hot-Start PCR workflow showing inhibition at room temperature and specific activation during initial denaturation
Experimental Protocols and Methodologies
Standard Hot-Start PCR Protocol
The following protocol is adapted from manufacturer recommendations for antibody-based Hot-Start polymerases [42]:
Reaction Setup (20-50 μL volume):
- Master Mix Preparation:
- 1X PCR Buffer (typically supplied with enzyme)
- 200 μM each dNTP (dATP, dCTP, dGTP, dTTP)
- 1.5-2.5 mM MgCl₂ (concentration may require optimization)
- 0.1-1 μM each forward and reverse primer
- 1.25 units Hot-Start DNA Polymerase
- 10-100 ng template DNA
- Sterile water to final volume
- Thermal Cycling Conditions:
- Initial Denaturation/Activation: 95°C for 2-10 minutes (duration depends on Hot-Start method)
- Amplification Cycles (25-35 cycles):
- Denaturation: 95°C for 30 seconds
- Annealing: 50-65°C for 30 seconds (temperature based on primer Tm)
- Extension: 72°C for 30-60 seconds per kb
- Final Extension: 72°C for 5-10 minutes
- Hold: 4°C indefinitely
Novel Hot-Start Implementation Using E. coli-Expressed Taq
Recent advancements have simplified Hot-Start PCR implementation. A 2023 study demonstrated that E. coli-expressing Taq DNA polymerase (EcoliTaq) can be used directly in PCR without purification [44]. The cellular membrane naturally separates the polymerase from other reaction components, creating an intrinsic Hot-Start effect until initial denaturation disrupts the membranes [44].
Optimized Buffer Composition for Direct PCR:
- 2% Tween 20 (enhances amplification efficiency)
- 0.4 M trehalose (counteracts PCR inhibitors in complex samples)
- High-pH buffer (improves performance with blood samples)
This approach provides a cost-effective alternative to commercial Hot-Start systems while maintaining comparable specificity and yield [44].
Research Reagent Solutions
Table 2: Essential Reagents for Hot-Start PCR Implementation
| Reagent Category | Specific Examples | Function in Hot-Start PCR | Optimization Tips |
|---|---|---|---|
| Hot-Start DNA Polymerases | AmpliTaq Gold (chemically modified) [23], Platinum Taq (antibody-based) [23], KOD Hot Start (antibody-based) [43], AptaTaq (aptamer-based) [43] | Catalyzes DNA synthesis; inhibited at room temperature | Select based on activation time, stringency requirements, and downstream applications [23] |
| Specialized Primers | OXP-modified primers [25], Hairpin primers [41] | 3' modifications block premature extension | Thermolabile groups (e.g., OXP) automatically convert to native form during heating [25] |
| Reaction Buffers | Mg²⁺-supplemented buffers [5], Additive-enhanced buffers [44] | Provides optimal ionic environment | Mg²⁺ concentration (0.5-5.0 mM) critically affects specificity and yield [5] |
| PCR Additives | DMSO (1-10%) [5], Formamide (1.25-10%) [5], BSA (10-100 μg/mL) [5], Tween 20 (1-2%) [44] | Enhances specificity, reduces secondary structure, counteracts inhibitors | DMSO and formamide help with GC-rich templates; BSA counters inhibitors in blood samples [5] [44] |
| Modified dNTPs | Hot-Start dNTPs with protecting groups [41] | Prevents incorporation until activation | Can be used with standard polymerases to implement Hot-Start [41] |
Applications and Benefits in Research and Diagnostics
Key Applications
Hot-Start PCR provides particular advantages in the following scenarios:
- Low-Copy Number Target Detection: Essential when template DNA is limited (<10⁴ copies) as it prevents primer-dimer formation that would otherwise dominate the reaction [42]
- Complex Template Amplification: Critical for mammalian genomic DNA and other complex templates where mispriming sites are abundant [42]
- Multiplex PCR Applications: Enables simultaneous amplification of multiple targets by preventing cross-primer interactions [43]
- High-Throughput Settings: Allows reaction setup at room temperature on automated liquid handling platforms without compromising specificity [23]
- Diagnostic Applications: Provides the reliability required for clinical diagnostics, forensics, and genetic testing where false results have significant consequences [25]
Quantifiable Benefits
The implementation of Hot-Start technology delivers measurable improvements in PCR performance:
- Increased Specificity: Reduction or elimination of non-specific bands and primer-dimers in gel electrophoresis [23]
- Enhanced Sensitivity: Up to 1000-fold improvement in detection limits for low-abundance targets [25]
- Improved Yield: Higher quantities of the desired amplicon due to reduced competition from non-specific products [23]
- Greater Reliability: More consistent results across replicates and between experiments [45]
Hot-Start PCR represents a fundamental optimization strategy that addresses the core limitation of conventional PCR: non-specific amplification during reaction setup. By employing antibody-based, chemical, or novel primer-directed inhibition mechanisms, researchers can significantly enhance assay specificity, sensitivity, and reliability. The continued evolution of Hot-Start technologies, including recent developments in direct PCR from complex samples and cost-effective enzyme production methods, ensures that this approach will remain essential for advancing molecular diagnostics, genetic research, and drug development. Implementation of the protocols and principles outlined in this guide will enable researchers to overcome common PCR challenges and generate more robust, reproducible results across diverse applications.
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology, yet a common challenge that compromises its efficacy is non-specific amplification. This occurs primarily through mispriming, where primers bind to off-target sequences with partial complementarity, leading to the amplification of unwanted products and background noise on gels. [46] For new researchers, optimizing PCR conditions, especially the annealing temperature, is a critical but often time-consuming step. The optimal annealing temperature is traditionally based on the calculated melting temperature (Tm) of the primers. However, the true Tm can be influenced by buffer components, template quality, and primer concentration, making any calculation an approximation. [46] Consequently, using a single, static annealing temperature can often yield suboptimal results, either from non-specific products at temperatures that are too low or poor yields at temperatures that are too high. [46] Touchdown (TD) PCR was developed as a powerful and efficient cycling strategy to overcome this optimization hurdle, enhancing both the specificity and sensitivity of PCR amplification without the need for extensive preliminary experiments or primer redesign. [47] [48]
The Core Principle of Touchdown PCR
The Basic Mechanism
Touchdown PCR is a thermal cycling modification designed to favor the amplification of the desired specific target over non-specific ones. The fundamental principle involves starting with an annealing temperature that is 5–10°C above the calculated Tm of the primers. [46] [49] This high, stringent temperature minimizes primer binding, permitting only the most perfectly matched primer-template duplexes to form. In subsequent cycles, the annealing temperature is systematically decreased by 1–2°C per cycle until it reaches, or "touches down," at a temperature that is a few degrees below the primer's optimal Tm. [46] [48] [49]
This step-down approach confers a significant competitive advantage to the desired amplicon. In the initial high-stringency cycles, only the specific target with perfect complementarity is efficiently amplified, generating a small amount of correct product. As the temperature becomes more permissive, this specific product, now present in the reaction, outcompetes non-specific targets for primer binding and polymerase resources. [46] [50] Any small difference in binding efficiency between the correct and incorrect targets produces an exponential advantage, as PCR amplification proceeds, ensuring the specific product dominates the final reaction mixture. [47]
Standard PCR vs. Touchdown PCR: A Comparative View
The table below summarizes the key differences between standard PCR and touchdown PCR.
Table 1: Comparison of Standard PCR and Touchdown PCR
| Feature | Standard PCR | Touchdown PCR |
|---|---|---|
| Annealing Temperature | Single, fixed temperature based on primer Tm. [48] | A decreasing gradient, starting high and stepping down to the target Tm. [46] [48] |
| Primary Advantage | Simplicity of protocol. | Increased specificity and sensitivity; reduces need for extensive optimization. [47] [48] |
| Specificity Control | Relies entirely on the accuracy of a single, pre-determined annealing temperature. | Empirically selects for the best-matched primers through stringency at the start of the reaction. [46] |
| Handling of Complex Templates | Can struggle with complex templates (e.g., genomic DNA), GC-rich sequences, or low-homology templates. [48] | Particularly useful for difficult templates, including GC-rich sequences, and when primer-template identity is not perfect. [48] |
| Optimization Workflow | Requires testing multiple fixed temperatures to find the optimal one, which can be time- and resource-intensive. [46] | The cycling program itself performs the optimization within a single run, saving time and reagents. [47] |
Visualizing the Touchdown PCR Workflow
The following diagram illustrates the logical workflow and the competitive advantage established during the touchdown PCR process.
Detailed Touchdown PCR Protocol and Experimental Methodology
A Standardized Two-Phase Protocol
A robust and widely cited protocol for touchdown PCR involves two distinct phases. [46] The following table outlines a detailed cycling program based on a primer pair with a calculated Tm of 57°C.
Table 2: Example Touchdown PCR Protocol Based on a Primer Tm of 57°C
| Step | Temperature (°C) | Time | Stage & Cycles | Purpose |
|---|---|---|---|---|
| 1. Initial Denaturation | 95 | 3:00 min | 1 cycle | Fully denature the complex template and activate hot-start polymerase. |
| 2. Denaturation | 95 | 0:30 min | Stage 1: Touchdown10-15 cycles | Separate DNA strands before each annealing step. |
| 3. Annealing | 67 (Tm +10°C) | 0:45 min | High-stringency annealing: Only perfect matches form. | |
| 4. Extension | 72 | 0:45 min | Polymerase extends the primed template. | |
| 5. Denaturation | 95 | 0:30 min | Stage 2: Amplification15-20 cycles | Separate DNA strands. |
| 6. Annealing | (Decreases by 1°C/cycle) | 0:45 min | Step-down annealing: Temperature decreases from 66°C to 57°C over Stage 1. | |
| 7. Extension | 72 | 0:45 min | Polymerase extends the primed template. | |
| 8. Final Extension | 72 | 5:00 min | 1 cycle | Ensure all amplicons are fully extended. |
Phase 1: Touchdown (Cycles 1-10) The annealing temperature starts at 67°C (Tm +10°C) and decreases by 1°C per cycle. Over these 10 cycles, the temperature "touches down" from 67°C to 58°C. This phase is critical for establishing specificity. [46]
Phase 2: Amplification (Cycles 11-30) The PCR continues for an additional 15-20 cycles using the final, permissive annealing temperature reached at the end of Phase 1 (e.g., 57-58°C). By this stage, the specific product is the dominant template and is efficiently amplified to high yield. [46]
Advanced Optimization and Troubleshooting
Even with a standardized protocol, some experiments may require further optimization. Here are key strategies from expert sources:
- Keep Reactions Cool: Until thermal cycling begins, keep all reaction components on ice to prevent non-specific priming at room temperature. [46]
- Employ Hot-Start PCR: Use a hot-start DNA polymerase, which remains inactive until the initial denaturation step. This prevents primer-dimer formation and mispriming during reaction setup. [46] [50]
- Consider PCR Additives: For persistently difficult templates (e.g., GC-rich sequences), combine touchdown PCR with additives like DMSO (2.5-5%), which helps denature complex secondary structures. [46] [51]
- Limit Total Cycle Number: To prevent the late-stage appearance of non-specific bands, keep the total number of amplification cycles (touchdown + amplification) below 35. [46]
- Adjust the Touchdown Gradient: If non-specific products persist or yield is low, adjust the touchdown phase to 2–3 cycles per 1°C drop in temperature, providing more amplification time at each stringent temperature. [46]
Applications and Advanced Variations
Key Applications in Modern Research
Touchdown PCR is not just a troubleshooting tool but a standard method in several advanced applications due to its inherent robustness.
- Amplification of Difficult Templates: It is particularly valuable for templates with high GC content, extensive secondary structure, or those that are present in low copy numbers. [48] [51]
- Reverse Transcription PCR (RT-PCR) and cDNA Library Generation: The enhanced specificity is crucial when working with complex RNA populations. [47]
- Single Nucleotide Polymorphism (SNP) Screening: The high stringency helps distinguish between alleles that differ by a single base pair. [47]
- Evolutionary PCR and Multigene Family Amplification: When primer sequences may not perfectly match the target (e.g., when amplifying homologous genes from different species), touchdown PCR can selectively amplify the intended target amidst closely related sequences. [48]
- Overlap Extension PCR: Touchdown cycling parameters have been successfully integrated into overlap extension PCR protocols to improve the efficiency and specificity of gene fusion and mutagenesis experiments without the need for separate optimization. [52]
Multiple Touchdown PCR (MT-PCR)
A significant advancement is Multiple Touchdown PCR (MT-PCR), which combines the principles of multiplex PCR and touchdown PCR. In MT-PCR, several primer sets for different DNA targets are used in a single reaction, while the annealing temperature is decreased from a high set point (e.g., 66°C) by 0.5–1°C per cycle. [53] This technique is powerful for applications like the simultaneous detection of multiple antibiotic resistance genes in clinical samples, as it prevents cross-amplification of non-target DNA and reduces false-positive results, which is a common challenge in standard multiplex PCR. [53]
The Scientist's Toolkit: Essential Reagents for Touchdown PCR
Successful implementation of touchdown PCR relies on high-quality reagents. The following table details key components and their optimized functions.
Table 3: Essential Research Reagent Solutions for Touchdown PCR
| Reagent / Solution | Function and Importance in Touchdown PCR |
|---|---|
| High-Fidelity or Standard DNA Polymerase | Catalyzes DNA synthesis. High-fidelity enzymes (e.g., Q5) offer superior accuracy for cloning. [52] |
| Hot-Start DNA Polymerase | Critical for specificity. It remains inactive until the initial denaturation step, preventing non-specific amplification and primer-dimer formation during reaction setup. [46] [50] |
| Optimized PCR Buffer | Provides the optimal chemical environment (pH, salts) for polymerase activity. Magnesium ion (Mg²⁺) concentration is a critical cofactor that may require optimization. [51] |
| dNTP Mix | The building blocks (dATP, dCTP, dGTP, dTTP) for new DNA strand synthesis. A balanced, high-quality mix is essential for efficient amplification. |
| Primers (Oligonucleotides) | Specifically designed to flank the target sequence. Their quality, concentration, and design (avoiding self-complementarity) are paramount for success. [54] |
| Template DNA | The DNA containing the target to be amplified. Integrity and concentration are crucial; for genomic DNA, 30-100 ng is typically sufficient. [51] |
| PCR Additives (e.g., DMSO) | Co-solvents that can help denature difficult templates, especially GC-rich sequences. Typical working concentration is 2.5-5%. [46] [51] |
Touchdown PCR represents a sophisticated yet accessible approach to PCR optimization. By employing a step-down annealing strategy, it actively selects for the desired amplicon in the early phases of amplification, leading to superior specificity and yield. For new researchers, mastering touchdown PCR is a valuable skill that can save considerable time and resources in the lab. Its utility extends from basic molecular biology to advanced clinical diagnostics, making it an indispensable technique in the modern scientist's arsenal. As with any technique, success hinges on careful primer design, high-quality reagents, and a willingness to engage in iterative troubleshooting to adapt the core protocol to one's specific experimental needs.
Multiplex PCR has revolutionized molecular diagnostics by enabling the simultaneous amplification of multiple target sequences in a single reaction tube using more than one pair of primers [55]. This powerful variant of conventional PCR offers substantial savings in time, labor, and reagents while conserving sometimes limited sample material [55]. In diagnostic laboratories, the technique has proven particularly valuable for identifying viruses, bacteria, fungi, and parasites that cause similar clinical symptoms, allowing for comprehensive syndromic testing [55] [56].
However, the development of robust multiplex PCR assays presents significant technical challenges. The presence of multiple primer pairs in a single reaction dramatically increases the probability of spurious amplification products, primarily through the formation of primer dimers [55]. Furthermore, preferential amplification of certain targets can occur due to phenomena known as PCR drift and PCR selection, leading to biased results [55]. Successful multiplex PCR therefore requires meticulous optimization, with primer design representing the most critical factor determining assay performance [55] [57]. This guide provides a comprehensive framework for designing and optimizing multiplex PCR primer sets to achieve sensitive and specific multi-target detection.
Core Principles and Challenges in Multiplex PCR Design
Key Obstacles to Successful Multiplexing
The fundamental challenge in multiplex PCR stems from the quadratic growth in potential primer dimer interactions as the number of primers increases. For an N-plex PCR primer set comprising 2N primers, there are (2N choose 2) possible primer dimer interactions [57]. In a 96-plex assay (192 primers), this translates to approximately 18,000 potential primer pair interactions that must be considered [57]. These nonspecific interactions consume reaction components and can be amplified more efficiently than the desired targets, compromising assay sensitivity and specificity [55].
Additionally, preferential amplification presents a major obstacle, where certain templates amplify more efficiently than others in the same reaction [55]. This bias can arise from stochastic fluctuations in early amplification cycles (PCR drift) or inherent template properties such as GC content, secondary structures, or gene copy number (PCR selection) [55]. The choice of primers significantly influences this bias, with some primer pairs driving reactions to plateau phase regardless of starting concentration, while others produce product in concentration-dependent manners [55].
Foundational Primer Design Parameters
Effective multiplex primer design begins with adherence to core principles that promote uniform amplification efficiency across all targets:
- Primer Length: 18-30 nucleotides or more to ensure specificity [55]
- GC Content: 35-60% to maintain appropriate melting temperatures [55] [5]
- Melting Temperature (Tm): All primers should have nearly identical optimum annealing temperatures, typically with Tm between 52-58°C and less than 5°C difference between forward and reverse primers [55] [5]
- 3' End Stability: Avoid complementary 3' ends between primers to prevent dimer formation [5]
- Specificity: Primers should not display significant homology to one another or to non-target sequences [55]
Primers designed with consistent thermodynamic properties, particularly standard free energy (ΔG°) of approximately -11.5 kcal/mol, demonstrate optimal tradeoffs between amplification efficiency and specificity [57].
A Systematic Workflow for Multiplex Primer Design
The following workflow outlines a methodical approach to designing highly multiplexed PCR primer sets that minimize dimer formation and ensure uniform target amplification.
Primer Candidate Generation
Begin by identifying "pivot" nucleotides in the target sequence that must be included in the amplicon, such as mutation hotspots or conserved regions [57]. Systematically generate proto-primers with 3' ends positioned just outside these critical regions, then truncate from the 3' end until achieving the target ΔG° between -10.5 and -12.5 kcal/mol [57]. Apply additional filters to remove candidates with GC content outside the 25-75% range or those prone to secondary structure formation [57].
Computational Optimization with SADDLE Algorithm
For highly multiplexed assays, computational optimization becomes essential. The Simulated Annealing Design using Dimer Likelihood Estimation (SADDLE) algorithm provides a robust framework for designing primer sets with minimal dimer formation [57]. This stochastic approach efficiently navigates the vast optimization space through six key steps:
Figure 1: SADDLE Algorithm Workflow
The algorithm evaluates potential primer sets using a Loss function L(S) that quantifies the severity of primer dimer formation by summing the "Badness" of interaction between every pair of primers in the set [57]. Through iterative refinement, SADDLE can reduce primer dimer fractions from 90.7% in naively designed sets to 4.9% in optimized 96-plex assays [57].
Experimental Validation and Optimization
Computationally designed primer sets require experimental validation through a systematic optimization process:
- Initial Testing: Verify individual primer pairs in uniplex reactions before combining
- Hot Start Implementation: Use hot start PCR methodology to prevent primer dimer formation during reaction setup [55]
- Annealing Temperature Optimization: Perform gradient PCR to identify optimal annealing temperature
- Primer Ratio Adjustment: Titrate primer concentrations to balance amplification efficiency
- Additive Screening: Test PCR enhancers including DMSO (1-10%), glycerol, bovine serum albumin (~400ng/μL), or betaine to overcome secondary structures or GC-rich challenges [55] [5]
Critical Reaction Components and Optimization Parameters
Successful multiplex PCR requires careful optimization of all reaction components beyond primer design. The table below summarizes key parameters and their optimal ranges for multiplex applications.
Table 1: Multiplex PCR Reaction Components and Optimization Guidelines
| Component | Function | Optimal Concentration | Multiplex-Specific Considerations |
|---|---|---|---|
| Primers | Target sequence recognition | 0.1-1μM each [5] | May require concentration balancing between targets; higher concentrations increase dimer risk [55] |
| DNA Polymerase | Enzymatic amplification | 1.25-2.5U/50μL reaction [5] | Use hot-start variants; may require 4-5x increase over uniplex for high plexity [55] |
| MgCl₂ | Polymerase cofactor | 1.5-2.5mM [5] | Critical optimization parameter; higher concentrations may be needed [55] |
| dNTPs | Building blocks for DNA synthesis | 20-200μM each [5] | Balanced equimolar concentration essential |
| Template DNA | Amplification target | 10-100ng human genomic DNA [5] | Higher complexity templates may require optimization |
| Additives | Enhance specificity/yield | DMSO: 1-10% [5]; BSA: ~400ng/μL [55] | Particularly helpful for GC-rich targets or complex templates [55] |
Thermal Cycling Conditions
Optimized thermal cycling parameters are essential for successful multiplex amplification:
- Initial Denaturation: 94-98°C for 1-5 minutes [5]
- Denaturation: 94-98°C for 10-60 seconds [5]
- Annealing: 5°C below average primer Tm for 30-60 seconds [5]
- Extension: 68-72°C for 30-60 seconds per kilobase [5]
- Cycle Number: 35-45 cycles, increased for low-abundance targets [5]
- Final Extension: 68-72°C for 5-10 minutes [5]
Advanced Multiplexing Strategies and Technologies
Digital PCR Multiplexing
Partition-based digital PCR (dPCR) technologies offer inherent advantages for multiplex detection by physically separating amplification reactions, thereby minimizing competition between targets [58]. dPCR demonstrates superior sensitivity and precision for quantifying low-abundance targets in complex backgrounds, with nanoplate-based dPCR showing lower intra-assay variability (median CV%: 4.5%) compared to qPCR [58]. Color combination multiplexing in Crystal Digital PCR enables detection of up to 21 targets using 7-color channels by assigning two fluorophores per target [59].
Color Cycle Multiplex Amplification
Color Cycle Multiplex Amplification (CCMA) represents a recent innovation that significantly expands multiplexing capacity on standard qPCR instruments [56]. This approach uses fluorescence permutations rather than combinations, where each target produces a predefined sequence of fluorescence signals across multiple channels [56]. Using oligonucleotide blockers to programmably delay amplification, CCMA can theoretically discriminate up to 136 distinct targets using just 4 fluorescence channels [56].
Figure 2: CCMA Fluorescence Permutation Principle
Troubleshooting Common Multiplex PCR Issues
Even with careful design, multiplex assays may require troubleshooting to achieve optimal performance. The table below addresses common challenges and recommended solutions.
Table 2: Multiplex PCR Troubleshooting Guide
| Problem | Possible Causes | Solutions |
|---|---|---|
| Preferential Amplification | Varying primer efficiencies, template differences [55] | Redesign primers with matched ΔG°; optimize primer ratios; use PCR enhancers [55] |
| Primer Dimer Formation | Complementary 3' ends; suboptimal annealing [55] | Implement hot start PCR; redesign primers; optimize annealing temperature; use SADDLE algorithm [57] |
| Low Signal for Specific Targets | inefficient primers; secondary structures [55] | Redesign primers; increase cycle number; add DMSO, BSA, or betaine [55] [5] |
| Non-specific Amplification | Low annealing temperature; excessive enzyme [55] | Increase annealing temperature; reduce polymerase amount; optimize Mg²⁺ concentration [5] |
| Inconsistent Results | PCR inhibitors; reaction component variability [55] | Purify template DNA; prepare master mixes; include appropriate controls [55] |
Table 3: Essential Research Reagents for Multiplex PCR Development
| Reagent/Category | Specific Examples | Function in Multiplex PCR |
|---|---|---|
| DNA Polymerase | Hot-start Taq, Pfu polymerase (high-fidelity) [5] | Enzymatic DNA amplification with reduced pre-cycling activity |
| PCR Buffer Systems | Mg²⁺-containing buffers with additives | Maintain optimal pH and cofactor concentration |
| Nucleotide Mix | dNTP sets (dATP, dCTP, dGTP, dTTP) [5] | Provide building blocks for DNA synthesis |
| PCR Enhancers | DMSO, betaine, BSA, glycerol [55] [5] | Reduce secondary structures, improve efficiency |
| Nucleic Acid Stain | SYBR Green, EvaGreen | Detect amplification in real-time or endpoint analysis |
| Probe Systems | TaqMan probes, molecular beacons | Enable target-specific detection in multiplex qPCR |
| Clean-up Kits | SPRI beads, enzymatic clean-up | Remove primers, enzymes, and contaminants post-amplification |
The development of robust multiplex PCR assays demands meticulous attention to primer design, reaction optimization, and validation. By adhering to the systematic approach outlined in this guide—incorporating computational design tools like SADDLE, optimizing reaction components, and implementing appropriate controls—researchers can successfully develop multiplex assays that deliver sensitive, specific, and reproducible detection of multiple targets. As PCR technologies continue to evolve, methods such as dPCR and CCMA will further expand multiplexing capabilities, enabling increasingly comprehensive analysis of complex samples across diverse research and diagnostic applications.
The Polymerase Chain Reaction (PCR) is a foundational tool in molecular biology, but standard protocols often fail when faced with technically challenging templates. For new researchers, optimizing PCR is a critical skill that bridges theoretical knowledge and practical application in the laboratory. This guide addresses three common yet complex challenges: amplifying GC-rich regions, performing long-range PCR, and implementing fast PCR protocols. These techniques are essential for advanced applications in gene cloning, next-generation sequencing library preparation, and rapid diagnostics [2] [60]. Success in these areas requires a systematic understanding of reaction components, cycling conditions, and specialized reagents. The following sections provide detailed methodologies and optimization strategies to overcome these technical hurdles, framed within the context of reproducible, robust experimental design for research and drug development applications.
Conquering GC-Rich PCR
GC-rich sequences (typically >60% GC content) present formidable challenges due to their high thermodynamic stability and propensity to form secondary structures. The strong hydrogen bonding between guanine and cytosine bases results in higher melting temperatures, while stable hairpins, knots, and tetraplexes can hinder polymerase progression and primer annealing [61] [62]. These factors often lead to PCR failure, characterized by absent or truncated products, mispriming, and overall poor amplification efficiency. Overcoming these issues requires a multipronged optimization strategy addressing both reaction composition and thermal cycling parameters.
Optimization Strategies and Reagents
Table 1: Optimization Strategies for GC-Rich PCR
| Optimization Approach | Specific Implementation | Mechanism of Action |
|---|---|---|
| Organic Additives | DMSO (1-10%), betaine (1-2.5 M), formamide (1.25-10%), 7-deaza-dGTP | Disrupts secondary structures, lowers template melting temperature, reduces base stacking interactions [61] [62] |
| Specialized Polymerases | AccuPrime GC-Rich DNA Polymerase, polymerases from Pyrococcus furiosus (Pfu) | Enhanced processivity and stability at high temperatures; better ability to unwind complex structures [61] [62] |
| Magnesium Concentration | Gradient testing from 0.5-5.0 mM; typically 1.5-2.0 mM for Taq | Essential cofactor for polymerase; optimal concentration crucial for primer annealing and fidelity [5] [63] |
| Temperature Adjustments | Higher denaturation temperatures (up to 95°C), touchdown PCR | Promotes complete separation of DNA strands and prevents secondary structure formation [61] |
| Modified PCR Methods | Slow-down PCR | Incorporates dGTP analogs and uses modified cycling with lowered ramp rates and additional cycles [61] |
Experimental Protocol: Amplifying GC-Rich Nicotinic Acetylcholine Receptor Subunits
The following optimized protocol successfully amplified challenging GC-rich nAChR subunits from Ixodes ricinus (Ir-nAChRb1, 65% GC) and Apis mellifera (Ame-nAChRa1, 58% GC) [62].
1. Reagent Setup:
- Polymerase: Platinum SuperFi or Phusion High-Fidelity DNA Polymerase
- Buffer: Use the manufacturer's provided GC buffer or enhancer if available.
- Additives: Prepare stock solutions of 100% DMSO and 5M betaine.
- Primers: Design primers with a length of 15-30 nucleotides and a GC content of 40-60%. The melting temperatures (Tm) for forward and reverse primers should be closely matched (within 5°C) [5].
2. Reaction Assembly (50 μL total volume):
- Assemble the reaction mixture on ice as detailed in Table 2.
- Gently mix the components and centrifuge briefly to collect the reaction mixture at the bottom of the tube.
Table 2: Reaction Mixture for GC-Rich PCR
| Component | Final Concentration/Amount |
|---|---|
| 10X Polymerase Reaction Buffer | 1X |
| dNTP Mix | 200 μM each |
| MgSO₄ (or MgCl₂) | 1.5 - 2.0 mM |
| Forward Primer | 0.1 - 1.0 μM |
| Reverse Primer | 0.1 - 1.0 μM |
| DNA Template | 10 - 100 ng |
| DMSO | 3 - 5% |
| Betaine | 1 - 1.5 M |
| DNA Polymerase | 1.0 - 1.5 U |
| Nuclease-free Water | To 50 μL |
3. Thermal Cycling Conditions:
- Initial Denaturation: 98°C for 2 minutes (1 cycle)
- Amplification Cycles (35 cycles):
- Denaturation: 98°C for 15 seconds
- Annealing: 68°C for 20 seconds (Temperature can be optimized using a gradient)
- Extension: 72°C for 1 minute per kb of amplicon length
- Final Extension: 72°C for 5 - 10 minutes (1 cycle)
- Hold: 4°C indefinitely
4. Analysis:
- Analyze 5-10 μL of the PCR product by agarose gel electrophoresis to verify amplification success and product size.
Mastering Long-Range PCR
Long-range PCR aims to amplify DNA fragments significantly longer than those achievable with standard protocols, typically exceeding 5 kb and reaching up to 20-30 kb. This technique is invaluable for sequencing large genomic regions, analyzing haplotypes, and constructing complex cloning vectors [64] [65]. The primary challenges include maintaining polymerase processivity over extended templates, minimizing replication errors, and reducing the formation of chimeric reads—artifacts where a single amplicon appears to be derived from two different biological templates [64]. Success hinges on selecting the appropriate enzyme and meticulously optimizing reaction conditions.
Enzyme Selection and Performance
Not all long-range polymerases perform equally. A comparative study of six commercial enzymes evaluated their ability to amplify three amplicons (5.8 kb, 9.7 kb, and 12.9 kb) under identical conditions [65].
Table 3: Performance Comparison of Long-Range DNA Polymerases
| DNA Polymerase | 5.8 kb Amplicon | 9.7 kb Amplicon | 12.9 kb Amplicon | Key Characteristics |
|---|---|---|---|---|
| PrimeSTAR GXL (TaKaRa) | Success | Success | Success | Robust performance across various sizes and Tm values under identical conditions [65] |
| SequalPrep (Invitrogen) | Success | Success | Success | Reliable for multiple targets [65] |
| AccuPrime (Invitrogen) | Success | Failure | Success | Requires condition optimization for specific amplicons [65] |
| LA Taq Hot Start (TaKaRa) | Success | Failure | Success | Requires condition optimization for specific amplicons [65] |
| KAPA Long Range HotStart | Success | Failure | Failure | Limited to smaller fragments in this evaluation [65] |
| QIAGEN LongRange | Success | Failure | Failure | Limited to smaller fragments in this evaluation [65] |
A more recent study (2025) found the UltraRun LongRange PCR Kit (Qiagen) to have a 90% success rate for amplifying targets up to 22 kb, demonstrating its utility for diagnostic workflows [64].
Experimental Protocol: Long-Range PCR for Targeted Nanopore Sequencing
This protocol, adapted from a 2025 clinical diagnostic workflow, is designed for phasing distantly separated variants (up to ~20 kb apart) using long-range PCR and Nanopore sequencing [64].
1. Reagent Setup:
- Polymerase: UltraRun LongRange PCR Kit (Qiagen) or PrimeSTAR GXL Polymerase (TaKaRa)
- Primers: Design primers using NCBI Primer-BLAST. For sequencing, you may add adapters for your sequencing platform (e.g., Nextera for Illumina, Native Barcodes for Nanopore).
2. Reaction Assembly (50 μL total volume):
- The reaction is assembled as per the manufacturer's instructions for the chosen kit. A representative mixture is shown in Table 4.
- A key consideration is to use a limited number of cycles (e.g., 26 cycles) to minimize the formation of chimeric reads, a common artifact in long-range PCR [64].
Table 4: Representative Long-Range PCR Reaction Mixture
| Component | Final Concentration/Amount |
|---|---|
| 5X Long-Range PCR Buffer | 1X |
| dNTP Mix | 200 μM each |
| Forward Primer | 0.5 μM |
| Reverse Primer | 0.5 μM |
| DNA Template (e.g., NA24385) | 150 ng |
| Long-Range DNA Polymerase | 1.5 - 2.5 U |
| Nuclease-free Water | To 50 μL |
3. Thermal Cycling Conditions:
- Initial Denaturation: 93°C for 3 minutes (1 cycle)
- Amplification Cycles (26 cycles):
- Denaturation: 93°C for 30 seconds
- Annealing & Extension: 68°C for 1 minute per kb of amplicon length (e.g., 15 minutes for a 15 kb product)
- Final Extension: 72°C for 10 minutes (1 cycle)
- Hold: 4°C indefinitely
4. Post-Amplification:
- Purification: Purify the PCR product using Agencourt AMPure XP beads or a similar system.
- Analysis: Check the concentration, purity, and integrity of the amplicon using a Tape Station System or agarose gel electrophoresis.
- Sequencing: Proceed with library preparation for your chosen sequencing platform (e.g., Oxford Nanopore Technologies' Flongle flow cells) [64].
Implementing Fast PCR Protocols
Fast PCR, or high-speed PCR, focuses on drastically reducing the total assay time, which is critical for point-of-care diagnostics, high-throughput screening, and rapid research feedback. Standard PCR can take 1-2 hours, but fast PCR aims to complete amplification in minutes by optimizing thermal cycler kinetics and reaction chemistry [10] [60]. The acceleration is achieved through rapid temperature transitions and optimized reaction components that allow efficient polymerization at faster rates.
Key Parameters and Commercial Technologies
The transition to high-speed PCR involves engineering and biochemical optimizations. Key parameters for microfluidic-based fast PCR include reactor geometry, materials with high thermal conductivity, and efficient heating/cooling methods such as Joule heating, thermoelectric heating, and plasmonic heating [10]. Commercial instruments have achieved run times ranging from 2 minutes to 1 hour [10]. Furthermore, the move towards integrated Point-of-Care Testing (POCT) platforms, termed QUICK-PCR (Quick, Ubiquitous, Integrated, Cost-efficient), represents the future of rapid diagnostics, though practical implementation faces hurdles in sample preparation, system integration, and clinical validation [60].
Experimental Protocol: Optimizing for Speed in Conventional Thermocyclers
While specialized equipment exists, researchers can significantly reduce PCR times using standard thermocyclers with the following optimizations.
1. Reagent Setup:
- Polymerase: Use specialized "fast" or "speed" polymerases that feature very high extension rates (e.g., 2-6 seconds per kb).
- Buffer: Use the matching proprietary buffer for the polymerase.
2. Reaction Assembly (20 μL total volume):
- The reaction is assembled on ice according to the manufacturer's instructions for the fast polymerase. A typical mixture is shown in Table 5.
- Reducing the total reaction volume can decrease heating/cooling times in some cyclers.
Table 5: Fast PCR Reaction Mixture
| Component | Final Concentration/Amount |
|---|---|
| 2X Fast Polymerase Mix | 1X |
| Forward Primer | 0.2 - 0.5 μM |
| Reverse Primer | 0.2 - 0.5 μM |
| DNA Template | 10 - 20 ng |
| Nuclease-free Water | To 20 μL |
3. Thermal Cycling Conditions:
- Use a 2-Step PCR Protocol (combining annealing and extension into one step) if primer design allows.
- Drastically shorten cycle steps:
- Initial Denaturation: 98°C for 30 seconds (1 cycle)
- Amplification Cycles (35-40 cycles):
- Denaturation: 98°C for 5-15 seconds
- Annealing/Extension: 68-72°C for 5-15 seconds per kb of amplicon length
- Final Extension: 68-72°C for 5-10 seconds (1 cycle)
- Hold: 4°C
4. Analysis:
- Analyze the product using standard gel electrophoresis or a faster, integrated readout method like real-time fluorescence.
The Scientist's Toolkit: Essential Research Reagents
Successful PCR optimization relies on a toolkit of specialized reagents and products. The following table details key solutions for overcoming the technical challenges discussed in this guide.
Table 6: Essential Research Reagent Solutions for Advanced PCR
| Reagent / Product Name | Function / Application | Example Use Case |
|---|---|---|
| DMSO (Dimethyl Sulfoxide) | Additive that disrupts secondary structures in GC-rich DNA, lowering Tm and improving amplification efficiency [5] [61] [62] | Added at 3-5% to PCR mixes to amplify challenging nAChR receptor genes [62] |
| Betaine | Organic additive that equalizes the contribution of GC and AT base pairs, homogenizing DNA melting behavior [62] | Used at 1-1.5 M concentration for amplification of GC-rich templates [62] |
| AccuPrime GC-Rich Polymerase | Specialized DNA polymerase engineered for high processivity through GC-stable structures and complex secondary formations [61] [63] | First-choice enzyme for templates with >65% GC content where standard polymerases fail [63] |
| Platinum SuperFi II / Phusion High-Fidelity | High-fidelity polymerases with proofreading (3'-5' exonuclease) activity for accurate amplification of long fragments [64] [62] | Used in long-range PCR (up to 20 kb) for clinical sequencing applications with high accuracy [64] |
| OneTaq GC Buffer & Enhancer | Specialized buffer system specifically formulated to enhance yield and specificity for GC-rich amplicons [61] | Supplied with NEB's OneTaq polymerase for use as a complete system for difficult amplifications |
| UltraRun LongRange PCR Kit | Optimized enzyme and buffer system for reliable amplification of very long DNA targets [64] | Demonstrated 90% success rate for fragments up to 22 kb in a clinical diagnostics workflow [64] |
| Agencourt AMPure XP Beads | Magnetic beads for post-PCR purification, cleaning up primers, enzymes, and salts before downstream sequencing [64] [65] | Standard clean-up step for long-range PCR amplicons prior to NGS library preparation [64] |
| Native Barcoding Kit (ONT) | Allows multiplexed sequencing of multiple long-range PCR amplicons on a single Nanopore flow cell [64] | Barcoding of up to 8 amplicons for sequencing on a Flongle flow cell for cost-effective targeted sequencing [64] |
Polymersse Chain Reaction (PCR) is a foundational technique in molecular biology, enabling the amplification of specific DNA sequences. Its effectiveness hinges on precise optimization of reaction components and conditions, which varies significantly depending on the downstream application. This guide details the optimized workflows for three critical applications: DNA cloning, quantitative PCR (qPCR), and Next-Generation Sequencing (NGS) library preparation, providing a structured framework for researchers.
PCR for Cloning
Core Optimization Parameters for Cloning
PCR for cloning demands high fidelity to ensure the amplified DNA fragment is an exact copy of the template, minimizing errors that could compromise downstream experiments. Key considerations include polymerase selection, primer design, and reaction condition optimization.
Table 1: Key Optimization Parameters for Cloning PCR
| Parameter | Recommendation | Rationale |
|---|---|---|
| DNA Polymerase | High-fidelity enzymes (e.g., Pfu, KOD) | Possess 3'→5' proofreading exonuclease activity, reducing error rates by up to 10-fold compared to standard Taq [37]. |
| Primer Design | 18-24 bases, Tm 55-65°C, GC content 40-60% | Ensures specific annealing; primers should have matched Tm (within 1-2°C) and a G or C at the 3' end (GC clamp) [37] [15]. |
| Annealing Temperature (Ta) | 3-5°C below the primer Tm | Optimizes specificity; can be determined empirically using a gradient PCR thermocycler [37] [63]. |
| Mg²⁺ Concentration | 1.5 - 2.0 mM (titrate if needed) | Essential polymerase cofactor; suboptimal levels reduce activity, while high levels promote non-specific binding [37] [63]. |
| Template Quantity | 1 ng (plasmid) to 1 μg (genomic DNA) | Excessive template can decrease specificity and promote non-specific amplification [63]. |
| Cycle Number | Keep to a minimum (e.g., 25-30 cycles) | Reduces the accumulation of stochastic errors during amplification [37]. |
Advanced Techniques: Touchdown PCR
Touchdown PCR is a highly effective method for increasing amplification specificity and is highly recommended for cloning applications. The method involves starting with an annealing temperature higher than the expected Tm and progressively decreasing it in subsequent cycles [63].
Quantitative PCR (qPCR)
Understanding qPCR Efficiency
In qPCR, amplification efficiency (E) is a critical metric that quantifies the rate at which a target sequence is amplified during each cycle. An efficiency of 100% (or E=2) represents a perfect doubling. Efficiencies between 90-110% are generally acceptable, but deviations indicate potential issues [66] [67]. Efficiency is calculated from the slope of a standard curve generated from a serial dilution of a template: E = 10^(-1/slope) - 1 [66].
Table 2: qPCR Quantification Methods and Applications
| Method | Principle | Advantages | Ideal Applications |
|---|---|---|---|
| Absolute Quantification (Standard Curve) | Unknown quantity is determined by interpolating its Cq value against a standard curve of known concentrations [68]. | Provides exact copy numbers; well-established. | Viral load quantification, determining gene copy number [68]. |
| Absolute Quantification (Digital PCR) | Sample is partitioned into many reactions; absolute count is derived from the ratio of positive to negative reactions [68]. | Highly precise; does not require a standard curve; tolerant to inhibitors. | Detection of rare alleles, copy number variation in complex mixtures [68]. |
| Relative Quantification (Comparative Cᴛ / ΔΔCᴛ) | Compares expression of a target gene to a reference (endogenous control) gene relative to a calibrator sample (e.g., untreated control) [68]. | High throughput; no standard curve needed; results expressed as fold-change. | Gene expression studies (e.g., response to drug treatment) [66] [68]. |
The ΔΔCᴛ Method for Relative Quantification
The comparative Cᴛ (ΔΔCᴛ) method is a common relative quantification approach. A key requirement is that the amplification efficiencies of the target and reference genes are approximately equal [66] [68]. The workflow is as follows:
NGS Library Preparation
Library Preparation Workflow
NGS library preparation involves fragmenting DNA and adding platform-specific adapter sequences to enable sequencing and sample multiplexing [69] [70]. The core steps are universal, though methods for fragmentation and adapter integration differ.
Table 3: DNA Fragmentation Methods for NGS
| Method | Process | Advantages | Disadvantages |
|---|---|---|---|
| Mechanical Shearing (Focused Acoustics) | Uses high-frequency sound waves to physically break DNA [69]. | Unbiased fragmentation; consistent fragment sizes; minimal sample loss [69]. | Requires specialized, costly equipment [69]. |
| Enzymatic Digestion | Uses non-specific nucleases to cleave DNA [69]. | Low input DNA requirement; streamlined, automatable workflow; cost-effective [69]. | Potential for sequence-specific bias [69]. |
| Tagmentation | Uses a transposase enzyme to simultaneously fragment DNA and insert adapter sequences [69] [70]. | Fastest workflow; combines fragmentation and adapter ligation into a single step [69]. | May exhibit insertion sequence bias. |
Core Steps in Library Construction
The following diagram outlines the general workflow for constructing a sequencing-ready library, incorporating the different fragmentation approaches.
Target Enrichment Strategies
For targeted sequencing (e.g., exome or gene panels), the library must be enriched for regions of interest. The two primary methods are hybrid capture and amplicon sequencing.
- Hybrid Capture: Biotinylated oligonucleotide probes complementary to the target regions are hybridized to the library. Probe-bound fragments are then captured using streptavidin-coated magnetic beads, while non-target fragments are washed away [69]. This method typically provides more uniform coverage but requires more input DNA and a longer workflow.
- Amplicon Sequencing: This method uses highly multiplexed PCR to amplify the target regions directly from the library [69]. It is faster and requires less input DNA but can be affected by amplification bias and PCR errors, potentially impacting coverage uniformity [69].
The Scientist's Toolkit: Essential Reagents
Table 4: Essential Reagents for Application-Driven PCR Workflows
| Reagent | Function | Application-Specific Notes |
|---|---|---|
| High-Fidelity Polymerase | DNA synthesis with proofreading for high accuracy. | Cloning: Essential to minimize mutation rates [37]. |
| Hot Start Polymerase | Enzyme activated only at high temperatures, preventing non-specific amplification during reaction setup. | Universal: Improves specificity in all PCR applications [37]. |
| dNTPs | Building blocks (A, dTTP, dGTP, dCTP) for new DNA strands. | qPCR: Consistent quality and concentration is vital for accurate Cq values [63]. |
| MgCl₂ | Essential cofactor for DNA polymerase activity. | Optimization: Concentration must be titrated for each new assay/primer set [37] [15]. |
| NGS Adapters | Short, double-stranded DNA containing sequences for flow cell binding and sample indexing (barcodes) [70]. | NGS: Enable cluster generation and multiplexing of samples in a single run [69] [70]. |
| Buffer Additives (DMSO, Betaine) | Destabilize DNA secondary structures and homogenize base-pair stability. | GC-Rich Targets: Critical for amplifying difficult templates with high GC content (>65%) [37]. |
Systematic PCR Troubleshooting and Optimization Strategies
Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology, yet even experienced researchers can encounter issues that compromise their results. For new researchers, systematically diagnosing and resolving these problems is a critical skill in PCR optimization. This guide provides an in-depth analysis of three common PCR challenges—no amplification, non-specific bands, and smeared gels—by exploring their root causes and presenting proven, detailed solutions. A structured troubleshooting approach ensures reliable, reproducible data and streamlines your experimental workflow.
Problem 1: No Amplification or Low Yield
A complete absence of PCR product or a faint band on an electrophoresis gel indicates a failure in the amplification process, often stemming from issues with reaction components or cycling conditions.
Diagnosis and Solutions
- Template DNA Issues: The DNA template may be degraded, contain inhibitors, or be present at an insufficient concentration.
- Solution: Check DNA concentration and purity using spectrophotometry (A260/A280 ratio). Re-isolate DNA if degraded or impure. For a standard PCR with 25-30 cycles, roughly 10⁴ copies of the template DNA are typically sufficient. Use 0.1–1 ng of plasmid DNA or 5–50 ng of genomic DNA in a 50 µL reaction [5] [13].
- Suboptimal Primer Design or Concentration: Primers with poor specificity, low melting temperatures (Tm), or incorrect concentrations can prevent amplification.
- Insufficient PCR Components or Incorrect Cycling:
- Solution: Ensure all reagents are fresh and added in correct proportions. Increase the number of cycles to 30-40 for low template concentrations, but avoid over-cycling (typically over 35 cycles) to prevent non-specific products and false positives [71] [1]. Verify that the extension time is appropriate for your polymerase (e.g., 1 minute per 1000 bp for standard Taq polymerase) [5].
Table 1: Troubleshooting No Amplification or Low Yield
| Possible Cause | Diagnostic Check | Solution |
|---|---|---|
| Template DNA | Low concentration, degradation, or inhibitors | Re-quantify and re-isolate DNA; use serial dilutions; ensure purity [71] [72] [1] |
| Primers | Incorrect design, concentration, or degradation | Redesign primers; check concentration (0.1-1.0 µM); use fresh aliquots [5] [13] [73] |
| dNTPs/Enzyme | Low concentration or inactive enzyme | Use fresh dNTPs (200 µM each); ensure correct polymerase amount (e.g., 1–2.5 units/50 µL) [5] [13] [73] |
| Mg²⁺ Concentration | Concentration too low | Optimize MgCl₂ concentration (typically 1.5-5.0 mM) [5] [73] |
| Cycling Conditions | Too few cycles; low annealing temperature | Increase cycle number (up to 30-40); optimize annealing temperature [71] [1] |
Problem 2: Non-Specific Bands and Primer Dimers
Non-specific amplification results in multiple unwanted bands on an electrophoresis gel, while primer dimers appear as a bright band near the bottom of the gel (~50 bp), both competing with the target amplicon.
Diagnosis and Solutions
- Primer-Related Issues: High primer concentration or primers with complementary sequences can lead to primer-dimer formation and mispriming.
- Low Reaction Stringency: An annealing temperature that is too low allows primers to bind to non-target sequences.
- Excessive Enzyme or Template: Too much polymerase or template DNA can increase the likelihood of amplifying non-target sequences.
Diagram 1: A systematic workflow for diagnosing and resolving non-specific bands in PCR.
Table 2: Troubleshooting Non-Specific Bands and Primer Dimers
| Possible Cause | Diagnostic Check | Solution |
|---|---|---|
| Annealing Temperature | Temperature too low | Increase annealing temperature; perform a gradient PCR [71] [5] |
| Primer Design | Self-complementarity; high concentration | Redesign primers; lower concentration (0.1-1.0 µM) [13] [72] [73] |
| Enzyme Activity | Non-specific binding during setup | Use a hot-start DNA polymerase [5] [72] |
| Mg²⁺ Concentration | Concentration too high | Optimize MgCl₂ concentration (test 1.5-5.0 mM) [5] [73] |
| Cycle Number | Too many cycles | Reduce number of cycles (keep within 20-35) [71] [73] |
Problem 3: Smeared Gels
A smear on an agarose gel appears as a diffuse, fuzzy background or laddering pattern, indicating the presence of DNA fragments of many different sizes. This can obscure your target band.
Diagnosis and Solutions
- Too Much Template DNA or Degraded Template: This is a common cause of smearing. Overloaded DNA can lead to non-specific amplification, while degraded DNA results in fragments of random lengths.
- Non-Optimal PCR Conditions: Excessive cycle numbers or extension times can cause accumulation of non-specific products and smearing.
- Primer Degradation or Contamination: Degraded primers can act as random primers, leading to random amplification. Contaminating DNA in reagents can also cause smearing.
- Solution: Use fresh primer aliquots and new reagents. Set up reactions in a clean, designated area to prevent cross-contamination. If smearing persists with a previously reliable primer set, it may be due to accumulated "amplifiable DNA contaminants," and designing new primers with different sequences can resolve the issue [75] [72].
- Gel Electrophoresis Issues: Problems with the gel itself or sample loading can create smears.
- Solution: Do not overload the gel well (recommended 0.1–0.2 µg DNA per mm of well width). Ensure the gel is the correct percentage for your fragment size and that the running buffer is fresh [74].
The Scientist's Toolkit: Essential Research Reagents
Successful PCR optimization relies on high-quality reagents. The table below details key components and their functions.
Table 3: Key PCR Reagents and Their Functions
| Reagent | Function | Key Considerations |
|---|---|---|
| Template DNA | The target DNA sequence to be amplified. | Quality and concentration are critical. Use 0.1-1 ng (plasmid) or 5-50 ng (genomic) per 50 µL reaction [5] [13]. |
| DNA Polymerase | Enzyme that synthesizes new DNA strands. | Select based on fidelity, processivity, and application. Use hot-start for specificity. Typical amount is 1-2.5 units/50 µL [5] [13] [73]. |
| Primers | Short DNA strands that define the start and end of the target. | Design for specificity (Tm 55-70°C). Use 0.1-1.0 µM final concentration [5] [13]. |
| dNTPs | Deoxynucleoside triphosphates (dATP, dCTP, dGTP, dTTP); the building blocks for DNA synthesis. | Use balanced concentrations of 20-200 µM each. Higher concentrations can inhibit PCR [5] [13]. |
| MgCl₂ | A cofactor essential for DNA polymerase activity. | Concentration is critical; optimize between 1.5-5.0 mM. It stabilizes DNA and dNTP binding [5] [13] [73]. |
| Reaction Buffer | Provides optimal pH and salt conditions for the reaction. | Usually supplied with the enzyme. May contain KCl and Tris-HCl [5] [76]. |
| PCR Additives | Assist with challenging templates (e.g., high GC content). | DMSO (1-10%), formamide (1.25-10%), or BSA can help reduce secondary structures [5]. |
Advanced Optimization and Final Protocol
For persistent problems, a systematic optimization strategy is required. This involves methodically testing one variable at a time while keeping others constant.
Experimental Protocol: Mg²⁺ and Annealing Temperature Optimization
Objective: To determine the optimal Mg²⁺ concentration and annealing temperature for a specific primer-template pair.
Materials:
- PCR master mix (without Mg²⁺)
- 25 mM MgCl₂ stock solution
- Template DNA and primers
- Thermal cycler
Method:
- Prepare Mg²⁺ Titration Series: Set up a series of PCR tubes with a final Mg²⁺ concentration ranging from 1.5 mM to 5.0 mM in 0.5 mM increments [73].
- Perform Annealing Temperature Gradient: Using the optimal Mg²⁺ concentration from step 1, run a PCR with an annealing temperature gradient spanning from 5°C below to 5°C above the calculated primer Tm [5].
- Analyze Results: Visualize all reactions on an agarose gel. The condition that produces a single, bright target band with the least background smearing or non-specific products is the optimal combination.
Diagram 2: A sequential workflow for advanced PCR optimization, illustrating the order in which key variables should be tested.
Mastering PCR troubleshooting is an essential component of molecular biology research. By understanding the underlying causes of no amplification, non-specific bands, and smeared gels, researchers can systematically diagnose and resolve issues. This guide provides a foundational framework, emphasizing the importance of methodical optimization of template quality, primer design, reagent concentrations, and cycling parameters. Applying this structured approach will significantly improve PCR reliability and efficiency, enabling robust and reproducible results in your scientific endeavors.
The polymerase chain reaction (PCR) is a foundational technique in molecular biology, enabling the amplification of specific DNA sequences from minimal starting material. The success of PCR hinges on the precise control of temperature and time within the thermal cycler, parameters that directly influence the efficiency, specificity, and yield of the amplification process [15]. For new researchers, understanding how to optimize these cycling conditions is critical for obtaining reliable and reproducible results in diverse applications, from basic gene cloning to advanced drug development diagnostics. This guide provides an in-depth examination of each thermal cycling step, offering detailed methodologies and structured data to empower scientists in systematically optimizing their PCR protocols.
The thermal cycling process comprises three fundamental steps—denaturation, annealing, and extension—each with distinct temperature requirements that govern DNA strand separation, primer binding, and enzymatic DNA synthesis [4]. These steps are typically repeated for 25-40 cycles, preceded by an initial denaturation and followed by a final extension [77]. Factors such as template complexity, primer characteristics, DNA polymerase properties, and buffer composition all interact to determine the optimal cycling parameters for any given reaction [4]. By methodically adjusting these variables and understanding their interplay, researchers can overcome common amplification challenges, including nonspecific products, poor yield, and amplification failure, particularly with difficult templates such as GC-rich regions or long amplicons.
Core Principles of Thermal Cycling
The PCR Cycle: A Three-Step Process
The fundamental PCR process relies on the precise cycling of three temperature-dependent steps that facilitate the exponential amplification of DNA. The denaturation step separates double-stranded DNA into single strands by breaking hydrogen bonds between complementary bases, typically occurring at 94-98°C [4] [5]. During annealing, the temperature is lowered to allow primers to bind complementary sequences on the single-stranded DNA templates; this temperature is primer-specific and generally ranges from 50-65°C [77]. The extension step then occurs at the optimal temperature for DNA polymerase activity (usually 68-72°C), during which the enzyme synthesizes new DNA strands by adding nucleotides to the 3' ends of the annealed primers [4] [78]. In some cases, when the primer annealing temperature is close to the extension temperature, a two-step PCR protocol can be employed where annealing and extension are combined into a single step [78].
The following diagram illustrates the sequential nature of these steps and their temperature relationships:
Essential PCR Components and Their Functions
Successful PCR optimization requires not only appropriate thermal cycling conditions but also a precise mixture of biochemical components that support the amplification reaction. The following table details the key reagents and their functions in a typical PCR reaction:
| Component | Function | Typical Concentration |
|---|---|---|
| DNA Template | Provides the target sequence for amplification | 1pg–1µg genomic DNA; 104–107 copies [77] [15] |
| Primers | Short DNA sequences that define the region to be amplified | 0.1–1µM each; 20–50 pmol per reaction [77] [15] |
| DNA Polymerase | Enzyme that synthesizes new DNA strands | 0.5–2.5 units per 50µl reaction [77] [15] |
| dNTPs | Building blocks (nucleotides) for DNA synthesis | 20–200µM each [5] [15] |
| MgCl₂ | Essential cofactor for DNA polymerase activity | 1.5–2.0 mM (may require optimization) [77] [39] |
| PCR Buffer | Provides optimal pH and salt conditions for enzyme activity | 1X concentration [5] [15] |
Each component must be carefully optimized as improper concentrations can lead to amplification failure. For instance, excessive primer concentrations may promote nonspecific binding, while insufficient magnesium can reduce polymerase activity [77] [39]. The template quality is equally critical, with degraded or contaminated DNA resulting in poor amplification efficiency [39].
Optimizing Denaturation Parameters
Temperature and Time Considerations
The denaturation step is critical for separating double-stranded DNA into single strands, making the template accessible for primer binding. The initial denaturation at the beginning of the PCR program is typically performed at 94–98°C for 1–3 minutes [4]. This extended initial denaturation ensures complete separation of complex DNA templates and may help inactivate heat-labile contaminants [4]. For subsequent cycles, shorter denaturation times of 15–30 seconds at 94–98°C are generally sufficient [77]. However, templates with high GC content (>65%) often require higher denaturation temperatures (up to 98°C) or longer times due to their increased thermal stability [4] [78].
Prolonged exposure to high temperatures should be avoided with some DNA polymerases (e.g., Taq) as it can lead to enzyme inactivation, though highly thermostable enzymes derived from Archaea (e.g., Pfu) can withstand extended high-temperature incubation [4]. The presence of buffer components with high salt concentrations may also necessitate higher denaturation temperatures to achieve efficient strand separation [4].
Denaturation Optimization Protocol
To optimize denaturation conditions for challenging templates:
- Start with standard conditions: Begin with 30 seconds at 95°C for cyclic denaturation and 2 minutes at 95°C for initial denaturation [77].
- Increase temperature incrementally: If amplification is inefficient, especially for GC-rich templates, increase the temperature to 98°C in 0.5°C increments [4].
- Extend denaturation time: If increasing temperature alone is insufficient, extend the denaturation time up to 2 minutes [4].
- Consider additives: Include DMSO (1-10%), formamide (1.25-10%), or betaine (0.5-2.5 M) to reduce DNA thermal stability and improve denaturation efficiency [4] [15].
The table below summarizes denaturation parameters for different template types:
| Template Type | Initial Denaturation | Cyclic Denaturation | Special Considerations |
|---|---|---|---|
| Standard DNA | 94–95°C for 1–2 minutes [77] [78] | 94–95°C for 15–30 seconds [77] | None |
| High GC Content | 98°C for 2–5 minutes [4] [78] | 98°C for 30–60 seconds [4] | Use additives like DMSO or betaine [78] |
| Long Amplicons | 94–98°C for 1 minute [78] | 94–98°C for 20–30 seconds [78] | Minimize time to reduce depurination [78] |
| Direct PCR | 98°C for 2 minutes [78] | 94–98°C for 10–30 seconds [78] | Required for some specialized polymerases |
Optimizing Annealing Parameters
Determining Annealing Temperature
The annealing temperature is perhaps the most critical parameter for PCR specificity, as it determines the stringency of primer-template binding. The annealing temperature is primarily determined by the melting temperature (Tm) of the primers, which is the temperature at which 50% of the primer-duplex is dissociated [4]. A general rule is to start with an annealing temperature 3–5°C below the calculated Tm of the primers [4]. For primers with significantly different Tms, the temperature should be based on the primer with the lower Tm, though ideally primer pairs should have Tms within 5°C of each other [77] [15].
Several methods exist for calculating Tm. The simplest formula is: Tm = 4(G + C) + 2(A + T), which considers only base composition [4]. More accurate calculations account for salt concentration using the formula: Tm = 81.5 + 16.6(log[Na+]) + 0.41(%GC) – 675/primer length [4]. For greatest accuracy, the Nearest Neighbor method considers the thermodynamic stability of every adjacent dinucleotide pair and is the basis for many online Tm calculation tools [4].
Annealing Optimization Protocol
To systematically optimize annealing conditions:
- Calculate primer Tm: Use multiple methods to estimate Tm, beginning with the basic formula: Tm = 4(G + C) + 2(A + T) [4].
- Start with recommended temperature: Begin with an annealing temperature 3–5°C below the calculated Tm [4].
- Perform gradient PCR: Use the thermal cycler's gradient function to test a range of temperatures (typically ±5°C from the starting temperature) [4].
- Evaluate results: Analyze PCR products by agarose gel electrophoresis. Specific amplification appears as a single band of expected size [77].
- Adjust temperature: If nonspecific products are observed, increase the temperature in 2–3°C increments. If yield is low, decrease the temperature in 2–3°C increments [4].
The following diagram illustrates this optimization workflow:
Special Annealing Considerations
Several factors can influence optimal annealing conditions. PCR additives such as DMSO (at 1-10% concentration) can lower the effective Tm by approximately 5.5–6.0°C, necessitating corresponding adjustments to annealing temperature [4] [5]. Some specialized reaction buffers are formulated to enable universal annealing temperatures (e.g., 60°C) regardless of primer Tm, reducing optimization needs [4]. For two-step PCR protocols, where annealing and extension are combined, the temperature should be set based on the extension requirements (typically 68-72°C) while ensuring primer binding can still occur [78].
Optimizing Extension Parameters
Temperature and Time Settings
The extension step allows the DNA polymerase to synthesize the complementary DNA strand from the annealed primers. The extension temperature is typically set to the optimal activity temperature of the specific DNA polymerase being used: 68–72°C for most thermostable enzymes [4] [78]. A lower extension temperature (68°C) is often preferred for two-step PCR and when amplifying longer templates (>4 kb) as it reduces the rate of depurination that can damage the DNA [78]. For standard three-step PCR and amplification of short fragments (<4 kb), 72°C is commonly used [78].
The extension time is primarily determined by the length of the amplicon and the synthesis rate of the DNA polymerase. General guidelines recommend 1 minute per kilobase for Taq DNA Polymerase and 2 minutes per kilobase for slower enzymes like Pfu DNA Polymerase [4]. However, "fast" enzymes such as PrimeSTAR GXL or SpeedSTAR HS can significantly reduce extension times to 5–20 seconds per kilobase [78].
Extension Optimization Protocol
To optimize extension conditions:
- Calculate theoretical time: Determine the base extension time using the polymerase's synthesis rate (e.g., 1 min/kb for Taq) multiplied by the amplicon length in kb [4].
- Adjust for enzyme properties: Modify the time based on the specific polymerase characteristics—reduce time for "fast" enzymes, increase for "slow" ones [4] [78].
- Consider template complexity: For complex genomic DNA or GC-rich regions, increase extension time by 20–30% [4].
- Evaluate results: If longer products are absent or show smearing on gels, increase extension time. If nonspecific products appear, consider reducing time [4].
The table below summarizes extension parameters for different scenarios:
| Scenario | Temperature | Time Calculation | Special Considerations |
|---|---|---|---|
| Standard PCR | 72°C [78] | 1 min/kb for Taq [4] | Suitable for most applications |
| Long Amplicons | 68°C [78] | 1–2 min/kb [4] | Lower temperature reduces depurination [78] |
| Fast Polymerases | 68–72°C [78] | 5–20 sec/kb [78] | Follow manufacturer's recommendations |
| High-Fidelity PCR | 72°C [4] | 2 min/kb for Pfu [4] | Slower enzymes with proofreading |
Final Extension Step
The final extension step after the last PCR cycle ensures that all nascent DNA strands are fully synthesized. This step typically lasts 5–15 minutes at the extension temperature [4]. For cloning applications using Taq polymerase, a 30-minute final extension is recommended to ensure proper 3'-dA tailing for TA cloning [4]. The duration of this step should be optimized to ensure full-length polymerization, particularly for GC-rich templates or long amplicons where incomplete extension can result in heterogeneous products or smeared bands on agarose gels [4].
Cycle Number and Special Applications
Determining Optimal Cycle Numbers
The number of PCR cycles significantly impacts product yield and specificity. Typically, 25–35 cycles are sufficient for most applications [4]. Fewer cycles (20-25) are preferable for high-template reactions or when maximizing fidelity is critical, as error frequency increases with cycle number [77]. For low-copy templates (<10 copies), up to 40 cycles may be necessary to generate detectable product [4]. Exceeding 45 cycles is generally not recommended as it can lead to increased nonspecific amplification and accumulation of reaction by-products, resulting in a plateau effect where product yield no longer increases [4].
The relationship between cycle number and product yield follows an exponential amplification pattern that eventually plateaus due to depletion of reagents, accumulation of inhibitors, and reduced enzyme activity. Monitoring this amplification curve is essential for optimizing cycle number for specific applications [4].
Special PCR Applications and Their Cycling Parameters
Different PCR applications require specific modifications to standard cycling conditions:
Long-Range PCR: For amplifying fragments >10 kb, use longer extension times (1-2 min/kb), combination of polymerases with high processivity and proofreading activity, and possibly reduced temperatures to maintain enzyme activity during prolonged cycling [4] [5]. Template quality is particularly critical for long-range PCR, as DNA damage significantly reduces amplification efficiency [78].
GC-Rich Templates: Templates with >65% GC content benefit from higher denaturation temperatures (98°C), shorter annealing times, primers with higher Tm (>68°C), and additives like DMSO (2.5-5%) or formamide that help disrupt secondary structures [4] [78]. Specific polymerases optimized for GC-rich templates are also available [78].
AT-Rich Templates: For templates with high AT content (>80%), lower extension temperatures (60-65°C) can improve results, as DNA replication remains reliable at these reduced temperatures [78]. Polymerases recommended for GC-rich templates often work well for AT-rich templates too [78].
Rapid PCR: Using "fast" polymerases and reduced step times can significantly shorten PCR protocols. With enzymes like SpeedSTAR HS, denaturation and annealing times can be reduced to 5-15 seconds, and extension to 10-20 seconds per kb [78].
Troubleshooting Common Thermal Cycling Issues
Problem Identification and Resolution
Even with careful optimization, PCR amplification can encounter issues. The following table outlines common thermal cycling-related problems, their probable causes, and solutions:
| Problem | Possible Causes | Solutions |
|---|---|---|
| No amplification | Denaturation temperature too low [4]Annealing temperature too high [4]Insufficient initial denaturation [4] | Increase denaturation temperature [4]Decrease annealing temperature in 2–3°C increments [4]Extend initial denaturation time [4] |
| Nonspecific bands | Annealing temperature too low [4]Excessive cycle number [4]Magnesium concentration too high [77] | Increase annealing temperature in 2–3°C increments [4]Reduce cycle number to 25–35 [4]Optimize Mg²⁺ concentration [77] |
| Smear of bands | Excessive template [77]Annealing temperature too low [4]Extension time too long [39] | Reduce template amount [77]Increase annealing temperature [4]Optimize extension time [39] |
| Low yield | Extension time too short [4]Denaturation time too short [4]Insufficient cycles for template amount [4] | Increase extension time [4]Increase denaturation time [4]Increase cycle number (up to 40) [4] |
Advanced Optimization Strategies
When basic troubleshooting fails, advanced strategies may be necessary:
Touchdown PCR: Start with an annealing temperature 10°C above the calculated Tm and gradually decrease it by 1–2°C per cycle until the target temperature is reached. This approach preferentially amplifies specific products early in the reaction when enzyme fidelity is highest [78].
Hot Start PCR: Incorporating hot-start DNA polymerases (activated by the initial denaturation step) prevents nonspecific amplification during reaction setup by inhibiting polymerase activity at lower temperatures [5].
Additive Optimization: Systematically test PCR enhancers including DMSO (1-10%), formamide (1.25-10%), betaine (0.5-2.5 M), or BSA (10-100 μg/mL) to improve amplification of difficult templates [4] [15].
Magnesium Titration: Optimize Mg²⁺ concentration in 0.5 mM increments from 1.0-4.0 mM, as Mg²⁺ concentration affects primer annealing, template denaturation, enzyme activity, and fidelity [77] [39].
Optimizing thermal cycler conditions is a systematic process that requires careful attention to the interplay between temperature, time, and reaction components. By methodically adjusting denaturation, annealing, and extension parameters based on template characteristics and amplification goals, researchers can achieve specific, efficient, and reproducible PCR results across diverse applications. The guidelines presented in this technical guide provide a foundation for developing optimized protocols, while the troubleshooting strategies offer pathways for resolving amplification challenges. As PCR technologies continue to evolve, with new polymerases and instrumentation offering enhanced capabilities, the fundamental principles of thermal cycling optimization remain essential knowledge for researchers in basic science, diagnostics, and drug development.
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology, yet successful amplification often requires meticulous optimization of the reaction environment, especially when dealing with challenging templates such as those with high GC-content, complex secondary structures, or low abundance [15] [79]. This guide focuses on two critical aspects of reaction optimization: the titration of magnesium ions (Mg2+), an essential cofactor, and the use of enhancing additives like DMSO, BSA, and betaine. Properly fine-tuning these parameters can mean the difference between a failed experiment and a specific, high-yield amplification, making this knowledge indispensable for new researchers and professionals in drug development [80].
The Critical Role of Magnesium Ions (Mg2+)
Magnesium ions (Mg2+) are a crucial cofactor for DNA polymerase enzymes. They are directly involved in the catalytic reaction of DNA synthesis and play a key role in maintaining the stability of the primer-template complex [81].
Mechanisms of Action
- Enzyme Activity Maintenance: Mg2+ binds to the active center of DNA polymerase, which is essential for the enzyme to maintain its catalytic function and facilitate the formation of phosphodiester bonds [81].
- dNTP Binding: Mg2+ is involved in the binding of deoxynucleoside triphosphates (dNTPs) to the DNA template, enabling the nucleophilic attack that extends the DNA strand [81].
- Specificity Regulation: The concentration of Mg2+ significantly influences reaction specificity. An optimal concentration promotes specific primer binding, while deviations can lead to non-specific amplification or primer-dimer formation [15] [81].
Experimental Protocol for Mg2+ Titration
A systematic approach to Mg2+ titration is fundamental for PCR optimization.
- Prepare a Stock Solution: Typically, MgCl2 is provided as a 25 mM stock solution with the PCR buffer [15].
- Set Up the Titration Series: In a series of identical PCR reactions, vary the MgCl2 concentration. A standard starting range is 0.5 mM to 4.0 mM, with increments of 0.5 mM or 1.0 mM [81] [79]. A sample reaction setup for a 50 µl final volume is detailed in Table 1.
- Analyze Results: Run the PCR products on an agarose gel. The optimal Mg2+ concentration will produce a single, intense band of the expected size with minimal to no non-specific products or primer-dimers [79].
Table 1: Sample Reaction Setup for a 50 µl Mg2+ Titration Experiment
| Reaction Component | Final Concentration/Amount | Volume to Add per Reaction |
|---|---|---|
| 10X PCR Buffer | 1X | 5 µl |
| dNTP Mix | 200 µM (50 µM each) | 1 µl of 10 mM stock |
| Forward Primer | 20 pmol | 1 µl of 20 µM stock |
| Reverse Primer | 20 pmol | 1 µl of 20 µM stock |
| DNA Template | 1-1000 ng | Variable |
| Taq DNA Polymerase | 0.5-2.5 Units | 0.5-1.0 µl |
| 25 mM MgCl₂ Stock | Variable (e.g., 1.0 - 4.0 mM) | 2-8 µl (to achieve desired final concentration) |
| Sterile Water | Q.S. to 50 µl | Variable |
PCR Additives: DMSO, BSA, and Betaine
When standard reaction conditions fail, chemical additives can be powerful tools to enhance amplification efficiency and specificity. Their mechanisms and applications are summarized in Table 2.
Table 2: Common PCR Additives and Their Optimization
| Additive | Primary Mechanism | Typical Final Concentration | Key Applications | Considerations & Optimization |
|---|---|---|---|---|
| DMSO | Reduces DNA secondary structure stability; lowers Tm [81] | 2% - 10% [81]; 5% is often optimal [82] [79] | GC-rich templates [79] [50]; ITS2 DNA barcodes [82] | Reduces Taq polymerase activity [81]; may require lower annealing temperature [50] |
| Betaine | Reduces DNA secondary structure; equalizes base-stacking stability [81] | 0.5 M - 2.5 M [81]; 1 M is often effective [82] | GC-rich templates [81]; alternative to DMSO for failed reactions [82] | Use betaine or betaine monohydrate, not hydrochloride [81]; not recommended with DMSO [82] |
| BSA | Binds inhibitors and impurities; stabilizes polymerase [81] | 10 - 100 µg/ml [15]; ~0.8 mg/ml (800 µg/ml) [81] | Reactions with inhibitors (e.g., from FFPE tissue, humic substances) | Can be tested broadly when sample purity is suspect |
Dimethyl Sulfoxide (DMSO)
DMSO enhances PCR primarily by reducing the formation of stable secondary structures in the DNA template, which is particularly beneficial for GC-rich sequences [81] [50]. A study amplifying the high-GC EGFR promoter region found that 5% DMSO was necessary for successful amplification, providing the desired amplicon yield without non-specific products [79]. Furthermore, research on plant ITS2 DNA barcodes demonstrated that 5% DMSO achieved a 91.6% PCR success rate in previously unamplifiable samples [82].
Betaine
Betaine (also known as trimethylglycine) functions as an osmoprotectant that can denature DNA and eliminate the dependence of melting temperature on base pair composition [81]. This makes it highly effective for amplifying GC-rich regions. In the same ITS2 barcoding study, 1 M betaine achieved a 75% success rate for samples that failed with standard conditions. Notably, the one sample that did not amplify with DMSO was successfully amplified with betaine, though combining DMSO and betaine in the same reaction was not beneficial [82].
Bovine Serum Albumin (BSA)
BSA acts as a "clean-up" agent in PCR. It binds to and neutralizes common inhibitors found in DNA preparations, such as phenolic compounds, salts, and other impurities, thereby protecting the DNA polymerase [81]. This is especially valuable when using suboptimal DNA templates, such as those extracted from formalin-fixed paraffin-embedded (FFPE) tissues [79].
Integrated Experimental Workflow and Visualization
The following diagram illustrates a logical workflow for systematically troubleshooting and optimizing a PCR reaction using Mg2+ titration and additives.
Diagram 1: A logical workflow for PCR optimization via Mg2+ titration and additive use.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for PCR Optimization
| Reagent / Tool | Function in Optimization |
|---|---|
| MgCl₂ Solution | Titration of the essential Mg²⁺ cofactor for DNA polymerase. |
| DMSO | Additive to disrupt DNA secondary structures in GC-rich templates. |
| Betaine | Additive to denature DNA and reduce secondary structure formation. |
| BSA | Additive to bind inhibitors and increase enzyme stability. |
| Hot-Start DNA Polymerase | Enzyme engineered to reduce non-specific amplification during reaction setup [50]. |
| Gradient Thermal Cycler | Instrument to empirically determine the optimal annealing temperature. |
Fine-tuning the PCR reaction environment through Mg2+ titration and the strategic use of additives like DMSO, BSA, and betaine is a critical skill for overcoming common amplification challenges. A methodical approach—beginning with primer design, followed by Mg2+ optimization, and then testing individual additives—systematically addresses the primary causes of PCR failure. By mastering these techniques, researchers can ensure robust, specific, and efficient DNA amplification, thereby advancing their work in genetic analysis, diagnostics, and drug development.
The Taguchi Method represents a systematic application of design-of-experiments (DoE) principles that enables rapid optimization of complex biochemical processes, including quantitative polymerase chain reaction (qPCR). Unlike conventional optimization strategies that rely on time-consuming and resource-intensive trial-and-error approaches, the Taguchi method utilizes balanced orthogonal arrays to investigate the effects of multiple factors simultaneously with a significantly reduced number of experimental runs [83] [84]. This methodology was developed by Dr. Genichi Taguchi and has been widely adopted for industrial process design before being successfully applied to molecular biology applications, particularly PCR optimization [85].
For researchers developing qPCR assays, the optimization process traditionally involves determining the optimal concentrations of multiple reaction components such as magnesium (Mg++), deoxynucleotides (dNTPs), primers, fluorescent dyes (e.g., SYBR Green), and polymerase enzyme [83]. The fundamental challenge lies in the interdependent nature of these components, where adjusting a single factor in isolation provides an incomplete picture of its optimal setting. The Taguchi method addresses this limitation by providing a structured framework for exploring these interactions efficiently, ultimately leading to assays with enhanced performance, sensitivity, and reproducibility [84].
Theoretical Foundation of the Taguchi Method
Core Principles and Advantages
The Taguchi method operates on several key principles that distinguish it from conventional optimization approaches. First, it employs pre-determined orthogonal arrays that distribute variables in a balanced manner across all experimental trials [84]. This design ensures that each level of every factor is tested an equal number of times against each level of all other factors, enabling comprehensive exploration of the experimental space with minimal runs.
A significant advantage of this approach is the dramatic reduction in experimental workload. For example, a conventional factorial design investigating five factors at four levels each would require 1,024 (4^5) individual experiments to test all possible combinations. In contrast, the Taguchi method can achieve comparable optimization using only 16 trials through its L16(4^5) orthogonal array [83]. This efficiency translates to substantial cost savings; one study reported reducing optimization costs from over A$26,000 using factorial design to just A$2,300 using the Taguchi method [84].
The method incorporates a unique signal-to-noise ratio (S/N) metric as an objective function to identify optimal conditions. The S/N ratio effectively distinguishes between desired signal (PCR product yield) and unwanted experimental variation (noise), with different S/N equations selected based on the optimization objective [83].
Mathematical Framework: Signal-to-Noise Ratios
The Taguchi method employs distinct signal-to-noise ratio equations depending on the performance metric being optimized:
"Smaller is better" (Equation 1): Used when optimizing for lower threshold cycle (Ct) values, where earlier Ct values indicate greater product accumulation: S/N = -10log₁₀[1/n ∑(y²)]
"Larger is better" (Equation 2): Applied when optimizing for PCR efficiency, where steeper amplification plot slopes (larger numerical values) indicate greater reaction efficiency: S/N = -10log₁₀[1/n ∑(1/y²)]
In these equations, 'y' represents the measured output (e.g., Ct value or reaction efficiency), and 'n' denotes the number of experimental replicates [83]. The negative sign ensures that higher S/N ratios always correspond to better performance, regardless of which equation is used.
Experimental Design and Implementation
Preliminary Considerations for qPCR Optimization
Before implementing the Taguchi method, researchers must establish clear optimization objectives. The definition of "optimal performance" varies depending on the specific application; for instance, diagnostic assays may prioritize sensitivity, while high-throughput screening might emphasize speed or cost-efficiency [83]. This context determines which performance metrics will be used to calculate S/N ratios.
Prior knowledge of the qPCR system is essential for selecting appropriate factors and levels. The Taguchi method requires researchers to identify which reaction components will be investigated and establish meaningful concentration ranges for testing. Common factors include Mg++ concentration, dNTP concentration, primer concentration, SYBR Green concentration, and polymerase concentration [83]. The selection of factors should be based on understanding of their potential impacts on reaction kinetics and efficiency.
Step-by-Step Taguchi Protocol for qPCR
Step 1: Factor and Level Selection Based on the orthogonal array structure, researchers typically select 4-5 critical factors to investigate, each at 3-4 different levels. The levels should span a reasonable range around standard concentrations to ensure the optimal combination is captured. The table below illustrates a typical factor-level configuration for qPCR optimization:
Table 1: Example Factors and Levels for Taguchi qPCR Optimization
| Factor | Level 1 | Level 2 | Level 3 | Level 4 |
|---|---|---|---|---|
| Mg++ (mM) | 1.0 | 1.5 | 2.0 | 2.5 |
| dNTPs (μM) | 100 | 150 | 200 | 250 |
| Primers (nM) | 100 | 200 | 300 | 400 |
| SYBR Green (X) | 0.5 | 1.0 | 1.5 | 2.0 |
| Polymerase (U) | 0.5 | 1.0 | 1.5 | 2.0 |
Step 2: Orthogonal Array Selection and Experimental Setup For five factors at four levels each, an L16(4^5) orthogonal array is appropriate, requiring only 16 experimental combinations rather than 1024 [83]. The orthogonal array distributes the factor levels in a balanced manner across all experiments, as shown in this representative layout:
Table 2: L16(4^5) Orthogonal Array Experimental Layout
| Experiment | Mg++ | dNTPs | Primers | SYBR Green | Polymerase |
|---|---|---|---|---|---|
| 1 | A | A | A | A | A |
| 2 | A | B | B | B | B |
| 3 | A | C | C | C | C |
| 4 | A | D | D | D | D |
| 5 | B | A | B | C | D |
| 6 | B | B | A | D | C |
| 7 | B | C | D | A | B |
| 8 | B | D | C | B | A |
| 9 | C | A | C | D | B |
| 10 | C | B | D | C | A |
| 11 | C | C | A | B | D |
| 12 | C | D | B | A | C |
| 13 | D | A | D | B | C |
| 14 | D | B | C | A | D |
| 15 | D | C | B | D | A |
| 16 | D | D | A | C | B |
Note: Letters A-D correspond to the factor levels defined in Table 1.
Step 3: Experimental Execution and Data Collection Each of the 16 experimental combinations should be performed in replicate (typically n=3) to account for experimental variation. For each reaction, relevant performance metrics are recorded, including Ct values, reaction efficiency, and end-point fluorescence [83]. These quantitative measurements serve as the 'y' values for S/N ratio calculations.
Diagram 1: Taguchi Method Workflow for qPCR Optimization (82 characters)
Data Analysis and Interpretation
Calculation of Signal-to-Noise Ratios
After completing the experimental runs, S/N ratios are calculated for each factor at each level. For example, to calculate the S/N ratio for dNTPs at level B, one would extract the performance data (y-values) from all experiments where dNTPs were tested at level B (experiments 2, 6, 10, and 14 in Table 2) and apply the appropriate S/N equation [83]. This process is repeated for each factor at each level, generating a comprehensive dataset that reveals how each component concentration affects assay performance.
The selection of which S/N equation to use depends on the optimization objective. If the goal is to maximize product accumulation (indicated by lower Ct values), the "smaller is better" equation is appropriate. Conversely, if maximizing reaction efficiency (indicated by steeper amplification slopes), the "larger is better" equation should be employed [83].
Graphical Analysis and Optimization
The calculated S/N ratios are then plotted against factor levels, with separate graphs generated for each reaction component. Data points are typically fitted to a curve (e.g., cubic curve fit represented by a third-degree polynomial equation) using graphing software [83]. The maximum S/N value on each graph, interpolated from the fitted curve, identifies the optimal concentration for that specific component.
Diagram 2: Taguchi Data Analysis Process (67 characters)
This graphical approach enables researchers to identify optimal concentrations even for levels not explicitly tested in the original experimental design, providing finer resolution than the initial factor levels would suggest.
Verification and Iterative Refinement
Experimental Confirmation
The optimal conditions identified through Taguchi analysis represent calculated predictions that must be verified experimentally. Researchers should prepare qPCR reactions using the optimized parameters and compare their performance against standard conditions [83]. This confirmation step is critical for validating the Taguchi predictions and ensuring robust assay performance.
Performance metrics for verification should include reaction efficiency (ideally 90-110%), linearity (R² ≥ 0.99), sensitivity (limit of detection), and specificity (assessed via melt curve analysis for SYBR Green assays) [86] [87]. The confirmed optimal conditions should demonstrate statistically significant improvement over pre-optimized parameters.
Iterative Optimization
If the initial Taguchi array identifies a promising region of the optimization space but requires finer resolution, a second iterative Taguchi array can be implemented with factor levels clustered more tightly around the optimal ranges identified in the first round [83] [84]. This iterative approach progressively narrows the optimal windows for each factor, further enhancing assay performance.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of the Taguchi method for qPCR optimization requires careful selection of reagents and materials. The following table summarizes key components and their functions:
Table 3: Research Reagent Solutions for Taguchi qPCR Optimization
| Reagent/Material | Function | Optimization Considerations |
|---|---|---|
| DNA Polymerase | Enzymatic amplification of target sequence | Hot-start variants reduce non-specific amplification; concentration affects yield and specificity [88] |
| MgCl₂ | Cofactor for polymerase activity; stabilizes DNA duplex | Critical optimization factor; affects primer annealing, enzyme processivity, and product specificity [15] |
| dNTPs | Building blocks for DNA synthesis | Concentration balanced with Mg²⁺; affects reaction efficiency and fidelity [83] |
| Primers | Sequence-specific amplification | Design parameters: length (18-25 bp), GC content (40-60%), Tm (55-65°C), avoid self-complementarity [30] |
| Fluorescent Detection System | Signal generation for real-time monitoring | SYBR Green (intercalating dye) vs. hydrolysis probes (TaqMan); choice affects cost and specificity requirements [86] |
| Buffer Components | Maintain optimal pH and ionic strength | May contain additives like DMSO, betaine, or BSA to enhance specificity and efficiency [15] |
| Passive Reference Dye | Normalize fluorescence signals between wells | ROX concentration must match instrument requirements (no, low, or high ROX) [86] |
Complementary qPCR Optimization Strategies
While the Taguchi method efficiently optimizes reaction chemistry, comprehensive qPCR development requires attention to additional parameters. Primer design is particularly critical, with optimal characteristics including length of 18-25 bases, GC content between 40-60%, and melting temperatures of 55-65°C for both forward and reverse primers within 5°C of each other [30]. The 3' ends should feature G or C bases (GC clamp) to increase priming efficiency [15].
Thermal cycling parameters also require optimization, particularly annealing temperature, which can be systematically tested using temperature gradient PCR [88]. For probe-based detection systems, probe design must be considered, with optimal probes having a Tm 10°C higher than primers and avoiding G repeats, especially at the 5' end [88].
Template quality and concentration significantly impact qPCR performance. RNA samples should undergo rigorous quality assessment, and cDNA synthesis should be optimized with appropriate controls (no-template controls and no-reverse-transcriptase controls) [86]. A serial dilution of template should be included to assess amplification efficiency and dynamic range [87].
Applications, Limitations, and Future Directions
The Taguchi method has been successfully applied to diverse PCR applications beyond conventional qPCR, including optimization of RAPD-PCR profiles [85], STR amplification for forensic analysis [84], and multiplex PCR systems. However, researchers should recognize its limitations, particularly for complex multiplex qPCR assays where multiple targets are amplified simultaneously.
A significant constraint is that the Taguchi method optimizes based on a single signal-to-noise ratio, making it challenging to balance multiple competing objectives in multiplex reactions [83]. In such cases, alternative DoE approaches such as response surface methodology may be more appropriate for finding optimal compromise conditions.
Future applications of the Taguchi method in molecular diagnostics may include optimization of digital PCR, isothermal amplification techniques, and CRISPR-based detection systems. As these technologies mature, the systematic optimization approach provided by Taguchi methods will remain valuable for developing robust, reproducible assays suitable for clinical and commercial applications.
The integration of Taguchi optimization with high-throughput instrumentation and automated liquid handling systems presents promising opportunities for further reducing optimization time while increasing experimental precision. By combining systematic experimental design with advanced laboratory automation, researchers can accelerate the development of next-generation molecular diagnostics with enhanced performance characteristics.
In the polymerase chain reaction (PCR), the template DNA is more than just a starting material; it is the foundational blueprint upon which the entire amplification process is built. The quality, quantity, and integrity of the template directly govern the sensitivity, efficiency, and ultimate success of the reaction [89]. For researchers embarking on PCR optimization, a thorough understanding of template management is crucial. Challenges such as the presence of PCR inhibitors, template degradation, or suboptimal concentrations can lead to complete amplification failure or misleading results [90] [91]. This guide provides an in-depth examination of these factors, offering evidence-based strategies to ensure robust and reliable PCR outcomes, forming a core component of a comprehensive introduction to PCR optimization.
Template Quantity: Determining the Optimal Amount
Fundamental Principles and Guidelines
Using the correct amount of template DNA is critical. Insufficient template may prevent primers from locating their complementary sequences, leading to failed or weak amplification [89]. Conversely, excessive template can increase mispriming events and promote non-specific amplification [89]. The optimal quantity is not a fixed value but depends on the complexity and copy number of the target sequence.
As a general rule, for a standard 25-30 cycle PCR, approximately 10^4 copies of the template DNA are sufficient to generate a detectable product [5]. In terms of mass, this typically translates to 10–100 ng of genomic DNA for most reactions, with the lower end of this range (e.g., 10 ng) being sufficient for abundant targets like housekeeping genes [5]. It is recommended to use no more than 1 µg of template DNA per PCR reaction to avoid issues related to overloading [89].
Table 1: Recommended Template Quantities for Various DNA Sources
| Template Source | Recommended Amount | Notes | Key References |
|---|---|---|---|
| Standard Genomic DNA | 30–100 ng | Optimal for most PCR reactions; adjust based on target abundance. | [5] |
| Low Copy Number Targets | <100–200 pg | Increase cycle number to 34 or more for enhanced sensitivity. | [5] |
| Plasmid DNA | ~1 pg–1 ng | Lower requirements due to low complexity and high copy number. | [89] |
| General Maximum | ≤1 µg | Prevents mispriming and non-specific amplification. | [89] |
Molar Conversions for Template Quantification
For precise experiment design, especially when comparing different template types, understanding molar conversions is essential.
Table 2: Molar Conversions for Various Nucleic Acid Templates
| Nucleic Acid Template | Size / Description | pmol/µg | Molecules/µg |
|---|---|---|---|
| 1 kb DNA Fragment | 1,000 bp | 1.52 | 9.1 x 10^11 |
| pUC19 Plasmid | 2,686 bp | 0.57 | 3.4 x 10^11 |
| Lambda DNA | 48,502 bp | 0.03 | 1.8 x 10^10 |
| Genomic DNA (E. coli) | 4.7 x 10^6 bp/haploid genome | 3.0 x 10^-4 | 1.8 x 10^8* |
| Genomic DNA (Human) | 3.3 x 10^9 bp/haploid genome | 4.7 x 10^-7 | 2.8 x 10^5* |
*For single-copy genes. [89]
Template Quality: Managing Purity and Integrity
The Impact of Template Degradation and Handling
Template DNA is susceptible to degradation over time, particularly if stored improperly. This can severely reduce PCR efficiency [1]. Regularly testing template concentration, especially for long-stored samples, is a fundamental best practice. Furthermore, specific sample types may require specialized handling techniques. For instance, when working with yeast, a protocol involving boiling cells for 5 minutes followed by freezing at -80°C for 3 minutes before thawing has been shown to drastically improve PCR yield [1]. Always using freshly prepared and accurately quantified template DNA is the first line of defense against quality-related failures.
Understanding and Managing PCR Inhibitors
PCR inhibitors are a heterogeneous class of substances that can originate from the biological sample itself or be introduced during sample preparation [91]. Their mechanisms of action are diverse, including:
- Degrading or denaturing the DNA polymerase (e.g., via proteases, detergents, or phenol) [91].
- Depleting essential cofactors like magnesium ions (Mg²⁺), which are critical for polymerase activity [5] [91].
- Binding directly to nucleic acids, preventing primer annealing and template denaturation [90] [91].
- Interfering with fluorescence in real-time PCR and sequencing applications, leading to inaccurate quantification [90].
Table 3: Common PCR Inhibitors and Their Sources
| Inhibitor Category | Example Inhibitors | Common Sources | Primary Mechanism of Inhibition |
|---|---|---|---|
| Blood Components | Hemoglobin, Immunoglobulin G (IgG), Lactoferrin | Blood, serum, plasma | IgG binds ssDNA; Hemoglobin and lactoferrin inhibit polymerase. [90] [91] |
| Soil and Plant Matter | Humic and Fulvic Acids, Polysaccharides, Polyphenols | Soil, plant tissues, roots | Bind to polymerase and template DNA; polysaccharides mimic nucleic acids. [92] [90] [91] |
| Sample Processing Reagents | Heparin, EDTA, Phenol, Ethanol, Ionic Detergents | Collection tubes, extraction kits, purification reagents | EDTA chelates Mg²⁺; phenol/ethanol denature enzymes; detergents disrupt activity. [90] [91] |
| Food and Microbial Components | Fats, Proteins, Calcium, Collagen, Melanin | Milk, fecal matter, meat, bacterial cells | Calcium competes with Mg²⁺; collagen inhibits polymerase; nucleases degrade template. [91] |
Strategies for Overcoming Inhibition and Quality Issues
Pre-PCR Strategies: Sample Preparation and Nucleic Acid Extraction
Robust sample collection and nucleic acid extraction are the most effective ways to preemptively remove PCR inhibitors.
- Sample Collection: Refine collection to minimize co-extraction of inhibitors. For plant materials, this may involve removing parts high in polysaccharides; for soil-coated samples, thorough washing is beneficial [92].
- Nucleic Acid Extraction: Silica-based column kits, many of which incorporate specific Inhibitor Removal Technology (IRT), are highly effective at eliminating potent inhibitors like humic acid [92]. Methods utilizing paramagnetic beads also provide efficient purification and are amenable to automation [92] [90]. For the highest purity, a two-stage DNA separation process (e.g., using a silicon dioxide suspension followed by paramagnetic particles) can be employed to concentrate the sample and drastically reduce inhibitors [92].
In-Reaction Strategies: Additives and Robust Reagents
When inhibitors persist, several strategies can be applied directly to the PCR setup:
- DNA Dilution: A simple tenfold dilution of the DNA extract can often reduce inhibitor concentration below a critical threshold, though this also dilutes the target DNA and may impact sensitivity [92].
- PCR Enhancers and Additives: Specific compounds can be added to the master mix to counteract inhibitors.
- Bovine Serum Albumin (BSA): Effective at mitigating inhibition from humic acid, tannic acid, and blood components by binding to the inhibitory substances [92] [5] [91]. A typical final concentration is 400 ng/µL (or 10–100 µg/mL) [5] [15].
- Dimethyl Sulfoxide (DMSO): Helps lower the melting temperature (Tm), which is particularly useful for amplifying GC-rich templates (>60% GC). Use at 1–10% final concentration [5] [15].
- Non-ionic Detergents: Tween 20, Triton X-100, or NP-40 (at 0.1–1%) can stabilize DNA polymerases and prevent secondary structures [5].
- Betaine and Glycerol: Used to reduce secondary structure formation in GC-rich templates, with betaine often used at 0.5 M to 2.5 M [15] [91].
- Inhibitor-Tolerant Polymerases: Selecting a robust DNA polymerase is a powerful solution. Many modern polymerases, including specialized blends and engineered versions of Taq, exhibit significantly higher resistance to inhibitors found in blood, soil, and complex samples [90] [91]. Hot-start polymerases are also recommended to prevent non-specific amplification at lower temperatures [1] [5].
PCR Inhibition Troubleshooting Workflow
Experimental Protocols for Quality Assessment and Inhibition Testing
Protocol: Testing for PCR Inhibition Using Exogenous DNA
This protocol is used to confirm the presence of inhibitors in a DNA extract.
- Obtain an Exogenous Control: Secure a DNA template and a corresponding primer set that is entirely absent from your sample DNA. A common approach is to spike in a plasmid, synthetic DNA fragment, or a non-pathogenic bacterium (e.g., a specific yeast strain) not found in the samples [92].
- Set Up PCR Plates:
- Well A (Control): Contains a fixed, known amount of the exogenous DNA alone.
- Well B (Test): Contains the same amount of exogenous DNA plus the sample DNA extract.
- Run Real-Time PCR: Perform qPCR using the assay specific for the exogenous target.
- Analyze Results: Compare the quantification cycle (Cq) values between the two wells. A statistically significant delay (higher Cq) in Well B compared to Well A indicates the presence of PCR inhibitors in the sample DNA extract [92].
Protocol: Post-Extraction Cleanup Using Paramagnetic Beads
This methodology provides a rapid and effective way to purify DNA extracts after initial preparation.
- Transfer DNA Extract: Place your DNA extract (in a suitable buffer) into a 1.5 mL microcentrifuge tube.
- Add Beads: Add a calibrated volume of paramagnetic bead suspension (e.g., AMPure XP) to the DNA. The bead-to-sample ratio is critical and often manufacturer-defined.
- Bind DNA: Incubate at room temperature to allow DNA to bind to the beads.
- Capture Beads: Place the tube on a magnetic stand until the solution clears. Carefully remove and discard the supernatant.
- Wash: While the tube is on the magnetic stand, add a freshly prepared 70–80% ethanol solution. Incubate for 30 seconds, then remove and discard the ethanol. Repeat this wash step once.
- Dry Pellet: Briefly air-dry the bead pellet to ensure all residual ethanol has evaporated. Do not over-dry.
- Elute DNA: Remove the tube from the magnetic stand and resuspend the beads in nuclease-free water or a low-salt elution buffer. Incubate to allow DNA to dissociate.
- Recover DNA: Place the tube back on the magnetic stand. Once clear, transfer the purified supernatant containing your DNA to a new tube [92].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Reagents for Managing Template Quality and Inhibitors
| Reagent / Kit | Function / Application | Key Characteristics |
|---|---|---|
| QIAGEN DNeasy PowerSoil Kit | DNA extraction from soil, plant, and difficult samples. | Contains Inhibitor Removal Technology (IRT) to remove humic acids and other common inhibitors. [92] |
| Hieff Ultra-Rapid II HotStart PCR Master Mix | PCR amplification of complex templates. | Designed for fast, efficient amplification of difficult templates (bacterial colonies, high GC content). [1] |
| Phusion Flash High-Fidelity PCR Master Mix | Direct PCR from impure samples. | High tolerance to PCR inhibitors, enabling faster workflows with minimal sample purification. [90] |
| Paramagnetic Beads (e.g., AMPure XP) | Post-extraction nucleic acid cleanup. | Used to purify and size-select DNA, effectively removing salts, primers, and other inhibitors. [92] |
| Bovine Serum Albumin (BSA) | PCR additive to counteract inhibitors. | Binds to inhibitory compounds like phenolics, humic acids, and components in blood. [5] [91] |
| Dimethyl Sulfoxide (DMSO) | PCR additive for complex templates. | Disrupts secondary structures, lowers Tm, and improves amplification of GC-rich regions. [5] [15] |
Managing Template Purity and Complexity
Effective management of template DNA quality and quantity is a non-negotiable aspect of robust PCR experimental design. By understanding the principles of template quantification, recognizing the sources and mechanisms of common PCR inhibitors, and implementing a strategic toolkit of purification methods, reagent additives, and inhibitor-tolerant enzymes, researchers can overcome the significant challenge of amplification failure. Diligent application of these protocols for quality assessment and inhibition testing ensures the integrity of molecular data, paving the way for reliable and reproducible results in diagnostic and research applications.
Validating PCR Results and Comparing Method Performance
For researchers embarking on PCR optimization, establishing robust assay performance is a critical step that bridges experimental development and reliable scientific discovery. Three fundamental parameters—Limit of Detection (LOD), precision, and specificity—form the cornerstone of assay validation, ensuring that results are both reproducible and meaningful. Within the context of molecular diagnostics, these parameters determine an assay's fitness for purpose, whether for basic research or clinical application [93]. The LOD defines the lowest concentration of an analyte that can be reliably distinguished from zero, establishing the sensitivity threshold of your assay. Precision quantifies the degree of reproducibility in measurements when the same sample is tested repeatedly under specified conditions. Specificity confirms that the assay detects only the intended target without cross-reacting with non-target sequences, ensuring the accuracy of your conclusions. Together, these parameters provide a comprehensive framework for evaluating assay performance, reducing the risk of erroneous results that could compromise research integrity or clinical decision-making [94].
Experimental Protocols for Determination
Determining the Limit of Detection (LOD)
The LOD represents the lowest quantity of an analyte that can be detected with stated probability, defining your assay's sensitivity frontier. A well-defined LOD protocol is essential for applications requiring high sensitivity, such as pathogen detection in early infection or minimal residual disease monitoring in oncology.
Probit Analysis Protocol:
- Sample Preparation: Prepare a dilution series of the target nucleic acid in a matrix that mimics the clinical or experimental sample (e.g., background DNA, transport medium). Use a standard of known concentration, ideally traceable to an international standard [95] [94].
- Replication and Testing: Test each dilution in a minimum of 20 replicates to establish a statistically significant detection rate at each concentration level. This replication is crucial for accounting for stochastic effects at low target concentrations [95].
- Data Analysis: Calculate the percentage of positive detections at each dilution. Apply probit regression analysis to this dataset to determine the concentration at which 95% of replicates test positive. This concentration represents the formal LOD of your assay [95].
- Verification: Confirm the determined LOD by testing 20 independent replicates at the calculated concentration to verify that ≥19/20 (95%) test positive [95].
Alternative approaches include the 2- or 3-standard deviation methods, but probit analysis is generally preferred for molecular assays due to its statistical rigor for binary (positive/negative) outcomes.
Establishing Assay Precision
Precision validation measures the agreement between repeated measurements and encompasses both repeatability (intra-assay precision) and reproducibility (inter-assay precision). This parameter is essential for understanding the expected variability in your results and establishing confidence intervals for quantitative measurements.
Precision Testing Protocol:
- Sample Selection: Prepare at least two different concentrations of the target analyte—one near the LOD (2× LOD) and one at a higher, clinically relevant level (5× LOD) [95].
- Intra-Assay Precision (Repeatability): For each concentration, perform a minimum of 5 replicate measurements within a single assay run using the same operator, reagents, and equipment. Calculate the coefficient of variation (CV) for the quantitative results (e.g., Ct values for qPCR or copies/μL for dPCR) [95].
- Inter-Assay Precision (Reproducibility): For each concentration, test 5 replicates across different assay runs conducted on different days by different operators. Maintain consistent sample aliquots but vary reagent lots and equipment if appropriate to your testing environment. Calculate the CV across these measurements [95].
- Acceptance Criteria: For quantitative PCR assays, intra-assay CVs should generally be ≤0.70% and inter-assay CVs ≤0.50% for melting temperature (Tm) values, though acceptance criteria should be established based on the assay's intended use [95]. For Ct values, CVs <5% are often considered acceptable [96].
Demonstrating Assay Specificity
Specificity testing confirms that an assay detects only the intended target(s) and does not cross-react with genetically similar or clinically relevant non-target organisms. This is particularly crucial for diagnostic assays where false positives could lead to inappropriate treatment decisions.
Specificity Testing Protocol:
- In Silico Analysis: Before wet-lab testing, perform computational specificity assessment using BLAST or similar tools to check primer and probe sequences against genetic databases. Verify that they align exclusively with the intended target and not with non-target sequences [95] [94].
- Wet-Lab Testing with Panels: Assemble a panel of non-target organisms that are genetically similar or likely to be present in the sample matrix. For respiratory pathogen assays, this might include other viruses or bacteria causing similar symptoms [95]. For parasitic assays like Spirometra mansoni detection, include other common parasites found in the same host species [96].
- Experimental Procedure: Test each non-target organism in replicates using the established assay protocol. Use samples with high concentrations of non-target organisms to challenge the assay's specificity stringently.
- Cross-Reactivity Assessment: Confirm that no amplification occurs with any non-target organism. Any observed cross-reactivity necessitates redesign of primers or probes to improve specificity [95] [96].
- Inclusivity Testing: As a complementary specificity parameter, verify that the assay detects all known variants, subtypes, or strains of the target organism to ensure no false negatives occur due to genetic diversity [94].
Quantitative Data Comparison
The following tables consolidate quantitative benchmarks for LOD, precision, and specificity from recent validation studies, providing reference points for assay development.
Table 1: Limits of Detection (LOD) Across PCR Technologies
| Assay Type | Target | LOD | Matrix | Reference |
|---|---|---|---|---|
| FMCA-based multiplex PCR | SARS-CoV-2 | 4.94-14.03 copies/μL | Nasopharyngeal swabs | [95] |
| qPCR | Spirometra mansoni | 100 copies/μL | Cat fecal DNA | [96] |
| PCR | Spirometra mansoni | 0.7 ng/μL | Egg-derived DNA | [96] |
| LAMP | Spirometra mansoni | 7.47 pg/μL | Cat fecal DNA | [96] |
Table 2: Precision Metrics from Validation Studies
| Assay Type | Precision Level | CV (%) | Measurement | Reference |
|---|---|---|---|---|
| FMCA-based multiplex PCR | Intra-assay | ≤0.70% | Tm value | [95] |
| FMCA-based multiplex PCR | Inter-assay | ≤0.50% | Tm value | [95] |
| qPCR | Intra-batch | <5% | Quantitative detection | [96] |
| qPCR | Inter-batch | <5% | Quantitative detection | [96] |
Table 3: Specificity Testing Outcomes
| Assay Type | Targets | Non-Targets Tested | Specificity Outcome | Reference |
|---|---|---|---|---|
| FMCA-based multiplex PCR | 6 respiratory pathogens | 10 respiratory viruses, 4 bacteria | No cross-reactivity observed | [95] |
| PCR/qPCR/LAMP | Spirometra mansoni | Common parasites in cat/dog feces | No cross-reactivity observed | [96] |
Workflow Visualization
LOD Determination Flow
Specificity Validation Flow
Research Reagent Solutions
Table 4: Essential Reagents for PCR Validation Studies
| Reagent/Kit | Function | Example Use Case | Reference |
|---|---|---|---|
| Automated Nucleic Acid Extraction System | Isolation and purification of RNA/DNA templates | Processing nasopharyngeal swabs in viral transport medium | [95] |
| RNA/DNA Extraction Kit | Concurrent extraction of both RNA and DNA | Multiplex respiratory panel detecting both RNA and DNA pathogens | [95] |
| One Step U* Mix & Enzyme Mix | Integrated reverse transcription and PCR amplification | Streamlined workflow for RNA virus detection | [95] |
| Fluorescent Probes with Modified Bases (e.g., THF residues) | Enhanced hybridization stability across variants | Robust detection of variable pathogen subtypes | [95] |
| Faecal Genomic DNA Extraction Kit | Optimized nucleic acid isolation from complex matrices | Detection of parasites in stool samples | [96] |
| Certified Reference Strains | Analytical specificity and inclusivity testing | Establishing assay detection breadth for diverse strains | [94] |
Within the framework of polymerase chain reaction (PCR) optimization, the accurate analysis of amplification products is a critical determinant of experimental success. This guide provides an in-depth technical examination of three cornerstone post-amplification techniques: gel electrophoresis, melting curve analysis, and sequencing. For new researchers, mastering the comparative strengths, limitations, and specific applications of these methods is fundamental to making informed decisions in experimental design, particularly in diagnostic and drug development contexts. While gel electrophoresis offers a foundational, low-cost separation technique, melting curve analysis enables rapid, closed-tube genotyping, and sequencing provides the ultimate resolution for base-pair level confirmation. The integration of artificial intelligence and novel microfluidic platforms is now revolutionizing these traditional methods, enhancing their accuracy, throughput, and accessibility for scientific and clinical professionals.
Method Comparison at a Glance
The following table summarizes the core characteristics, outputs, and applications of the three primary analysis techniques to guide method selection.
Table 1: Comparative overview of PCR analysis methods
| Feature | Gel Electrophoresis | Melting Curve Analysis (HRM) | Sequencing |
|---|---|---|---|
| Primary Principle | Size-based separation via gel matrix and electric field [97] | Discrimination based on DNA melting temperature (Tm) in a gradual denaturation [8] [98] | Determination of the exact nucleotide sequence [8] |
| Key Output | Band pattern on a gel image [97] | Melting curve (fluorescence vs. temperature) and derivative melt peaks [98] [99] | Chromatogram and text file of A, T, G, C sequence [8] |
| Information Gained | Amplicon presence/absence and approximate size [97] | Genotype, sequence variation (SNPs), species identification [8] [99] | Exact nucleotide composition, mutation identification, haplotype confirmation [8] |
| Typical Resolution | ~10 base pairs (for standard agarose) [8] | Single nucleotide polymorphism (SNP) [8] [100] | Single base pair [8] |
| Throughput | Medium | High | Low to Medium |
| Cost | Low | Medium | High |
| Ease of Use | Simple, requires post-PCR handling | Simple, closed-tube method | Complex, requires specialized facilities |
| Primary Application | Qualitative/semi-quantitative confirmation of amplification | Mutation screening, species differentiation, SNP genotyping [8] [98] [99] | Gold standard validation, novel mutation discovery, phylogenetic analysis [8] |
Gel Electrophoresis
Core Principles and Workflow
Gel electrophoresis remains a ubiquitous laboratory method for the separation and semi-quantitative analysis of DNA fragments by size [97]. The core principle involves suspending DNA molecules within wells in a gel matrix (commonly agarose or polyacrylamide) and applying an electric field. The negatively charged DNA migrates toward the positive anode, with smaller fragments moving through the gel pores more rapidly than larger ones, resulting in a characteristic pattern of "bands" [97]. These bands are visualized using fluorescent intercalating dyes, such as Ethidium Bromide or safer alternatives like SYBR Safe, under UV light.
The standard workflow for analysis has remained largely unchanged for decades, typically involving manual or semi-automated steps: digitally defining lanes, identifying bands within each lane, and quantifying band intensity to estimate DNA mass or concentration [97].
Detailed Experimental Protocol
Protocol: Agarose Gel Electrophoresis for PCR Product Analysis
- Gel Preparation: Prepare a 1-2% agarose solution in 1X TAE or TBE buffer. Microwave until the agarose is completely dissolved. Allow the solution to cool slightly (to ~60°C), then add a fluorescent nucleic acid stain (e.g., Ethidium Bromide or SYBR Safe) as per manufacturer's instructions. Pour the solution into a casting tray with a well comb and allow it to solidify at room temperature.
- Sample Loading: Mix a small aliquot of your PCR reaction (typically 5-10 µL) with a 6X DNA loading dye. Carefully load the mixture into the wells of the solidified gel. Include an appropriate DNA molecular weight ladder (e.g., 100 bp ladder) in one well to enable size estimation of the amplicons.
- Electrophoresis: Place the gel in an electrophoresis chamber filled with 1X TAE or TBE buffer. Run the gel at a constant voltage (e.g., 80-120 V) for 30-60 minutes, or until the dye front has migrated sufficiently.
- Visualization & Analysis: Image the gel using a UV or blue light transilluminator. Analyze the resulting image to confirm the presence of a band at the expected size and assess its intensity and purity (i.e., absence of non-specific bands or primer dimers).
Advanced Innovation: AI-Powered Analysis
A significant modern advancement is the integration of artificial intelligence (AI) to overcome the limitations of manual and traditional algorithmic analysis. AI-powered systems like GelGenie can now automatically identify gel bands in seconds across a wide range of experimental conditions [97].
This approach uses a U-Net model trained on a vast dataset of manually labelled gels to perform segmentation—classifying each pixel in a gel image as either 'band' or 'background' [97]. This method eliminates the oversimplification of reducing a lane to a 1D intensity profile, allowing for accurate band identification even under sub-optimal conditions like warped bands, high background, or diffuse bands [97]. This technology enables a single-click, highly consistent analysis pipeline that can be seamlessly integrated into researcher workflows.
Melting Curve Analysis
Core Principles and Workflow
High-Resolution Melting (HRM) curve analysis is a powerful, closed-tube technique that operates following the amplification phase of a real-time PCR. Its principle is based on the gradual denaturation (melting) of double-stranded DNA amplicons while monitoring the loss of fluorescence from a saturating DNA-binding dye, such as SYBR Green or EvaGreen [8] [98] [100]. The resulting plot of fluorescence as a function of temperature is the melting curve. The point of inflection of this curve, or the negative derivative, reveals the melting temperature (Tm), which is highly dependent on the amplicon's length, GC content, and most importantly, its sequence composition [98]. This allows HRM to discriminate between sequences that differ by even a single nucleotide polymorphism (SNP), making it invaluable for species identification, mutation detection, and genotyping [8] [99].
A key advantage of HRM over probe-based qPCR (e.g., TaqMan) is its cost-effectiveness, as it eliminates the need for expensive, sequence-specific fluorogenic probes and requires only a single detection channel present on most conventional real-time cyclers [98].
Detailed Experimental Protocol
Protocol: High-Resolution Melting (HRM) Curve Analysis
- Reaction Setup: Prepare a real-time PCR master mix containing all standard components: reaction buffer, dNTPs, MgCl₂, forward and reverse primers, DNA template, a thermostable DNA polymerase (e.g., HotStart Taq), and a saturating double-stranded DNA binding dye like SYBR Green or EvaGreen [8] [98].
- Amplification Phase: Run a standard real-time PCR protocol on a instrument capable of HRM, such as a Light Cycler 96 (Roche). This typically includes an initial denaturation, followed by 40-45 cycles of denaturation, annealing, and extension [8].
- High-Resolution Melting Phase: Immediately after amplification, the instrument performs the HRM step. The protocol is:
- Denature the amplicons at 95°C for a short period (e.g., 1 minute).
- Cool rapidly to a temperature below the expected Tm (e.g., 60°C) to re-form heterogenous double-stranded DNA.
- Gradually increase the temperature in small increments (e.g., 0.1-0.2°C) while continuously measuring the fluorescence. The temperature range should span from below the expected Tm to above the complete denaturation point (e.g., 60-95°C) [8].
- Data Analysis: Use the instrument's software to generate normalized and temperature-shifted melting curves or derivative melt peaks. Clustering of samples with identical sequences will be apparent, allowing for clear differentiation from variants [8] [99].
Advanced Innovation: Digital Microfluidics for HRM
Recent innovations are making HRM faster and more accessible. A prominent example is the development of a digital microfluidics (DMF) platform that uses a trapezoidal aluminum substrate to create a near-perfect linear temperature gradient [100].
This platform allows for spatial melting curve analysis, where a single droplet of PCR product is stretched into a thread across this predefined temperature gradient. Fluorescence is then captured in a single exposure, enabling the determination of Tm in seconds rather than minutes, and fully integrating nucleic acid amplification with subsequent MCA on a single chip to avoid contamination [100]. This technology has been successfully used to distinguish single-nucleotide mutations in KRAS gene targets with high accuracy [100].
Sequencing
Core Principles and Workflow
DNA sequencing is the definitive method for determining the precise nucleotide order of a DNA fragment. In the context of PCR analysis, it is most often Sanger sequencing that is used to validate amplicons. The principle involves using a single primer, dNTPs, and a small proportion of fluorescently labeled chain-terminating dideoxynucleotides (ddNTPs) in a PCR-like reaction. This generates a nested set of DNA fragments, each terminating at a specific base. These fragments are separated by capillary electrophoresis, and a laser detects the fluorescent label to produce a chromatogram, which is computationally translated into the DNA sequence.
Sequencing provides the highest level of resolution and is considered the gold standard for confirming the identity of a PCR product, identifying unknown mutations, or constructing phylogenetic trees [8].
Detailed Experimental Protocol
Protocol: Sanger Sequencing of PCR Amplicons
- PCR Product Purification: The initial PCR product must be purified to remove excess primers, dNTPs, and enzymes that can interfere with the sequencing reaction. This is typically done using enzymatic clean-up (e.g., ExoSAP-IT) or bead-based purification kits.
- Sequencing Reaction Setup: Set up the sequencing reaction using a kit such as BigDye Terminator. The reaction mix includes the purified PCR product, sequencing primer (typically one of the PCR primers), BigDye terminators, and buffer. The thermal cycling conditions involve rapid heating and cooling to generate the chain-terminated fragments.
- Post-Reaction Clean-up: Purify the sequencing reaction products to remove unincorporated dye terminators, usually via ethanol/sodium acetate precipitation or column-based methods.
- Capillary Electrophoresis: Load the purified samples into a sequencer. The machine injects the fragments into a capillary array, where they are separated by size. A laser excites the fluorescent dyes as fragments pass a detector.
- Data Analysis: The instrument's software generates an electropherogram (chromatogram) and the base-called sequence. This sequence can be analyzed using software like BLAST for identification or aligned with reference sequences for variant calling.
The Scientist's Toolkit
Table 2: Essential research reagents and materials for PCR analysis
| Item | Function/Application |
|---|---|
| HotStart DNA Polymerase | Reduces non-specific amplification and primer-dimer formation by inhibiting polymerase activity at low temperatures, improving assay specificity and yield [1] [5]. |
| Saturating DNA Binding Dye (e.g., SYBR Green, EvaGreen) | Essential for HRM analysis; fluoresces when bound to double-stranded DNA, allowing monitoring of DNA denaturation during melting [98]. |
| MgCl₂ | An essential cofactor for thermostable DNA polymerases; its concentration (typically 1.5-2.5 mM) must be optimized as it directly impacts enzyme activity and fidelity [5]. |
| dNTPs | The building blocks (dATP, dTTP, dCTP, dGTP) for DNA synthesis; all four should be present in equivalent concentrations (usually 200 µM each) for efficient amplification [5]. |
| PCR Additives (DMSO, BSA) | DMSO helps denature secondary structures in GC-rich templates. BSA can alleviate the effects of PCR inhibitors present in biological samples [5]. |
| DNA Molecular Weight Ladder | A mix of DNA fragments of known sizes, run alongside samples on a gel to estimate the size of unknown amplicons [97]. |
| Synthetic Plasmid Controls | Used as positive controls in species-specific PCR assays; contain cloned target sequences to ensure primer specificity and reaction efficiency [98]. |
Integrated Workflow and Method Selection
The relationship between the three analytical methods and their role in a typical experimental pipeline can be visualized as a logical workflow. This guides researchers from initial verification to ultimate confirmation.
Diagram 1: Logical workflow for PCR analysis methods
This workflow demonstrates how the methods are often used in concert. Gel electrophoresis serves as the initial, low-cost quality control check. A successful amplification then proceeds to HRM for rapid screening of sequence variations. If HRM indicates a potential variant or a mixed profile, the sample can be sent for sequencing, which provides definitive, base-pair level confirmation [8]. This integrated approach balances efficiency, cost, and resolution.
Gel electrophoresis, melting curve analysis, and sequencing form a complementary triad of techniques for the analysis of PCR products. Gel electrophoresis provides a foundational, accessible method for size-based separation. High-resolution melting curve analysis offers a powerful, cost-effective solution for rapid genotyping and variant scanning in a closed-tube system. Sequencing remains the unequivocal gold standard for definitive sequence validation. For new researchers, understanding the principles, protocols, and comparative value of each method is essential for designing robust and reliable experiments. The ongoing integration of AI and novel microfluidic platforms promises to further enhance the precision, speed, and accessibility of these core analytical techniques, solidifying their role in the future of molecular diagnostics and biomedical research.
For researchers embarking on molecular biology experiments, the polymerase chain reaction (PCR) serves as a fundamental technique for amplifying specific DNA sequences. However, not all PCR enzymes are created equal. The choice between standard and high-fidelity DNA polymerases can significantly impact the accuracy and success of downstream applications. This guide provides new researchers with a comprehensive framework for understanding PCR fidelity, offering detailed protocols for assessing error rates, and presenting actionable strategies for selecting the appropriate polymerase based on experimental goals. Within the broader context of PCR optimization, recognizing the trade-offs between speed, yield, and accuracy becomes paramount for designing robust and reproducible experiments.
Fundamental Differences Between Standard and High-Fidelity PCR
At the core of the distinction between standard and high-fidelity PCR is the biochemical mechanism by which the DNA polymerase ensures accurate DNA synthesis.
Standard PCR (e.g., Taq DNA Polymerase): This enzyme, isolated from Thermus aquaticus, is renowned for its robust amplification capability and high speed. However, it lacks a proofreading domain, which is a dedicated 3'→5' exonuclease activity that can excise misincorporated nucleotides during DNA synthesis. Consequently, Taq polymerase exhibits a relatively high error rate, typically in the range of 1x10⁻⁴ to 2x10⁻⁵ errors per base per duplication [37]. While sufficient for routine applications like genotyping or diagnostic assays where minor sequence variations are inconsequential, this error rate makes it unsuitable for cloning, protein expression studies, or sequencing where sequence integrity is critical.
High-Fidelity PCR (e.g., Pfu, KOD): High-fidelity polymerases are engineered for accuracy. They incorporate a proofreading function that actively scans and removes mismatched nucleotides immediately after incorporation. This proofreading capability drastically reduces the error rate by 10 to 50-fold compared to standard Taq polymerase, resulting in error rates as low as 1x10⁻⁶ to 4.5x10⁻⁷ errors per base per duplication [37]. A prime example is the PfuUltra II Fusion HS DNA polymerase, which features a genetically engineered Pfu DNA polymerase mutant, ArchaeMaxx polymerase enhancement factors, and a hot-start formulation to maximize accuracy and specificity for demanding downstream applications [101].
Table 1: Quantitative Comparison of Standard and High-Fidelity DNA Polymerases
| Feature | Standard Taq Polymerase | High-Fidelity Polymerase |
|---|---|---|
| Proofreading Activity | No | Yes (3'→5' exonuclease) |
| Typical Error Rate | 1x10⁻⁴ to 2x10⁻⁵ | 1x10⁻⁶ to 4.5x10⁻⁷ |
| Relative Fidelity | 1X | 10X to 50X higher |
| Primary Applications | Routine screening, genotyping, diagnostic assays | Cloning, sequencing, protein expression, gene therapy |
| Amplification Speed | Fast | Generally slower |
| Representative Enzymes | Taq DNA Polymerase | Pfu, KOD, PfuUltra II Fusion [37] [101] |
The following diagram illustrates the fundamental difference in the mechanism of action between a non-proofreading and a proofreading polymerase, highlighting the critical step that confers high fidelity.
Experimental Design for Assessing PCR Error Rates
Accurately determining the error rate of a PCR polymerase is a critical step in protocol validation, especially for applications requiring high sequence integrity. The following provides a detailed methodology for a classic lacI-based mutation assay, a reliable and widely accepted approach.
LacI PCR-based Mutation Assay Protocol
This assay measures the frequency of mutations introduced during PCR amplification of the lacI gene. Functional loss of the gene product serves as a selectable marker for mutations.
1. Materials and Reagents
- Template DNA: A plasmid containing the entire lacI gene.
- Test Polymerases: The standard and high-fidelity polymerases to be compared (e.g., Taq vs. PfuUltra II).
- Primers: Specifically designed to flank and amplify the full-length lacI gene.
- dNTPs: High-quality, purified deoxynucleotide triphosphate mix.
- PCR Buffers: Use the manufacturer's recommended buffers for each polymerase.
- Competent Cells: E. coli strains suitable for transformation and plating.
- Selection Plates: LB agar plates containing X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) and IPTG (isopropyl β-D-1-thiogalactopyranoside).
2. Step-by-Step Workflow
- Step 1: PCR Amplification. Amplify the lacI gene from the plasmid template using the two different polymerases under comparison. The reaction conditions (number of cycles, template amount, reaction volume) must be strictly identical for both to ensure a fair comparison. Use a minimum of 10 independent PCR reactions per polymerase to obtain statistically significant data.
- Step 2: Purification. Purify all PCR products to remove enzymes, salts, and unused dNTPs that could inhibit downstream steps. Use a reliable purification system, such as the PureLink PCR Purification Kit [102].
- Step 3: Ligation & Transformation. Ligate the purified PCR products back into an appropriate vector backbone. Transform the ligation products into highly competent E. coli cells. The transformation should be performed in duplicate or triplicate for accuracy.
- Step 4: Plating and Selection. Plate the transformed cells onto selection plates containing X-Gal and IPTG. Incubate the plates overnight at 37°C.
- Step 5: Data Collection and Analysis. Count the colonies the next day. Blue colonies contain a functional lacI gene (no inactivating mutations). White colonies contain a mutated, non-functional lacI gene.
- Step 6: Calculation. The mutation frequency is calculated as (Number of white colonies) / (Total number of colonies). The error rate can then be derived from the mutation frequency using established statistical models that account for the number of detectable sites in the lacI gene and the number of duplication events during PCR.
The workflow for this quantitative assay is summarized below:
Optimization Strategies for High-Fidelity PCR
Achieving the theoretical fidelity of a high-fidelity polymerase requires careful optimization of the reaction conditions. Meticulous adjustment of chemical, thermal, and design parameters is non-negotiable for robust and reproducible results [37].
Primer Design for Specificity and Fidelity
The quality of the oligonucleotide primers is the most significant determinant of reaction specificity and efficiency. Poorly designed primers lead directly to non-specific products and decreased fidelity [37].
- Primer Length: Optimal performance is typically observed with primers between 18 and 24 bases.
- Melting Temperature (Tm): The ideal Tm for primers should fall between 55°C and 65°C, with the forward and reverse primers closely matched (within 1-2°C).
- GC Content: Aim for a GC content between 40% and 60%.
- 3' End Stability: The last five bases at the 3' end should be rich in G and C bases to enhance stability and ensure efficient polymerase extension initiation.
- Avoid Secondary Structures: Utilize design software like Primer Express to predict and avoid primer-dimers and hairpins [103].
Calibration of the Annealing Temperature
The annealing temperature (Ta) is perhaps the most critical thermal parameter. Proper Ta calibration is the main tool used to minimize non-specific binding.
- Relationship to Tm: For most protocols, the optimal annealing temperature is 3–5°C below the calculated Tm of the primers.
- Gradient PCR: The most efficient method for determining the optimal Ta is to perform a gradient PCR, running the same reaction across a range of annealing temperatures (e.g., 50–68°C) and analyzing the results by gel electrophoresis for the strongest, most specific band.
Magnesium and Buffer Optimization
- Mg²⁺ Concentration: Magnesium ions are an essential cofactor for all DNA polymerases. The typical optimal concentration ranges from 1.5 to 2.5 mM for a standard reaction, but this must be optimized for each primer-template system. Fine-tuning the Mg²⁺ concentration, often by titrating a MgCl₂ solution, is a fundamental step [37].
- Buffer Additives: For challenging templates, such as those with high GC content (>65%), additives can be crucial.
- DMSO (2–10%) helps resolve strong secondary structures.
- Betaine (1–2 M) homogenizes the thermodynamic stability of GC- and AT-rich regions.
Essential Reagents for High-Fidelity PCR
A successful high-fidelity PCR experiment relies on a suite of optimized reagents. The following toolkit lists key materials and their critical functions.
Table 2: Research Reagent Solutions for High-Fidelity PCR
| Reagent / Kit | Function / Application | Key Characteristics |
|---|---|---|
| PfuUltra II Fusion HS DNA Polymerase [101] | High-fidelity amplification for cloning, sequencing. | Proofreading activity, hot-start, enhanced processivity. |
| PureLink PCR Purification Kit [102] | Post-amplification cleanup of DNA. | Removes enzymes, salts, dNTPs; essential for downstream steps. |
| MagMAX Nucleic Acid Extraction Kits [102] | Automated purification of high-quality template DNA. | Superior binding capacity; removes PCR inhibitors. |
| Dynabeads [102] | Target capture and purification (e.g., for NGS). | Low non-specific binding; high reproducibility. |
| Primer Express Software [103] | In-silico design of specific primers and probes. | Predicts Tm, secondary structures; optimizes for multiplexing. |
| dNTPs, MgCl₂ Solution [37] | Building blocks and essential cofactor for DNA synthesis. | High-purity; concentration must be optimized. |
| DMSO / Betaine [37] | Buffer additives for difficult templates (GC-rich). | Improves yield and specificity by resolving secondary structures. |
The strategic selection between standard and high-fidelity PCR is a fundamental decision that directly dictates the success of downstream applications. Standard Taq polymerase offers speed and robustness for analytical and diagnostic workflows where ultimate sequence accuracy is not critical. In contrast, high-fidelity polymerases, with their intrinsic proofreading activity, are indispensable for cloning, protein expression, and next-generation sequencing, where even a single mutation can compromise experimental results. By integrating the principles of primer design, reaction optimization, and a rigorous assessment of error rates as outlined in this guide, researchers can make informed, strategic choices. This systematic approach to PCR optimization ensures the generation of high-quality, reliable data, forming a solid foundation for advanced molecular biology research and drug development.
Respiratory infections represent a significant global health burden, causing substantial morbidity and mortality worldwide [95]. The clinical presentation of respiratory infections caused by pathogens such as SARS-CoV-2, influenza viruses, and respiratory syncytial virus (RSV) shows significant overlap, making syndromic diagnosis challenging based on symptoms alone [104]. The COVID-19 pandemic further complicated the epidemiological landscape of respiratory viruses, disrupting their predictable seasonal patterns and leading to unusual circulation peaks in subsequent periods [105].
Traditional diagnostic methods for respiratory pathogens, including viral culture and serological tests, have limitations in sensitivity, specificity, and turnaround time [95]. These challenges are particularly pronounced during seasonal outbreaks when multiple pathogens co-circulate simultaneously, a phenomenon dubbed the "tripledemic" when involving SARS-CoV-2, influenza, and RSV [104]. Molecular diagnostic techniques, especially polymerase chain reaction (PCR), have revolutionized respiratory pathogen detection by offering high sensitivity, specificity, and speed [95].
This case study focuses on the clinical validation of a novel fluorescence melting curve analysis-based (FMCA) multiplex PCR assay for simultaneous detection of six respiratory pathogens, framed within the broader context of PCR optimization for new researchers. We present comprehensive analytical validation, clinical performance metrics, and implementation considerations to guide researchers in developing robust molecular diagnostics for respiratory infections.
Materials and Methods
Assay Design and Principle
The novel multiplex PCR assay was designed to simultaneously detect six major respiratory pathogens: SARS-CoV-2, influenza A virus (IAV), influenza B virus (IBV), respiratory syncytial virus (RSV), human adenovirus (hADV), and Mycoplasma pneumoniae (MP) [95]. These pathogens were selected based on their clinical significance and frequent co-circulation during respiratory infection seasons.
The assay employs fluorescence melting curve analysis (FMCA), which leverages the unique melting temperatures (Tm) of specific hybridization probes bound to their complementary DNA sequences during PCR [95]. By analyzing distinct Tm profiles, the FMCA platform can differentiate between multiple pathogens in a single reaction tube, providing a streamlined approach for multiplex pathogen detection. To enhance robustness across pathogen subtypes, probes were modified with base-free tetrahydrofuran (THF) residues at positions corresponding to known or potential base mismatches, minimizing their impact on probe melting temperature and improving hybridization stability across variant strains [95].
Table 1: Target Pathogens and Genetic Markers for Multiplex PCR Assay
| Pathogen | Target Genes | Clinical Significance |
|---|---|---|
| SARS-CoV-2 | Envelope protein (E) and nucleocapsid phosphoprotein (N) genes | Primary causative agent of COVID-19 with continued seasonal circulation |
| Influenza A Virus (IAV) | Matrix protein (M) gene | Seasonal epidemics with potential for pandemic spread |
| Influenza B Virus (IBV) | Nonstructural protein 1 (NS1) gene | Seasonal influenza causing significant morbidity |
| Respiratory Syncytial Virus (RSV) | Matrix protein (M) gene | Major cause of bronchiolitis and pneumonia in children and elderly |
| Human Adenovirus (hADV) | Hexon gene | Causes respiratory illnesses ranging from mild to severe |
| Mycoplasma pneumoniae | CARDS toxin gene | Atypical bacterium causing "walking pneumonia" |
Primer and Probe Design
Specific primers and probes were designed based on conserved genomic regions of each target pathogen using Primer Premier 5 and Primer Express 3.0.1 software [95]. Sequences were verified for specificity using the NCBI BLAST tool against the comprehensive database. The primer and probe design process adhered to fundamental PCR optimization principles:
- Primer length was maintained at 15-30 nucleotides [15]
- GC content was optimized to 40-60% for appropriate melting temperatures [15]
- The 3' ends of primers included G or C bases to increase priming efficiency and prevent "breathing" of ends [15] [106]
- Melting temperatures (Tm) for primer sets were designed to differ by no more than 5°C [15]
- Di-nucleotide repeats and single base runs were avoided to prevent secondary structure formation [15]
Laboratory Validation Methods
Analytical Sensitivity and Limit of Detection
The limit of detection (LOD) was determined using probit analysis, defined as the lowest concentration detectable with ≥95% probability [95]. Each dilution was assessed in 20 replicates to ensure statistical robustness. The LOD was established using quantified reference materials and international standards where available.
For comparative assays, analytical sensitivity was evaluated using digital PCR-based standards and reference materials to establish the lower limit of detection (LoD) with 95% probability [107]. This approach provides absolute quantification without requiring standard curves, enhancing measurement precision.
Analytical Specificity
Cross-reactivity testing was performed using a panel of non-target respiratory pathogens, including 10 respiratory viruses and 4 bacteria, to confirm assay specificity [95]. Additionally, 47 reference strains of different subtypes of the target pathogens were obtained from the National Institutes for Food and Drug Control (NIFDC) and BeNa Culture Collection (BNCC) to evaluate assay inclusivity across genetic variants.
Precision and Reproducibility
Assay precision was evaluated through both intra-assay (repeatability) and inter-assay (reproducibility) testing [95]. Two concentrations of mixed plasmids containing viral target fragment sequences (5× LOD and 2× LOD for each target) were used for precision assessment. Intra-assay variability was determined by analyzing each concentration five times in a single run, while inter-assay variability was assessed by testing each concentration five times in separate runs conducted by different operators on different days.
Clinical Performance Evaluation
Clinical validation was conducted through a prospective single-center study at Shaanxi Provincial Hospital of Tuberculosis Prevention and Treatment Hospital from October 2023 to February 2024 [95]. A total of 1005 nasopharyngeal swabs were collected from patients presenting with symptoms of acute respiratory infections. All samples were transported in viral transport media and processed using automated nucleic acid extraction systems.
For comparison, two commercial RT-qPCR kits (Sansure Biotech, China) approved by China's National Medical Products Administration (NMPA) were used as reference methods [95]. Discordant results between the novel FMCA assay and reference methods were resolved through Sanger sequencing, providing a robust mechanism for assessing true positive and negative results.
Workflow Visualization
Figure 1: Experimental Workflow for Multiplex PCR Assay. The process begins with sample collection, followed by automated nucleic acid extraction, PCR setup with asymmetric primer design, thermocycling, melting curve analysis, and final pathogen identification based on specific melting temperatures (Tm).
Results
Analytical Performance
The novel FMCA-based multiplex PCR assay demonstrated high analytical sensitivity with limits of detection (LOD) ranging between 4.94 and 14.03 copies/μL across the six target pathogens [95]. This sensitivity is comparable to conventional single-plex RT-PCR assays while providing the advantage of multiplex detection.
The assay showed exceptional precision with intra-assay and inter-assay coefficients of variation (CVs) ≤0.70% and ≤0.50%, respectively [95]. These low CV values indicate high reproducibility, essential for clinical implementation where consistent performance across operators and testing days is critical.
Specificity testing revealed no cross-reactivity with non-target respiratory pathogens, confirming the primer and probe design specificity [95]. The assay successfully detected all tested subtypes of the target pathogens, demonstrating robust inclusivity across genetic variants.
Table 2: Analytical Performance Metrics of Multiplex PCR Assay
| Performance Parameter | Result | Method of Assessment |
|---|---|---|
| Limit of Detection (LOD) | 4.94-14.03 copies/μL | Probit analysis (≥95% detection) |
| Intra-assay Precision | CV ≤ 0.70% | 5 replicates at 2 concentrations in single run |
| Inter-assay Precision | CV ≤ 0.50% | 5 replicates at 2 concentrations across separate runs |
| Analytical Specificity | No cross-reactivity | Testing against 10 respiratory viruses and 4 bacteria |
| Inclusivity | Detected all subtypes | 47 reference strains of target pathogens |
| Turnaround Time | 1.5 hours | Sample collection to result |
| Cost per Sample | $5 | Reagent and consumable calculation |
Clinical Validation
Clinical evaluation using 1005 samples demonstrated 98.81% overall agreement with reference RT-qPCR methods [95]. The assay identified pathogens in 51.54% of cases, reflecting the high prevalence of respiratory infections during the study period (October 2023 to February 2024). Notably, the assay detected co-infections in 6.07% of positive cases, highlighting the importance of multiplex testing in identifying complex infection patterns that might be missed with single-plex assays.
Resolution of discordant results through Sanger sequencing confirmed the superior sensitivity of the FMCA assay in low viral load scenarios, where it correctly identified several true positive cases that were missed by the reference methods [95]. This enhanced sensitivity at low pathogen concentrations is particularly valuable for early infection detection and in immunocompromised patients who may present with lower viral loads.
Comparative studies with other multiplex platforms have shown similar high performance. One evaluation of three PCR-based platforms demonstrated 100% concordance between laboratory-developed tests and FDA-approved comparator methods (BioFire RP2.1) for detecting SARS-CoV-2, influenza A/B, and RSV [104].
Comparative Assay Performance
Multiplex PCR panels for respiratory pathogens have demonstrated superior detection rates compared to traditional methods. One study comparing the BioFire FilmArray Pneumonia Panel with bacterial culture found a significantly higher positivity rate (60.3% vs. 52.8%) while showing substantial concordance (77.2%) with culture results [108]. This enhanced detection capability is particularly valuable for identifying viral pathogens and bacterial organisms that are difficult to culture.
Another laboratory-developed modular multiplex-PCR panel for 16 respiratory viruses demonstrated excellent analytical sensitivity, with LoDs ranging from 9.4 cp/mL for hCoV-NL63 to 21,419 cp/mL for HPIV-2 [107]. The assay showed strong linearity over 4-7 log steps and high precision, with inter- and intra-run variability ranging between 0.13-0.74 Ct [107].
Table 3: Comparison of Multiplex PCR Platforms for Respiratory Pathogen Detection
| Platform/Assay | Targets | Clinical Sensitivity | Clinical Specificity | Turnaround Time | Key Features |
|---|---|---|---|---|---|
| FMCA-based Multiplex PCR [95] | 6 pathogens | 98.81% agreement with reference PCR | 100% by sequencing | 1.5 hours | Cost-effective ($5/sample) |
| BioFire RP2.1 Panel [104] | 4 pathogens | 100% concordance with LDTs | 100% concordance with LDTs | ~1 hour | FDA-approved, ease of use |
| TrueMark Select Panel [104] | 4 pathogens | 100% concordance with other methods | 100% concordance with other methods | ~1 hour | Laboratory-developed test |
| OpenArray Platform [104] | 32 targets | 100% concordance for key pathogens | 100% concordance for key pathogens | ~2 hours | High-throughput expanded panel |
| Modular Lab-developed Panel [107] | 16 viruses | 99.4% for SARS-CoV-2, 95% for influenza A | 96.3-100% | ~2 hours | Automated high-throughput |
Discussion
Technical Optimization Considerations
The successful development and validation of the FMCA-based multiplex PCR assay exemplifies several key principles in PCR optimization. The use of asymmetric PCR with unequal primer ratios favored the production of single-stranded DNA, reducing competition from complementary strands and facilitating more efficient probe-target hybridization during melting curve analysis [95]. This technical refinement enhanced the resolution of melting peaks, crucial for accurate differentiation of multiple pathogens in a single reaction.
Primer and probe design played a critical role in assay performance. The incorporation of base-free tetrahydrofuran (THF) residues at positions corresponding to potential mismatches minimized the impact of genetic variation on probe melting temperatures, ensuring robust detection across pathogen subtypes [95]. This approach aligns with advanced primer design strategies that account for sequence diversity while maintaining hybridization efficiency.
Reaction optimization followed established PCR principles, including careful adjustment of primer concentrations (typically 0.4-0.5 μM) to balance amplification efficiency with specificity [1]. The use of hot-start DNA polymerase prevented non-specific amplification during reaction setup, while optimized magnesium concentrations supported efficient amplification across all targets despite their sequence differences.
Applications in Respiratory Infection Management
The implementation of multiplex PCR assays for respiratory pathogen detection has transformed clinical management of respiratory infections. During the COVID-19 pandemic, SARS-CoV-2 dominated respiratory infections, accounting for 65.5% of positive cases [105]. However, in the post-pandemic period, viral circulation has shifted notably toward other pathogens, particularly Rhinovirus/Enterovirus (71.5% of positive samples) [105]. This epidemiological transition underscores the importance of comprehensive testing beyond SARS-CoV-2.
Multiplex PCR panels enable accurate pathogen identification in cases with overlapping clinical presentations, guiding appropriate therapy selection and antimicrobial stewardship. The detection of viral co-infections (6.07% in the FMCA assay validation) is particularly important for understanding disease severity and tailoring management strategies [95]. Studies have shown that co-infections occur more frequently in children (14.1%) than adults (2.7%), highlighting the value of comprehensive testing in pediatric populations [105].
Practical Implementation Considerations
The FMCA-based assay offers significant cost advantages at $5 per sample, representing an 86.5% reduction compared to commercial multiplex PCR kits [95]. This cost-effectiveness makes sophisticated multiplex testing accessible in resource-limited settings where respiratory infection burden is often highest.
The rapid turnaround time of 1.5 hours enables timely clinical decision-making for infection control measures and treatment selection [95]. This represents a substantial improvement over traditional culture methods that require 24-48 hours for results and separate tests for different pathogen types.
High-throughput capability allows processing of large sample volumes during seasonal outbreaks when testing demand peaks. Automated nucleic acid extraction and standardized analysis pipelines further enhance implementation feasibility in busy clinical laboratories [95] [107].
Research Reagent Solutions
Table 4: Essential Research Reagents for Multiplex PCR Assay Development
| Reagent/Category | Specific Examples | Function in Assay | Optimization Considerations |
|---|---|---|---|
| DNA Polymerase | One Step U* Enzyme Mix [95], Hieff Ultra-Rapid II HotStart PCR Master Mix [1] | Catalyzes DNA amplification; hot-start prevents non-specific amplification | Thermostability, processivity, fidelity; hot-start modification critical for multiplex reactions |
| Fluorescent Probes | THF-modified probes [95], TaqMan probes [109] | Target detection through hybridization and fluorescence | Tm optimization, dye selection, modification for mismatch tolerance |
| Primers | Custom-designed primers [95] | Sequence-specific amplification | Length (15-30 nt), Tm (52-58°C), GC content (40-60%), avoid dimers and secondary structures |
| Sample Collection Media | Viral Transport Medium (VTM) [95] [109] | Preserves sample integrity during transport | Compatibility with downstream extraction, stability characteristics |
| Nucleic Acid Extraction Kits | MPN-16C RNA/DNA extraction kit [95], MagMAX Viral/Pathogen kits [104] | Isolate nucleic acids from clinical samples | Yield, purity, removal of inhibitors, automation compatibility |
| Reference Materials | ATCC strains [110], international standards [107] | Assay validation and quality control | Well-characterized concentration, commutability with clinical samples |
This case study demonstrates the successful development and validation of a novel FMCA-based multiplex PCR assay for detection of six respiratory pathogens. The assay exhibits high sensitivity, specificity, and precision while offering significant advantages in cost-effectiveness and turnaround time compared to commercial alternatives.
The clinical validation with 1005 samples confirmed excellent agreement with reference methods (98.81%) and identified a substantial rate of co-infections (6.07%), highlighting the clinical value of comprehensive multiplex testing. The resolution of discordant results by Sanger sequencing established the assay's superior performance in low viral load scenarios.
For researchers entering the field of PCR optimization, this case study illustrates critical success factors: meticulous primer and probe design, incorporation of technical enhancements such as asymmetric PCR and base modifications, and rigorous analytical validation using international standards. The FMCA platform represents a flexible foundation that can be adapted to include emerging variants and additional pathogens, strengthening global preparedness for future respiratory epidemics.
As respiratory virus epidemiology continues to evolve in the post-pandemic era, multiplex PCR assays will play an increasingly important role in clinical diagnostics, infection control, and public health surveillance. The optimization principles and validation approaches detailed in this case study provide a roadmap for developing robust molecular diagnostics that meet the challenges of respiratory pathogen detection.
The choice between implementing laboratory-developed tests (LDTs) and commercial kits represents a critical decision point in molecular diagnostics, particularly in the context of PCR optimization. This technical guide provides a comprehensive cost-benefit analysis framed within the broader thesis of introducing new researchers to PCR optimization. We examine key performance metrics, validation requirements, and practical considerations for both testing approaches, drawing on recent evidence from infectious disease diagnostics. The analysis reveals that while commercial kits offer standardized, regulatory-compliant solutions, LDTs provide flexibility and cost advantages that remain essential for responding to emerging threats and specialized testing needs. This whitepaper equips researchers and drug development professionals with a structured framework for selecting the optimal testing approach based on their specific diagnostic context, resource constraints, and performance requirements.
The emergence of novel pathogens and the continuous evolution of existing ones necessitate robust, adaptable diagnostic solutions. In response, two primary testing paradigms have emerged: commercial diagnostic kits and laboratory-developed tests (LDTs). Commercial kits are in vitro diagnostic products developed, manufactured, and sold by commercial entities, typically featuring standardized protocols and regulatory approvals (e.g., CE marking or FDA authorization). In contrast, LDTs are laboratory-developed tests designed, validated, and implemented within individual laboratory settings to address specific, often localized, diagnostic needs [111].
The COVID-19 pandemic highlighted the complementary roles of both approaches. While commercial kits enabled rapid diagnostic rollout during the initial emergency response, LDTs provided crucial early detection capabilities when commercial options were unavailable or unsuitable [111]. For instance, the first SARS-CoV-2 LDT was published just weeks after the initial genome sequence was deposited in public databases, demonstrating the agility of laboratory-developed solutions [111].
Understanding the cost-benefit tradeoffs between these approaches is fundamental to PCR optimization, affecting everything from assay design and validation to implementation and quality assurance. This analysis provides new researchers with a structured framework for evaluating these competing testing platforms across multiple dimensions, including analytical performance, development timeline, regulatory compliance, and economic considerations.
Comparative Analysis: Key Performance Metrics
Analytical Performance
Independent evaluations demonstrate that both properly validated LDTs and commercial kits can achieve high analytical performance, though significant variability exists within both categories.
A large-scale evaluation of 185 commercial SARS-CoV-2 antigen lateral flow devices revealed a 35% overall pass rate through a rigorous three-phase evaluation process, with only 5 of the 1017 initially screened kits ultimately being procered for the UK National COVID-19 Testing Programme [112]. This highlights the importance of independent verification even for commercially marketed products. Similarly, a comparison of seven commercial RT-PCR kits found that while all demonstrated acceptable performance for routine diagnostics, the estimated 95% limit of detection varied within a 6-fold range, and detection rates using clinical samples showed some inter-kit variations [113].
For LDTs, performance is highly dependent on validation rigor. One study of extraction-free SARS-CoV-2 RT-PCR protocols found that optimized LDTs could achieve 84.26% sensitivity compared to standard extraction-based methods, with a mean increase in cycle threshold value of +3.8 [114]. The same study demonstrated that methodological choices significantly impact performance, with sample dilution combined with heat treatment increasing sensitivity by 38% compared to heat treatment alone [114].
Table 1: Analytical Performance Comparison of Commercial SARS-CoV-2 Detection Methods
| Evaluation Metric | Commercial Kits (SARS-CoV-2) | LDTs (SARS-CoV-2) |
|---|---|---|
| Overall Pass Rate | 35% (185/1017 kits) [112] | N/A |
| Sensitivity Range | Variable between kits [113] | 84.26% (optimized extraction-free protocol) [114] |
| Specificity | High when properly validated [113] | Protocol-dependent [114] |
| Limit of Detection | 6-fold variation between kits [113] | Dependent on optimization [114] |
| Cross-reactivity | Properly designed kits show minimal cross-reactivity [113] | Must be rigorously validated during development [111] |
Development Timeline and Responsiveness
A critical advantage of LDTs is their potential for rapid deployment in response to emerging threats. The first SARS-CoV-2 LDT was published on January 23, 2020, just weeks after the initial genome sequence was made publicly available [111]. This rapid response capability is particularly valuable during the early stages of disease outbreaks when commercial kits may not yet be available or sufficiently validated.
Commercial kit development typically follows a more protracted timeline due to manufacturing scaling, quality control processes, and regulatory compliance requirements. However, once established, commercial kits can be rapidly distributed and implemented across multiple laboratory settings, providing a standardized approach to large-scale testing initiatives.
The evaluation pipeline for commercial tests can be streamlined through structured approaches. The UK's rapid evaluation programme for SARS-CoV-2 antigen tests established a three-phase process including desktop review, laboratory testing with cultured virus, and clinical sample validation, enabling evidence-based procurement decisions [112]. Such structured approaches facilitate the timely assessment of commercial tests during public health emergencies.
Regulatory and Quality Assurance Considerations
Regulatory oversight represents a fundamental distinction between commercial kits and LDTs. Commercial kits typically carry CE marking or FDA approval, providing a baseline level of performance validation [111]. However, it is important to note that "CE marking is only a declaration of compliance with European legislative requirements; it does not necessarily guarantee the rigorous validation of the assay" [111].
For LDTs, regulatory requirements vary significantly by jurisdiction. In the United States, CLIA regulations require laboratories to verify manufacturer's performance specifications when implementing FDA-cleared tests, while modified tests or LDTs require establishments of analytical sensitivity and specificity [111]. In Europe, the IVD Regulations (EU) 2017/746 impose specific requirements for laboratory-developed tests [111].
Quality assurance should follow established guidelines regardless of test type. The MIQE guidelines (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) provide a comprehensive framework for ensuring PCR assay quality and transparency [111]. Similarly, the STARD initiative (Standards for Reporting of Diagnostic Accuracy) improves reporting standards for diagnostic assays [111].
Economic Considerations
Direct and Indirect Costs
The economic analysis of LDTs versus commercial kits extends beyond simple per-test cost comparisons to include both direct and indirect expenditures across the test lifecycle.
Table 2: Cost Structure Comparison Between Commercial Kits and LDTs
| Cost Component | Commercial Kits | LDTs |
|---|---|---|
| Per-test Reagent Cost | Higher (includes manufacturer markup) | Lower (direct reagent cost only) |
| Development Cost | Borne by manufacturer | Borne by laboratory |
| Validation Cost | Primarily verification | Comprehensive validation required |
| Equipment Cost | Platform-specific | Potentially more flexible |
| Personnel Cost | Lower (standardized protocols) | Higher (expertise-intensive) |
| Quality Control | Structured by manufacturer | Laboratory-designed and implemented |
| Regulatory Compliance | Pre-market approval | Laboratory responsibility |
Commercial kits typically involve higher per-test reagent costs but lower personnel requirements due to standardized protocols and manufacturer support. For example, one study noted that commercial kits are "more expensive than LDTs" but "enable the rapid introduction of assays" [111]. The same source noted that the cost advantage of LDTs is particularly pronounced for "small-scale, specialist tests targeting rarely occurring infectious pathogens" [111].
For LDTs, the significant development, validation, and quality control costs must be amortized across the total number of tests performed. One study of extraction-free SARS-CoV-2 testing noted that "the average cost incurred per sample using extraction kits falls within the range of 6–12 USD, while the cost associated with the application of specific reagents such as Proteinase K and RNase inhibitors in the conducted experiments amounts to merely 1.15 USD per sample" [114]. This substantial cost differential highlights the potential economic advantage of well-designed LDTs.
Cost-Benefit Optimization Strategies
Several strategies can optimize the cost-benefit ratio regardless of test platform selection:
Extraction-free protocols: For LDTs, simplified sample processing can significantly reduce both cost and turnaround time. One optimized extraction-free protocol incorporating heat treatment, sample dilution, and reagent additions reduced per-test costs by approximately 80% compared to commercial extraction kits [114].
Process efficiency: For commercial kits, leveraging manufacturer training resources and technical support can reduce personnel costs and implementation time [115].
Preventive quality control: Implementing rigorous initial validation and continuous monitoring processes reduces costly errors and retesting for both platforms [111].
Strategic platform selection: Using commercial kits for high-volume, standardized testing while reserving LDTs for specialized, low-volume applications can optimize resource allocation [111].
Methodological Protocols
Protocol 1: LDT Development and Validation
The development and validation of laboratory-developed tests requires a systematic approach to ensure reliability and accuracy [111].
Figure 1: LDT Development and Validation Workflow. This process ensures laboratory-developed tests meet performance requirements before clinical implementation.
Step 1: Define Purpose and Requirements
Clearly establish the clinical or research purpose of the assay, including target pathogens, sample types, and required performance characteristics. This foundational step guides all subsequent development decisions [111].
Step 2: Develop Validation Plan
Create a comprehensive plan outlining the validation approach, including sample requirements, acceptance criteria, and methods for resolving discrepant results. The plan should address "the biological, technical and operator-related factors that affect the assay's ability to detect the target in the specific sample-type" [111].
Step 3: Verify Individual Components
Assess each assay component (primers, probes, enzymes, buffers) against established performance requirements. For PCR assays, this includes optimizing primer sequences, concentrations, and thermal cycling conditions [111].
Step 4: Analytical Validation
Establish analytical performance characteristics including:
- Analytical sensitivity (LoD): Determine the lowest concentration of analyte that can be reliably detected using serial dilutions of reference materials [111].
- Analytical specificity: Evaluate cross-reactivity with related pathogens or interfering substances [111].
- Precision and reproducibility: Assess intra-assay and inter-assay variability [111].
- Reportable range: Establish the quantitative range of the assay [111].
Step 5: Clinical Validation
Validate assay performance using well-characterized clinical samples. "Typically, 100 samples of 50-80 positive and 20-50 negative specimens are used" [111]. When genuine clinical samples are limited, "it may be necessary to construct test samples by spiking various concentrations of the analyte into a suitable matrix" [111].
Step 6: Implementation and Quality Monitoring
Implement the validated assay with continuous quality monitoring, including internal controls, proficiency testing, and periodic revalidation to detect assay drift or emerging interferences [111].
Protocol 2: Commercial Kit Verification
Even for CE-marked or FDA-approved commercial tests, laboratories must verify manufacturer performance claims under local conditions [111].
Figure 2: Commercial Kit Verification Workflow. This process ensures manufacturer claims are met under local laboratory conditions.
Step 1: Verify Accuracy
Compare results from the commercial kit to a reference method or established comparator using well-characterized samples. For modified commercial assays, also verify "analytical sensitivity to include inhibitory substances" [111].
Step 2: Verify Precision
Assess intra-assay and inter-assay precision using multiple replicates across different runs, operators, and equipment when applicable [111].
Step 3: Establish Reportable Range
Confirm the range of analyte concentrations that can be reliably measured, verifying the manufacturer's claims across the entire measuring interval [111].
Step 4: Verify Reference Intervals
Validate any reference intervals or cut-off values provided by the manufacturer using local population samples when appropriate [111].
Step 5: Implementation with Quality Control
Implement the verified test with appropriate quality control measures, including "continually monitoring the levels of internal and external positive controls to ensure the validation status of the assay is maintained" [111].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Molecular Test Development and Optimization
| Reagent/Category | Function in Test Development | Application Examples |
|---|---|---|
| Proteinase K | Digests proteins and inactivates nucleases | Improves viral nucleic acid accessibility in extraction-free protocols [114] |
| RNase Inhibitors | Protects RNA from degradation | Preserves RNA integrity in extraction-free methods [114] |
| Primer/Probe Sets | Target-specific sequence recognition | Ultra-conserved element targeting for variant-resistant assays [116] |
| Taq Polymerase | DNA amplification | PCR core enzyme; requires optimization of concentration and buffer conditions [117] |
| dNTPs | Building blocks for DNA synthesis | Essential PCR component; concentration affects efficiency and specificity [117] |
| Buffer Systems | Maintain optimal reaction conditions | Composition affects specificity, efficiency, and inhibitor resistance [111] |
| Internal Controls | Monitor reaction efficiency and inhibition | Distinguish true negatives from assay failures [111] |
| Standard Reference Materials | Calibration and quality assurance | Enables quantitative measurements and inter-laboratory comparisons [111] |
The cost-benefit analysis between laboratory-developed tests and commercial kits reveals a nuanced landscape where neither approach universally dominates. Rather, the optimal choice depends on specific application requirements, resource constraints, and performance expectations.
Commercial kits offer significant advantages in settings requiring standardization, rapid implementation, and regulatory compliance. Their pre-validated nature, technical support, and quality-controlled manufacturing make them particularly valuable for high-volume routine testing and laboratories with limited method development expertise. However, their higher per-test costs and potential lag in responding to emerging threats represent significant limitations.
LDTs provide compelling benefits in scenarios demanding specialized applications, rapid response capabilities, and cost-sensitive high-volume testing. Their flexibility enables customization for specific research questions or local pathogen variants, while their lower reagent costs offer significant economic advantages at scale. These benefits come with substantial requirements for technical expertise, comprehensive validation, and continuous quality management.
For new researchers embarking on PCR optimization, this analysis suggests a contingency-based approach to test selection: leveraging commercial kits for standardized, high-throughput applications while developing LDT capabilities for specialized needs and emergency response. This dual-capability model ensures both operational efficiency and strategic flexibility in an evolving diagnostic landscape.
Future developments in molecular diagnostics, including point-of-care testing, multiplexed pathogen detection, and artificial intelligence-assisted assay design, will continue to reshape the cost-benefit calculus between these platforms. However, the fundamental tradeoffs between standardization and flexibility, and between convenience and cost, will likely remain central to test selection decisions for the foreseeable future.
Conclusion
Mastering PCR optimization is a critical skill that bridges fundamental molecular biology and advanced clinical diagnostics. By understanding core principles, applying specialized methods for challenging templates, systematically troubleshooting failures, and rigorously validating results, researchers can ensure their data is both reliable and reproducible. The future of PCR lies in the development of more robust, multiplexed, and cost-effective assays, as demonstrated by their pivotal role in infectious disease surveillance. The strategies outlined in this guide provide a solid foundation for new researchers to contribute to these advancements, driving progress in drug development, personalized medicine, and global public health.
References
- 1. Free Guide: Top 5 PCR Optimization Tips - Yeasen [https://www.yeasenbio.com/blogs/molecular-biology/free-guide-top-5-pcr-optimization-tips]
- 2. Polymerase Chain Reaction (PCR) - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK589663/]
- 3. Polymerase Chain Reaction (PCR) Process Steps [https://www.coleparmer.com/tech-article/pcr-process-steps-explained]
- 4. PCR Cycling Parameters—Six Key Considerations for ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html]
- 5. Optimizing PCR: Proven Tips and Troubleshooting Tricks [https://www.the-scientist.com/optimizing-pcr-proven-tips-and-troubleshooting-tricks-71660]
- 6. Polymerase Chain Reaction [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/pcr/polymerase-chain-reaction?srsltid=AfmBOorHIABf9RHrCXcTa4jLsv8KsXHSyjTLrFD0wmNT-tO0SZbYm0fV]
- 7. PCR/qPCR Data Analysis [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/genomics/qpcr/data-analysis?srsltid=AfmBOoq_74uuwXJ6sSk6dxIoRNP-hxF-aLYX1CPRJm20P3yi5pz2apDk]
- 8. Optimization of the real-time PCR platform method using ... [https://www.nature.com/articles/s41598-025-16972-9]
- 9. Integrating biotechnology into diagnostic labs: Innovations ... [https://www.sciencedirect.com/science/article/pii/S2590262825000565]
- 10. A roadmap to high-speed polymerase chain reaction (PCR) [https://www.sciencedirect.com/science/article/pii/S0956566323007728]
- 11. Polymerase Chain Reaction (PCR) - NCBI - NIH [https://www.ncbi.nlm.nih.gov/probe/docs/techpcr/]
- 12. Polymerase chain reaction [https://en.wikipedia.org/wiki/Polymerase_chain_reaction]
- 13. PCR Setup—Six Critical Components to Consider [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-component-considerations.html]
- 14. What are PCR primers? [https://www.minipcr.com/what-are-pcr-primers/]
- 15. Polymerase Chain Reaction: Basic Protocol Plus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4846334/]
- 16. dNTPs and Primers in PCR [https://www.bocsci.com/resources/dntps-and-primers-in-pcr.html?srsltid=AfmBOop9-Vw7h7JBwuIyN5JYaJ8N9EWrhprv6OUWeIMiyCHnTIytHuS6]
- 17. dNTPs: Structure, Role & Applications [https://www.baseclick.eu/science/glossar/dntps/]
- 18. PCR Additives & Buffer Selection [https://www.aatbio.com/resources/application-notes/pcr-additives-buffer-selection]
- 19. Using dNTP in Polymerase Chain Reaction (PCR) [https://www.biochain.com/blog/using-dntp-in-polymerase-chain-reaction-pcr/]
- 20. DNA Polymerase—Four Key Characteristics for PCR [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/dna-polymerase-characteristics.html]
- 21. What does DNA polymerase fidelity mean? [https://www.neb.com/en-us/tools-and-resources/feature-articles/polymerase-fidelity-what-is-it-and-what-does-it-mean-for-your-pcr?srsltid=AfmBOop4ZWHN4mvjT2ZMwJB5VYyi6345lt9H88051-7a88gMCqXrl9CZ]
- 22. Error Rate Comparison during Polymerase Chain Reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4150459/]
- 23. How is Hot-Start Technology Beneficial For Your PCR [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/hot-start-technology-benefits-PCR.html]
- 24. Hot-Start PCR: Higher specificity, higher yield, ... [https://www.genaxxon.com/shop/en/blog/hot-start-pcr-higher-specificity-higher-yield-higher-sensitivity]
- 25. Hot Start PCR with heat-activatable primers [https://pmc.ncbi.nlm.nih.gov/articles/PMC2582603/]
- 26. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOop6w39lPOTNuqoOC5Ff1hlGpAh5Vmj713UhiLKzMko-2bWiJ_6P]
- 27. PCR Primer Design Tips - Behind the Bench [https://www.thermofisher.com/blog/behindthebench/pcr-primer-design-tips/]
- 28. Proven tips for PCR primer design [https://www.neb.com/en-us/nebinspired-blog/proven-tips-for-pcr-primer-design?srsltid=AfmBOorswnbbsQMPLeDoyqW-33v4uz0Vopbrkej48avEgcgM_0VMkheX]
- 29. How to design primers for PCR - Integra Biosciences [https://www.integra-biosciences.com/united-states/en/blog/article/how-design-primers-pcr]
- 30. How to design PCR primers [https://www.minipcr.com/how-to-design-pcr-primers/]
- 31. Rules and Tips for PCR & qPCR Primer Design | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 32. Archived | DNA Amplification | GC Content Percentage [https://nij.ojp.gov/nij-hosted-online-training-courses/dna-amplification/overview/primer-design/gc-content-percentage]
- 33. What is the role of magnesium in PCR, and what ... [https://help.takarabio.com/36168/kb/article/110034/what-is-the-role-of-magnesium-in-pcr-and-what-is-the-optimal-concentration]
- 34. What Is the Role of MgCl2 in PCR Amplification Reactions? [https://www.excedr.com/resources/what-is-the-role-of-mgcl2-in-pcr]
- 35. Investigating concentration effects on reaction efficiency ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269725001472]
- 36. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOopI0sNFvB1BA9Wm35L82ySgh4AC28HI_b766ljzzmGus6SpVVpQ]
- 37. Optimizing PCR Reaction Conditions for High Fidelity and ... [https://www.labx.com/resources/optimizing-pcr-reaction-conditions-for-high-fidelity-and-yield/5579]
- 38. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOorIVrOo0p2rSfLH8eQhIrrEUQFVYPU8NWbSkL-B8ywTXz5bCuWZ]
- 39. PCR Optimization: Factors Affecting Reaction Specificity ... [https://www.mybiosource.com/learn/pcr-optimization-factors-affecting-reaction-specificity-and-efficien/]
- 40. Mathematical Modeling and Thermodynamic Integration for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12620865/]
- 41. Hot start PCR [https://en.wikipedia.org/wiki/Hot_start_PCR]
- 42. Hot Start PCR: Definition, Protocol and Application [https://www.bocsci.com/resources/hot-start-pcr-definition-protocol-and-application.html?srsltid=AfmBOoryDJw35SSIspgdPQUaLp7hrgYSnlDE0tnGJJjkb0jxWX4AXvVA]
- 43. Hot Start PCR [https://www.merckmillipore.com/HN/es/technical-documents/technical-article/genomics/pcr/hot-start-pcr]
- 44. Development of a Simple Direct and Hot-Start PCR Using ... [https://www.mdpi.com/1422-0067/24/14/11405]
- 45. Why hot start is so hot? [https://solisbiodyne.com/EN/why-hot-start-is-so-hot/]
- 46. Touchdown PCR: Easy Intro Plus 5 Expert Tips [https://bitesizebio.com/2203/touchdown-pcr-a-primer-and-some-tips/]
- 47. Touchdown PCR for increased specificity and sensitivity in ... [https://pubmed.ncbi.nlm.nih.gov/18772872/]
- 48. Standard vs. Touchdown PCR [https://www.aatbio.com/resources/application-notes/standard-vs-touchdown-pcr]
- 49. How is "Touchdown PCR" used to increase PCR specificity? [https://www.qiagen.com/us/resources/faq/75]
- 50. PCR Methods - Top Ten Strategies [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-methods.html]
- 51. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOoqYw9YEIW1CC25X56tJ4Z6XM1pxwwuEfuMsn-6mH32ku3ZpD4q9]
- 52. Optimization of overlap extension PCR for efficient ... [https://www.sciencedirect.com/science/article/pii/S2215016119303358]
- 53. Multiple Touchdown PCR (MT-PCR): A New Application of ... [https://www.blue-raybio.com/en/blog/Multiple-Touchdown-PCR-MT-PCR-A-New-Application-of-PCR-for-Better-Precision-and-Stability]
- 54. : Strategies To Minimize Primer Dimer PCR Optimization [https://genemod.net/blog/pcr-optimization]
- 55. Multiplex PCR: Optimization and Application in Diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC88949/]
- 56. Advancing quantitative PCR with color cycle multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417387/]
- 57. Designing highly multiplex PCR primer sets with Simulated ... [https://www.nature.com/articles/s41467-022-29500-4]
- 58. Digital PCR outperforms quantitative real-time PCR for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288185/]
- 59. How to multiplex with color combination in Crystal Digital ... [https://www.stillatechnologies.com/digital-pcr/naica-system-analysis/how-to-multiplex-with-color-combination-in-crystal-digital-pcr/]
- 60. Towards practical point-of-care quick, ubiquitous ... [https://www.nature.com/articles/s41378-025-01057-4]
- 61. 5 easy tips to address problems amplifying GC-rich regions [https://bitesizebio.com/24002/problems-amplifying-gc-rich-regions-problem-solved/]
- 62. Optimizing PCR amplification of GC-rich nicotinic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12145843/]
- 63. Faster PCR Optimization [https://bitesizebio.com/31937/optimizing-your-pcr-faster/]
- 64. Long-range PCR and Nanopore sequencing for localisation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12628991/]
- 65. Long-range PCR in next-generation sequencing [https://www.nature.com/articles/srep05737]
- 66. PCR efficiency | Determining amplification efficiencies [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/pcr-quantification/determining-amplification-efficiencies]
- 67. Understanding qPCR Efficiency and Why It Exceeds 100% [https://biosistemika.com/blog/qpcr-efficiency-over-100/]
- 68. Absolute vs. Relative Quantification for qPCR [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/absolute-vs-relative-quantification-real-time-pcr.html]
- 69. Preparation of DNA Sequencing Libraries for Illumina ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/next-generation-sequencing/dna-sequencing-preparation-illumina.html]
- 70. Master NGS library preparation for high-quality sequencing [https://www.idtdna.com/pages/technology/next-generation-sequencing/library-preparation]
- 71. Weak PCR Band Results or Smearing - What To Do [https://www.goldbio.com/blogs/articles/troubleshooting-pcr-part-three-solutions-for-weak-bands-and-smearing?srsltid=AfmBOooaoBWEONoKGPlcr-0We0GbrIFocNdECTKbPOBlRGInA01tvgIA]
- 72. Troubleshooting: Common Problems and PCR Solutions [https://www.mybiosource.com/learn/pcr-troubleshooting-common-problems-and-solutions/]
- 73. Why do I get smeared PCR products? [https://www.qiagen.com/us/resources/faq/87]
- 74. Nucleic Acid Gel Electrophoresis Troubleshooting [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/na-electrophoresis-education/na-electrophoresis-troubleshooting.html]
- 75. Troubleshooting Non-Specific Amplification [https://bento.bio/resources/bento-lab-advice/troubleshooting-non-specific-amplification-with-bento-lab/]
- 76. PCR: A Comprehensive Guide [https://biopathogenix.com/pcr-a-comprehensive-guide/]
- 77. Guidelines for PCR Optimization with Taq DNA Polymerase [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/guidelines-for-pcr-optimization-with-taq-dna-polymerase?srsltid=AfmBOoo0vnyPNtlUo2sFXWuahTHKtp8MEu-xBCS0JWTAv7Gk5-NPk0ck]
- 78. Optimizing your PCR [https://www.takarabio.com/learning-centers/pcr/faq/optimization?srsltid=AfmBOoqqkpI2eqe0s51gd7QAy3c7IIfV0TIVfN86rvMe7Nvu8oDe7Ygz]
- 79. Optimization of PCR Conditions for Amplification of GC‐ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6807403/]
- 80. Optimizing PCR: One Scientist's Not So Fond Memories [https://www.promegaconnections.com/optimizing-pcr-one-scientists-not-so-fond-memories/]
- 81. Enhancement of DNA amplification efficiency by using ... [https://www.bocsci.com/blog/enhancement-of-dna-amplification-efficiency-by-using-pcr-additives/?srsltid=AfmBOopho0FbkoDY3LxUejzdOlkIF4sUFy_Z9J8rCNV2MefaRmvKSudd]
- 82. DMSO and betaine significantly enhance the PCR ... [https://pubmed.ncbi.nlm.nih.gov/32433893/]
- 83. Optimizing qPCR Using the Taguchi Method [https://bitesizebio.com/46365/optimizing-qpcr-using-the-taguchi-method/]
- 84. Reduce optimisation time and effort: Taguchi experimental ... [https://www.sciencedirect.com/science/article/abs/pii/S1875176808000784]
- 85. A simple procedure for optimising the polymerase chain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC308365/]
- 86. qPCR optimization in focus: tips and tricks for excellent ... [https://www.genaxxon.com/shop/en/blog/qpcr-optimization-in-focus-tips-and-tricks-for-excellent-analyses]
- 87. An optimized protocol for stepwise optimization of real-time ... [https://www.nature.com/articles/s41438-021-00616-w]
- 88. How to optimize qPCR in 9 steps? [https://westburg.eu/knowledge/pcr_qpcr/hate_optimizing_qpcr]
- 89. What should the starting template DNA quality and quantity ... [https://www.qiagen.com/us/resources/faq/74]
- 90. PCR inhibition in qPCR, dPCR and MPS—mechanisms and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7072044/]
- 91. Inhibit inhibition: PCR inhibition and how to prevent it [https://www.bioecho.com/blog/inhibit-inhibition-pcr-inhibition-and-how-to-prevent-it]
- 92. Tips for coping with PCR inhibitors [https://www.npdn.org/newsletter/2020/12/article/3]
- 93. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 94. Quantitative PCR validation for research scientists: Part 1 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 95. Development and clinical validation of a novel multiplex ... [https://www.nature.com/articles/s41598-025-16434-2]
- 96. Development of PCR, qPCR and LAMP methods for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12508883/]
- 97. GelGenie: an AI-powered framework for gel ... [https://www.nature.com/articles/s41467-025-59189-0]
- 98. Development of Melting-Curve-Based Real-Time PCR for ... [https://www.mdpi.com/1422-0067/26/19/9411]
- 99. Development of Multiplex PCR and Melt–Curve Analysis for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10217914/]
- 100. High-resolution melting curve analysis with linear ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400525013292]
- 101. 安捷伦- PfuUltra II Fusion 高保真DNA 聚合酶 [https://www.agilent.com/zh-cn/product/polymerase-chain-reaction-(pcr)/pcr-enzymes-reagents/high-fidelity-gc-rich-target-dna-polymerases-for-pcr/pfuultra-ii-fusion-high-fidelity-dna-polymerase-785916?srsltid=AfmBOoqm2bFcx-ixmAxOw7vBAfFTEC11UX-tZi-kIwj8J5D0w-zvn0hX]
- 102. 核酸世家IN有尽有| Thermo Fisher Scientific - CN - 赛默飞 [https://www.thermofisher.cn/cn/zh/home/promo/invitrogen-product-families.html]
- 103. 【2025最新】Primer Express 3.0.1安装教程:PCR实验设计必 ... [https://blog.csdn.net/echoarts/article/details/153966688]
- 104. Performance Assessment of a Multiplex Real-Time PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11545397/]
- 105. Temporal dynamics and forecasting of respiratory viral ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507870/]
- 106. Mastering PCR: A Comprehensive Guide to Polymerase ... [https://www.labx.com/resources/mastering-pcr-a-comprehensive-guide-to-polymerase-chain-reaction-techniques-optimizatio/89]
- 107. Analytical and clinical validation of a novel, laboratory ... [https://www.sciencedirect.com/science/article/pii/S1386653224000556]
- 108. Pathogen detection by multiplex PCR: A comparative study ... [https://www.sciencedirect.com/science/article/pii/S2212534525001212]
- 109. Laboratory Validation of a Fully Automated Point-of-Care ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523385/]
- 110. Development and validation of multiplex real-time PCR for ... [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-024-09028-2]
- 111. Validating Real-Time Polymerase Chain Reaction (PCR) Assays [https://pmc.ncbi.nlm.nih.gov/articles/PMC7834634/]
- 112. Laboratory evaluation of 185 commercial assays for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12496213/]
- 113. Comparison of seven commercial RT-PCR diagnostic kits ... [https://www.sciencedirect.com/science/article/pii/S1386653220301542]
- 114. Optimization of extraction-free protocols for SARS-CoV-2 ... [https://www.nature.com/articles/s41598-023-47645-0]
- 115. Guidance for SARS-CoV-2 Rapid Testing in Point-of-Care ... [https://www.cdc.gov/covid/php/lab/point-of-care-testing.html]
- 116. New RT-PCR Assay for the Detection of Current and Future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9863853/]
- 117. Mitochondrial DNA cox1 [https://bio-protocol.org/exchange/minidetail?id=4017308&type=30]