Mastering Long-Range PCR: A Comprehensive Protocol for High-Fidelity Genomic DNA Amplification in Research
This detailed guide provides researchers, scientists, and drug development professionals with a complete framework for successful long-range PCR amplification of genomic DNA.
Mastering Long-Range PCR: A Comprehensive Protocol for High-Fidelity Genomic DNA Amplification in Research
Abstract
This detailed guide provides researchers, scientists, and drug development professionals with a complete framework for successful long-range PCR amplification of genomic DNA. It covers the foundational principles, offering a clear understanding of enzyme selection, template quality, and buffer chemistry. We present a robust, step-by-step methodological protocol optimized for challenging templates. A dedicated troubleshooting section addresses common pitfalls, from non-specific bands to complete amplification failure, providing actionable solutions. Finally, we discuss validation strategies, including fragment analysis and sequencing verification, and compare long-range PCR with alternative technologies like NGS and cloning. This article aims to empower users to reliably generate long, accurate amplicons for applications in gene mapping, mutation analysis, and next-generation sequencing library construction.
Understanding Long-Range PCR: Principles, Challenges, and Applications for Genomic DNA
What is Long-Range PCR? Defining Amplicon Lengths Beyond Standard PCR
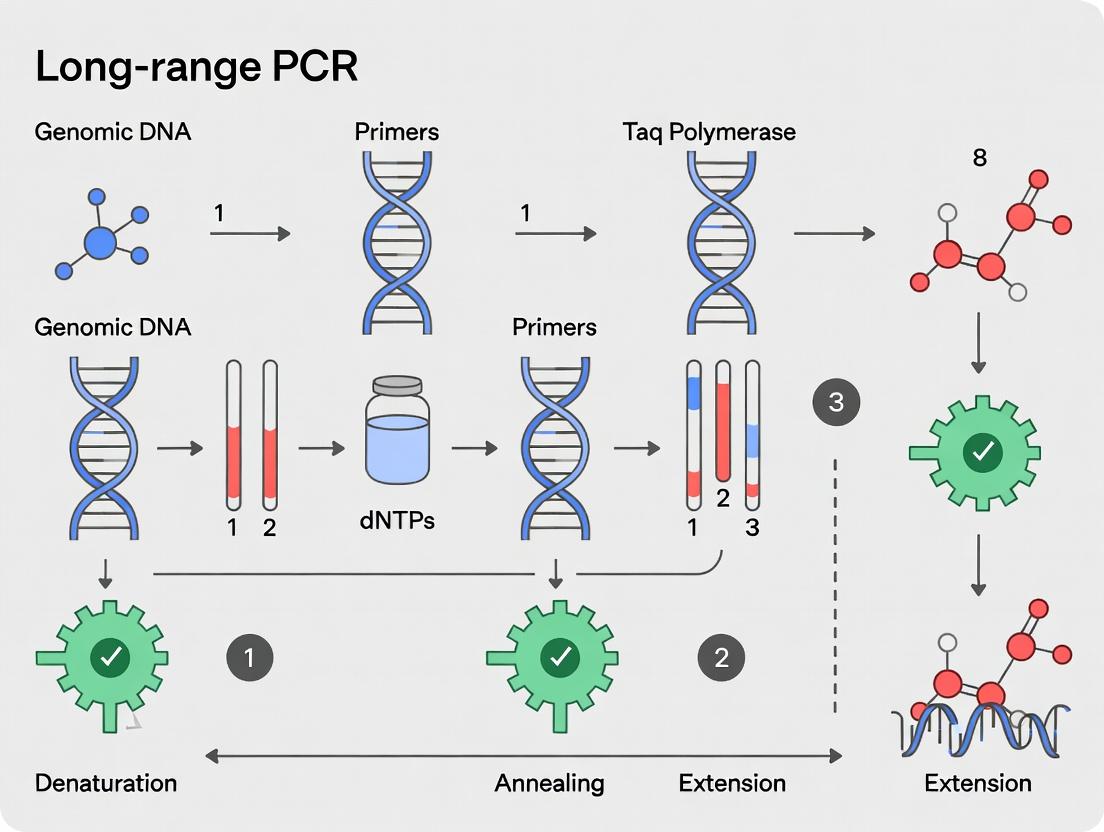
Long-Range PCR (LR-PCR) is a specialized Polymerase Chain Reaction technique optimized to amplify DNA fragments significantly longer than those achievable with standard PCR protocols. While conventional PCR typically amplifies targets up to 3-5 kilobases (kb), LR-PCR can amplify fragments from 5 kb up to over 40 kb. This capability is crucial for applications like genome mapping, cloning, sequencing, and structural variant analysis, where large, contiguous DNA segments are required.
Key Principles and Innovations
The success of LR-PCR hinges on two primary innovations:
- Polymerase Blends: The use of a thermostable DNA polymerase with high processivity (e.g., a modified Taq polymerase) combined with a proofreading polymerase (e.g., Pfu or Pwo). The proofreading enzyme corrects incorporation errors, preventing the accumulation of mutations that would prematurely terminate synthesis over long extensions.
- Buffer Optimization: Specially formulated buffers that enhance polymerase stability and processivity. These often include additives like DMSO, glycerol, or betaine to reduce secondary structures (e.g., GC-rich regions) and minimize template denaturation, facilitating the amplification of complex genomic regions.
Quantitative Comparison: Standard PCR vs. Long-Range PCR
Table 1: Comparative Performance Metrics of Standard and Long-Range PCR
| Parameter | Standard PCR | Long-Range PCR |
|---|---|---|
| Typical Amplicon Length | 0.1 - 3 kb | 5 - 40+ kb |
| Polymerase Type | Single non-proofreading (e.g., Taq) | Blend (Proofreading + non-proofreading) |
| Extension Time | 1 min/kb | 1-3 min/kb (protocol-dependent) |
| Typical Cycle Number | 25-35 | 25-35 |
| Template Quality Requirement | Moderate | High (Intact, high-molecular-weight DNA) |
| Primary Application | Short target amplification, genotyping | Genomic cloning, sequencing, structural analysis |
Table 2: Impact of Polymerase Blends on Fidelity and Yield
| Polymerase Composition | Processivity | Fidelity (Error Rate) | Optimal Fragment Length |
|---|---|---|---|
| Standard Taq | Moderate | Low (~1 x 10⁻⁴) | < 3 kb |
| Proofreading Only (e.g., Pfu) | Lower | High (~1.3 x 10⁻⁶) | < 5 kb |
| LR Blend (Taq + Pfu) | High | Medium-High (~5 x 10⁻⁶) | 5 - 40+ kb |
Application Notes: Long-Range PCR in Genomic Research
Within the context of a thesis on LR-PCR for genomic DNA amplification, this technique serves as a foundational tool for:
- Generating Probes for FISH: Amplifying large genomic segments for use as fluorescent in situ hybridization probes.
- Sequencing Template Preparation: Amplifying large exons or multi-gene clusters for downstream Sanger or next-generation sequencing.
- Detecting Genomic Rearrangements: Identifying deletions, duplications, or translocations that span many kilobases.
- Cloning and Expression Vector Construction: Amplifying large gene constructs with native regulatory elements.
Detailed Experimental Protocol for Long-Range PCR
Title: Amplification of a 15 kb Genomic Locus from Human DNA
Objective: To reliably amplify a 15-kilobase target region from high-quality human genomic DNA for downstream sequencing analysis.
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for LR-PCR
| Reagent/Material | Function & Rationale |
|---|---|
| High-Fidelity LR-PCR Enzyme Mix | A proprietary blend of a high-processivity polymerase and a proofreading enzyme. Essential for accurate, long-fragment synthesis. |
| 5X Specialized LR-PCR Buffer | Contains optimized salts, additives (e.g., betaine), and enhancers to stabilize polymerase over long cycles and melt secondary structures. |
| High-Purity dNTP Mix (10 mM each) | Provides balanced nucleotide substrates for error-free, efficient elongation. |
| Target-Specific Primers (10 µM) | Designed with stringent criteria (Tm ~68°C, 25-35 bases, minimal secondary structure) for specific, high-temperature annealing. |
| High-Molecular-Weight Genomic DNA | Purified using gentle methods (e.g., column-based or phenol-chloroform) to ensure fragment integrity >40 kb. |
| Nuclease-Free Water | Prevents enzymatic degradation of reaction components. |
| Thermal Cycler with Ramp Control | Allows precise control of temperature transition rates, critical for primer annealing and enzyme binding to long templates. |
Methodology:
Reaction Setup (50 µL Total Volume):
- On ice, combine the following in a sterile, thin-walled PCR tube:
- Nuclease-Free Water: 30.5 µL
- 5X LR-PCR Buffer: 10 µL
- dNTP Mix (10 mM each): 1 µL
- Forward Primer (10 µM): 2.5 µL
- Reverse Primer (10 µM): 2.5 µL
- Template Genomic DNA (100-200 ng): 3 µL
- LR-PCR Enzyme Mix: 0.5 µL
- Mix gently by pipetting. Centrifuge briefly.
- On ice, combine the following in a sterile, thin-walled PCR tube:
Thermal Cycling Conditions:
- Initial Denaturation: 94°C for 2 minutes (complete denaturation of long DNA).
- Cycling (30 cycles):
- Denaturation: 94°C for 15 seconds.
- Annealing: 68°C for 30 seconds (use primers with high, specific Tm).
- Extension: 68°C for 12 minutes (calculated at 45-60 seconds/kb for the enzyme blend used).
- Final Extension: 68°C for 10 minutes.
- Hold: 4°C.
Post-Amplification Analysis:
- Analyze 5-10 µL of the product by pulsed-field or standard agarose gel electrophoresis (0.6-0.8% gel) alongside a high-molecular-weight DNA ladder.
- Purify the remaining product using a PCR clean-up kit designed for large fragments for downstream applications.
Troubleshooting Notes:
- No Product/Smear: Optimize template quality/quantity, increase extension time, or adjust Mg²⁺ concentration (if buffer allows).
- Non-Specific Bands: Increase annealing temperature, use a hot-start enzyme mix, or optimize primer design.
- Short Products Only: Check template integrity, ensure polymerase blend is active, and verify that extension time is sufficient.
Visualizing the Workflow and Principles
Diagram 1 Title: Polymerase Synergy & LR-PCR Workflow
Diagram 2 Title: Overcoming PCR Challenges for Long Targets
Application Notes and Protocols
Within the context of developing a robust long-range PCR (LR-PCR) protocol for genomic DNA amplification—a critical step in genome analysis, variant discovery, and downstream applications in drug development—understanding the enzymatic core is paramount. Success hinges on moving beyond standard Taq polymerase to employ specialized high-fidelity polymerases and optimized enzyme blends. These systems balance high processivity, proofreading activity, and the ability to navigate complex, GC-rich, or long genomic templates.
Core Principles and Quantitative Performance Data
High-fidelity polymerases for LR-PCR are typically family B polymerases (e.g., Pfu, Pwo) or engineered chimeric enzymes. They possess a 3'→5' exonuclease (proofreading) activity that corrects misincorporated nucleotides, yielding significantly lower error rates than non-proofreading enzymes. However, this activity can also degrade primers and single-stranded templates. For long amplicons (>10 kb), processivity—the number of nucleotides added per binding event—is critical. Single high-fidelity enzymes often lack the necessary speed and processivity for efficient long-range amplification.
This limitation is solved by enzyme blends, which synergistically combine a high-fidelity polymerase with a processive, strand-displacing polymerase (often a modified Taq). The blend leverages the rapid extension and strong binding of one enzyme with the proofreading capability of another.
Table 1: Quantitative Comparison of PCR Polymerase Systems for Long-Range Amplification
| Polymerase System | Example Enzymes | Avg. Error Rate (mutations/bp) | Optimal Amplicon Length Range | Processivity | Key Characteristic for LR-PCR |
|---|---|---|---|---|---|
| Standard Taq | Taq DNA Pol | 2.0 x 10⁻⁵ | < 3 kb | Moderate | Fast but error-prone; insufficient for long targets. |
| Proofreading-Only | Pfu, Pwo | 1.3 x 10⁻⁶ | 1 - 5 kb | Low | High fidelity but slow and low yield for long amplicons. |
| Engineered High-Fidelity | Phusion, Q5, Kapa HiFi | ~4.4 x 10⁻⁷ | up to 20 kb | High | Optimized fusion enzymes; best single-enzyme option for length/fidelity. |
| Optimized Enzyme Blend | Taq + Pfu blend, Platinum SuperFi II | ~1.6 x 10⁻⁶ | 5 - 40+ kb | Very High | Superior processivity and yield on complex, long templates; balanced fidelity. |
Table 2: Impact of Enzyme Blends on Long-Range PCR Success Rate*
| Template (Human gDNA) | Target Amplicon Size | Single High-Fidelity Pol Success | Enzyme Blend Success | Critical Blend Component Function |
|---|---|---|---|---|
| GC-rich promoter region | 15 kb | 40% | 95% | Strand displacement through secondary structures. |
| Repetitive element region | 12 kb | 25% | 85% | Reduced pausing and primer displacement. |
| Standard coding region | 20 kb | 60% | 98% | Sustained polymerization over entire length. |
*Success defined as a single, specific band of correct size on agarose gel electrophoresis.
Detailed Experimental Protocol: Long-Range PCR of Genomic DNA Using an Enzyme Blend
Protocol: Amplification of a 15-20 kb Genomic Locus from Human gDNA
I. The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Function in LR-PCR |
|---|---|
| High-Quality, High-MW Genomic DNA (e.g., from blood or cell culture) | Intact template is non-negotiable; avoid sheared DNA. |
| Optimized LR-PCR Enzyme Blend (e.g., Platinum SuperFi II, LA Taq with GC buffer) | Provides processivity, fidelity, and robustness. |
| dNTP Mix (10 mM each) | Nucleotide substrates; stable concentration is vital for long extensions. |
| Betaine (5M stock) | Additive that equalizes strand melting, crucial for GC-rich regions. |
| DMSO | Additive that reduces secondary structure; use judiciously (2-4%). |
| High-Fidelity PCR Buffer (often supplied with enzyme) | Typically contains Mg²⁺, salts, and stabilizers optimized for the blend. |
| Target-Specific Primers (20-30 nt, 40-60% GC) | Long amplicons require high-Tm, specific primers; design using LR-PCR guidelines. |
| Nuclease-Free Water | Reaction integrity. |
| Thermal Cycler with Extended Ramp Speed Control | Precise temperature transitions improve specificity for long targets. |
II. Step-by-Step Methodology
Template Preparation: Dilute high-molecular-weight human gDNA to a working concentration of 10-50 ng/µL in nuclease-free water. Keep on ice.
Master Mix Assembly (50 µL reaction):
- Always include a no-template control (NTC).
- Assemble on ice in a sterile, thin-walled 0.2 mL PCR tube. *If using a system with separate polymerase and buffer, follow manufacturer's blend ratio.
Thermal Cycling Conditions:
- Use a "Hot Start" protocol if the enzyme blend supports it. Determine optimal annealing temperature based on primer Tm. *Use the polymerase's recommended extension temperature.
Post-Amplification Analysis:
- Analyze 5-10 µL of product by pulsed-field gel electrophoresis (PFGE) or standard agarose gel electrophoresis (0.6-0.8% agarose) run at low voltage (2-3 V/cm) for several hours to resolve long amplicons.
- Purify the remaining product using a column-based kit designed for long DNA fragments.
Mandatory Visualizations
Diagram Title: Enzyme Blend Workflow in Long-Range PCR
Diagram Title: Strategy for Overcoming LR-PCR Challenges
Long-range PCR for genomic DNA amplification is a cornerstone of modern genetic research, enabling the study of large genes, haplotype phasing, and next-generation sequencing library construction. Within the context of a broader thesis on optimizing long-range PCR protocols, this document addresses three critical technical challenges: amplification through GC-rich regions, secondary structures, and complex templates. Successfully overcoming these hurdles is essential for researchers, scientists, and drug development professionals working with difficult genomic targets.
Understanding the Challenges: Quantitative Impact
Table 1: Impact of Sequence Complexity on PCR Success Rates
| Challenge | Typical Sequence Feature | Failure Rate in Standard PCR* | Primary Consequence |
|---|---|---|---|
| GC-Rich Regions | >65% GC content | 60-80% | Premature polymerase dissociation, primer misfolding, nonspecific amplification. |
| Secondary Structures | Hairpins, G-quadruplexes | 40-70% | Polymerase stalling, incomplete extension, reduced yield. |
| Complex Template | High repeats, long size (>10 kb) | 50-90% | Mispriming, truncated products, amplification bias. |
*Data synthesized from current literature and manufacturer application notes.
Application Notes & Protocols
Optimizing for GC-Rich Regions
GC-rich sequences exhibit high melting temperatures and strong inter-strand associations, leading to inefficient denaturation and primer annealing.
Key Reagent Solutions:
- Betaine (5 M stock): A chemical chaperone that equalizes the contribution of GC and AT base pairs to DNA stability, promoting uniform melting.
- DMSO (1-10% v/v): Reduces DNA secondary structure and lowers the melting temperature.
- 7-deaza-dGTP: Partially replaces dGTP to reduce hydrogen bonding in GC pairs, weakening strand association.
- Specialized Polymerase Blends: Engineered enzymes with enhanced processivity and strand displacement activity.
Detailed Protocol: GC-Rich Long-Range PCR
- Primer Design: Design primers with melting temperatures (Tm) of 68-72°C. Avoid 3'-GC clamps. Consider placing primers in flanking, lower-GC areas if possible.
- Reaction Setup (50 µL):
- Template DNA: 100-500 ng genomic DNA
- Long-range PCR buffer (commercial, 1X final)
- dNTPs: 0.4 mM each (consider 0.2 mM dGTP + 0.2 mM 7-deaza-dGTP)
- Primers: 0.4 µM each
- Additives: Betaine (1 M final), DMSO (3-5% v/v)
- Polymerase blend: 2.5 units (e.g., a mix of a high-fidelity and a processive polymerase)
- Cycling Conditions:
- Initial Denaturation: 98°C for 2 min.
- 35 Cycles:
- Denaturation: 98°C for 20 sec (shorter, high-temp denaturation is key).
- Annealing: 70°C for 30 sec (use a higher, more stringent temperature).
- Extension: 68°C for 1 min/kb.
- Final Extension: 72°C for 10 min.
- Analysis: Run 5-10 µL on a 0.8% agarose gel. Expect a single, sharp band of correct size.
Disrupting Secondary Structures
Intramolecular structures like hairpins can block polymerase progression. G-quadruplexes in promoter regions are particularly problematic.
Key Reagent Solutions:
- DMSO and Betaine: As above, to destabilize structures.
- Single-Stranded DNA Binding Proteins (SSBs): E. coli SSB or T4 gp32 can be added (50-200 ng/reaction) to bind and unwind secondary structures during elongation.
- Modified dNTPs: 7-deaza-dGTP helps disrupt G-quadruplexes.
- Enhanced Denaturation: Use temperature-controlled ramping or a "touchdown" start.
Detailed Protocol: PCR with SSB Additive
- Prepare a standard long-range PCR mix as in 2.1, including betaine/DMSO.
- Add SSB: Include E. coli SSB at a final concentration of 10-40 nM (added after buffer but before polymerase).
- Use a slow ramp rate (0.5-1°C/sec) from annealing to extension to allow SSB binding.
- Consider a "Hot Start" with polymerase activation at 98°C for 1 min before cycling to prevent nonspecific activity during setup.
Managing Complex and Long Templates
Long templates and those with repeat sequences demand maximum polymerase fidelity and processivity.
Key Reagent Solutions:
- High-Fidelity, Processive Polymerase Blends: Essential for accurate synthesis over many kilobases.
- Optimized Buffer Systems: Proprietary buffers often contain pH stabilizers and processivity enhancers.
- Touchdown PCR: To increase specificity at the start of amplification.
- Gradient PCR: For empirically determining optimal annealing temperatures.
Detailed Protocol: Long-Range (>15 kb) Amplification
- Template Integrity: Use high-quality, high-molecular-weight DNA (check on pulsed-field gel). Minimize pipetting shear.
- Reaction Setup (50 µL):
- Template: 300-1000 ng genomic DNA.
- Commercial Long-Range Buffer (1X).
- dNTPs: 0.4 mM each.
- Primers: 0.3 µM each (lower concentration increases specificity for long targets).
- Additives: Betaine (1 M final). Avoid DMSO if polymerase is sensitive to it.
- Polymerase blend: 3.5 units.
- Cycling Conditions (Two-Step PCR):
- Initial Denaturation: 98°C for 2 min.
- 10 Cycles of Touchdown:
- Denaturation: 98°C for 20 sec.
- Annealing/Extension: 68°C for 1 min/kb + 30 sec. (High, combined step).
- 25 Cycles:
- Denaturation: 98°C for 20 sec.
- Annealing/Extension: 68°C for 1 min/kb + 30 sec.
- Final Extension: 72°C for 12 min.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Challenging Long-Range PCR
| Item | Function/Application | Example (Brand/Type) |
|---|---|---|
| Specialized Polymerase Blend | Combines high processivity (for length) with high fidelity (for accuracy). | PrimeSTAR GXL, KAPA HiFi HotStart, LongAmp Taq |
| Betaine (5M) | GC-clamp destabilizer; homogenizes DNA melting temperature. | Sigma-Aldrich Betaine Solution |
| DMSO | Reduces secondary structure; lowers DNA Tm. | Molecular biology grade DMSO |
| 7-deaza-dGTP | Reduces hydrogen bonding in GC-rich regions and G-quadruplexes. | Roche 7-deaza-2'-deoxyguanosine 5'-triphosphate |
| Single-Stranded Binding Protein (SSB) | Binds and melts DNA secondary structures during elongation. | NEB E. coli SSB, Thermo Scientific T4 gp32 |
| High-Fidelity Buffer System | Optimized pH, salt, and co-factors for long, accurate synthesis. | Provided with polymerase blends |
| High-Quality dNTPs | Ensure high purity and correct concentration for error-free synthesis. | PCR-grade dNTP mix |
| Low-Binding Tubes & Tips | Minimize adsorption of precious template and enzyme. | PCR tubes with polymer coating |
Visualization of Strategies and Workflows
Title: Strategy for GC-Rich PCR
Title: Overcoming Secondary Structures
Title: Long-Range PCR Workflow
Integrated Troubleshooting Table
Table 3: Problem-Shooting Guide for Challenging PCRs
| Symptom | Possible Cause (GC/Structure/Complexity) | Recommended Solution |
|---|---|---|
| No Product | Excessive secondary structure, poor denaturation. | Increase denaturation temp/time; add DMSO + Betaine; try SSB protein. |
| Smear or Multiple Bands | Mispriming in complex or repeat regions; low specificity. | Use Touchdown PCR; lower primer concentration; increase annealing temperature. |
| Product Shorter Than Expected | Polymerase stalling at GC-rich zones or structures. | Include 7-deaza-dGTP; use a more processive polymerase blend; add Betaine. |
| Inconsistent Results | Variable template quality/quantity; inhibitor presence. | Repurify DNA; use a gradient PCR for optimization; include a positive control. |
Within the broader thesis on Long-range PCR (LR-PCR) protocols for genomic DNA amplification research, the optimization of reaction components is critical. Amplifying long fragments (≥5 kb) from complex genomic templates presents challenges including secondary structure formation, premature polymerase dissociation, and spurious priming. This application note details the use of essential reagents—DMSO, betaine, and optimized buffer systems—to overcome these obstacles, enabling robust and reliable amplification of targets up to 40 kb.
Role of Essential Reagents in Long-range PCR
Dimethyl Sulfoxide (DMSO)
DMSO (typically used at 1-10% v/v) is a polar aprotic solvent that enhances LR-PCR by disrupting base pairing, particularly in GC-rich regions. It reduces the melting temperature (Tm) of DNA, helping to denature secondary structures that can block polymerase progression. Excessive DMSO can inhibit polymerase activity; thus, titration is required.
Betaine (Trimethylglycine)
Betaine (0.5-2.5 M) is a zwitterionic osmolyte that equalizes the contribution of GC and AT base pairs to duplex stability. It promotes DNA strand separation by reducing the Tm difference across heterogeneous sequences, preventing polymerase pausing, and minimizing template reannealing. It is especially beneficial for high-GC content and complex genomic targets.
Optimized Buffer Systems
Commercial long-range PCR buffers are specifically formulated with:
- Enhanced pH buffering (often Tris-based at pH 8.5-9.0) to counteract pH drops during extended cycling.
- Supplemental magnesium (Mg2+, 1.5-3.0 mM) as a critical cofactor for high-fidelity polymerases.
- Stabilizers (e.g., ammonium sulfate, glycerol) to maintain polymerase stability and processivity over long extension times.
- dNTPs balanced at higher concentrations (e.g., 400 µM each) to support synthesis of long amplicons.
Table 1: Quantitative Summary of Reagent Roles & Concentrations
| Reagent | Primary Function | Typical Working Concentration | Key Consideration in LR-PCR |
|---|---|---|---|
| DMSO | Disrupts DNA secondary structure; reduces Tm. | 1-10% (v/v) | Optimize by 2% increments; >10% often inhibits polymerase. |
| Betaine | Homogenizes base pair stability; reduces template reannealing. | 0.5-2.5 M (often 1.0-1.3 M) | Can be combined with DMSO; effective for GC-rich targets (>70%). |
| Mg2+ | Essential polymerase cofactor. | 1.5-3.0 mM (varies by system) | Concentration is critical; must be optimized with dNTPs. |
| dNTPs | Substrates for DNA synthesis. | 200-400 µM each | Higher concentrations support long extensions but increase error rate if unbalanced. |
| PCR Buffer | Maintains pH, ionic strength, stability. | 1X (commercial blend) | Often contains (NH4)2SO4, proprietary enhancers. |
Application Notes & Protocols
Protocol 1: Titration of DMSO and Betaine for GC-Rich LR-PCR
Objective: Determine the optimal concentration of DMSO and/or betaine for amplifying a 15 kb GC-rich (72% GC) genomic target. Materials:
- High-fidelity LR-PCR enzyme mix (e.g., Q5, KAPA HiFi, or specialty mixes).
- Genomic DNA (human, mouse, etc., >50 ng/µL, high integrity).
- 10 mM dNTP mix.
- DMSO (molecular biology grade).
- 5 M Betaine solution (filter sterilized).
- Target-specific primers (20 µM stock).
- Nuclease-free water.
Methodology:
- Prepare a master mix for n+1 reactions containing:
- 1X Commercial LR-PCR Buffer
- 400 µM each dNTP
- 2.0 mM MgCl2 (or as recommended for polymerase)
- 0.5 µM each primer
- 1 unit/µL high-fidelity polymerase
- 50 ng/µL genomic DNA template
- Aliquot the master mix into 8 PCR tubes.
- Add DMSO and Betaine to create the following conditions (final volume adjusted with water):
- Tube 1: Control (No DMSO, No Betaine)
- Tube 2: 2% DMSO
- Tube 3: 4% DMSO
- Tube 4: 6% DMSO
- Tube 5: 1.0 M Betaine
- Tube 6: 1.5 M Betaine
- Tube 7: 1.0 M Betaine + 2% DMSO
- Tube 8: 1.5 M Betaine + 4% DMSO
- Perform PCR cycling:
- Initial Denaturation: 98°C for 30 sec.
- 35 cycles: [98°C for 10 sec, 68°C for 12 min].
- Final Extension: 72°C for 10 min.
- Hold at 4°C.
- Analyze 5 µL of each product by 0.8% agarose gel electrophoresis.
Protocol 2: Optimized Buffer System Comparison for Ultra-Long Amplification
Objective: Compare the performance of three commercial LR-PCR buffer systems for amplifying a 30-40 kb genomic fragment. Materials: As in Protocol 1, plus three commercial LR-PCR kits (e.g., System A, B, C). Methodology:
- Set up three reactions, each following the manufacturer's recommended protocol for a 50 µL reaction. Use identical template (100 ng), primers (0.3 µM), and cycling conditions (optimized for the longest fragment).
- Use a "two-step" cycling protocol typical for very long targets:
- Initial Denaturation: 94°C for 2 min.
- 10 cycles: [94°C for 10 sec, 62°C for 30 sec, 68°C for 10 min].
- 25 cycles: [94°C for 10 sec, 62°C for 30 sec, 68°C for 10 min + 20 sec/cycle].
- Final Extension: 72°C for 10 min.
- Include a reagent mix containing 1 M betaine in a parallel set of reactions.
- Analyze products via pulsed-field gel electrophoresis (PFGE) or a high-percentage agarose gel (0.6%).
Table 2: Expected Outcome Comparison for Protocol 2
| Buffer System | Additive | Yield (ng/µL) * | Specificity (Non-specific Bands) | Max Reliable Length (kb) * |
|---|---|---|---|---|
| System A | None | Medium | Low | 25 |
| System A | 1 M Betaine | High | Low | 35 |
| System B | None | High | Medium | 30 |
| System B | 1 M Betaine | Very High | Low | 40 |
| System C | None | Low | Very Low | 20 |
| System C | 1 M Betaine | Medium | Very Low | 30 |
Hypothetical data based on typical kit performances.
Visualizations
Diagram: Mechanism of Betaine & DMSO in LR-PCR
Title: How Betaine and DMSO Enable Long-Range PCR
Diagram: LR-PCR Optimization Workflow
Title: Stepwise Optimization of Long-Range PCR Reagents
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Long-Range PCR Optimization
| Item | Function in LR-PCR | Example Product/Specification |
|---|---|---|
| High-Fidelity DNA Polymerase | Engineered for high processivity and low error rate over long extensions. | Q5 Hot Start (NEB), KAPA HiFi, PrimeSTAR GXL. |
| Optimized 10X LR-PCR Buffer | Proprietary blend of salts, buffering agents, and stabilizers. | Supplied with enzyme; may contain (NH4)2SO4. |
| Molecular Biology Grade DMSO | Reduces secondary structure; must be sterile and nuclease-free. | Sigma D8418, Invitrogen. |
| 5M Betaine Solution | Homogenizes template melting; filter-sterilized. | Sigma B0300, supplied in some PCR kits. |
| High-Purity dNTP Mix | Balanced 10mM solution of each dNTP; critical for fidelity. | ThermoFisher Scientific, NEB. |
| MgCl2 Solution (25-50 mM) | Separate solution for fine-tuning polymerase activity. | Supplied with most polymerase systems. |
| High-Integrity Genomic DNA | Intact, high molecular weight template. | Purified via column/CTAB; A260/280 ~1.8. |
| LR-PCR Validated Primers | Designed for high Tm and specificity for long targets. | 20-30 bases, Tm matched, HPLC purified. |
| Nuclease-Free Water | Reaction assembly; ensures no RNase/DNase contamination. | Ultra-pure, PCR-grade (e.g., ThermoFisher). |
This Application Note contextualizes three pivotal downstream applications—Gene Cloning, Mutation Detection, and Next-Generation Sequencing (NGS) Library Preparation—within a research thesis focused on developing and optimizing a Long-range PCR (LR-PCR) protocol for high-fidelity genomic DNA amplification. Successful LR-PCR, which amplifies targets from 5 kb to over 40 kb, provides the high-quality, high-molecular-weight DNA template essential for these advanced applications, enabling critical studies in functional genetics, variant analysis, and comprehensive genomic profiling.
Application Note 1: Gene Cloning of Long-Range PCR Products
Objective: To clone large, LR-PCR-amplified gene fragments into suitable vectors for functional expression studies, mutagenesis, or stable cell line generation.
Protocol:
- LR-PCR Amplification: Perform LR-PCR on genomic DNA using a high-fidelity polymerase blend (e.g., mixture of Taq and proofreading polymerases). Typical 50 µL reaction: 100-500 ng genomic DNA, 0.3 µM each primer, 1x LR-PCR buffer, 350 µM dNTPs, 2-3 mM MgSO₄, and 2-3 units of enzyme blend. Cycling: Initial denaturation 94°C for 2 min; 30 cycles of 94°C for 15 sec, 60-68°C for 30 sec, 68°C for 1-6 min/kb; final extension 68°C for 10 min.
- Product Purification: Clean the amplicon using a magnetic bead-based purification system (e.g., SPRI beads) to remove primers, enzymes, and salts. Elute in nuclease-free water.
- Vector Preparation: Linearize and dephosphorylate a cloning vector (e.g., pUC19, Gateway entry vector) appropriate for large inserts. For TA-cloning of A-tailed LR-PCR products, use ready-prepared T-vectors.
- Ligation: Incubate purified LR-PCR product and prepared vector with DNA ligase. For blunt-end or seamless cloning, use a 3:1 to 5:1 (insert:vector) molar ratio. Incubate at 16°C for 1-16 hours.
- Transformation: Transform competent E. coli cells (high-efficiency >10⁸ cfu/µg) with 2-5 µL of the ligation mix via heat shock or electroporation. Plate onto selective agar.
- Screening: Pick colonies for PCR screening or restriction digest to confirm insert presence and size.
Key Research Reagent Solutions:
| Reagent/Material | Function in Experiment |
|---|---|
| High-Fidelity LR-PCR Enzyme Mix | Amplifies long genomic fragments with minimal error rate. |
| Magnetic Bead Purification Kit | Efficiently cleans PCR products without size bias or ethanol carryover. |
| Seamless/TA Cloning Kit | Facilitates efficient, directional insertion of PCR products into vectors. |
| High-Efficiency Competent Cells | Essential for achieving viable transformants with large plasmid constructs. |
Diagram: Workflow for Cloning Long-Range PCR Products
Application Note 2: Mutation Detection from LR-PCR Amplicons
Objective: To identify and characterize sequence variants (SNPs, indels) within large genomic regions amplified by LR-PCR.
Protocol:
- Template Preparation: Amplify target region from test and control samples via optimized LR-PCR. Purify amplicons thoroughly.
- Sanger Sequencing: For targeted mutation screening.
- Fragmentation & Purification: For amplicons >1kb, perform internal primer walking or fragment the LR-PCR product via shearing or restriction digest. Re-purify.
- Cycle Sequencing: Set up reactions with purified amplicon (10-100 ng), 3.2 pmol primer, and BigDye Terminator v3.1 mix. Cycling: 25 cycles of 96°C for 10 sec, 50°C for 5 sec, 60°C for 4 min.
- Clean-up & Capillary Electrophoresis: Remove unincorporated dyes using column- or bead-based methods. Run on genetic analyzer.
- High-Resolution Melting (HRM) Analysis: For scanning unknown variants.
- Set up real-time PCR on LR-PCR product (diluted 1:10-1:100) with saturating dsDNA dye (e.g., EvaGreen) and primers for the sub-region of interest.
- Perform precise melting curve analysis (0.1°C increments). Compare curve shapes to controls.
- Data Analysis: For Sanger, align sequences to reference using tools like SnapGene or BLAST. For HRM, use instrument software to group samples by melting profile differences.
Quantitative Data Table: Mutation Detection Methods Comparison
| Method | Effective Amplicon Input Size | Approx. Sensitivity | Time to Result (Post-PCR) | Key Application |
|---|---|---|---|---|
| Sanger Sequencing | 0.5 - 5 kb (per read) | ~15-20% allele frequency | 1-2 days | Definitive variant identification, known mutation confirmation. |
| HRM Analysis | 0.1 - 0.5 kb (amplicon) | ~1-5% allele frequency (for heterozygotes) | 2-3 hours | Rapid scanning for unknown variants in a defined region. |
| Restriction Fragment Length Polymorphism (RFLP) | Up to full LR-PCR product | ~1-5% allele frequency | 1 day | Detection of specific variants that alter restriction sites. |
Diagram: Mutation Detection Pathways from LR-PCR
Application Note 3: NGS Library Preparation Using LR-PCR Amplicons
Objective: To convert large, LR-PCR-amplified genomic regions into sequencer-ready libraries for targeted resequencing or custom panel analysis.
Protocol (Illumina-compatible, Tagmentation-based):
- LR-PCR Amplification & QC: Generate and purify amplicon as in Application Note 1. Quantify using fluorometry (e.g., Qubit). Assess integrity via agarose gel or FEMTO Pulse system.
- Tagmentation: Use a kit like Nextera XT. Combine purified LR-PCR amplicon (up to 1 ng in 5 µL) with ATM (Amplicon Tagmentation Mix). Incubate at 55°C for 5-10 minutes. Immediately neutralize with NT (Neutralization Tagment) Buffer.
- Limited-Cycle PCR Amplification: Add unique dual-index (i5 and i7) primers and a PCR master mix to the tagmented DNA. Cycle: 72°C for 3 min; 95°C for 30 sec; 12-15 cycles of [95°C for 10 sec, 55°C for 30 sec, 72°C for 30 sec]; final extension 72°C for 5 min.
- Library Clean-up & Size Selection: Purify the PCR product using SPRI beads. For optimal size selection (e.g., removal of very short fragments), perform a dual-sided bead cleanup (e.g., 0.5x and 1.0x bead ratios).
- Library QC & Pooling: Quantify final library by fluorometry. Assess size distribution using a Bioanalyzer or TapeStation (expected peak: 300-800 bp). For multiplexing, pool equimolar amounts of uniquely indexed libraries.
- Sequencing: Dilute pool to final loading concentration (typically 1.2-1.8 pM for MiSeq) and denature with NaOH. Sequence on appropriate Illumina platform (MiSeq, NextSeq).
Key Research Reagent Solutions:
| Reagent/Material | Function in Experiment |
|---|---|
| Nextera XT or Flex Kit | Enzymatically fragments (tagments) DNA and adds adapter sequences in a single step. |
| Fluorometric dsDNA Assay Kit | Accurately quantifies low-concentration DNA for library input normalization. |
| SPRI Magnetic Beads | Performs clean-up and size selection of libraries without column loss. |
| High-Sensitivity DNA Analysis Kit | Precisely assesses library fragment size distribution prior to sequencing. |
Diagram: NGS Library Prep from LR-PCR Amplicons
Step-by-Step Protocol: Optimized Workflow for Reliable Long-Range Amplification
Within the context of developing and optimizing a Long-range PCR (LR-PCR) protocol for genomic DNA amplification, the preparation of template DNA is the single most critical pre-analytical factor. Success in amplifying fragments exceeding 5 kb, often up to 20-40 kb, is exceptionally sensitive to the integrity, concentration, and purity of the starting template. This application note details the stringent requirements and validated protocols for template DNA preparation to ensure robust and reproducible LR-PCR outcomes for genomic research and downstream applications in drug target validation.
Quantitative Requirements for LR-PCR Template DNA
The following table summarizes the optimal and acceptable ranges for template DNA parameters specific to long-range amplification.
Table 1: Template DNA Specifications for Long-Range PCR
| Parameter | Optimal Range | Acceptable Range | Measurement Method | Rationale for LR-PCR |
|---|---|---|---|---|
| Concentration | 50 - 200 ng/µL | 10 - 500 ng/µL | Fluorometry (Qubit) | Ensures sufficient target molecules without inhibitor carryover. |
| Purity (A260/A280) | 1.8 - 1.9 | 1.7 - 2.0 | Spectrophotometry (Nanodrop) | Ratios outside range indicate protein/phenol contamination which inhibit Taq and proof-reading polymerases. |
| Purity (A260/A230) | 2.0 - 2.2 | 1.8 - 2.4 | Spectrophotometry (Nanodrop) | Low values indicate chaotropic salt, EDTA, or carbohydrate contamination, disrupting polymerization. |
| Molecular Weight Integrity | > 50 kb average size | > 30 kb average size | Pulse-field or 0.4-0.6% agarose gel electrophoresis | Full-length template is essential for priming across long distances. Sheared DNA yields partial or no products. |
| Total Amount per 50 µL rxn | 100 - 500 ng | 50 - 1000 ng | Calculated from concentration | Balance between detection sensitivity and inhibition risk. |
Detailed Protocols for Template DNA Preparation
Protocol 3.1: High-Molecular-Weight (HMW) Genomic DNA Isolation from Cultured Cells (Column-Based)
Objective: To obtain high-integrity, ultra-pure genomic DNA suitable for LR-PCR amplification of targets >10 kb.
Materials: Cell pellet (1-5 x 10^6 cells), PBS, Proteinase K, RNase A, Lysis Buffer (with chaotropic salts), Wash Buffers (ethanol-based), Elution Buffer (10 mM Tris-HCl, pH 8.5), HMW DNA purification columns.
Procedure:
- Cell Lysis: Resuspend cell pellet in PBS and centrifuge. Thoroughly resuspend in lysis buffer containing Proteinase K. Incubate at 56°C for 1-2 hours until completely lysed.
- RNase Treatment: Add RNase A (final conc. 20 µg/mL) to the lysate. Incubate at room temperature for 2-5 minutes.
- Binding: Add ethanol (or isopropanol) to the lysate and mix thoroughly. Transfer the mixture to a specialized HMW DNA binding column. Centrifuge at ≥ 6000 x g for 1 minute. Note: Do not exceed recommended g-force to prevent shearing.
- Washing: Wash the column membrane twice with the provided wash buffers, centrifuging to dry the membrane completely after the second wash.
- Elution: Place the column in a clean 1.5 mL microcentrifuge tube. Apply 50-100 µL of pre-warmed (65°C) Elution Buffer directly onto the center of the membrane. Incubate at room temperature for 2-5 minutes. Centrifuge at 6000 x g for 2 minutes to elute the DNA. Repeat elution with a second aliquot for higher yield.
- Quality Control: Quantify DNA using a fluorometric assay. Assess integrity by running 100 ng on a 0.5% agarose gel at 2-3 V/cm for 2-3 hours alongside a high-molecular-weight ladder.
Protocol 3.2: Assessment of DNA Integrity by Gel Electrophoresis
Objective: To visually confirm the average size of genomic DNA exceeds 30 kb.
Procedure:
- Prepare a 0.5-0.6% agarose gel in 1X TAE buffer. Use a wide-tooth comb.
- Mix 100 ng of DNA sample with 6X loading dye (do not use dyes containing harsh denaturants). Load alongside a HMW DNA ladder (e.g., Lambda HindIII, or dedicated 50 kb ladder).
- Run the gel in 1X TAE buffer at 2-3 volts per cm (e.g., 50V for a 20 cm gel) for 2.5-3 hours with active cooling (4°C) if possible.
- Stain the gel with a fluorescent intercalating dye (e.g., SYBR Safe, GelRed) and image. Intact HMW DNA should appear as a tight, high-molecular-weight band with minimal smearing downward.
Protocol 3.3: Purification and Concentration of Existing DNA Samples
Objective: To clean up and concentrate degraded-quality or dilute DNA samples for LR-PCR.
Materials: DNA sample, AMPure XP or SPRI beads (PEG/NaCl solution), 80% ethanol, TE buffer, magnetic stand.
Procedure (SPRI Bead Cleanup):
- Binding: Vortex bead solution. Add a 0.7X volume of room-temperature beads to 1 volume of DNA sample. Mix thoroughly by pipetting. Incubate at room temperature for 5 minutes.
- Capture: Place the tube on a magnetic stand for 5 minutes or until the supernatant is clear. Carefully remove and discard the supernatant.
- Washing: With the tube on the magnet, add 200 µL of freshly prepared 80% ethanol without disturbing the bead pellet. Incubate for 30 seconds, then remove the ethanol. Repeat the wash a second time. Air-dry the pellet for 2-5 minutes until cracks appear. Do not over-dry.
- Elution: Remove the tube from the magnet. Resuspend the bead pellet in 20-50 µL of TE buffer or nuclease-free water. Incubate at room temperature for 2 minutes. Place back on the magnet until clear. Transfer the eluted DNA (supernatant) to a clean tube.
Visualizations
Title: Template DNA Parameters Impact on LR-PCR Outcome
Title: HMW Genomic DNA Isolation and QC Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Kits for Template DNA Preparation
| Item | Function in Template Prep | Key Consideration for LR-PCR |
|---|---|---|
| HMW DNA Isolation Kit (e.g., Qiagen Genomic-tip, MagAttract HMW) | Gentle lysis and purification designed to preserve DNA strand length. | Select kits specifically validated for fragments >50 kb. Avoid vortexing during protocol. |
| Fluorometric DNA Assay (e.g., Qubit dsDNA BR/HS Assay, Picogreen) | Accurate quantification of double-stranded DNA, unaffected by common contaminants. | Critical for precise dosing of template (ng/rxn). More reliable than A260 for LR-PCR. |
| Pulse-Field Gel Electrophoresis System | Definitive analysis of ultra-high molecular weight DNA integrity (>50 kb). | Gold-standard for assessing template suitability for very long-range (>20 kb) targets. |
| SPRI Magnetic Beads (e.g., AMPure XP, CleanNA) | Size-selective purification and concentration of DNA; can remove short fragments. | Use a 0.7X or 0.8X ratio to retain large fragments while removing primers, dNTPs, and salts. |
| Proteinase K (Molecular Grade) | Efficient digestion of nucleases and chromatin proteins during lysis. | Essential for complete lysis and prevention of DNA degradation during isolation. |
| RNase A (DNase-free) | Removal of contaminating RNA which can skew quantification and inhibit PCR. | Required step to ensure accurate DNA concentration measurement via fluorometry or spectroscopy. |
| Low-EDTA TE Buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0-8.5) | Long-term storage buffer for DNA. Minimal EDTA chelates Mg2+ less. | Preferred over water or high-EDTA TE to prevent degradation while avoiding Mg²⁺ sequestration in PCR. |
| Wide-Bore/Filter Pipette Tips | Aspiration and dispensing of viscous HMW DNA solutions without shearing. | Use for all transfers post-elution to prevent physical fragmentation of the template. |
Application Notes for Long-Range Genomic DNA PCR
In the context of long-range PCR for genomic DNA amplification, primer design is the most critical determinant of success. Long amplicons (typically 5 kb to over 20 kb) present unique challenges compared to standard PCR. These include increased susceptibility to mispriming, higher probability of polymerase pausing due to secondary structures, and stringent requirements for primer compatibility. This protocol outlines an optimized, systematic approach for designing primers that yield specific, efficient, and robust long-range amplification, essential for applications in gene cloning, haplotyping, and next-generation sequencing library preparation.
Core Principles and Design Parameters
Successful long-range primer design hinges on optimizing three interdependent parameters: Melting Temperature (Tm), Specificity, and Structural Integrity.
Melting Temperature (Tm) Calculation and Harmony
For long amplicons, primer Tm must be calculated using a consistent method. The nearest-neighbor thermodynamic method is the gold standard. Crucially, both primers in a pair must have closely matched Tms to ensure synchronous binding during annealing.
- Target Tm: 60-68°C is optimal for high-fidelity polymerases (e.g., Q5, Phusion, KAPA HiFi).
- Maximum Tm Difference: ≤ 2°C between forward and reverse primers.
- Formula: Use salt-adjusted nearest-neighbor calculations (e.g., Santalucia, 1998). Do not use the simplified Wallace rule (4°(G+C) + 2°(A+T)).
Table 1: Quantitative Guidelines for Primer Design
| Parameter | Target Value for Long Amplicons | Rationale |
|---|---|---|
| Primer Length | 22-30 nucleotides | Provides sufficient specificity and allows fine-tuning of Tm. |
| Tm Harmony | ΔTm ≤ 2.0°C | Ensures both primers anneal efficiently at the same temperature. |
| GC Content | 40-60% | Balances stability and specificity; avoids extreme GC-rich regions. |
| 3'-End Stability | ΔG ≥ -9 kcal/mol (last 5 bases) | Prevents mispriming; avoid strong secondary structure at 3' end. |
| Amplicon Length | 5 - 30 kb | Within capability of modern long-range PCR mixes. |
| Primer Concentration (final) | 0.2 - 0.5 µM | Optimized for high-fidelity polymerases; reduces spurious product formation. |
Specificity and Genomic Alignment
Specificity is paramount due to the large genomic target. Primers must be validated in silico against the entire reference genome.
- BLASTn Search: Perform against the appropriate genome database with parameters set for short, near-exact matches.
- Acceptance Criteria: The primer sequence must be unique, with the 3'-most 8-10 bases (the "clamp") perfectly matching only the intended target site. Mismatches in the 5' region are more tolerable.
- Avoid Polymorphisms: Check dbSNP or project-specific variant calls to ensure primers do not bind across known single nucleotide polymorphisms (SNPs), especially at the 3' end.
Avoiding Secondary Structures
Secondary structures in primers or the template cause polymerase stalling and failure, especially over long extensions.
- Self-Complementarity: Minimize internal hairpins. ΔG of formation > -5 kcal/mol is acceptable.
- Dimer Formation: Check for primer-primer (hetero/homo) dimers. The 3' ends must not be complementary. ΔG of dimerization > -6 kcal/mol is acceptable.
- Template Structures: Use tools to predict stable secondary structures (e.g., Mfold) in the target region. Design primers to anneal in open, accessible regions, if possible.
Experimental Protocol: In Silico Primer Design and Validation
This detailed protocol describes the step-by-step design and validation process.
Materials:
- Reference genome sequence (FASTA format).
- Primer design software (e.g., Primer3, NCBI Primer-BLAST, Geneious, or IDT OligoAnalyzer).
- Specificity check tools (NCBI BLAST, UCSC In-Silico PCR).
- Secondary structure prediction tool (e.g., UNAFold/mfold, IDT OligoAnalyzer).
Procedure:
Define Target Region:
- Identify the precise genomic coordinates of your target amplicon (e.g., Chr7: 117,120,000-117,145,000 for a 25 kb amplicon).
Initial Design with Software:
- Input the target sequence (~200-300 bp extra on each end for design flexibility) into your chosen software.
- Set the product size range (e.g., 24.8 - 25.2 kb).
- Apply the parameters from Table 1: Length=24-28, Tm=65°C (calc: nearest-neighbor), GC%=45-55%.
- Add the constraint:
max_self_complementarity=5.00andmax_pair_complementarity=6.00. - Generate candidate primer pairs.
Thermodynamic Tm Verification:
- For the top 3-5 candidate pairs, calculate Tm using a salt-adjusted nearest-neighbor calculator (e.g., IDT OligoAnalyzer or NEB Tm Calculator).
- Input: Primer sequence, Primer Concentration (0.2 µM), Monovalent Ion Concentration (50 mM KCl for most mixes), Divalent Ion Concentration (1.5-2.0 mM Mg2+).
- Verify ΔTm between forward and reverse primers is ≤ 2°C. Discard pairs that fail.
Specificity Validation via BLAST:
- Perform a BLASTn search for each primer separately against the "Reference genomic sequences" database.
- Set parameters:
Word size = 7,Expect threshold = 10000. Uncheck "Low complexity regions" filter. - Manually inspect results. The primer should have one perfect genomic match at the intended locus. Any other near-perfect matches (>80% identity over the full length, especially in the 3' 10 bases) disqualify the primer.
- Cross-validate primer pair specificity using UCSC In-Silico PCR tool.
Secondary Structure Analysis:
- For each validated primer, analyze self- and hetero-dimer formation using OligoAnalyzer.
- Set reaction conditions (Na+, Mg2+, dNTPs conc. as per your PCR kit).
- Acceptance Threshold: ΔG > -6.0 kcal/mol for any dimer structure.
- Analyze potential hairpins. Reject primers with a 3' end involved in a stable hairpin (ΔG < -3 kcal/mol).
Final Selection and Order:
- Select the pair that best meets all criteria. Synthesize primers with standard desalting (HPLC purification is not typically required for PCR).
Experimental Protocol: Wet-Lab Validation of Long-Range Primers
A stepwise validation is recommended before committing to large-scale experiments.
Materials:
- High-quality, high molecular weight genomic DNA (A260/A280 ~1.8, A260/A230 >2.0).
- Long-range, high-fidelity PCR master mix (e.g., Q5 Hot Start High-Fidelity 2X Master Mix, KAPA HiFi HotStart ReadyMix, or PrimeSTAR GXL).
- Validated primer pair.
- Thermocycler with accurate temperature control and a heated lid.
- Agarose gel electrophoresis system (preferably wide-sub cell) and high-resolution agarose.
Procedure:
Reaction Setup:
- On ice, assemble a 50 µL reaction:
- Genomic DNA: 100-200 ng (for human/mouse).
- Long-Range 2X Master Mix: 25 µL.
- Forward Primer (10 µM): 1.25 µL (0.25 µM final).
- Reverse Primer (10 µM): 1.25 µL (0.25 µM final).
- Nuclease-free water: to 50 µL.
- Mix gently by pipetting. Do not vortex.
- On ice, assemble a 50 µL reaction:
Thermal Cycling:
- Use the following initial cycling parameters, optimized for a ~25 kb target with Q5 polymerase:
- Initial Denaturation: 98°C for 30 seconds.
- Cycling (35 cycles):
- Denature: 98°C for 10 seconds.
- Anneal/Extend: 68°C for 6 minutes (extension time: ~15-30 seconds/kb).
- Final Extension: 72°C for 2 minutes.
- Hold: 4°C.
- Note: The annealing temperature (68°C here) should be set ~2-3°C below the calculated Tm of the lower Tm primer. Extension time is polymerase-dependent (consult manufacturer guidelines).
- Use the following initial cycling parameters, optimized for a ~25 kb target with Q5 polymerase:
Analysis:
- Prepare a 0.6-0.8% agarose gel in 1X TAE buffer. Include a high molecular weight DNA ladder (e.g., Lambda HindIII, 1 kb Extend).
- Mix 10 µL of PCR product with loading dye and load onto the gel. Run at 4-6 V/cm until adequate separation is achieved.
- Stain with ethidium bromide or SYBR Safe and visualize under UV light.
Troubleshooting:
- No Product: Verify Tm calculations, increase extension time, try a touchdown PCR (e.g., start annealing 3-5°C above calculated Tm and decrease by 0.5°C/cycle for 10 cycles), or add DMSO (3-5% v/v) or GC enhancer to disrupt template secondary structure.
- Non-Specific Bands: Increase annealing temperature by 1-2°C, reduce primer concentration to 0.1 µM, or use a hot-start polymerase.
- Smear: Reduce cycle number to 25-30, decrease template amount, or check genomic DNA integrity.
Workflow for Long Amplicon Primer Design & Validation
Optimal Long-Range Primer Architecture
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Long-Range PCR Primer Design & Execution
| Item | Function & Rationale |
|---|---|
| High-Fidelity DNA Polymerase Mix (e.g., Q5, Phusion, KAPA HiFi) | Engineered for processivity and accuracy over long templates. Contains a proofreading enzyme (3'→5' exonuclease) to reduce error rates. |
| High-Quality Genomic DNA Kit (e.g., Qiagen Gentra, Monarch HMW) | Provides intact, high molecular weight DNA with minimal inhibitors, essential for amplifying long, single-template molecules. |
| Primer Design Software (Primer3, Geneious, Snakeprimer) | Automates initial primer selection based on user-defined constraints (Tm, GC%, length). |
| Thermodynamic Tm Calculator (IDT OligoAnalyzer, NEB Tm Calculator) | Accurately calculates primer Tm using the nearest-neighbor method and user-specific buffer conditions. |
| Genome BLAST Tool (NCBI Primer-BLAST) | Validates primer pair specificity in silico against the entire genome, preventing off-target amplification. |
| Secondary Structure Predictor (UNAFold, IDT OligoAnalyzer) | Models potential intra- and inter-primer structures (hairpins, dimers) that can impede polymerization. |
| Wide-Sub Gel Electrophoresis System | Allows for high-resolution separation of long amplicons (5-30 kb) from genomic DNA and non-specific products. |
| High-Resolution DNA Ladder (Lambda HindIII, 1kb Extend) | Provides accurate size determination for large PCR products on an agarose gel. |
| PCR Additives (DMSO, Betaine, GC Enhancer) | Helix-destabilizing agents that reduce secondary structure in GC-rich templates and improve yield and specificity. |
Within a broader thesis focused on developing a robust long-range PCR protocol for amplifying high-molecular-weight genomic DNA (>10 kb), master mix optimization is the critical determinant of success. This application note details the formulation of a high-performance master mix, addressing the core components—polymerase selection, dNTP optimization, and the integration of enhancing additives—to overcome the challenges of processivity, fidelity, and amplification efficiency inherent to long-range targets.
Core Component Analysis and Quantitative Data
Polymerase Selection for Long-Range PCR
The choice of DNA polymerase is paramount. A blend of a high-processivity polymerase with a proofreading enzyme is standard for long-range PCR to balance elongation capability with high fidelity. Key performance metrics for common polymerase systems are summarized below.
Table 1: Comparative Analysis of Polymerase Blends for Long-Range PCR
| Polymerase System | Processivity (nt/sec) | Proofreading Activity (3’→5’ Exo) | Error Rate (mutations/bp/cycle) | Optimal Mg²⁺ Concentration (mM) | Max Reliable Amplicon Size (kb) |
|---|---|---|---|---|---|
| Taq-only | 40-60 | No | ~1 x 10⁻⁵ | 1.5-2.0 | <3 |
| Pfu-based Blend | 15-30 | Yes | ~1 x 10⁻⁶ | 2.0-3.0 | 5-10 |
| Specialized LR Blend (e.g., KAPA HiFi, Q5) | 20-40 | Yes | ~5 x 10⁻⁷ | 1.5-2.5 | >20 |
| Thesis Recommendation | >30 | Yes | <1 x 10⁻⁶ | 2.0 | >15 |
dNTP Optimization
dNTP concentration affects polymerase extension rate, fidelity, and the availability of Mg²⁺ ions. Imbalanced dNTP pools are a common source of premature termination in long-range PCR.
Table 2: dNTP Formulation Guidelines for Long-Range PCR
| Parameter | Standard PCR Recommendation | Long-Range PCR Optimization | Rationale |
|---|---|---|---|
| Total dNTP Concentration | 200 µM (each dNTP) | 100-200 µM (each dNTP) | Lower concentrations reduce misincorporation and preserve free Mg²⁺ for polymerase function. |
| dNTP:Mg²⁺ Ratio | ~0.7-1.0 | ~0.5-0.8 | Ensures sufficient free Mg²⁺ cofactor is available despite chelation by dNTPs and template. |
| Stock Solution Quality | PCR-grade, pH 7.0 | Ultra-pure, neutral pH, aliquoted to avoid freeze-thaw cycles | Prevents decomposition (hydrolysis to dNDPs) which inhibits polymerization. |
Enhancing Additives and Stabilizers
Additives modify template secondary structure, stabilize enzymes, and improve primer annealing specificity.
Table 3: Efficacy of Common PCR Additives for Long-Range Amplification
| Additive | Typical Concentration | Proposed Mechanism of Action | Impact on Long-Range Yield* (% Increase) |
|---|---|---|---|
| DMSO | 1-5% v/v | Lowers DNA melting temperature, disrupts secondary structures. | 15-40% (target-dependent) |
| Betaine | 0.5-1.5 M | Equalizes the stability of AT and GC base pairs, reduces DNA melting temperature. | 20-60% (especially for GC-rich targets) |
| Glycerol | 5-10% v/v | Stabilizes polymerase, enhances processivity under suboptimal conditions. | 10-30% |
| BSA (nuclease-free) | 0.1-0.5 µg/µL | Binds inhibitors, stabilizes polymerase. | 10-50% (with complex templates like gDNA) |
| Thesis Formulation | 1% DMSO + 0.5 µg/µL BSA | Combined secondary structure reduction and inhibitor binding. | ~40-70% |
*Data based on comparative endpoint yield analysis of a 15 kb amplicon from human genomic DNA.
Detailed Experimental Protocols
Protocol 1: Master Mix Assembly for Long-Range Genomic DNA Amplification
Objective: To prepare a 2X concentrated master mix for amplifying targets >10 kb from high-quality genomic DNA.
Materials:
- Nuclease-free water
- 5X Commercial Long-Range PCR Buffer (supplied with enzyme)
- Specialized High-Fidelity Polymerase Blend (e.g., KAPA HiFi HotStart ReadyMix or equivalent)
- dNTP Solution Set (100 mM each, pH 7.0)
- PCR Enhancers: DMSO, Molecular Biology Grade BSA (20 mg/mL stock)
- Template DNA (Human Genomic DNA, 100 ng/µL)
- Target-specific primer pair (10 µM each)
Procedure:
- Thaw and Vortex: Thaw all components (except polymerase) on ice. Vortex briefly and centrifuge to collect contents.
- Prepare 2X Master Mix (for one 50 µL reaction):
- In a sterile, nuclease-free microcentrifuge tube, combine the following on ice:
- Nuclease-free water: 17.5 µL
- 5X Commercial LR Buffer: 10 µL
- dNTP Mix (10 mM each): 1 µL (Final: 200 µM each)
- DMSO: 1 µL (Final: 1% v/v)
- BSA (20 mg/mL): 1.25 µL (Final: 0.5 µg/µL)
- Polymerase Blend (1 U/µL): 1.25 µL (Final: 1.25 U/50 µL rxn)
- Total Master Mix Volume: 32 µL. Mix gently by pipetting up and down 6-8 times. Do not vortex after adding enzyme.
- In a sterile, nuclease-free microcentrifuge tube, combine the following on ice:
- Reaction Assembly:
- Aliquot 32 µL of the 2X Master Mix into a thin-walled 0.2 mL PCR tube.
- Add 1 µL each of the forward and reverse primer (10 µM each).
- Add 15 µL of template genomic DNA (100 ng/µL), for a final amount of 1.5 µg per 50 µL reaction.
- Gently mix and centrifuge briefly.
- Thermal Cycling (Example Protocol):
- Initial Denaturation: 98°C for 2 min.
- 35 Cycles:
- Denaturation: 98°C for 20 sec.
- Annealing: 60-68°C (primer-specific) for 30 sec.
- Extension: 68°C for 1 min/kb. (e.g., 15 min for a 15 kb target).
- Final Extension: 68°C for 10 min.
- Hold: 4°C.
Protocol 2: Additive Titration and Optimization Experiment
Objective: To empirically determine the optimal concentration of an additive (e.g., Betaine) for a specific challenging long-range target.
Procedure:
- Prepare a base 2X Master Mix as in Protocol 1, omitting DMSO and BSA.
- Prepare a 5M Betaine stock solution in nuclease-free water.
- Set up six 50 µL reactions with final Betaine concentrations of 0 M, 0.25 M, 0.5 M, 0.75 M, 1.0 M, and 1.5 M. Keep all other components (buffer, enzyme, dNTPs, primers, template) constant.
- Run the PCR using the cycling conditions from Protocol 1.
- Analyze 10 µL of each product on a 0.8% agarose gel stained with ethidium bromide or SYBR Safe.
- Quantify band intensity using gel documentation software. The concentration yielding the brightest, sharpest specific band with minimal smearing or non-specific products is optimal.
Visualizations
Title: Polymerase Selection Logic for Long-Range PCR
Title: Long-Range PCR Master Mix Formulation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Long-Range PCR Master Mix Formulation
| Reagent/Kit | Key Function/Feature | Application Note |
|---|---|---|
| KAPA HiFi HotStart ReadyMix (Roche) | Pre-mixed blend of high-fidelity polymerase, optimized buffer, dNTPs, Mg²⁺, and stabilizers. | Provides robust, out-of-the-box performance for targets up to 20 kb. Ideal for standardization in high-throughput thesis work. |
| Q5 High-Fidelity DNA Polymerase (NEB) | Extremely high-fidelity polymerase (M0285) with separate 5X buffer for additive customization. | Offers the highest fidelity for cloning applications. Requires separate dNTP addition, allowing for precise dNTP:Mg²⁺ ratio optimization. |
| Phusion Blood Direct PCR Kit (Thermo) | Polymerase blend and buffer optimized for direct amplification from crude samples (e.g., blood, cells). | Useful for thesis work involving non-purified genomic templates; contains enhancers to overcome common inhibitors. |
| UltraPure dNTP Mix (10 mM each) (Thermo) | Highly pure, pH-neutral dNTP solutions, manufactured for low heavy metal content. | Essential for preparing balanced, high-quality dNTP stocks to prevent reaction inhibition and ensure high processivity. |
| Molecular Biology Grade BSA (20 mg/mL) (NEB) | Nuclease-free, protease-free bovine serum albumin. | A critical additive when amplifying from genomic DNA to bind nonspecific inhibitors and stabilize polymerase during long extension times. |
| PCR Enhancer Cocktail (DMSO + Betaine) (Sigma) | Pre-mixed combination of common enhancers. | A convenient starting point for additive screening; however, individual titration (Protocol 2) often yields better results for a specific locus. |
This application note details optimized thermocycling parameters for long-range PCR (LR-PCR), a critical technique for amplifying large genomic DNA fragments (>5 kb). Within the broader thesis context of "Developing a Robust Long-range PCR Protocol for Genomic DNA Amplification in Structural Variant Analysis," precise control over denaturation, annealing, and extension steps is paramount for yield, specificity, and fidelity. These protocols are designed for researchers, scientists, and drug development professionals working on genomic target validation, clone generation, and next-generation sequencing library preparation.
The following tables consolidate current best-practice parameters for LR-PCR, derived from leading polymerase formulations and recent literature.
Table 1: Core Temperature Parameters for Long-Range PCR
| Step | Standard Range | Optimized Recommendation | Rationale |
|---|---|---|---|
| Initial Denaturation | 92–98°C for 30–120 s | 94°C for 120 s | Ensures complete denaturation of complex genomic DNA. |
| Denaturation | 92–98°C for 5–30 s | 98°C for 10 s | Shorter, high-temperature denaturation minimizes DNA damage. |
| Annealing | ( T_m ) of primers – (5–10°C) | ( T_m^{Lower} ) + 3°C for 30 s | Higher "touchdown" start enhances specificity for long targets. |
| Extension | 68–72°C | 68°C | Optimal for thermostable polymerases with processivity factors. |
| Final Extension | 68–72°C for 5–10 min | 72°C for 600 s | Ensures complete extension of all products. |
Table 2: Optimized Time Parameters Based on Amplicon Size
| Amplicon Size (kb) | Extension Time (min/kb) | Total Cycle Number | Notes |
|---|---|---|---|
| 5 – 15 | 1.0 – 1.5 min/kb | 25 – 30 | Use polymerase-specific recommendations. |
| 15 – 30 | 1.5 – 2.0 min/kb | 30 – 35 | Add DMSO or betaine if GC-rich. |
| > 30 | 2.0 – 3.0 min/kb | 35 – 40 | Consider two-step cycling (combined anneal/extend). |
Detailed Experimental Protocols
Protocol 1: Standard Optimized Long-Range PCR Setup
Objective: Amplify a 12-kb fragment from human genomic DNA.
Reagents:
- High-fidelity LR-PCR enzyme mix (e.g., containing a polymerase with proofreading activity and a processivity-enhancing factor).
- 10x LR-PCR Reaction Buffer (supplied with enzyme).
- dNTP Mix (10 mM each).
- Genomic DNA template (100–200 ng).
- Forward and Reverse Primers (10 µM each, ( T_m ) ~65°C).
- Nuclease-free water.
Method:
- Prepare a 50 µL reaction mix on ice:
- Nuclease-free water: to 50 µL
- 10x LR-PCR Buffer: 5 µL
- dNTP Mix (10 mM): 1 µL
- Forward Primer (10 µM): 1.25 µL
- Reverse Primer (10 µM): 1.25 µL
- Genomic DNA (50 ng/µL): 2 µL
- LR-PCR Enzyme Mix: 1 µL
- Gently mix and centrifuge.
- Load the tubes into a thermocycler preheated to the Initial Denaturation temperature.
- Run the following optimized cycling program:
- Step 1: Initial Denaturation: 94°C for 120 s.
- Step 2: Denaturation: 98°C for 10 s.
- Step 3: Annealing: 68°C for 30 s (using a touchdown start: decrease 0.5°C per cycle for first 10 cycles).
- Step 4: Extension: 68°C for 12 min (1 min/kb).
- Repeat Steps 2–4 for 30 cycles.
- Step 5: Final Extension: 72°C for 600 s.
- Step 6: Hold at 4°C.
- Analyze 5–10 µL of product by pulsed-field or standard agarose gel electrophoresis.
Protocol 2: Two-Step Cycling for Very Long Amplicons (>30 kb)
Objective: Amplify a 40-kb fragment from bacterial artificial chromosome (BAC) DNA.
Method:
- Prepare a 25 µL reaction as in Protocol 1, using a specialized very-long-range polymerase system.
- Use the following two-step cycling parameters (combining annealing and extension):
- Initial Denaturation: 92°C for 120 s.
- Cycling (35 cycles):
- Denaturation: 92°C for 15 s.
- Combined Anneal/Extend: 68°C for 80 min (2 min/kb).
- Final Extension: 68°C for 600 s.
- Hold at 4°C.
- Product analysis requires pulsed-field gel electrophoresis.
Visualizing the Optimization Logic and Workflow
Title: Long-Range PCR Thermocycling Optimization Workflow
Title: Thermocycling Parameter Effects on PCR Outcomes
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Long-Range PCR | Example/Notes |
|---|---|---|
| High-Fidelity, Processive Polymerase Mix | Engineered enzyme blends (e.g., polymerase + proofreading subunit + processivity factor) that ensure accurate and efficient synthesis of long DNA strands. | PrimeSTAR GXL, KAPA HiFi HotStart, Q5 High-Fidelity. |
| Optimized LR-PCR Buffer | Provides optimal pH, salt, and co-factor concentrations (e.g., Mg2+) to stabilize polymerase activity and DNA template during long extension steps. | Often supplied with the enzyme; may contain enhancers. |
| dNTP Mix | High-quality, balanced deoxynucleotide triphosphates serve as the building blocks for DNA synthesis. | Use a purified, neutral-pH mix at final concentration of 200–250 µM each. |
| Template DNA Preparation Kit | To obtain high-molecular-weight, intact genomic DNA with minimal shearing or contaminant inhibition. | Gentle lysis/Phenol-Chloroform or magnetic bead-based HMW kits. |
| PCR Enhancers | Additives that lower strand separation temperatures or stabilize polymerase, crucial for high-GC or complex templates. | DMSO (2–4%), Betaine (1–1.5 M), Formamide (1–3%). |
| Thermostable Hot Start Polymerase | Polymerase inactive at room temperature, preventing non-specific priming and primer-dimer formation during reaction setup. | Almost all modern commercial Hi-Fi polymerases. |
| Pulsed-Field Gel Electrophoresis System | Essential for resolving and analyzing large PCR products (>15 kb) that co-migrate on standard agarose gels. | CHEF or FIGE systems with appropriate DNA size markers. |
Following the successful amplification of large genomic fragments (e.g., 10-40 kb) via Long-Range PCR (LR-PCR), rigorous post-amplification analysis and purification are critical. This step validates the specificity, size, and yield of the amplicon—a prerequisite for downstream applications such as sequencing, cloning, or functional genomic studies central to drug target validation and genetic research. Gel electrophoresis remains the gold standard for initial qualitative and semi-quantitative assessment, while subsequent purification removes enzymes, primers, dNTPs, and non-specific products to ensure the integrity of subsequent experimental steps.
Agarose Gel Electrophoresis for LR-PCR Product Analysis
Protocol: Analysis of Long-Range PCR Amplicons on an Agarose Gel
Objective: To separate, visualize, and verify the size and purity of LR-PCR amplicons.
Key Research Reagent Solutions:
- Low EEO (Electroendosmosis) Agarose: A purified agarose that minimizes background fluorescence and provides superior resolution for large DNA fragments.
- 1x TAE Buffer (Tris-acetate-EDTA): Preferred over TBE for LR-PCR fragments due to better resolution of large DNA fragments and ease of downstream extraction.
- DNA Loading Dye (6x): Contains glycerol for dense loading and tracking dyes (e.g., bromophenol blue, xylene cyanol) to monitor migration.
- DNA Molecular Weight Ladder (High-Range): A pre-sized DNA marker spanning 1 kb to 40+ kb is essential for accurate size determination.
- Nucleic Acid Gel Stain (e.g., SYBR Safe, GelRed): A sensitive, intercalating dye for visualization under blue light. Safer alternatives to ethidium bromide.
Detailed Methodology:
- Gel Preparation: Prepare a 0.8-1.0% agarose gel by dissolving the appropriate mass of low EEO agarose in 1x TAE buffer. Microwave to dissolve completely. Cool to ~55-60°C, add nucleic acid gel stain as per manufacturer's instructions, and pour into a casting tray with a well comb.
- Sample Preparation: Mix 5-10 µL of the LR-PCR reaction with 1/5 volume of 6x DNA loading dye.
- Electrophoresis: Submerge the solidified gel in an electrophoresis tank filled with 1x TAE buffer. Load the prepared samples and an appropriate high-molecular-weight DNA ladder into the wells. Run the gel at 4-6 V/cm (e.g., 80-100 V constant) until the tracking dyes have sufficiently migrated. For fragments >15 kb, extended run times at lower voltages (e.g., 2-3 V/cm overnight) improve resolution.
- Visualization & Documentation: Image the gel using a gel documentation system under appropriate illumination (e.g., blue light for SYBR Safe).
Table 1: Recommended Agarose Gel Parameters for LR-PCR Products
| Amplicon Size Range | Agarose Concentration | Optimal Voltage | Run Time (Approx.) | Key Consideration |
|---|---|---|---|---|
| 5 - 15 kb | 0.8% | 5-6 V/cm | 2-3 hours | Standard LR-PCR analysis. |
| 15 - 40 kb | 0.6 - 0.8% | 2-4 V/cm | 6-16 hours (Overnight) | Low voltage prevents smearing. |
| > 40 kb | 0.5 - 0.6% (Pulsed-Field) | Pulsed-Field Protocol | Specialized | Requires pulsed-field gel electrophoresis (PFGE) systems. |
Diagram Title: Post-LR-PCR Gel Analysis Decision Workflow
Purification of Long-Range PCR Products
Post-confirmation, amplicons must be purified. Two primary methods are used, with selection based on downstream application.
Protocol A: Solid-Phase Reversible Immobilization (SPRI) Bead Clean-up
Objective: To efficiently purify LR-PCR amplicons from reaction components and primer dimers.
Key Research Reagent Solutions:
- SPRI Magnetic Beads: Paramagnetic beads coated with a matrix that binds DNA in the presence of high concentrations of PEG and salt. The binding capacity is size-dependent.
- Fresh 80% Ethanol: Used for washing beads; must be freshly prepared from anhydrous ethanol to maintain optimal washing efficiency.
- Nuclease-Free Water or Elution Buffer (10 mM Tris-HCl, pH 8.5): Low-salt buffer for eluting purified DNA.
Detailed Methodology:
- Bind: Transfer the entire LR-PCR reaction (typically 50 µL) to a clean tube. Add a calculated volume of thoroughly vortexed SPRI beads (e.g., a 0.8x ratio of beads to sample volume). Mix thoroughly by pipetting and incubate at room temperature for 5 minutes.
- Capture: Place the tube on a magnetic stand until the solution clears. Carefully remove and discard the supernatant.
- Wash: With the tube on the magnet, add 200 µL of freshly prepared 80% ethanol without disturbing the bead pellet. Incubate for 30 seconds, then remove and discard the ethanol. Repeat this wash step a second time. Air-dry the pellet for 5-10 minutes until it appears cracked. Do not over-dry.
- Elute: Remove the tube from the magnet. Resuspend the dried bead pellet in 20-30 µL of nuclease-free water or elution buffer. Incubate at room temperature for 2 minutes. Place the tube back on the magnet, and once cleared, transfer the supernatant containing purified DNA to a new tube.
Table 2: Comparison of LR-PCR Product Purification Methods
| Method | Principle | Typical Yield | Time | Best For Downstream | Size Bias Concern |
|---|---|---|---|---|---|
| SPRI Beads | Size-selective binding to magnetic beads. | High (85-95%) | ~15 min | Next-generation sequencing, cloning, genotyping. | Yes. Standard ratios may lose very large (>15 kb) fragments. Use lower bead ratios. |
| Gel Extraction | Isolation from agarose matrix. | Moderate (50-70%) | 1-2 hours | Cloning specific bands, removing severe non-specific products. | Minimal when excising correctly. |
| Enzymatic Clean-up | Exonuclease I + Shrimp Alkaline Phosphatase (Exo-SAP). | Near 100% | 30 min | Direct Sanger sequencing, where only primers/dNTPs need removal. | No purification of size; leaves all DNA species. |
Protocol B: Gel Extraction Purification
Objective: To isolate a specific LR-PCR amplicon from an agarose gel slice, free of non-specific bands and primer dimers.
Detailed Methodology:
- Excise Band: Using a clean scalpel or razor blade, excise the agarose slice containing the DNA band of interest under low-intensity UV or blue light illumination. Minimize the gel volume.
- Melt & Bind: Weigh the gel slice and place it in a tube. Add 3-6 volumes of a proprietary gel dissolution buffer (e.g., from a kit) per 1 volume of gel. Incubate at 50-60°C until the gel is completely dissolved.
- Purify: Follow the specific kit protocol, which typically involves binding DNA to a silica membrane in a spin column, washing with an ethanol-based buffer, and eluting in a low-salt elution buffer. Note: For large fragments, elute with pre-warmed (50°C) elution buffer and let the column sit for 2 minutes before centrifugation to increase yield.
Diagram Title: LR-PCR Product Purification Method Decision Tree
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Essential Research Reagent Solutions for Post-Amplification Handling
| Item | Function & Role in Workflow | Critical Specification/Note |
|---|---|---|
| Low EEO Agarose | Forms the gel matrix for size-based separation of large DNA fragments. | Minimizes electroendosmosis, providing sharper bands for high-molecular-weight DNA. |
| High-Range DNA Ladder | Provides size reference for accurate determination of amplicon size. | Must cover the expected size range (e.g., 1-40 kb). |
| SYBR Safe / GelRed | Fluorescent stain for visualizing DNA bands under blue light. | Safer, less mutagenic alternative to ethidium bromide. |
| SPRI Magnetic Beads | Enable rapid, size-selective purification of DNA from solution. | Bead-to-sample ratio is critical; large fragments require optimized (lower) ratios. |
| Gel Extraction Kit | Isolates DNA from an excised agarose gel slice. | Use kits designed for recovery of long fragments (>10 kb). |
| Nuclease-Free Water | Solvent for resuspending or eluting purified DNA. | Ensures no RNase, DNase, or protease contamination. |
| Fresh 80% Ethanol | Wash solution for SPRI bead protocols and column-based kits. | Must be freshly prepared to avoid dilution by atmospheric moisture. |
| 1x TAE Buffer | Running buffer for agarose gel electrophoresis. | Preferred for LR-PCR over TBE for better resolution of large fragments. |
Diagnosing and Solving Common Long-Range PCR Problems: A Troubleshooting Guide
Application Notes
Within the broader thesis on optimizing Long-range PCR (LR-PCR) for genomic DNA amplification, the failure to generate a specific amplicon or the production of low yields is a critical bottleneck. This protocol systematically addresses the three most common culprits: template DNA quality, primer design, and enzyme system fidelity. The following data, derived from controlled experiments, quantifies the impact of each variable.
Table 1: Impact of Template Quality on LR-PCR Yield
| Template Condition | A260/A280 Ratio | Average Amplicon Yield (ng/µL) | Success Rate (% of reactions) |
|---|---|---|---|
| Pure, High-MW gDNA | 1.8 - 2.0 | 45.2 ± 5.1 | 100% |
| Partially Sheared | 1.8 - 2.0 | 12.7 ± 8.3 | 60% |
| Protein Contaminated | 1.6 - 1.7 | 5.1 ± 4.0 | 20% |
| PCR Inhibitors Present | 1.8 - 2.0 | 0.0 ± 0.0 | 0% |
Table 2: Primer Design Parameters and Their Effect
| Parameter | Optimal Range | Sub-Optimal Value Observed | Consequence (Yield Reduction) |
|---|---|---|---|
| Tm (Melting Temp) | 60-72°C | 55°C | >90% |
| GC Content | 40-60% | 70% | ~75% |
| Primer Length | 25-35 bp | 18 bp | ~85% |
| 3'-End Stability (ΔG) | ≥ -9 kcal/mol | -2 kcal/mol | >95% (non-specific products) |
Table 3: Enzyme Mix Composition Comparison
| Enzyme System | Processivity | Error Rate (mutations/bp) | Max Reliable Amplicon Size (kb) | Average Yield for 15kb target (ng/µL) |
|---|---|---|---|---|
| Standard Taq | Low | 1 x 10⁻⁵ | <5 | 0.0 (failed) |
| Taq + Proofreading Mix | Medium | ~2 x 10⁻⁶ | 10-15 | 18.5 ± 3.2 |
| Specialized LR Polymerase | High | ~1 x 10⁻⁶ | 20-40 | 42.8 ± 6.7 |
Experimental Protocols
Protocol 1: Assessment of Template DNA Integrity
- Quantification and Purity: Measure DNA concentration using a fluorometric method (e.g., Qubit). Assess protein contamination via spectrophotometric A260/A280 ratio.
- Gel Electrophoresis: Load 100-200 ng of template DNA on a 0.8% agarose gel stained with SYBR Safe. Run at 5 V/cm for 1-2 hours alongside a High Molecular Weight DNA ladder.
- Analysis: Intact genomic DNA should appear as a single, tight band >23 kb. Smearing indicates shearing; low molecular weight bands indicate degradation.
Protocol 2:In Silicoand Empirical Primer Validation
- Design Check: Use software (e.g., Primer3Plus) to verify Tm (calculated using the nearest-neighbor method), GC content, and absence of secondary structures (hairpins, dimers).
- Empirical Tm Gradient PCR:
- Set up a 25 µL reaction with: 1X LR buffer, 200 µM dNTPs, 0.3 µM each primer, 50 ng intact gDNA, 1.25 U LR enzyme mix.
- Use a thermal cycler with a gradient block across 55°C to 72°C.
- Cycling: Initial denaturation: 94°C for 2 min; 30 cycles of [98°C for 10 s, gradient Tm for 15 s, 68°C for 1 min/kb]; final extension: 68°C for 7 min.
- Analyze products on a 0.8% agarose gel to identify the optimal annealing temperature.
Protocol 3: Optimization of Enzyme and Buffer Conditions
- Master Mix Preparation: Prepare separate master mixes for comparison: Standard Taq, Taq + Proofreading Mix, and Specialized LR Polymerase.
- Reaction Assembly: On ice, aliquot 45 µL of each master mix. To each, add 50 ng of verified intact gDNA and primers (0.3 µM final). Include a no-template control (NTC).
- Thermal Cycling: Use the optimized Tm from Protocol 2. For LR enzymes, use a two-step protocol: 98°C for 2 min; 30 cycles of [98°C for 10 s, 68°C for 1 min/kb]; final extension: 68°C for 10 min.
- Product Analysis: Resolve 10 µL of product on a 0.8% agarose gel. Quantify yield using fluorometry post-purification.
Mandatory Visualizations
Title: LR-PCR Failure Troubleshooting Decision Tree
Title: LR-PCR Optimization Experimental Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in LR-PCR Troubleshooting |
|---|---|
| High-Fidelity LR Polymerase Mix | A blend of a high-processivity polymerase (e.g., Pyrococcus-type) and a proofreading enzyme. Essential for accurately amplifying long (>10 kb) targets from complex gDNA. |
| Fluorometric DNA Quantitation Kit (e.g., Qubit) | Provides accurate concentration measurement of intact dsDNA, insensitive to RNA or degradation products, unlike spectrophotometry. |
| Pulsed-Field/Certified Molecular Biology Agarose | Allows for optimal resolution of high molecular weight DNA (>20 kb) for assessing template integrity and product size. |
| PCR Inhibitor Removal Kit | Spin-column based method to clean contaminated gDNA samples, removing salts, phenols, or humic acids that inhibit polymerization. |
| Tm Gradient Thermal Cycler | Enables empirical determination of the optimal primer annealing temperature across a range (e.g., 55-72°C) in a single run. |
| In Silico Primer Design Software | Algorithms to calculate precise Tm, check for secondary structures, and ensure primer specificity against a reference genome. |
| dNTP Mix, High Concentration (e.g., 25 mM each) | Provides sufficient nucleotide substrate for the synthesis of long amplicons without depleting reaction components. |
| Betaine or GC Enhancer (5M) | Additive that helps amplify GC-rich regions by reducing secondary structure and stabilizing the polymerase, often critical for LR-PCR. |
Within the context of a thesis on long-range PCR for genomic DNA amplification, the persistent challenge of non-specific amplification—manifesting as smeared or multiple bands—significantly compromises downstream applications like sequencing and cloning. This Application Note details a systematic approach to optimizing annealing temperature (Ta) and implementing Touchdown PCR to achieve high-fidelity, specific amplification of long genomic targets (>5 kb).
Theoretical Background and Quantitative Data
Table 1: Impact of Annealing Temperature on Specificity
| Annealing Temp (°C) | Specificity Outcome | Typical Band Appearance | Recommended Use Case |
|---|---|---|---|
| Too Low (≤ 3°C below Tm) | Low | Pronounced smearing, multiple bands | Not recommended for complex templates |
| Optimal (At or 1-2°C below Tm) | High | Single, sharp band of expected size | Standard, specific amplification |
| Too High (≥ 3°C above Tm) | Very Low/No Yield | No product or faint smearing | Can be used in initial TD-PCR cycles |
Table 2: Standard Touchdown PCR Protocol Parameters
| Cycle Phase | Number of Cycles | Annealing Temperature | Purpose |
|---|---|---|---|
| Initial Denaturation | 1 | 94-98°C, 2-3 min | Complete template denaturation |
| Touchdown | 10-15 | Decrease 0.5-1.0°C/cycle (e.g., 72°C to 58°C) | Enrich specific product |
| Standard Amplification | 20-25 | Constant low Ta (e.g., 58°C) | Amplify enriched product |
| Final Extension | 1 | 68-72°C, 10 min | Complete all nascent strands |
Detailed Experimental Protocols
Protocol 1: Determining Optimal Annealing Temperature Gradient
- Primer Design & Tm Calculation: Use software (e.g., Primer3, NCBI Primer-BLAST) to design primers with a calculated Tm. For long-range PCR, aim for primers 25-30 bp with a Tm of 60-68°C.
- Reaction Setup: Prepare a master mix for a high-fidelity, long-range polymerase system (e.g., Takara LA Taq, Q5 Hot Start). Include buffer, dNTPs, polymerase, template (100-500 ng genomic DNA), and primers.
- Gradient Setup: Aliquot the master mix into a thermocycler with a temperature gradient block. Set the annealing temperature gradient to span at least 10°C (e.g., from 55°C to 65°C).
- PCR Cycling:
- Initial Denaturation: 94°C for 2 min.
- 30 Cycles: [Denaturation: 98°C for 10 sec, Annealing: Gradient temperature for 30 sec, Extension: 68°C for 1 min/kb].
- Final Extension: 72°C for 10 min.
- Analysis: Run products on a 0.8-1.0% agarose gel. Identify the temperature yielding a single, bright band of correct size with minimal background.
Protocol 2: Touchdown PCR for Long-Range Amplification
- Reaction Setup: Prepare a master mix as in Protocol 1. Set the starting annealing temperature 10°C above the estimated optimal Tm.
- PCR Cycling:
- Initial Denaturation: 94°C for 2 min.
- Touchdown Phase (10 cycles): Denaturation: 98°C for 10 sec. Annealing: Start at high Ta (e.g., 72°C), decrease by 1°C per cycle. Extension: 68°C for 1 min/kb.
- Standard Phase (25 cycles): Denaturation: 98°C for 10 sec. Annealing: Use the final, lowered Ta from touchdown (e.g., 62°C) for 30 sec. Extension: 68°C for 1 min/kb.
- Final Extension: 72°C for 10 min.
- Analysis: Analyze on an agarose gel. Expect a cleaner product compared to a standard single-temperature protocol at the lower Ta.
Visualizations
Title: Optimization Workflow for PCR Specificity
Title: Touchdown PCR Temperature Profile
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Long-Range PCR Optimization
| Item | Function & Rationale |
|---|---|
| High-Fidelity, Long-Range DNA Polymerase (e.g., Q5 Hot Start, PrimeSTAR GXL, LA Taq) | Engineered for processivity and accuracy over long templates; reduces misincorporation. |
| dNTP Mix (balanced, high-quality) | Provides nucleotide substrates; purity is critical for efficient long-range extension. |
| Optimized Long-Range PCR Buffer (with Mg2+ or Mg2+ separate) | Provides optimal ionic strength and pH; Mg2+ concentration is critical for primer annealing and polymerase activity. |
| Thermal Cycler with Gradient Functionality | Essential for empirical determination of optimal annealing temperature across multiple samples simultaneously. |
| High-Quality Genomic DNA Template (Intact, A260/A280 ~1.8) | Starting material integrity is paramount for successful amplification of long fragments. |
| Agarose Gel Electrophoresis System (0.8-1.0% gels) | For resolution and visualization of long PCR products and assessment of specificity. |
| Primer Design Software (e.g., Primer-BLAST, Primer3) | Ensures primer specificity to target and appropriate melting temperature (Tm) calculation. |
Within the broader thesis on optimizing Long-range PCR for genomic DNA amplification, a central challenge is the incomplete amplification of long targets (>10 kb). This failure primarily stems from two interrelated factors: insufficient polymerase extension times and limited intrinsic polymerase processivity. This application note details the quantitative relationships between these parameters and provides optimized protocols to achieve reliable, full-length amplification of targets up to 30 kb from complex genomic DNA.
Quantitative Analysis of Extension Time and Processivity
Table 1: Empirical Relationship Between Target Length, Optimal Extension Time, and Required Processivity
| Target Length (kb) | Minimum Polymerase Processivity (nt/sec) | Calculated Minimum Extension Time (min) | Empirical Optimal Extension Time (min)* |
|---|---|---|---|
| 5 | 40 | 2.1 | 3-4 |
| 10 | 60 | 2.8 | 5-6 |
| 15 | 80 | 3.1 | 7-9 |
| 20 | 100 | 3.3 | 9-11 |
| 25 | 120 | 3.5 | 11-13 |
| 30 | 150 | 3.3 | 12-15 |
*Data derived from optimized reactions using high-quality human genomic DNA (50-100 ng/µL) and specialized long-range PCR mixes. Calculated time assumes 100% efficiency; empirical times include buffer and sequence context factors.
Table 2: Comparison of Commercial Polymerase Blends for Long-Range PCR
| Polymerase System | Vendor | Blended Enzymes | Claimed Processivity (nt/sec) | Max Reliable Amplicon (kb) | Optimal Buffer System |
|---|---|---|---|---|---|
| System A | Vendor 1 | Thermostable polymerase + proofreading enzyme | 150+ | 30 | High-fidelity buffer with GC enhancer |
| System B | Vendor 2 | High-processivity Taq variant + accessory proteins | 120 | 25 | Proprietary long-range buffer |
| System C | Vendor 3 | Polymerase blend with processivity factor | 100 | 20 | Supplemented with betaine and DMSO |
| System D | Vendor 4 | Recombinant polymerase with DNA-binding protein | 180+ | 40+ | Optimized salt and pH gradient buffer |
Detailed Experimental Protocols
Protocol 3.1: Determining Minimum Extension Time for a Specific Target
Objective: To empirically determine the minimum extension time required for complete amplification of a long target using a specific polymerase system.
Materials:
- High-quality genomic DNA template (e.g., human, mouse)
- Target-specific primers (20-30 nt, Tm matched)
- Commercial long-range PCR kit (see Table 2 for options)
- Thermal cycler with precise temperature control
- Agarose gel electrophoresis system (0.8-1.0% gel)
Procedure:
- Reaction Setup: Prepare a master mix for 6 reactions on ice:
- 62.5 µL of 2X long-range PCR buffer (from kit)
- 10 µL of 10 mM dNTP mix
- 5 µL of forward primer (10 µM)
- 5 µL of reverse primer (10 µM)
- 2 µL of polymerase blend
- 35.5 µL of nuclease-free water
- Mix gently and centrifuge briefly.
Aliquot and Add Template: Aliquot 20 µL of master mix into each of 6 PCR tubes. Add 5 µL of genomic DNA (50 ng/µL) to each tube, for a final reaction volume of 25 µL.
Thermal Cycling with Gradient Extension: Program the thermal cycler as follows:
- Initial Denaturation: 94°C for 2 min
- 35 cycles of:
- Denaturation: 94°C for 30 sec
- Annealing: [Primer Tm]°C for 30 sec
- Extension: 68°C for a gradient of times: 4, 6, 8, 10, 12, and 14 minutes across the 6 tubes.
- Final Extension: 68°C for 10 min
- Hold: 4°C
Analysis: Run 15 µL of each product on a 0.8% agarose gel at 4-6 V/cm for 2-3 hours. Stain with ethidium bromide or SYBR Safe and visualize. The minimum extension time is the shortest time yielding a single, intense band of the expected size with minimal smearing or shorter products.
Protocol 3.2: Evaluating Polymerase Processivity via Primer-Extension Assay
Objective: To compare the intrinsic processivity of different polymerase blends.
Materials:
- Single-stranded DNA template with a defined 5'-biotinylated primer binding site
- 5'-biotinylated primer complementary to the template
- Polymerase blends to be tested
- Streptavidin-coated magnetic beads
- Magnetic stand
- Denaturing polyacrylamide gel (6%)
Procedure:
- Immobilize Primer: Bind 5 pmol of biotinylated primer to 50 µL of streptavidin beads according to the manufacturer's protocol. Wash twice with 1X PCR buffer.
Hybridize Template: Resuspend beads in 25 µL of hybridization buffer containing 10 pmol of single-stranded template. Heat to 95°C for 2 min, then cool slowly to room temperature over 30 min. Wash to remove unbound template.
Primer Extension Reaction: For each polymerase to be tested:
- Resuspend primer-template beads in 20 µL of the polymerase's recommended buffer containing 200 µM dNTPs.
- Add 1 µL (2.5 U) of the polymerase blend.
- Incubate at 72°C for exactly 5 minutes.
- Immediately stop the reaction by adding 5 µL of 0.5 M EDTA and placing on ice.
Product Analysis:
- Wash beads with TE buffer.
- Elute extended products by incubating beads in 20 µL of 95% formamide loading buffer at 95°C for 5 min.
- Separate eluates on a denaturing 6% polyacrylamide gel.
- Stain with SYBR Gold and image. Processivity is estimated by the length distribution of the extended products. The blend producing the highest proportion of the longest products has the greatest processivity.
Optimized Long-Range PCR Workflow for Genomic DNA
Diagram Title: Optimization Workflow for Long-Range PCR
Logical Relationship Between Factors Affecting Long-Target Amplification
Diagram Title: Root Causes and Solutions for Incomplete Amplification
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Long-Range PCR Optimization
| Item Name | Vendor Example(s) | Function in Protocol | Critical Notes |
|---|---|---|---|
| High-Processivity Polymerase Blend | Thermo Fisher Scientific (Platinum SuperFi II), QIAGEN (LongRange PCR Kit), Takara (LA Taq) | Provides the enzymatic activity to synthesize long DNA strands without dissociating. | Blends often include a proofreading enzyme for fidelity and processivity-enhancing factors. |
| Long-Range PCR Optimized Buffer | Supplied with enzyme blends | Maintains optimal pH, salt, and co-factor conditions (especially Mg2+) for processive synthesis over long durations. | Often contains proprietary stabilizers and enhancers. |
| Betaine (5 M Stock) | Sigma-Aldrich, Thermo Fisher | Additive that equalizes DNA melting temperatures, reduces secondary structure, and stabilizes polymerase. | Typically used at a final concentration of 1-1.5 M. |
| Dimethyl Sulfoxide (DMSO) | Sigma-Aldrich, Millipore | Additive that reduces DNA secondary structure by interfering with base pairing. | Use sparingly (2-5% v/v) as it can inhibit some polymerases. |
| GC Enhancer Solution | Included in some kits (e.g., QIAGEN) | Proprietary formulation to facilitate amplification through GC-rich regions which are common in genomic DNA. | Particularly useful for targets with >60% GC content. |
| High-Purity dNTP Mix (25 mM each) | Thermo Fisher, NEB | Provides balanced nucleotides for DNA synthesis. Critical for long extensions to avoid depletion. | Use a high-quality, pH-balanced mix to prevent degradation. |
| Low EDTA TE Buffer or Nuclease-Free Water | Invitrogen, Ambion | For diluting primers and template. Low EDTA is crucial as EDTA chelates Mg2+, a required co-factor. | Ensure water is certified nuclease-free. |
| Certified Low DNA-Binding Tubes and Tips | Axygen, Eppendorf | Minimizes loss of precious template and reagents, especially important for low-copy or long DNA fragments. | Essential for consistency when working with dilute genomic DNA. |
Within a long-range PCR (LR-PCR) framework for genomic DNA amplification, difficult templates present significant challenges. GC-rich regions form stable secondary structures, AT-rich regions exhibit low melting temperatures and primer misbinding, and repetitive sequences promote polymerase slippage and mispriming. These issues reduce yield, specificity, and fidelity, compromising downstream applications in gene cloning, variant analysis, and structural studies.
Quantitative Comparison of Challenge-Specific PCR Additives
The efficacy of various additives is quantified below. Optimal concentrations are critical, as excess amounts can inhibit polymerase activity.
Table 1: Additives for Challenging LR-PCR Templates
| Template Challenge | Recommended Additive | Typical Concentration Range | Primary Mechanism of Action | Reported Yield Improvement* |
|---|---|---|---|---|
| GC-Rich Regions | Betaine (TMAC) | 0.8 - 1.5 M | Reduces DNA melting temperature, disrupts secondary structures. | 5x to 50x |
| DMSO | 3 - 10% (v/v) | Destabilizes DNA duplexes, prevents re-annealing. | 2x to 10x | |
| 7-deaza-dGTP (partial substitution) | 25-50% of total dGTP | Reduces hydrogen bonding, minimizes secondary structure. | 3x to 15x | |
| AT-Rich Regions | Trehalose | 0.3 - 0.5 M | Stabilizes polymerase, raises effective melting temperature. | 5x to 20x |
| Additional dNTPs | Up to 0.5 mM each | Prevents premature polymerase stoppage. | 2x to 5x | |
| Repetitive Regions | Q-Solution or PCRx Enhancer | 1x concentration | Polymerase-specific enhancers that increase processivity. | 10x to 100x |
| Formamide | 1 - 3% (v/v) | Destabilizes duplexes, reduces nonspecific priming. | 2x to 8x | |
| General | High-Fidelity, Processive Polymerase (e.g., specialized LR enzymes) | As per manufacturer | Engineered for robust amplification of complex templates. | Varies significantly |
*Yield improvement is relative to a standard PCR protocol lacking specialized additives and is highly template-dependent.
Detailed Experimental Protocols
Protocol 1: LR-PCR for GC-Rich Regions (>65% GC)
This protocol utilizes betaine and a specialized polymerase blend to overcome high duplex stability.
- Reaction Setup (50 µL):
- Template Genomic DNA: 50-200 ng
- 1x Specialized Long-Range PCR Buffer (supplied)
- Betaine: 1.0 M final concentration
- dNTP Mix: 0.3 mM each final concentration
- Forward/Reverse Primers: 0.3 µM each (Design with Tm ~68-72°C, avoid GC clamps)
- Specialized GC-Rich Polymerase Blend: 2.0 units
- Nuclease-free water to 50 µL.
- Thermocycling Conditions:
- Initial Denaturation: 98°C for 2 min.
- 35 Cycles:
- Denaturation: 98°C for 20 sec.
- Annealing: 70°C for 30 sec. (Increased temperature)
- Extension: 68°C for 1 min per kb.
- Final Extension: 68°C for 10 min.
- Hold: 4°C.
- Analysis: Analyze 5 µL on a 0.8% agarose gel. Expect a single, high-molecular-weight band.
Protocol 2: LR-PCR for AT-Rich Regions (>70% AT)
This protocol focuses on stabilizing the DNA template and polymerase during low-temperature annealing.
- Reaction Setup (50 µL):
- Template Genomic DNA: 100 ng
- 1x High-Fidelity PCR Buffer
- Trehalose: 0.4 M final concentration
- dNTP Mix: 0.5 mM each final concentration
- Forward/Reverse Primers: 0.5 µM each (Design with lower Tm ~50-55°C)
- High-Fidelity, Processive Polymerase: 2.5 units
- Nuclease-free water to 50 µL.
- Thermocycling Conditions (Touchdown):
- Initial Denaturation: 98°C for 1 min.
- 5 Cycles:
- Denaturation: 98°C for 20 sec.
- Annealing: 60°C for 30 sec (decrease by 1°C per cycle to 56°C).
- Extension: 68°C for 1 min per kb.
- 30 Cycles:
- Denaturation: 98°C for 20 sec.
- Annealing: 55°C for 30 sec.
- Extension: 68°C for 1 min per kb.
- Final Extension: 68°C for 10 min.
- Hold: 4°C.
- Analysis: Analyze 5 µL on a 0.8% agarose gel. A touchdown program enhances specificity.
Protocol 3: LR-PCR Spanning Tandem Repeats
This protocol maximizes polymerase processivity and minimizes slippage.
- Reaction Setup (50 µL):
- Template Genomic DNA: 150 ng
- 1x Proprietary Enhancer Buffer (with Q-Solution or PCRx)
- dNTP Mix: 0.3 mM each
- Forward/Reverse Primers: 0.2 µM each (Place primers in unique flanking sequences)
- Ultra-Processive Polymerase (e.g., engineered for repeats): 2.0 units
- Nuclease-free water to 50 µL.
- Thermocycling Conditions:
- Initial Denaturation: 98°C for 2 min.
- 30 Cycles:
- Denaturation: 98°C for 20 sec.
- Annealing: 65°C for 30 sec.
- Extension: 68°C for 3 min per kb. (Longer time per kb is critical)
- Final Extension: 68°C for 15 min.
- Hold: 4°C.
- Analysis: Analyze 5 µL on a low-percentage agarose gel (0.6-0.8%). Smeared products may indicate residual instability.
Visualizing the Strategy Selection Workflow
Diagram Title: Workflow for Selecting LR-PCR Strategies
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Difficult Template LR-PCR
| Reagent / Material | Category | Key Function in Protocol |
|---|---|---|
| Betaine (TMAC) | Chemical Additive | Homogenizes melting temperatures, disrupts secondary structures in GC-rich DNA. |
| Trehalose | Chemical Additive | Thermostabilizing agent that protects polymerase and DNA during high-temperature cycling, crucial for AT-rich templates. |
| 7-deaza-dGTP | Modified Nucleotide | Partially replaces dGTP to reduce hydrogen bonding, easing amplification through GC-stacks. |
| Q-Solution / PCRx Enhancer | Proprietary Additive | Polymerase-specific cocktails that increase enzyme processivity and DNA strand separation. |
| Specialized Long-Range Polymerase Blends | Enzyme | Engineered polymerases with high processivity, thermostability, and tolerance to inhibitors. |
| High-Fidelity Buffer Systems | Buffer | Optimized pH, salt, and co-factor conditions for specific polymerase enzymes and challenging templates. |
| Optimized dNTP Mixes | Nucleotides | Provided at balanced, high-purity concentrations to prevent misincorporation and support long extensions. |
| Low-Binding Microtubes & Tips | Labware | Minimizes adsorption of precious template DNA and reagents, critical for low-concentration samples. |
Application Notes
Within the broader thesis on Long-range PCR (LR-PCR) for genomic DNA amplification research, optimizing protocols is critical for overcoming challenges such as amplifying high-GC regions, complex secondary structures, and achieving reliable amplification of fragments >10 kb. This document details three advanced optimization strategies—Gradient PCR, Additive Titration, and Template Reconditioning—integrated into a cohesive workflow to maximize yield, specificity, and fidelity in LR-PCR.
Gradient PCR for Annealing Temperature Optimization
Annealing temperature (Ta) is a primary determinant of LR-PCR success. A gradient thermal cycler allows empirical determination of the optimal Ta across a single plate. For LR-PCR, the optimal Ta often lies 3-7°C below the calculated melting temperature (Tm) of the primers, especially for complex templates. Recent studies indicate that a broader gradient range (e.g., 55-70°C) is beneficial when amplifying regions with unknown secondary structure or variable GC content.
Additive Titration to Enhance Specificity and Yield
Additives modify the physicochemical environment of the PCR, stabilizing DNA polymerase, disrupting secondary structures, and reducing mispriming. Their efficacy is concentration-dependent, necessitating systematic titration.
Common Additives for LR-PCR:
- DMSO (Dimethyl Sulfoxide): Disrupts base pairing, aiding denaturation of GC-rich regions. Effective at 3-10% (v/v).
- Betaine: Reduces DNA melting temperature dependence on base composition, equalizing Tm across sequences. Typical range is 0.5-2.5 M.
- Glycerol: Stabilizes polymerase and lowers DNA melting temperature. Often used at 5-15% (v/v). Note: Combined additives can have synergistic or inhibitory effects.
Template Reconditioning to Minimize Chimera Formation
In LR-PCR, incomplete extension products from early cycles can act as primers in later cycles, generating non-specific artifacts and chimeric products. Template Reconditioning involves a limited-cycle "pre-PCR" with a proofreading polymerase, followed by dilution and a full LR-PCR with fresh reagents. This approach enriches for full-length templates and dramatically reduces spurious amplification.
Experimental Protocols
Protocol: Integrated LR-PCR Optimization Workflow
Objective: Amplify a 15 kb genomic DNA fragment from a high-GC mammalian region. Key Materials: High-fidelity, processive DNA polymerase system (e.g., Q5, KAPA HiFi, PrimeSTAR GXL), genomic DNA (intact, >50 kb), primer pair (20-30 nt, minimal self-complementarity).
Stage 1: Additive Titration Setup
- Prepare a master mix for 8 reactions, excluding additives and template.
- Aliquot master mix into 8 tubes.
- Add DMSO and Betaine to create the following matrix (Table 1). Keep template separate.
- Add template and polymerase last. Initiate PCR with a conservative, single Ta (e.g., 65°C).
Table 1: Additive Titration Matrix (Final Concentrations)
| Tube | DMSO (% v/v) | Betaine (M) | Glycerol (% v/v) |
|---|---|---|---|
| 1 | 0 | 0 | 0 |
| 2 | 3 | 0 | 0 |
| 3 | 5 | 0 | 0 |
| 4 | 0 | 1.0 | 0 |
| 5 | 0 | 1.5 | 0 |
| 6 | 3 | 1.0 | 0 |
| 7 | 5 | 1.5 | 0 |
| 8 | 5 | 1.0 | 8 |
Stage 2: Gradient PCR
- Using the best additive condition from Stage 1, set up a gradient PCR.
- Set the thermal cycler gradient to span 58°C to 68°C across a 96-well plate.
- Run the LR-PCR program:
- 98°C for 30 sec (initial denaturation)
- 30 cycles: 98°C for 10 sec, Gradient (Ta) for 30 sec, 68°C for 12 min (extension)
- 72°C for 10 min (final extension)
- Analyze products via 0.8% agarose gel electrophoresis. Identify the Ta giving the strongest, single band.
Stage 3: Template Reconditioning Protocol
- Pre-PCR: Assemble a 25 µL reaction with the optimized additives and Ta, but run for only 10 cycles.
- Dilution: Dilute the pre-PCR reaction 1:50 in sterile TE buffer or nuclease-free water.
- Full LR-PCR: Use 2-5 µL of the dilution as template for a fresh 50 µL reaction with all new reagents (polymerase, dNTPs, buffer). Run for an additional 25-30 cycles using the optimized program from Stage 2.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for LR-PCR Optimization
| Item | Function in LR-PCR Optimization |
|---|---|
| High-Fidelity Proofreading Polymerase (e.g., Q5, KAPA HiFi) | Essential for accurate amplification of long fragments; low error rate and high processivity. |
| Gradient Thermal Cycler | Enables empirical determination of optimal annealing temperature across multiple samples in one run. |
| DMSO (Molecular Biology Grade) | Additive that destabilizes DNA secondary structure, crucial for GC-rich template amplification. |
| Betaine (5M Stock Solution) | Homogenizing agent that reduces Tm dependence on GC content, improving amplification efficiency. |
| Molecular Biology Grade Glycerol | Stabilizes polymerase activity and can lower DNA melting temperature in reaction mix. |
| Long-Range PCR dNTP Mix | Balanced, high-quality dNTPs at appropriate concentration (e.g., 200 µM each) for fidelity and yield. |
| High Purity Genomic DNA | Intact, high molecular weight template (>50 kb) is critical for successful long-range amplification. |
| Optimized LR-PCR Buffer | Often provided with polymerase; contains salts and pH stabilizers optimized for long extensions. |
Visualizations
Diagram 1: Integrated LR-PCR Optimization Workflow (79 chars)
Diagram 2: Mechanism of PCR Additives in LR-PCR (58 chars)
Ensuring Fidelity and Choosing the Right Tool: Validation Methods and Technology Comparison
Within the context of a thesis focused on Long-range PCR (LR-PCR) for genomic DNA amplification, validating the success and specificity of the amplification reaction is a critical step. Agarose gel electrophoresis remains the fundamental, first-line analytical technique for this purpose. It provides a rapid, visual assessment of PCR product presence, approximate size, and purity, confirming that the intended genomic fragment has been amplified without significant non-specific products or primer-dimer formation. This protocol details the steps for preparing, running, and analyzing agarose gels to verify LR-PCR amplicons, which are typically in the range of 5 kb to over 20 kb.
Experimental Protocols
Protocol: Agarose Gel Electrophoresis for LR-PCR Product Analysis
Objective: To separate, visualize, and verify the size of LR-PCR amplicons.
Materials:
- TAE (Tris-acetate-EDTA) or TBE (Tris-borate-EDTA) buffer, 1x working concentration.
- Molecular biology grade agarose.
- DNA loading dye (6x).
- DNA molecular weight ladder (appropriate for long fragments, e.g., 1 kb DNA Ladder, Lambda HindIII digest).
- GelRed or SYBR Safe nucleic acid stain.
- Gel casting tray, comb, and electrophoresis chamber.
- Power supply.
- Blue-light or UV transilluminator and imaging system.
Methodology:
Gel Preparation: Prepare a 0.8-1.0% agarose solution by dissolving the appropriate mass of agarose in 1x TAE buffer. Microwave until fully dissolved. Cool to approximately 55-60°C. Add the intercalating stain (e.g., GelRed) as per manufacturer's instructions. Pour into a sealed gel tray with a comb inserted and allow to solidify for 30-45 minutes.
Sample Preparation: Mix 5-10 µL of each LR-PCR reaction product with 1-2 µL of 6x DNA loading dye.
Gel Loading: Place the solidified gel into the electrophoresis chamber, submerged in 1x TAE buffer. Carefully remove the comb. Load the prepared samples and an appropriate DNA ladder into the wells.
Electrophoresis: Run the gel at 4-8 V/cm (distance between electrodes) for 45-90 minutes. Lower voltage (e.g., 4-5 V/cm) is preferred for better resolution of long fragments.
Visualization & Analysis: Image the gel using a blue-light or UV transilluminator. Compare the migration distance of the sample bands to the ladder to estimate amplicon size. A single, sharp band at the expected size indicates successful amplification.
Protocol: Fragment Length Verification via Sizing Software
Objective: To obtain a quantitative size estimate of the amplified fragment.
Methodology:
- Image Acquisition: Capture a high-quality digital image of the stained gel with the ladder and samples.
- Software Analysis: Import the image into gel analysis software (e.g., ImageJ with appropriate plugin, or manufacturer-specific software).
- Calibration: Define the known molecular weights of the ladder bands to create a standard curve (log10(bp) vs. migration distance).
- Measurement: Measure the migration distance of the sample band. The software will interpolate its size from the standard curve.
- Validation: Confirm that the calculated size is within 5-10% of the expected in silico amplicon length.
Data Presentation
Table 1: Expected vs. Observed Fragment Sizes for LR-PCR Validation
| Sample ID | Target Locus | Expected Size (bp) | Observed Size (bp) | % Deviation | Gel Purity Assessment |
|---|---|---|---|---|---|
| LR-01 | Gene A | 12,450 | 12,200 | -2.0% | Single, sharp band |
| LR-02 | Gene A | 12,450 | 12,550 | +0.8% | Single, sharp band |
| LR-03 | Gene B | 8,750 | ~5,000 & smear | N/A | Non-specific/partial |
| LR-NTC | N/A | 0 | 0 | N/A | No bands |
Table 2: Optimal Agarose Gel Conditions for Different Amplicon Sizes
| Target Amplicon Size Range | Agarose Concentration | Recommended Voltage | Approximate Run Time (for 8 cm gel) | Optimal Stain |
|---|---|---|---|---|
| 1 - 5 kb | 1.2 - 1.5% | 8-10 V/cm | 30-45 min | SYBR Safe |
| 5 - 15 kb | 0.8 - 1.0% | 5-8 V/cm | 60-75 min | GelRed |
| 15 - 30 kb | 0.6 - 0.8% | 4-5 V/cm | 75-90+ min | GelRed |
Visualizations
Gel Analysis Workflow for LR-PCR
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Agarose Gel Validation
| Item | Function & Rationale |
|---|---|
| Low EEO Agarose | Provides a uniform pore matrix with minimal electroendosmosis (EEO), crucial for the sharp resolution of large DNA fragments. |
| 1x TAE Buffer | The preferred running buffer for long fragment separation; its lower ionic strength and pH vs. TBE reduce gel heating and improve large DNA mobility. |
| High-Range DNA Ladder | Contains a series of DNA fragments of known sizes (e.g., 1-20 kb) to create a standard curve for accurate size estimation of the LR-PCR amplicon. |
| GelRed / SYBR Safe | Safer, sensitive fluorescent nucleic acid stains that are less mutagenic than ethidium bromide, used for visualizing DNA bands under blue/UV light. |
| 6x DNA Loading Dye | Contains glycerol to sink samples into wells and tracking dyes (e.g., bromophenol blue) to monitor electrophoresis progress. |
| PCR Clean-up / Gel Extraction Kit | Essential for purifying the correct band from the gel for downstream applications (sequencing, cloning) following successful verification. |
Within the context of optimizing a Long-range PCR (LR-PCR) protocol for genomic DNA amplification, assessing the fidelity of the amplified product is paramount. Even with high-fidelity polymerases, amplification errors can occur, potentially leading to erroneous conclusions in downstream applications such as cloning, functional studies, or variant analysis. This Application Note details two orthogonal, gold-standard strategies for fidelity assessment: Restriction Digest Analysis and Sanger Sequencing. These post-amplification validation techniques are critical for researchers, scientists, and drug development professionals who require high confidence in their amplified DNA sequences.
Research Reagent Solutions
The following table lists essential materials and reagents for performing fidelity assessment experiments.
| Item | Function |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion, PrimeSTAR GXL) | Amplifies long genomic targets with low error rates. Essential for generating the initial LR-PCR product. |
| Target-Specific LR-PCR Primers | Designed with stringent specificity for long-range amplification of the genomic region of interest. |
| Purified Genomic DNA Template | High-quality, high-molecular-weight DNA from the organism of interest. Input quality directly affects amplification fidelity. |
| Restriction Endonucleases | Enzymes that cut DNA at specific recognition sequences. Used to generate a predictable fragment pattern for validation. |
| Agarose & Electrophoresis System | For size-based separation of DNA fragments (LR-PCR products and digest fragments). |
| DNA Ladder (e.g., 1 kb Plus, 100 bp) | Molecular weight standard for accurate size determination of separated DNA bands. |
| PCR Purification Kit / Gel Extraction Kit | For purifying LR-PCR products away from primers, dNTPs, and polymerase prior to sequencing or digestion. |
| Sanger Sequencing Primers | Primers designed to sequence from the ends of the LR-PCR product or internal regions for comprehensive coverage. |
| Cycle Sequencing Reaction Mix | Contains dye-terminator chemistry for generating fluorescently labeled sequencing fragments. |
| Capillary Electrophoresis Sequencer | Instrument for separating and detecting labeled sequencing fragments (e.g., ABI 3730xl). |
Protocol 1: Restriction Digest Analysis for Rapid Structural Validation
Principle
This method verifies the gross structural integrity of the LR-PCR amplicon by comparing its restriction fragment length polymorphism (RFLP) pattern to the in silico digested reference sequence. A correct pattern confirms the presence of expected restriction sites and the overall approximate length, providing a rapid, cost-effective initial fidelity check.
Detailed Methodology
Step 1: In Silico Digest Simulation
- Using bioinformatics software (e.g., NEBcutter, SnapGene), analyze the reference genomic sequence for the amplified region.
- Select 2-3 restriction enzymes that generate a distinctive, easily resolvable pattern of fragments (typically 3-5 fragments between 0.5 kb and 5 kb).
- Record the expected number and sizes of the digestion products.
Step 2: Purification of LR-PCR Product
- Purify the LR-PCR reaction using a PCR purification kit according to the manufacturer's protocol to remove primers, dNTPs, and salts.
- Elute the DNA in nuclease-free water or the provided elution buffer.
- Quantify the DNA concentration using a spectrophotometer (e.g., Nanodrop).
Step 3: Restriction Digest Reaction
- Set up the following reaction in a 0.2 mL tube:
| Component | Volume | Final Amount/Concentration |
|---|---|---|
| Purified LR-PCR Product | X µL | 500 - 1000 ng total DNA |
| 10X Restriction Enzyme Buffer | 2.0 µL | 1X |
| Restriction Enzyme A | 1.0 µL | 5-10 units |
| Restriction Enzyme B (if double digest) | 1.0 µL | 5-10 units |
| Nuclease-free Water | to 20 µL | - |
- Mix gently and centrifuge briefly.
- Incubate at the optimal temperature for the selected enzyme(s) (typically 37°C) for 1-2 hours.
Step 4: Analysis by Agarose Gel Electrophoresis
- Prepare a 0.8% - 1.2% agarose gel in 1X TAE buffer with a DNA-intercalating dye.
- Load the following samples into separate wells:
- Lane 1: DNA Ladder (e.g., 1 kb Plus).
- Lane 2: Undigested, purified LR-PCR product (~50-100 ng).
- Lane 3: Digested LR-PCR product (load entire 20 µL reaction if possible).
- Run the gel at 5-8 V/cm until bands are adequately separated.
- Visualize under UV/blue light and compare the observed fragment sizes to the expected in silico pattern. A match confirms structural fidelity.
Table 1: Example In Silico vs. Observed Digest Data for a Hypothetical 7.5 kb LR-PCR Product
| Restriction Enzymes | Expected Fragment Sizes (bp) | Observed Fragment Sizes (bp) | Validation |
|---|---|---|---|
| EcoRI + BamHI | 3200, 2450, 1850 | 3150, 2400, 1850 | PASS (Sizes match within gel resolution limits) |
| HindIII Only | 4200, 3300 | 4200, 3100 | FAIL (One fragment is ~200 bp smaller, indicating a potential deletion or missing restriction site) |
Diagram 1: Restriction digest analysis workflow for LR-PCR product validation.
Protocol 2: Sanger Sequencing for Base-by-Base Fidelity Assessment
Principle
Sanger sequencing provides the definitive method for assessing amplification fidelity by directly determining the nucleotide sequence of the LR-PCR product. It identifies point mutations, small insertions, or deletions introduced by the polymerase, allowing for calculation of error rates and confirmation of specific sequences (e.g., edited sites, SNP locations).
Detailed Methodology
Step 1: Sequencing Strategy Design (Primer Walking)
- For amplicons > 1 kb, a single forward or reverse primer is insufficient. Design internal sequencing primers ("walking primers") every 500-800 bases to ensure overlapping sequence coverage across the entire LR-PCR product.
- Primer design criteria: 18-24 bp, Tm ~50-60°C, minimal secondary structure. Ensure primers are specific to the target amplicon.
Step 2: Purification and Quantification for Sequencing
- Purify the LR-PCR product using a gel extraction kit if non-specific bands are present; otherwise, use a PCR purification kit.
- Precisely quantify the DNA using a fluorometric assay (e.g., Qubit) for highest accuracy, as sequencing success is highly concentration-dependent.
- Dilute the DNA to a working concentration of 10-30 ng/µL in nuclease-free water.
Step 3: Cycle Sequencing Reaction Setup
- For each sequencing primer (forward, reverse, and internal walkers), set up a separate reaction.
- A typical 10 µL reaction in a 96-well plate format is:
- Template DNA (10-30 ng/µL): 1-3 µL (~50 ng total ideal)
- Sequencing Primer (3.2 µM): 1 µL
- BigDye Terminator v3.1 Ready Reaction Mix: 2 µL
- 5X Sequencing Buffer: 2 µL
- Nuclease-free water: to 10 µL
- Seal the plate, mix, and centrifuge briefly.
Step 4: Thermal Cycling and Cleanup
- Run the following cycle sequencing program:
- 96°C for 1 min (initial denaturation)
- 25 cycles of: 96°C for 10 sec, 50°C for 5 sec, 60°C for 4 min.
- 4°C hold.
- Purify the extension products to remove unincorporated dye terminators using a recommended method (e.g., EDTA/EtOH precipitation, magnetic bead cleanup, or column filtration).
Step 5: Capillary Electrophoresis and Analysis
- Resuspend the purified sequencing reactions in Hi-Di formamide and denature at 95°C for 3-5 minutes.
- Load onto a capillary electrophoresis sequencer (e.g., ABI 3730xl).
- Analyze the resulting chromatograms (trace files) using software such as Sequencing Analysis Software, FinchTV, or SnapGene Viewer.
- Manually inspect the entire trace for double peaks (indicating heterogeneity) or sequence deviations from the reference.
- Align the sequences from all primers to assemble a contiguous sequence of the entire amplicon and compare it to the reference sequence using alignment tools (e.g., Clustal Omega, BLAST).
Table 2: Sanger Sequencing Coverage and Fidelity Data for a 10 kb LR-PCR Product
| Sequencing Primer | Target Region (bp) | Read Length (bp) | Quality Score (QV20) | Sequence Variation Found vs. Reference |
|---|---|---|---|---|
| LR_F1 | 1 - 800 | 750 | 98% | None |
| LR_R1 | 9250 - 10000 | 700 | 99% | None |
| Walk_Int2 | 3500 - 4300 | 800 | 95% | Single T>C substitution at bp 4012 (potential polymerase error) |
| Walk_Int5 | 6000 - 6800 | 800 | 97% | None |
| Combined Coverage | 1 - 10000 | >99% | Average: 97% | Error Rate: 1 in 10,000 bp |
Diagram 2: Sanger sequencing workflow for comprehensive LR-PCR fidelity assessment.
For rigorous validation within a long-range PCR research thesis, a combined approach is recommended. Restriction digest analysis serves as a rapid, economical first-pass screen for structural integrity, catching major amplification failures. Subsequently, Sanger sequencing with a primer-walking strategy provides the necessary base-resolution data to quantify polymerase error rates and confirm the precise sequence of the amplicon, especially critical for applications in functional genomics and drug development. Together, these protocols form an essential quality control pipeline, ensuring the fidelity of amplified genomic DNA templates for all downstream investigations.
This application note provides a framework for selecting and implementing standard and long-range PCR techniques for genomic DNA amplification. It is situated within a broader thesis research project focused on optimizing long-range PCR protocols for challenging genomic targets, particularly in the context of structural variant analysis and gene cloning.
Table 1: Quantitative Comparison of Standard vs. Long-Range PCR
| Parameter | Standard PCR | Long-Range PCR |
|---|---|---|
| Typical Amplicon Size Range | 0.1 - 5 kb | 5 kb - 40+ kb |
| Polymerase Type | Taq or similar, non-proofreading | High-fidelity, proofreading (e.g., blend of Taq and Pfu) |
| Extension Time (per kb) | 0.5 - 1 minute | 1 - 4 minutes (varies by enzyme) |
| Cycle Number | 25-35 | 25-35 (often with longer cycles) |
| Typical Denaturation Temp/Time | 94-98°C, 10-30 sec | 98°C, 10-30 sec (for GC-rich targets) |
| Typical Annealing Temp | 50-65°C, 15-60 sec | 55-68°C, 15-60 sec (touchdown often used) |
| Template DNA Quantity | 1-100 ng (genomic) | 100-500 ng (high-quality genomic) |
| Primary Application | Short target genotyping, cloning, sequencing | Large fragment cloning, gap filling, structural analysis |
Table 2: Decision Matrix for Protocol Selection
| Target Characteristic | Recommended Method | Rationale |
|---|---|---|
| Size < 4 kb | Standard PCR | Faster, simpler, more cost-effective. |
| Size > 5 kb | Long-Range PCR | Standard polymerases lack processivity for long targets. |
| High GC Content (>65%) | Either, with additives | Both benefit from DMSO, betaine, or GC enhancer. |
| Complex, repetitive regions | Long-Range PCR with optimized buffers | Proofreading polymerases + enhancers improve fidelity and yield. |
| Routine genotyping/diagnostics | Standard PCR | Sufficient for most SNP and short indel detection. |
| Cloning large gene constructs | Long-Range PCR | Amplify entire genes with regulatory regions. |
| Template quality is degraded | Standard PCR | Short targets are more tolerant of fragmented DNA. |
Detailed Experimental Protocols
Protocol 1: Standard PCR for Genomic Targets (0.5-4 kb)
This protocol is optimized for robust amplification of short genomic segments from human or mouse DNA.
Materials:
- Genomic DNA template.
- Standard Taq DNA Polymerase (e.g., 5 U/µL).
- 10X Standard PCR Buffer (with 15 mM MgCl₂).
- dNTP Mix (10 mM each).
- Forward and Reverse Primers (10 µM each).
- Nuclease-free water.
- Thermocycler.
Procedure:
- Reaction Setup (50 µL total volume):
- Nuclease-free water: 36.5 µL
- 10X Standard PCR Buffer: 5 µL
- dNTP Mix (10 mM each): 1 µL
- Forward Primer (10 µM): 2 µL
- Reverse Primer (10 µM): 2 µL
- Genomic DNA (50-100 ng): 1-2 µL
- Taq DNA Polymerase (5 U/µL): 0.5 µL
- Mix gently and centrifuge briefly.
- Thermocycling Conditions:
- Initial Denaturation: 95°C for 3 min.
- 35 Cycles:
- Denaturation: 95°C for 30 sec.
- Annealing: Tm of primers (often 55-60°C) for 30 sec.
- Extension: 72°C for 1 min per kb.
- Final Extension: 72°C for 5 min.
- Hold: 4°C.
- Analysis: Run 5-10 µL of product on a 1% agarose gel.
Protocol 2: Long-Range PCR for Genomic Targets (5-20 kb)
This protocol is designed for amplifying large genomic fragments, critical for thesis research on gene cluster analysis.
Materials:
- High-quality, intact genomic DNA (e.g., extracted from fresh blood or cells).
- Long-Range PCR Enzyme System (e.g., LA Taq or similar proofreading blend).
- 2X Long-Range PCR Buffer (often provided with enzyme).
- dNTP Mix (higher concentration, e.g., 25 mM each).
- Forward and Reverse Primers (10-20 µM each, designed for long PCR).
- PCR enhancer (e.g., DMSO, betaine, or commercial GC enhancer) if required.
- Thermocycler with precise temperature control.
Procedure:
- Reaction Setup (50 µL total volume):
- Nuclease-free water: Variable, to 50 µL.
- 2X Long-Range PCR Buffer: 25 µL
- dNTP Mix (25 mM each): 0.4 µL (final 200 µM each)
- Forward Primer (20 µM): 1 µL
- Reverse Primer (20 µM): 1 µL
- Genomic DNA (200-500 ng): 2-5 µL
- PCR Enhancer (e.g., 5% DMSO): Optional, 2.5 µL
- Long-Range Enzyme Mix: 1-2 units (typically 0.5-1 µL).
- Mix gently, avoid bubbles. Centrifuge briefly.
- Thermocycling Conditions (Two-Step Protocol often preferred):
- Initial Denaturation: 94°C for 1 min.
- 10-20 Cycles (Touchdown Phase):
- Denaturation: 98°C for 10 sec.
- Annealing & Extension: 68°C for 1 min per kb. (Annealing temp may start higher and decrease incrementally.)
- Subsequent 15-20 Cycles:
- Denaturation: 98°C for 10 sec.
- Annealing & Extension: 65°C for 1 min per kb + 15-20 sec added per cycle.
- Final Extension: 72°C for 10 min.
- Hold: 4°C.
- Analysis: Run 5-10 µL of product on a 0.6-0.8% agarose gel at low voltage for optimal resolution.
Diagrams
PCR Selection Logic Workflow
Long-Range PCR Experimental Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for PCR-Based Genomic Amplification
| Reagent | Function in Standard PCR | Function in Long-Range PCR | Example Product Types | |
|---|---|---|---|---|
| DNA Polymerase | Catalyzes DNA synthesis. Lacks 3'→5' exonuclease activity. | Critical: Requires high-processivity, thermostable enzyme with proofreading (3'→5' exo) activity to reduce errors over long extensions. | Taq DNA Polymerase. | Enzyme blends (e.g., LA Taq, KAPA HiFi, Q5). |
| PCR Buffer | Provides optimal pH, salt, and Mg²⁺ conditions for Taq activity. | Often specialized; contains additives to stabilize polymerase over long runs and enhance processivity. Higher Mg²⁺ may be used. | Standard KCl-based buffer. | Proprietary buffers with betaine, (NH₄)₂SO₄, or other enhancers. |
| dNTPs | Building blocks for DNA synthesis. | Higher final concentrations (200-500 µM each) are often used to sustain synthesis over long templates. | 10 mM each dNTP stock. | 25-100 mM dNTP blend stocks. |
| PCR Enhancers | Optional, used for difficult templates (high GC). | Frequently essential to overcome secondary structure and promote amplification of long, complex targets. | DMSO, betaine, formamide. | Commercial GC enhancers, specialized additive cocktails. |
| Template DNA | Quality important, but shorter targets tolerate mild degradation. | Critical: Must be high molecular weight and intact. Degraded DNA yields no product or smearing. | Standard mini-prep genomic DNA. | Phenol-chloroform extracted DNA, column-purified high MW DNA. |
| Primers | Standard design (18-25 bases, 40-60% GC). | Often longer (25-35 bases), with higher Tm (65-72°C) to promote specificity during long extensions. | Desalted oligos. | HPLC- or PAGE-purified primers. |
Application Notes
This analysis, framed within a thesis on Long-Range PCR (LR-PCR) protocol development for genomic DNA amplification, compares three historical and contemporary methods for isolating large DNA fragments (5-40 kb): Long-Range PCR, traditional Cloning (plasmid-based), and Cosmid Library construction. The choice of method impacts project timelines, cost, fidelity, and technical accessibility.
Key Comparative Insights:
- Speed & Throughput: LR-PCR is the fastest method, enabling amplification of a specific target in hours. Cloning and Cosmid library construction are multi-day processes involving library generation, screening, and isolation.
- Template Requirement: LR-PCR requires prior sequence knowledge for primer design. Cloning and Cosmid methods are suitable for unknown sequences, with cosmids excelling in cloning very large, contiguous genomic regions.
- Fidelity & Error Rate: Modern high-fidelity LR-PCR polymerases have error rates rivaling that of bacterial cloning (~1 x 10⁻⁶ errors/bp). However, in vivo cloning allows for natural proofreading by bacterial machinery.
- Handling of Complex Regions: Cloning and Cosmid libraries can accommodate regions high in GC content or secondary structure that may challenge PCR amplification.
- Modern Application: LR-PCR is optimal for targeted amplification of known large exons, regulatory regions, or for rapid construction of recombinant vectors. Cosmid libraries remain a gold standard for physical mapping and sequencing of complex genomes, while traditional plasmid cloning is now largely reserved for subcloning smaller fragments or when PCR-independent methods are critical.
Quantitative Comparison Table
| Parameter | Long-Range PCR | Traditional Cloning (Plasmid) | Cosmid Library |
|---|---|---|---|
| Typical Insert Size | 5 - 40 kb | 0.1 - 10 kb | 30 - 45 kb |
| Time to Isolate Target | 3 - 6 hours | 3 - 7 days | 1 - 3 weeks |
| Sequence Knowledge Required | Yes (for primers) | No (for library) | No (for library) |
| Throughput (Targeted) | High (single target) | Low to Moderate | Very Low (requires screening) |
| Estimated Error Rate | ~1-3 x 10⁻⁶ errors/bp (High-Fidelity Polymerase) | ~1 x 10⁻⁶ errors/bp (Host repair) | ~1 x 10⁻⁶ errors/bp (Host repair) |
| Primary Modern Use Case | Targeted amplification, recombinant DNA construction | Subcloning, expression constructs, cloning difficult-to-amplify fragments | Genomic mapping, large contiguous DNA isolation, BAC library alternative |
Detailed Protocols
Protocol 1: Long-Range PCR for Genomic DNA Amplification
Objective: Amplify a 20-kb target fragment from human genomic DNA.
Materials: See "Research Reagent Solutions" below.
Method:
- Primer Design: Design 18-30 nt primers with melting temperatures (Tm) of 68-72°C. Avoid secondary structures. Calculate elongation time: 1-4 minutes per kb, depending on polymerase.
- Reaction Setup (50 µL):
- Genomic DNA (high-quality): 100-300 ng
- Long-Range PCR Buffer (with Mg²⁺): 1X final concentration
- dNTP Mix: 200 µM each
- Forward & Reverse Primer: 0.2 µM each
- High-Fidelity LR Polymerase Mix: 1-2 units
- Add PCR-grade water to 50 µL.
- Thermal Cycling:
- Initial Denaturation: 94°C for 2 min.
- 30-35 Cycles:
- Denaturation: 98°C for 10 sec.
- Annealing: 68°C for 30 sec.
- Extension: 68°C for 20 min (for 20 kb). Use a 10-15 sec/kb ramping time.
- Final Extension: 68°C for 10 min.
- Hold: 4°C.
- Analysis: Verify product size and integrity by pulsed-field gel electrophoresis (PFGE) or standard agarose gel electrophoresis with appropriate molecular weight markers.
Protocol 2: Construction of a Cosmid Genomic Library
Objective: Create a representative library of 35-45 kb genomic fragments.
Method:
- Genomic DNA Partial Digestion: Digest 50 µg of high-molecular-weight genomic DNA with a restriction enzyme (e.g., Sau3AI) under partial digest conditions (vary enzyme amount or time). Size-fractionate fragments (35-45 kb) by sucrose gradient centrifugation or pulsed-field gel excision.
- Cosmid Vector Preparation: Digest 10 µg of a cosmid vector (e.g., pWEB) with a compatible enzyme (e.g., BamHI). Dephosphorylate the linearized vector with alkaline phosphatase to prevent self-ligation.
- Ligation: Ligate size-selected genomic inserts to the prepared cosmid vector at a 3:1 (insert:vector) molar ratio using T4 DNA Ligase at 16°C for 12-16 hours.
- In Vitro Packaging: Mix the ligation reaction with commercial phage packaging extracts (containing phage head/tail proteins and terminase). Incubate at room temperature for 2-3 hours. The cos sites on the vector allow the recombinant DNA to be packaged into phage particles.
- Transduction & Library Amplification: Mix packaging reaction with an appropriate E. coli host strain (e.g., EC1000). Plate for single colonies on selective media (e.g., containing antibiotic). Pool colonies to create the amplified library.
- Screening: Screen library by colony hybridization with labeled probes or PCR.
Visualization
Diagram 1: Method Selection Workflow for Large DNA Fragment Isolation
Diagram 2: Cosmid Library Construction Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Context |
|---|---|
| High-Fidelity LR-PCR Enzyme Mix | Specialized polymerase blend (e.g., Taq + proofreading enzyme) for accurate amplification of long targets. Provides processivity and fidelity. |
| Cosmid Vector (e.g., pWEB) | Engineered plasmid containing bacteriophage lambda cos sites, origin of replication, and selectable marker for cloning large inserts and in vitro packaging. |
| In Vitro Phage Packaging Extract | Commercial extract containing pre-assembled phage heads, tails, and enzymes to package cosmid DNA into infectious phage particles for efficient bacterial delivery. |
| Pulsed-Field Gel Electrophoresis System | Apparatus for separating large DNA fragments (>20 kb) by alternating electric fields, essential for analyzing LR-PCR products and sizing cosmid inserts. |
| Size-Selective DNA Purification Kit | Utilizes column or bead-based methods to isolate DNA fragments within a specific size range (e.g., 35-45 kb for cosmid inserts) from agarose gels or solutions. |
| Competent Cells for Large Constructs | Specialized E. coli strains (e.g., EC1000, Stbl4) with high transformation efficiency and reduced recombination for stable maintenance of large plasmids/cosmids. |
Within the framework of a thesis on Long-range PCR (LR-PCR) for genomic DNA amplification, the ultimate value of amplified products lies in their effective integration with downstream applications. This protocol details the critical post-amplification steps required to prepare high-quality LR-PCR amplicons for Next-Generation Sequencing (NGS) and functional assays, ensuring data integrity and experimental success.
Post-Amplification Processing: Purification and Quantification
Prior to any downstream application, amplicons must be purified and accurately quantified to remove enzymes, primers, dNTPs, and non-specific products.
2.1. Purification Protocol
- Method: Solid-Phase Reversible Immobilization (SPRI) Bead Cleanup.
- Procedure:
- Transfer the LR-PCR reaction to a low-binding microcentrifuge tube.
- Vortex SPRI bead mixture thoroughly and add at a 1.0x sample volume:bead volume ratio for size selection (removes primers and fragments <100 bp). For stringent removal of primer dimers, a 0.6x ratio can be used, noting potential yield loss.
- Mix thoroughly by pipetting. Incubate for 5 minutes at room temperature.
- Place tube on a magnetic rack until supernatant is clear (~2 minutes). Carefully remove and discard supernatant.
- With tube on magnet, add 200 µl of freshly prepared 80% ethanol. Incubate for 30 seconds, then remove ethanol. Repeat wash.
- Air-dry beads for ~5 minutes until cracks appear. Do not over-dry.
- Remove from magnet. Elute DNA in 20-30 µl of nuclease-free water or 10 mM Tris-HCl (pH 8.0). Mix well. Incubate for 2 minutes at room temperature.
- Place on magnet. Transfer purified eluate to a new tube.
2.2. Quantification & Quality Assessment
- Fluorometric Quantification: Use a dsDNA-binding fluorescent dye assay (e.g., Qubit). This is preferred over absorbance (A260) for specificity.
- Fragment Analysis: Use capillary electrophoresis (e.g., Bioanalyzer, TapeStation) to confirm amplicon size and purity. This is critical for NGS library preparation.
Table 1: Post-Amplification QC Metrics and Target Values
| Parameter | Assessment Method | Target Value / Profile |
|---|---|---|
| DNA Concentration | Fluorometry (e.g., Qubit) | > 5 ng/µl (post-cleanup) |
| Purity (Salt/Protein) | Spectrophotometry (A260/A280) | 1.8 - 2.0 |
| Purity (Solvent Cont.) | Spectrophotometry (A260/A230) | 2.0 - 2.2 |
| Size Integrity | Capillary Electrophoresis | Single, dominant peak at expected LR-PCR product size (±5%) |
| Primer Dimer Cont. | Capillary Electrophoresis | Peak area < 1% of total |
Preparation for Next-Generation Sequencing
LR-PCR amplicons are ideal for targeted sequencing. The primary method is tagmentation-based library preparation, which is efficient for fragmented DNA.
3.1. Tagmentation-Based Library Prep Protocol
- Principle: The enzyme transposase simultaneously fragments the amplicon and adds adapter sequences.
- Procedure:
- Tagmentation Reaction: Combine 50-100 ng of purified LR-PCR amplicon with tagmentation buffer and enzyme. Incubate at 55°C for 10-15 minutes.
- Neutralization: Add neutralization buffer and incubate at room temp for 5 min. The product is now "tagmented" DNA.
- PCR Amplification with Indexes: Add a PCR master mix containing unique dual-index (UDI) primer sets to the tagmented DNA. Perform limited-cycle PCR (typically 12 cycles):
- 98°C for 45s.
- Cycle (12x): 98°C for 15s, 60°C for 30s, 72°C for 30s.
- Final extension: 72°C for 1 min.
- Library Purification: Clean up the final library using SPRI beads at a 1.0x ratio. Elute in 20 µL.
- Final QC: Quantify via fluorometry. Assess library size distribution using capillary electrophoresis (expected broad peak ~300-800 bp).
Workflow: From LR-PCR Amplicon to NGS Library
Preparation for Functional Studies: Cellular Transfection
LR-PCR amplicons containing a gene of interest and its regulatory elements can be used for direct functional assays in cell culture.
4.1. Mammalian Cell Transfection Protocol (for ~24-well plate)
- Transfection Method: Lipofection, suitable for large amplicon constructs.
- Procedure:
- Day 0: Seed mammalian cells (e.g., HEK293) in 500 µl complete growth medium per well to reach 70-90% confluency at transfection.
- Day 1 (Transfection): a. Dilute 500 ng of purified LR-PCR amplicon in 50 µl of serum-free/antibiotic-free medium (Tube A). b. Dilute 1.5 µl of lipofection reagent in 50 µl of the same medium (Tube B). Incubate 5 min. c. Combine Tube A and B. Mix gently. Incubate for 15-20 min at RT to form DNA-lipid complexes. d. Add the 100 µl complex dropwise to the cell well. Gently swirl plate.
- Day 2: Replace medium with fresh complete growth medium.
- Day 3-5: Assay for gene expression (e.g., microscopy, luciferase, western blot, flow cytometry).
Functional Study Pathways After Amplicon Transfection
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Downstream Amplicon Processing
| Reagent/Material | Function/Application | Key Considerations |
|---|---|---|
| SPRI Magnetic Beads | Size-selective purification of amplicons and NGS libraries. | Ratio determines size cutoff. Essential for removing primers and primer dimers. |
| Fluorometric DNA Dye Assay | Accurate quantification of dsDNA concentration. | Specific for dsDNA; unaffected by RNA or contaminants. Critical for input normalization. |
| Capillary Electrophoresis System | Assess amplicon/library size distribution and purity. | Provides digital gel-like data and molar concentration estimates. |
| Tagmentation Library Prep Kit | Rapid, efficient conversion of amplicons to sequencing libraries. | Integrates fragmentation and adapter addition in a single step. |
| Unique Dual Index (UDI) Primers | Multiplex samples during NGS by adding unique barcodes. | Enables sample pooling and prevents index hopping artifacts. |
| Lipofection Reagent | Forms complexes with large amplicon DNA for cellular delivery. | Optimized for large DNA fragments and high cell viability. |
| Cell Line with High Transfection Efficiency | Functional testing of amplicons (e.g., HEK293, HeLa). | Ensures robust expression for reporter or genome editing assays. |
| Nuclease-Free Water & Buffers | Resuspension and dilution of nucleic acids. | Prevents degradation of purified amplicons and libraries. |
Conclusion
Mastering long-range PCR requires a synergistic understanding of foundational biochemistry, a meticulous optimized protocol, systematic troubleshooting, and rigorous validation. By integrating the principles outlined—from selecting the right high-fidelity polymerase blend to designing specific primers and employing strategic additives—researchers can reliably amplify complex genomic regions up to 20-40 kb. This capability remains a cornerstone technique for constructing genetic maps, analyzing structural variants, and preparing templates for sequencing. As genomic research progresses towards more complex and personalized analyses, the robust amplification of long, specific DNA fragments will continue to be an indispensable skill. Future developments in enzyme engineering and reaction chemistry promise to further push the boundaries of amplicon length and fidelity, solidifying long-range PCR's role in advanced diagnostics, gene therapy vector construction, and comprehensive genomic research.