Microbiological Method Verification vs Validation: A Strategic Guide for Researchers and Drug Developers
This article provides a comprehensive guide for researchers, scientists, and drug development professionals on navigating the critical distinctions between method verification and validation in microbiological analysis.
Microbiological Method Verification vs Validation: A Strategic Guide for Researchers and Drug Developers
Abstract
This article provides a comprehensive guide for researchers, scientists, and drug development professionals on navigating the critical distinctions between method verification and validation in microbiological analysis. It clarifies foundational definitions, outlines step-by-step methodological protocols based on CLSI, ISO, and pharmacopoeial standards, addresses common troubleshooting scenarios, and offers a comparative framework for strategic decision-making. By synthesizing current regulatory requirements and practical applications, this resource aims to ensure data integrity, regulatory compliance, and operational efficiency in biomedical and clinical research.
Verification vs Validation: Defining the Cornerstones of Microbiological Quality
In laboratory environments, particularly within pharmaceutical development, clinical diagnostics, and food safety, the demand for accurate and reliable testing is non-negotiable [1]. Two cornerstone processes ensure the fitness of analytical methods: method validation and method verification. While both aim to confirm that a method is suitable for its intended purpose, they fulfill distinct roles within the product and method lifecycle [1]. A clear understanding of these concepts is critical for researchers, scientists, and drug development professionals to ensure regulatory compliance, data integrity, and ultimately, product safety and efficacy.
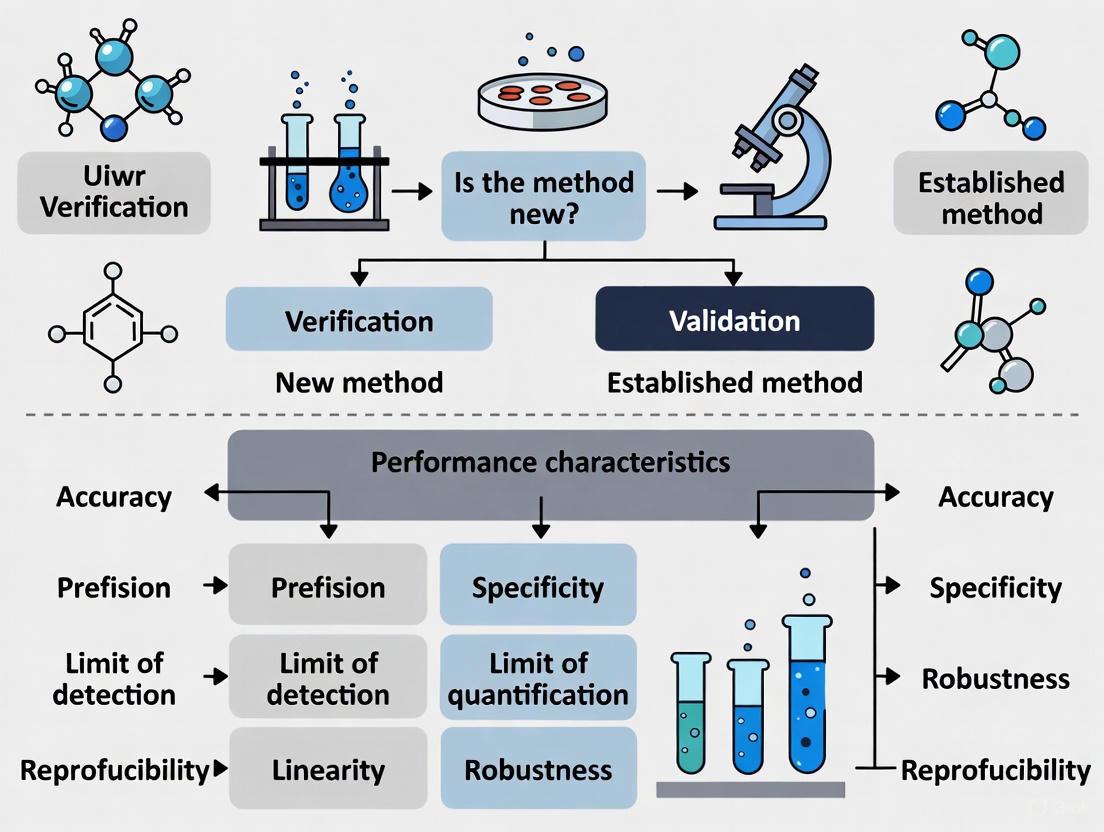
Method validation is the comprehensive process of proving that an analytical method is acceptable for its intended use, typically during development or when transferring methods between labs [1]. In contrast, method verification is the process whereby a laboratory confirms that a previously validated method performs as expected under its specific conditions [1]. This distinction is crucial in regulated environments where reproducibility and compliance are paramount. The International Organization for Standardization (ISO) 16140 series provides a standardized framework for these processes in microbiological testing, detailing protocols for both the initial validation of alternative methods and their subsequent verification in user laboratories [2].
Defining Method Validation
Core Concept and Purpose
Method validation is a documented process that proves an analytical method is reliable and acceptable for its intended purpose [1]. It is a comprehensive exercise involving rigorous testing and statistical evaluation to demonstrate that the method's performance characteristics meet predefined acceptance criteria. In the context of microbiology, this process is extensively detailed in standards such as ISO 16140-2, which provides the base protocol for the validation of alternative (proprietary) methods against a reference method [2]. The essential question validation answers is: "Is this method fundamentally fit-for-purpose?" [1].
When is Validation Required?
Validation is not a perpetual state; it is required in specific scenarios [1]:
- Development of a new analytical method.
- Significant modification of an existing method.
- Transfer of a method between laboratories or instruments.
- When required by regulatory bodies for novel methods or submissions.
Key Validation Parameters and Experimental Protocols
A full validation assesses multiple performance characteristics through defined experimental protocols. The table below summarizes the core parameters typically evaluated for both qualitative and quantitative microbiological methods, many of which are outlined in the ISO 16140 series [2].
Table 1: Key Parameters Assessed During Method Validation
| Parameter | Description | Experimental Approach |
|---|---|---|
| Accuracy | Closeness of agreement between a test result and the accepted reference value [1]. | Analysis of samples with known microbial concentrations (e.g., using certified reference materials) and comparison of results to the reference value. |
| Precision | Closeness of agreement between independent test results obtained under stipulated conditions [1]. | Repeatability: Multiple analyses of the same sample by the same analyst, same day. Intermediate Precision: Multiple analyses by different analysts, different days, different equipment. |
| Specificity | Ability to assess the analyte unequivocally in the presence of other components [1]. | Testing the method with samples containing likely interfering substances or non-target microorganisms to demonstrate no impact on detection. |
| Limit of Detection (LOD) | Lowest quantity of the analyte that can be detected [1]. | For qualitative methods, testing serial dilutions of low-level inocula to determine the point at which the target microorganism is consistently detected. |
| Limit of Quantitation (LOQ) | Lowest quantity of the analyte that can be quantified with acceptable precision and accuracy [1]. | For quantitative methods, determining the lowest level at which the count can be accurately enumerated, with defined precision and accuracy. |
| Linearity | Ability of the method to obtain test results proportional to the analyte concentration. | Testing a range of samples with different concentrations of the target microorganism to demonstrate a direct, proportional relationship. |
| Robustness | Capacity of the method to remain unaffected by small, deliberate variations in method parameters. | Introducing minor changes in procedural parameters (e.g., incubation temperature, time, reagent lots) to evaluate the method's reliability. |
Defining Method Verification
Core Concept and Purpose
Method verification is the process of confirming that a previously validated method performs as expected in a specific laboratory setting [1]. It is typically employed when a laboratory adopts a standard method—such as a compendial method from the USP, EP, or AOAC—that has already undergone full validation. The process provides assurance that the method functions correctly with the laboratory's specific instruments, analysts, and environmental conditions. As per the ISO 16140-3 standard, verification involves two key stages: implementation verification and (food) item verification [2].
When is Verification Required?
Verification is a more targeted process than validation and is applicable in specific situations [1]:
- Implementing a compendial or previously validated method in a laboratory for the first time.
- Demonstrating competency in performing a standardized method for accreditation standards like ISO/IEC 17025.
- Confirming that a validated method works for specific sample matrices under local conditions.
Key Verification Parameters
Verification does not re-assess every validation parameter. Instead, it focuses on confirming critical performance characteristics to ensure the method works as intended in the user's laboratory. The ISO 16140-3 standard provides a structured protocol for this process [2].
Table 2: Typical Parameters Assessed During Method Verification
| Parameter | Verification Focus |
|---|---|
| Accuracy | Confirm published accuracy claims are achievable under the lab's specific conditions [1]. |
| Precision | Demonstrate that the laboratory can achieve the method's stated precision with its personnel and equipment [1]. |
| Specificity | Confirm the method correctly identifies and/or detects the target microorganism in the presence of normal sample flora. |
| LOD/LOQ | Verify that the published detection or quantification limits can be achieved in the lab [1]. |
Comparative Analysis: Validation vs. Verification
Understanding the differences between validation and verification is a strategic necessity for efficient laboratory operation and regulatory compliance.
Table 3: Summary Comparison: Method Validation vs. Method Verification
| Comparison Factor | Method Validation | Method Verification |
|---|---|---|
| Objective | Prove the method is fit-for-purpose [1]. | Confirm the lab can perform the validated method correctly [1]. |
| Context | Method development, transfer, or significant change [1]. | Adoption of an existing, standardized method [1]. |
| Scope | Comprehensive assessment of all performance parameters [1]. | Limited assessment of critical parameters under local conditions [1]. |
| Resource Intensity | High (time, cost, personnel) [1]. | Moderate to low [1]. |
| Regulatory Driver | Required for new method submissions [1]. | Required for using compendial methods [1]. |
| Result Applicability | Broad (method can be used universally) [1]. | Specific to the performing laboratory [1]. |
The relationship between validation and verification, and their place in the method lifecycle, can be visualized as a sequential workflow.
The Regulatory and Standards Framework: ISO 16140
The ISO 16140 series, "Microbiology of the food chain - Method validation," is the internationally recognized standard for the validation and verification of microbiological methods [2]. This series provides detailed protocols for different scenarios, guiding laboratories, test kit manufacturers, and regulatory authorities.
The series consists of multiple parts, each addressing a specific aspect of validation or verification [2]:
- ISO 16140-1: Vocabulary - Defines key terms.
- ISO 16140-2: Protocol for the validation of alternative (proprietary) methods against a reference method. This is the core validation standard.
- ISO 16140-3: Protocol for the verification of reference and validated alternative methods in a single laboratory.
- ISO 16140-4: Protocol for method validation in a single laboratory.
- ISO 16140-5: Protocol for factorial interlaboratory validation for non-proprietary methods.
- ISO 16140-6: Protocol for the validation of alternative methods for microbiological confirmation and typing procedures.
- ISO 16140-7: Protocol for the validation of identification methods of microorganisms.
The Pathway from Validation to Verification
The ISO 16140 framework clearly delineates the journey from a new method to its routine use in a laboratory. The following diagram illustrates the decision process for selecting the appropriate ISO standard based on the method's status and the laboratory's goal.
Essential Reagents and Materials for Method Assessment
The execution of robust validation and verification studies relies on well-characterized biological materials and reagents. These tools are fundamental for generating reliable and reproducible data.
Table 4: Key Research Reagent Solutions for Validation and Verification
| Reagent/Material | Function in Validation/Verification |
|---|---|
| Certified Reference Materials (CRMs) | Well-characterized materials used for determining method accuracy, precision, and linearity. They provide a known, traceable value for comparison [3]. |
| Quality Control (QC) Organisms | Well-characterized microbial strains with defined profiles. Used for growth promotion testing, monitoring test methodologies, and serving as positive controls [3]. |
| In-House Isolates | Microbial strains isolated from the laboratory's own environment or products. Critical for demonstrating that a method can detect relevant, "wild" contaminants [3]. |
| Stressed Microorganisms | Cultures that have been subjected to sub-lethal stress (e.g., heat, desiccation). Used to challenge the method and ensure it can detect microorganisms that may be injured in a process [4]. |
| Selective and Non-Selective Media | Culture media used for the reference method and for resuscitating microorganisms. Their performance is crucial for a fair method comparison [2]. |
In the highly regulated world of microbiological testing, a precise understanding of method validation and method verification is not merely academic—it is a practical necessity for ensuring product safety and regulatory compliance. Validation is the comprehensive process of establishing that a method is fundamentally fit-for-purpose, while verification is the targeted process of confirming that a laboratory can successfully implement that validated method.
The structured framework provided by standards such as the ISO 16140 series offers a clear pathway for laboratories to follow, from the initial validation of a new method to its routine verification and use. By adhering to these principles and utilizing appropriate reagents and controls, researchers, scientists, and drug development professionals can ensure the reliability, accuracy, and defensibility of their microbiological data, ultimately supporting the development and release of safe and effective products.
In the highly regulated fields of clinical diagnostics and pharmaceutical development, robust regulatory frameworks are essential for ensuring the quality, safety, and efficacy of products and services. For researchers and drug development professionals, navigating the complex landscape of these standards is particularly crucial when establishing the reliability of microbiological methods. The distinction between method verification (confirming that a method works as intended in a specific laboratory) and validation (providing objective evidence that a method meets the requirements for its intended use) forms the cornerstone of analytical quality. This technical guide provides an in-depth analysis of four key regulatory frameworks—CLIA, ICH, ISO 17025, and Pharmacopoeial requirements—focusing on their specific implications for microbiological method verification and validation practices within research and development settings.
Scope, Focus, and Legal Status
Various regulatory standards govern laboratory testing, each with distinct primary focuses, from technical competence to patient safety and product quality.
Table 1: Key Characteristics of Laboratory and Quality Standards
| Standard | Primary Focus & Scope | Legal Status & Application | Governance Body |
|---|---|---|---|
| CLIA | Patient testing accuracy and reliability in clinical diagnostics [5]. | U.S. federal regulation; legally mandatory for clinical laboratories testing human specimens in the U.S. [6] [7]. | Centers for Medicare & Medicaid Services (CMS) [7]. |
| ISO/IEC 17025 | Technical competence and impartiality of testing and calibration labs; general requirements for all types of labs [8] [9]. | International standard; voluntary accreditation demonstrating operational competence [5] [9]. | International Organization for Standardization (ISO)/International Electrotechnical Commission (IEC) [9]. |
| ISO 15189 | Quality and competence specific to medical laboratories; focuses on the continuum of care and patient safety [6]. | International standard; voluntary accreditation that incorporates QMS and technical competence [5] [6]. | International Organization for Standardization (ISO) [6]. |
| ICH Guidelines | Pharmaceutical product quality, safety, and efficacy, including stability testing (Q1) and analytical validation (Q2) [10]. | International technical standards; adopted by regulatory authorities in different countries/regions (e.g., Europe, Japan, U.S.) [10]. | International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). |
| USP | Quality, purity, and identity of medicines and dietary supplements; specifies microbiological test methods and acceptance criteria [11]. | Recognized in U.S. law; compliance is often required by national regulations for marketing authorization [11]. | U.S. Pharmacopeia (USP) [11]. |
Core Requirements and Relevance to Method Assurance
Table 2: Core Requirements and Application to Method Verification & Validation
| Standard | Core Technical & Quality Requirements | Relevance to Microbiological Method V&V |
|---|---|---|
| CLIA | Personnel qualifications, quality control, proficiency testing, inspection and accreditation [5] [7]. | Mandates that laboratories must verify the performance specifications of non-FDA approved methods (laboratory-developed tests) before reporting patient results. |
| ISO/IEC 17025 | Impartiality, confidentiality, personnel competence, method validation, measurement uncertainty, equipment calibration, and reporting [12] [9]. | Requires labs to validate non-standard and laboratory-developed methods, and verify their ability to correctly perform standard methods. |
| ISO 15189 | Quality management system, technical competence, pre-and post-examination processes, personnel, equipment, and assay validation [5] [6]. | Emphasizes the validation of examination procedures, including accuracy, precision, measurement uncertainty, and biological reference intervals. |
| ICH Q2(R2) | Validation of analytical procedures for pharmaceuticals, defining key validation characteristics like specificity, accuracy, and robustness [10]. | Provides the definitive framework for validating analytical methods (including microbiological assays) during pharmaceutical development and registration. |
| USP | Standardized test methods, acceptance criteria, and quality attributes for compendial articles [11]. | Methods like <61>, <62>, and <71> must be verified under actual conditions of use. Chapters like <1223> and <1227> provide guidance on validation of alternative and microbiological recovery methods [11]. |
Implementation and Workflow
Pathways to Compliance and Accreditation
The process for implementing these standards and achieving compliance or accreditation varies significantly. CLIA compliance is a legal requirement for clinical laboratories in the U.S., enforced through inspections and sanctions [7]. For voluntary standards like ISO 15189, laboratories typically follow a structured journey towards accreditation.
Figure 1: The ISO 15189 accreditation process involves multiple stages from initial commitment to ongoing surveillance over a three-year cycle [6].
Method Validation and Verification Workflow
A risk-based approach is central to modern quality systems like ISO/IEC 17025:2017 [12]. The decision to verify or validate a method depends on its source and standardization.
Figure 2: A risk-based decision workflow for method verification versus validation, crucial for regulatory compliance.
Essential Research Reagents and Materials
Successful method validation and verification require specific, high-quality reagents and materials.
Table 3: Key Research Reagent Solutions for Microbiological V&V
| Reagent/Material | Critical Function in V&V | Application Examples |
|---|---|---|
| Reference Standards | Serves as the benchmark for calibrating equipment and validating methods; ensures metrological traceability [11]. | USP Endotoxin Reference Standard for LAL tests; Certified microbial cultures for identification assays [11]. |
| Competency Testing Kits | Verifies technician proficiency and method performance in routine practice; required for ongoing quality assurance [11]. | Enverify Viable Surface Sampling Kits for environmental monitoring competency [11]. |
| Characterized Microbial Strains | Used to establish accuracy, precision, and specificity of microbiological methods during validation [11]. | Instant Inoculator reference microorganisms for antimicrobial effectiveness testing (USP <51>) [11]. |
| Quality Control Cultures | Monures the ongoing performance and robustness of the method with each run or at defined intervals. | Using ATCC strains for daily QC of microbial enumeration tests (USP <61>) [11]. |
| Growth Media & Supplements | Supports microbial recovery and growth; formulation and performance are critical to method accuracy. | Validated soybean-casein digest medium for sterility testing (USP <71>); selective media for specified microorganisms (USP <62>) [11]. |
Experimental Protocols for Microbiological Method Validation
The following protocols outline core experiments for validating microbiological methods according to ICH, USP, and ISO principles.
Protocol for Specificity and Selectivity
1. Objective: To demonstrate that the method can accurately distinguish and detect the target microorganism(s) in the presence of other potentially interfering components, such as the product formulation, resident flora, or related species [11].
2. Materials:
- Test Strains: Pure cultures of target microorganism(s) (e.g., E. coli, S. aureus, C. albicans).
- Interfering Strains: Cultures of non-target microorganisms likely to be present.
- Sample Matrix: The product formulation without antimicrobial activity (or neutralized) and a placebo if available.
- Culture Media: Appropriate non-selective and selective media as required by the method.
3. Methodology:
- Inoculum Preparation: Prepare separate inocula of each test and interfering strain to a defined density (e.g., 10^1-10^2 CFU/mL for enrichment tests, 10^4-10^5 CFU/mL for enumeration).
- Spiking Experiment: Spike the sample matrix and a negative control (e.g., saline or buffer) with the target and non-target strains, both individually and in combination.
- Testing and Analysis: Perform the test method on all spiked samples and controls. The method should detect the target organism in all relevant samples with no detection in the negative controls. For enumeration methods, recovery of the target from the product matrix should be statistically equivalent to recovery from the control.
Protocol for Accuracy and Recovery
1. Objective: To quantify the closeness of agreement between the value found by the test method and the known accepted reference value, often expressed as percent recovery [11].
2. Materials:
- Test Microorganism: A qualified working stock of a relevant microorganism.
- Sample Matrix: The product under test, neutralized if antimicrobial.
- Reference Material: Same as used for specificity.
3. Methodology:
- Sample Preparation: Prepare a homogeneous sample of the product matrix. Spike the sample with a known, low level of the microorganism (e.g., 50-150% of the specification limit). Perform this in multiple replicates (n≥3).
- Control Preparation: Prepare an identical inoculum in a neutralizer or diluent without the product matrix to represent the "theoretical" 100% recovery.
- Testing and Analysis: Perform the test method on all spiked samples and controls. Calculate the percent recovery for each replicate: (Count from spiked sample / Count from control) × 100. The mean recovery and acceptance criteria should be established based on product type and method requirements (e.g., 70-130% for enumeration methods).
Protocol for Precision (Repeatability and Intermediate Precision)
1. Objective: To measure the degree of scatter between a series of measurements obtained from multiple sampling of the same homogeneous sample under defined conditions. Repeatability is assessed under same conditions (same analyst, day, equipment), while intermediate precision includes variations (different days, analysts) [11].
2. Materials: As per the Accuracy protocol.
3. Methodology:
- Repeatability: One analyst prepares a single homogeneous sample spiked with a target microorganism at a specific level (e.g., near the specification limit). The analyst tests this sample in multiple replicates (n≥6) in one session using the same equipment and reagents.
- Intermediate Precision: A second analyst, and/or the first analyst on a different day, repeats the repeatability experiment using a newly prepared inoculum and reagents.
- Statistical Analysis: Calculate the mean, standard deviation (SD), and relative standard deviation (%RSD) for each set of results. The %RSD for the intermediate precision set should be comparable to or only slightly higher than the repeatability %RSD, demonstrating method robustness against minor operational variations.
Navigating the regulatory landscape of CLIA, ICH, ISO 17025, and pharmacopoeial requirements is a fundamental requirement for ensuring data integrity and patient safety in research and drug development. While these frameworks originate from different sectors—clinical diagnostics, international standardization, and pharmaceutical quality—they converge on a common principle: the critical importance of demonstrated analytical reliability. A thorough understanding of the specific requirements for method verification and validation under each standard enables scientists to design efficient, compliant studies. By integrating these principles into the laboratory's quality management system, organizations can effectively bridge the gap between research and regulation, ensuring that microbiological methods are not only scientifically sound but also meet the stringent demands of global regulatory bodies.
In the rigorous world of drug development and microbiological research, the processes of method validation and method verification are critical pillars of quality assurance and regulatory compliance. Though often confused, they serve distinct purposes. Verification confirms that a laboratory can correctly perform a pre-existing, validated method, answering the question, "Are we performing the test correctly?" [13] [14]. In contrast, Validation establishes, through extensive objective evidence, that a method is fit for its intended purpose, answering the question, "Does the test method work as intended?" [13] [15] [1].
This distinction is not merely semantic; it is foundational to data integrity, patient safety, and the successful approval of new therapeutics. This guide provides researchers and scientists with a structured framework for making this critical decision, complete with experimental protocols and data presentation standards.
Core Concepts and Decision Framework
Fundamental Definitions and Distinctions
The core difference lies in the purpose and scope of each activity:
- Method Validation is the process of proving that an analytical method is acceptable for its intended use [1]. It is a comprehensive exercise performed to demonstrate that the method's performance characteristics—such as accuracy, precision, and specificity—meet predefined criteria for a specific analyte and matrix [13]. Validation is typically required for new methods, laboratory-developed tests (LDTs), or when an existing method is significantly modified [15] [1].
- Method Verification is the process of confirming that a previously validated method performs as expected in a specific laboratory [13] [1]. It demonstrates that the laboratory's personnel, equipment, and environment can successfully replicate the method's validated performance characteristics. Verification is performed when a laboratory adopts an unmodified, FDA-cleared or compendial method (e.g., from USP or AOAC) for the first time [15] [14].
A useful analogy from systems and software engineering further clarifies this distinction: Verification checks "Are we building the product right?" (i.e., according to specifications), while Validation checks "Are we building the right product?" (i.e., fulfilling user needs) [16] [17] [18].
Decision Logic for Microbiological Methods
The following workflow provides a step-by-step guide for determining whether validation or verification is required for a given microbiological method in a drug development context.
When is Full Method Validation Required?
Method validation is a foundational activity in research and development. It is mandated in scenarios where the reliability of the method itself has not been previously established for its new context.
Key Application Scenarios
- Development of Novel Methods or Assays: When a new microbiological method is created from scratch, such as a novel PCR assay for a specific pathogen or a new biomarker detection method, full validation is required to establish all its performance characteristics [1].
- Laboratory-Developed Tests (LDTs): Any test that is developed and performed within a single laboratory is considered an LDT. Since these are not commercially distributed or reviewed by the FDA, they require intensive internal validation before they can be used for patient testing or critical research data [15].
- Significant Modification of an Existing Method: If a laboratory makes a change to an FDA-cleared or validated method that falls outside the manufacturer's acceptable parameters—such as using a different specimen type, changing sample dilutions, or altering critical test parameters like incubation times—the modified method must be validated [15].
- Method Transfer to a New Matrix or Category: When a validated method is applied to a new type of sample (matrix) that it was not originally validated for, a validation study (often called a "fitness-for-purpose" or matrix extension study) is required. For instance, using a method validated for raw meat to test cooked chicken requires validation to account for potential inhibitors or microbial load differences [13].
- Regulatory Submissions for New Products: Data submitted to regulatory bodies like the FDA or EMA in support of new drug applications (NDAs), investigational new drugs (INDs), or medical devices must be generated using fully validated methods [1].
Validation Experimental Protocol and Parameters
A full method validation requires a multi-parameter experimental approach. The following table summarizes the key performance characteristics, their definitions, and common experimental methodologies, particularly for qualitative/semi-quantitative microbiological assays.
Table 1: Core Parameters for Microbiological Method Validation
| Parameter | Definition | Experimental Protocol Summary |
|---|---|---|
| Accuracy | Agreement between the test result and the true value [14]. | Test a minimum of 20 positive and negative samples (e.g., clinical isolates, spiked samples) against a reference method. Calculate as: (Number of agreements / Total tests) × 100 [15]. |
| Precision | Closeness of agreement between independent test results under specified conditions (repeatability and reproducibility) [14]. | Test a minimum of 2 positive and 2 negative samples in triplicate over 5 days by 2 different operators. Calculate agreement as for accuracy [15]. |
| Specificity | Ability to measure the analyte accurately in the presence of interfering substances [14]. | Test samples with known interfering substances or non-target microorganisms to demonstrate no cross-reactivity. |
| Sensitivity | Proportion of true positives correctly identified [14]. | Calculated as: a / (a + b), where a = true positives, b = false negatives [14]. |
| Detection Limit (LOD) | Lowest amount of analyte that can be detected. | Test serial dilutions of the target microorganism to determine the lowest concentration that yields a positive result ≥95% of the time. |
| Reportable Range | Range of analyte values over which the method provides accurate results. | Verify using a minimum of 3 samples with values at the upper and lower limits of the expected range [15]. |
| Robustness | Capacity of the method to remain unaffected by small, deliberate variations in method parameters. | Deliberately alter parameters like incubation temperature, time, or reagent lot to assess impact on results. |
When is Method Verification Sufficient?
Method verification is an efficiency-focused process that leverages prior validation work. It is sufficient in scenarios where the fundamental soundness of the method is already established, and the goal is to confirm its proper implementation in a new setting.
Key Application Scenarios
- Implementation of a Standard Compendial Method: When a laboratory adopts a well-established, published method from a recognized authority such as the US Pharmacopeia (USP), European Pharmacopoeia (EP), AOAC International, or a standard EPA method for the first time, verification is typically sufficient [1].
- Adoption of an Unmodified FDA-Cleared Test: Introducing a commercially available, FDA-cleared test kit (e.g., a new commercial cartridge-based molecular test) into the laboratory's test menu requires verification, not full validation, provided it is used exactly as specified by the manufacturer [15].
- Routine Re-confirmation in Quality Systems: While not a direct replacement for the initial verification, ongoing checks as part of a quality management system (e.g., after instrument servicing) are based on the principles of verification.
Verification Experimental Protocol and Parameters
Verification is a more targeted process than validation. The CLIA regulations require verification of several key performance characteristics for non-waived tests [15]. The scope is narrower, focusing on confirming that the lab can meet the manufacturer's or method's stated claims.
Table 2: Core Parameters for Microbiological Method Verification
| Parameter | Verification Protocol Summary | Acceptance Criteria |
|---|---|---|
| Accuracy | Test a minimum of 20 positive and negative samples and compare results to a reference method or known values [15]. | Results must meet the manufacturer's stated claims or lab-defined criteria (e.g., ≥95% agreement) [15]. |
| Precision | Test a minimum of 2 positive and 2 negative samples in triplicate, typically over 3-5 days. If the system is not fully automated, include a second operator [15]. | The results should demonstrate acceptable variance as per manufacturer's claims or lab-defined criteria [15]. |
| Reportable Range | Verify the upper and lower limits by testing samples with values near these limits [15]. | The method should correctly identify samples within the reportable range. |
| Reference Range | Test a minimum of 20 samples representative of the laboratory's patient population to confirm the normal or expected result [15]. | The established reference range must be appropriate for the lab's specific patient population. |
The Scientist's Toolkit: Essential Research Reagent Solutions
The execution of robust validation and verification studies relies on high-quality, traceable materials. The following table details essential reagents and their functions in these processes.
Table 3: Key Research Reagent Solutions for Method V&V
| Reagent/Material | Function in V&V | Critical Quality Attributes |
|---|---|---|
| Certified Reference Materials (CRMs) | Serves as the primary standard for assigning a value to an analyte. Used for establishing accuracy and calibrating equipment. | Purity, stability, and metrological traceability to an international standard. |
| Quality Control (QC) Strains | Used to verify precision (repeatability/reproducibility) and monitor ongoing method performance. | Genetically defined, viability, purity, and known reactivity in the test system. |
| Characterized Clinical Isolates | Provide real-world samples for accuracy and specificity studies. Help confirm the method works with diverse strain variants. | Well-characterized identity and phenotype (e.g., antibiotic resistance profile). |
| Inhibitor/Interference Substances | Used in specificity and robustness studies to challenge the method and ensure it is not affected by common interferents (e.g., pectin, fats, blood) [13]. | High purity, prepared at clinically relevant concentrations. |
| Molecular Grade Water | Serves as a negative control and a diluent for preparing samples and standards. | Confirmed to be nuclease-free and sterile to prevent false results. |
| Palmitoleyl palmitate | Palmitoleyl palmitate, MF:C32H62O2, MW:478.8 g/mol | Chemical Reagent |
| 9Z,12Z,15Z-octadecatrienoyl-CoA | 9Z,12Z,15Z-octadecatrienoyl-CoA, MF:C39H64N7O17P3S, MW:1028.0 g/mol | Chemical Reagent |
In the highly regulated and scientifically rigorous field of drug development and microbiology, understanding the distinction between method validation and verification is not optional—it is essential. The choice between them hinges on a simple but critical question: Are you establishing that a method works at all, or confirming that it works in your hands?
To ensure compliance, data integrity, and patient safety, scientists must adhere to the following principle: Validation is required for novel, modified, or laboratory-developed methods, while Verification is sufficient for implementing standard, unmodified methods. By applying the decision framework, experimental protocols, and reagent standards outlined in this guide, researchers can navigate these requirements with confidence, ensuring their methods are both scientifically sound and regulatorily compliant.
Standardizing Concepts like Method Transfer and Suitability Testing
In the highly regulated pharmaceutical and biopharmaceutical industry, the precision of analytical terminology is not merely academic—it is a fundamental requirement for ensuring product quality, patient safety, and regulatory compliance. Concepts such as method validation, method verification, method transfer, and method suitability testing form the backbone of analytical quality control. However, inconsistent application of these terms can lead to significant compliance risks, operational inefficiencies, and miscommunication during regulatory inspections. This guide provides a standardized framework for these core concepts, specifically contextualized within microbiological quality control, to equip researchers, scientists, and drug development professionals with the clarity needed to navigate this complex landscape. A foundational understanding begins with distinguishing between the two most frequently contrasted processes: method validation and method verification.
Core Terminology: Validation, Verification, and Suitability
Method Validation vs. Method Verification
Method Validation is the comprehensive, documented process of establishing through laboratory studies that an analytical method's performance characteristics are suitable for its intended purpose [19] [20]. It provides proof that a newly developed method is reliable, accurate, and robust for a specific application.
Method Verification, in contrast, is the documented evidence that a laboratory can competently execute a previously validated method (e.g., a compendial method from USP, Ph. Eur., or JP) under its specific conditions [1] [19]. It confirms that the method performs as expected for a specified product in a given laboratory, without repeating the entire validation exercise.
Table 1: Key Differences Between Method Validation and Method Verification
| Comparison Factor | Method Validation | Method Verification |
|---|---|---|
| Objective | To prove a method is fit-for-purpose [20] | To confirm a lab can perform a pre-validated method [19] |
| Typical Context | Newly developed in-house methods; significant modifications [19] | Adoption of compendial (USP, Ph. Eur.) or transferred methods [21] [19] |
| Scope | Full assessment of all relevant performance characteristics [20] | Limited assessment of critical parameters (e.g., precision, accuracy) [1] [15] |
| Regulatory Basis | ICH Q2(R2), USP <1225> [19] [20] | USP <1226>, CLIA regulations [15] [20] |
| Resource Intensity | High (time, cost, personnel) [1] | Moderate to low [1] |
Method Suitability Testing
Method Suitability Testing is a compendial concept, often serving as the verification of a pharmacopoeial method for a specific product [22]. It demonstrates that the compendial method is suitable under actual conditions of use and that the product to be tested does not interfere with the method [22]. A prime example is the Sterility Test Method Suitability Test (MST), which proves that the product itself does not possess antimicrobial properties that would lead to false-negative results [22]. In a microbiological context, this test is the verification that the specific product being tested does not inhibit the growth of microorganisms, thereby ensuring the reliability of the sterility test result.
Method Suitability Testing: A Deep Dive into the Sterility Test
Purpose and Principle
The Sterility Test Method Suitability Test is performed to verify under the actual test conditions that the product to be examined does not inhibit the growth of microorganisms. In simple terms, it ensures that any contaminating microorganisms present in the product would be detected and not masked by the product's inherent antimicrobial properties [22]. Without a successful suitability test, a passing sterility test result is invalid, as it could be a false negative [22].
Experimental Protocol
The MST is performed according to pharmacopoeial chapters (e.g., Ph. Eur. 2.6.1, USP <71>, JP 4.06) and must be conducted under aseptic conditions [22]. The following workflow and protocol detail the process for the membrane filtration method, which is preferred when the product is filterable.
Step-by-Step Procedure:
- Test Preparation: The test is performed under aseptic conditions, such as in a sterile test isolator. A filtration unit with a membrane pore size of ≤ 0.45 µm is used [22].
- Filtration and Rinsing: The membrane is wetted with an appropriate buffer. The product to be tested is filtered, and the membrane is rinsed with a suitable buffer (typically three times with 100 mL) to remove any antimicrobial properties of the product [22].
- Inoculation with Challenge Microorganisms: During the last rinsing step, the membrane is inoculated with a defined inoculum (not more than 100 CFU) of each of the following compendial microorganisms: Staphylococcus aureus, Bacillus subtilis, Pseudomonas aeruginosa, Clostridium sporogenes, Candida albicans, and Aspergillus brasiliensis [22]. These represent potential contaminants from the manufacturing environment.
- Culture Media Addition: Two different liquid culture media are added to the filtration units: Tryptic Soy Broth (TSB) for aerobes and fungi, and Fluid Thioglycollate Medium (FTM) for aerobes and anaerobes [22].
- Incubation and Examination: The units are incubated at temperatures specified by the pharmacopoeia (e.g., 20-25°C or 30-35°C) for a maximum of 3 days for bacteria and 5 days for yeasts and molds. They are examined daily for turbidity, which indicates microbial growth [22].
- Interpretation of Results: The growth in the test preparation (with product) is compared to a positive control (inoculated media without product). The test is successful only if the culture media with the product shows growth comparable to the positive control, demonstrating that the product does not inhibit growth [22]. If growth is inhibited, the test conditions (e.g., rinsing volume, use of neutralizing agents) must be modified.
Table 2: Key Reagents and Materials for Sterility Test Suitability
| Reagent/Material | Function/Description | Critical Attribute |
|---|---|---|
| Test Microorganisms | Challenge strains (e.g., S. aureus, B. subtilis, P. aeruginosa, C. sporogenes, C. albicans, A. brasiliensis) to demonstrate growth recovery [22]. | Viable count must be ≤ 100 CFU per strain. |
| Culture Media | TSB and FTM to support the growth of a wide spectrum of bacteria and fungi [22]. | Must have proven growth-promoting properties and be sterile. |
| Membrane Filter | Retains any microorganisms present in the product solution during filtration. | Pore size ≤ 0.45 µm with proven bacterial retention. |
| Rinsing Fluid | Buffered solutions (e.g., Fluid A, D, or K) to remove residual product from the membrane [22]. | Must effectively neutralize or remove antimicrobial properties without harming potential microbes. |
Analytical Method Transfer
Definition and Scope
Analytical Method Transfer is the documented process that qualifies a receiving laboratory (which could be internal or external) to use an analytical method that originated in a transferring laboratory [20] [23]. The goal is to ensure the method produces equivalent results in the receiving lab's environment, with its specific analysts, equipment, and reagents. It applies to previously validated, non-compendial methods. For compendial methods, a method verification is typically performed upon transfer [23].
Approaches and Protocol
The transfer process is governed by a pre-approved protocol that clearly defines objectives, materials, procedures, and acceptance criteria [24] [20]. Common transfer approaches include:
- Comparative Testing: The most common strategy, where the sending and receiving laboratories each conduct replicate testing of the same homogeneous sample(s), and the results are statistically compared for equivalence [24] [23].
- Co-validation: The receiving laboratory participates in the original validation study, performing a part of the validation (e.g., intermediate precision) [21].
- Transfer Waiver: Under specific, justified circumstances (e.g., the receiving lab has a proven history with a very similar method), a full transfer might be waived [20].
A successful transfer requires meticulous planning and assessment of the receiving lab's capabilities, including equipment qualification, analyst training, and availability of reference standards [24].
Regulatory Frameworks and Practical Applications
Guidance and Standards
Adherence to regulatory guidelines is non-negotiable. Key documents include:
- ICH Q2(R2): Provides the international standard for the validation of analytical procedures [19] [20].
- USP General Chapters <1225> (Validation), <1226> (Verification), <1224> (Transfer): Provide detailed requirements for compendial methods [19].
- ISO 16140 Series: Dedicated to the validation and verification of microbiological methods in the food and feed chain, offering a robust model for methodological rigor [2].
Application in Microbiological Testing
The concepts of verification and suitability testing are particularly relevant for established microbiological methods. For instance:
- Bacterial Endotoxin Testing (BET): As a compendial method, transferring the LAL test to a new lab or changing a key reagent (e.g., lysate supplier) does not require full validation. Instead, a comparative testing is performed to verify that the laboratory can construct a satisfactory standard curve and that the product can be tested without interference at the established dilution [24].
- Compendial Verification in Clinical Microbiology: For unmodified, FDA-cleared tests in a clinical lab, verification is required per CLIA regulations. This involves demonstrating accuracy, precision, reportable range, and reference range for the lab's specific patient population [15].
In the rigorous world of pharmaceutical and biopharmaceutical development, standardized terminology is the linchpin of quality and compliance. Method Validation is the foundational act of proving a method works for its purpose, while Method Verification confirms a laboratory's ability to implement a pre-validated method. The Method Suitability Test is a critical, product-specific verification for compendial methods, ensuring the product does not interfere with the test. Finally, Method Transfer is the structured process of moving a validated method between laboratories. By adhering to these precise definitions and their associated experimental protocols, organizations can ensure data integrity, streamline regulatory submissions, and ultimately safeguard patient health.
Implementing compliant Protocols: A Step-by-Step Guide to Verification and Validation
Method validation is a documented process that proves an analytical method is acceptable for its intended use [1]. It is a comprehensive exercise involving rigorous testing and statistical evaluation, typically required when developing new methods or transferring methods between labs and instruments [1]. For microbiological methods, validation specifically tests a method's ability to detect target organisms under a particular range of conditions and for particular matrix categories [13]. Unlike method verification, which confirms that a previously validated method performs as expected in a specific laboratory, validation establishes the fundamental performance characteristics of the method itself [1] [13].
Core Validation Parameters and Experimental Protocols
A robust validation protocol systematically assesses key performance parameters. The following table summarizes the essential characteristics, their definitions, and experimental approaches for microbiological methods.
Table 1: Core Parameters for Microbiological Method Validation
| Parameter | Definition | Recommended Experimental Approach |
|---|---|---|
| Accuracy | The agreement of results between the new method and a comparative method [15]. | Use a minimum of 20 clinically relevant isolates [15]. For qualitative assays, use a combination of positive and negative samples. Calculate as (number of results in agreement / total number of results) × 100 [15]. |
| Precision | The acceptable within-run, between-run, and operator variance [15]. | Use a minimum of 2 positive and 2 negative samples tested in triplicate for 5 days by 2 operators [15]. For fully automated systems, user variance may not be needed. |
| Specificity | The ability to detect the target organism without interference from other microorganisms or matrix components [1] [13]. | Test against a panel of related and unrelated non-target strains to demonstrate no cross-reactivity. Include samples with potential matrix interferents (e.g., high fat, acidity, or inhibitors like pectin) [13]. |
| Detection Limit | The lowest number of microorganisms that can be reliably detected by the method [1]. | Perform serial dilutions of the target organism to determine the lowest level that yields a positive result ≥95% of the time. |
| Quantitation Limit | The lowest number of microorganisms that can be quantified with acceptable accuracy and precision [1]. | For quantitative methods, determine the lowest level at which precision and accuracy targets are met. |
| Linearity | The ability of the method to obtain results directly proportional to the analyte concentration in the sample. | Test across a specified range using reference materials. |
| Robustness | A measure of the method's capacity to remain unaffected by small, deliberate variations in method parameters [1]. | Introduce small changes in critical parameters (e.g., incubation temperature ±1°C, time ±10%, reagent lot variations) and assess impact on results. |
| Reportable Range | The acceptable upper and lower limits of the test system that can be reported [15]. | Verify using a minimum of 3 samples. For qualitative assays, use known positive samples; for semi-quantitative, use samples near the manufacturer's cutoffs [15]. |
Detailed Protocol: Accuracy and Precision Studies
Accuracy Experimental Methodology:
- Sample Selection: Obtain a minimum of 20 clinically relevant bacterial isolates. For a qualitative assay, this should include a combination of strains known to be positive and negative for the target analyte [15].
- Source Material: Acceptable specimens can be sourced from certified reference materials, proficiency test samples, or de-identified clinical samples that have previously been tested with a validated comparator method [15].
- Testing Procedure: Test all samples using the new method (test method) and the validated comparative method in parallel.
- Data Analysis: Calculate the percentage agreement: (Number of results in agreement / Total number of results) × 100. The acceptance criteria should meet the manufacturer's stated claims or what the laboratory director determines is acceptable [15].
Precision Experimental Methodology:
- Sample Preparation: Select a minimum of 2 positive and 2 negative control samples. For semi-quantitative assays, use samples with a range of values from high to low [15].
- Testing Schedule: Test each sample in triplicate, over the course of 5 non-consecutive days, using 2 different qualified operators to incorporate between-run and operator variance [15].
- Data Analysis: Calculate the percent agreement for each sample type across all replicates and days. The results should meet the pre-defined acceptance criteria for variance.
Method Validation Workflow
The following diagram illustrates the logical sequence and key decision points in the method validation process.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful method validation relies on high-quality, well-characterized materials. The following table details key reagents and their functions.
Table 2: Essential Reagents for Microbiological Method Validation
| Reagent / Material | Function in Validation |
|---|---|
| Certified Reference Strains | Provide a traceable, reliable source of target microorganisms for accuracy, precision, and detection limit studies. |
| Inactivated Clinical Isolates | Broaden the assessment of method specificity by testing against a diverse panel of related and non-target organisms. |
| Matrix-Matched Controls | Assess the impact of the sample background (e.g., food, clinical specimen) on assay performance, crucial for fitness-for-purpose. |
| Selective & Non-Selective Growth Media | Used for culture-based comparator methods and to ensure the method can detect viable organisms in the presence of inhibitors. |
| Sample Preparation Buffers & Reagents | Critical for liberating microorganisms from complex matrices and neutralizing potential interferents like enzymes or antibiotics. |
| Molecular-Grade Water & Negative Controls | Establish the baseline for specificity and ensure no false positives arise from contaminated reagents or environmental background. |
| Anteisopentadecanoyl-CoA | Anteisopentadecanoyl-CoA, MF:C36H64N7O17P3S, MW:991.9 g/mol |
| 10-MethylHexadecanoyl-CoA | 10-MethylHexadecanoyl-CoA, MF:C38H68N7O17P3S, MW:1020.0 g/mol |
Navigating Regulatory and Matrix Considerations
Method validation is required by international regulatory bodies for new drug submissions, diagnostic test approvals, and environmental monitoring protocols [1]. Guidelines from organizations like the FDA, ICH, USP, AOAC, and ISO provide frameworks for validation protocols [1] [13].
A critical concept in microbiological validation is "fitness-for-purpose," which demonstrates that the method delivers expected results in a specific sample matrix [13]. If a method has been validated for a particular matrix, it is considered fit-for-purpose. If not, the laboratory must evaluate whether a matrix extension study is needed [13]. The first step is to consider the food matrix grouping, where products are categorized based on similar characteristics (e.g., AOAC guidelines consider eight food categories divided into 92 subcategories) [13]. The extent of additional testing required depends on the public health risk and the detection risk associated with the new matrix [13].
In the context of microbiological testing, the implementation of a compendial method or a method validated by a third party requires a laboratory to perform method verification. This process is distinct from method validation; verification confirms that a pre-validated method performs as expected in your specific laboratory environment, with your analysts, equipment, and reagents [25] [19]. It is a targeted assessment to demonstrate suitability under actual conditions of use, not a repeat of the full validation process [26]. Framed within a broader methodology, verification is a critical step that ensures the ongoing reliability and regulatory compliance of established methods upon their adoption into a new setting.
Method Verification vs. Validation: A Foundational Distinction
Understanding the distinction between verification and validation is crucial for selecting the correct protocol. The following table summarizes the key differences.
Table 1: Core Differences Between Method Verification and Method Validation
| Aspect | Method Verification | Method Validation |
|---|---|---|
| Definition | Confirming a previously validated method performs suitably in a specific laboratory [1] [26]. | A comprehensive process to establish and document that a method is fit for its intended purpose [19] [1]. |
| Core Question | "Can we use this existing method successfully in our lab?" | "Is this new method reliable and fit for its purpose?" |
| Scope | Limited assessment of critical performance characteristics [19]. | Full assessment of all relevant performance characteristics [19]. |
| When Performed | When adopting a compendial (e.g., USP, Ph. Eur.) or a transferred method [19] [1]. | When developing a new method or significantly modifying an existing one [19] [26]. |
| Regulatory Basis | USP General Chapter <1226> "Verification of Compendial Procedures" [25] [19]. | ICH Q2(R2) "Validation of Analytical Procedures" [19] [1]. |
The relationship between these processes and other related activities can be visualized in the method lifecycle workflow below.
Designing the Verification Protocol
A robust verification protocol is a pre-approved document that specifies the objectives, methodology, acceptance criteria, and responsibilities for the study.
Determining the Verification Scope
The extent of verification is not one-size-fits-all. It depends on factors such as the complexity of the procedure, the training and experience of the analyst, the type of equipment, and the specific article being tested [25]. For a standard compendial microbiological method like sterility testing or bioburden determination, the verification typically focuses on demonstrating precision and specificity under the new laboratory's conditions [25] [19]. Accuracy may also be assessed, depending on the specific situation of the sample being tested [25].
Key Performance Characteristics for Verification
The following characteristics are typically evaluated during verification, with the selection tailored to the method's intended use.
Table 2: Key Performance Characteristics for Method Verification
| Characteristic | Definition | Typical Verification Approach in Microbiology |
|---|---|---|
| Precision | The closeness of agreement between a series of measurements from multiple sampling of the same homogeneous sample. | Analyze multiple aliquots (e.g., n=6) of a homogeneous sample. Calculate the standard deviation and relative standard deviation. |
| Specificity | The ability to assess the analyte unequivocally in the presence of components that may be expected to be present. | Demonstrate that the method can detect the target microorganism in the presence of the product's matrix and normal flora. |
| Accuracy/Trueness | The closeness of agreement between the accepted reference value and the value found. | Use spiked samples with known concentrations of the target microorganism and calculate recovery. |
| Limit of Detection (LOD) | The lowest amount of analyte in a sample that can be detected, but not necessarily quantified. | Confirm the method can detect a low-level inoculum of the target microorganism that is near the expected detection limit. |
Experimental Methodology: The Comparison Study
A core experiment in many verification protocols is the comparison of method results against a reference or the documented method performance.
Protocol for a Quantitative Comparison Study
This methodology is critical for estimating the systematic error, or inaccuracy, of the method when implemented in your lab [27].
- Sample Preparation: Select a minimum of 40 different patient specimens or samples [27]. These should be selected to cover the entire working range of the method and represent the expected sample matrix [27].
- Testing Period: The experiment should be performed over a minimum of 5 different days to account for day-to-day variability [27].
- Analysis: Analyze each specimen using the new method (test method) and the comparative method. Common practice is to analyze each specimen singly by both methods, but duplicate measurements are advantageous for identifying discrepancies [27].
- Data Analysis:
- Graph the Data: Create a difference plot (test result minus comparative result vs. comparative result) or a comparison plot (test result vs. comparative result) to visually inspect the data for patterns and outliers [27].
- Calculate Statistics: For data covering a wide analytical range, use linear regression analysis to determine the slope, y-intercept, and standard deviation about the regression line (s~y/x~). The systematic error (SE) at a critical decision concentration (X~c~) is calculated as: SE = (a + bX~c~) - X~c~, where 'a' is the y-intercept and 'b' is the slope [27].
The workflow for executing this core experiment is detailed below.
The Scientist's Toolkit: Essential Reagents and Materials
The following table outlines key materials required for a typical microbiological method verification study.
Table 3: Essential Research Reagent Solutions for Microbiological Verification
| Item | Function in Verification |
|---|---|
| Reference Strains | Certified microbial strains (e.g., from ATCC) used to demonstrate specificity, precision, and accuracy by serving as a known positive control. |
| Culture Media | Growth media specified in the compendial method. Used to support the growth and detection of microorganisms; performance must be verified. |
| Neutralizers/Inactivators | Critical for antimicrobial effectiveness testing and bioburden methods to neutralize disinfectants or antimicrobial agents in the sample. |
| Sample Matrix (Placebo) | The product or material without the active ingredient. Used to prepare negative controls and to spike with reference strains for recovery studies. |
| Validated Spiking Inoculum | A suspension of reference microorganisms with a known and verified concentration, used in accuracy/recovery experiments. |
| 10(Z)-Heptadecenyl acetate | 10(Z)-Heptadecenyl acetate, MF:C19H36O2, MW:296.5 g/mol |
| (10Z,13Z,16Z)-3-oxodocosatrienoyl-CoA | (10Z,13Z,16Z)-3-oxodocosatrienoyl-CoA, MF:C43H70N7O18P3S, MW:1098.0 g/mol |
Data Analysis and Acceptance Criteria
The data generated from verification experiments must be evaluated against pre-defined, scientifically justified acceptance criteria. These criteria should be based on the intended use of the method and any regulatory guidance.
Table 4: Example Acceptance Criteria for Key Verification Parameters
| Parameter | Example Acceptance Criterion for a Quantitative Method |
|---|---|
| Precision (Repeatability) | Relative Standard Deviation (RSD) ≤ 15% for biological methods. |
| Accuracy (Recovery) | Mean recovery of the target microorganism between 70% and 130%. |
| Specificity | No interference from the sample matrix; clear detection of the target organism. |
| Linearity | Correlation coefficient (r) ≥ 0.98 over the specified range. |
For the comparison of methods experiment, the estimated systematic error at a medically or functionally critical decision level should be less than the allowable total error based on the method's intended use [27] [28].
A well-executed method verification protocol is a cornerstone of quality assurance in a microbiological laboratory. It provides documented evidence that a pre-validated method is under control and suitable for its intended use within a specific laboratory's environment. By following a structured approach—defining the scope, executing targeted experiments, and analyzing data against strict acceptance criteria—researchers and drug development professionals can ensure the generation of reliable, high-quality data that supports product safety and regulatory compliance.
Within pharmaceutical development and clinical diagnostics, the reliability of microbiological testing is paramount for ensuring drug safety and patient health. This reliability hinges on two distinct but complementary laboratory processes: method validation and method verification [13] [1]. Method validation is the comprehensive process of proving that an analytical method is acceptable for its intended use, establishing its performance characteristics during development or transfer [1]. Method verification, by contrast, is the process whereby a laboratory confirms that a previously validated method performs as expected in its specific environment, with its personnel and equipment [13] [29].
The distinction, while subtle, is critical. Validation asks, "Does this method work for its intended purpose in general?" while verification asks, "Can our laboratory perform this method correctly?" [14]. For researchers and drug development professionals, understanding this difference is a strategic necessity for regulatory compliance and scientific integrity [1].
This guide provides an in-depth examination of the four core performance parameters—Accuracy, Precision, Detection Limit, and Reportable Range—within the context of method verification, offering a practical framework for their assessment in a laboratory setting.
Core Performance Parameters in Method Verification
When a laboratory implements a new, previously validated method (e.g., an FDA-cleared or compendial method), it must verify key performance characteristics as required by standards such as the Clinical Laboratory Improvement Amendments (CLIA) [29]. The following parameters are central to this verification process for qualitative and semi-quantitative microbiological assays.
Accuracy
Accuracy measures the agreement between the results from the new method and those from a comparative method, confirming the method's ability to correctly identify the target analyte [29]. It is a fundamental indicator of a method's freedom from error.
Experimental Protocol for Verifying Accuracy:
- Sample Number and Type: A minimum of 20 clinically relevant isolates or samples should be used. These should include a combination of positive and negative samples for qualitative assays, or a range of samples with high to low values for semi-quantitative assays [29].
- Sample Sources: Acceptable specimens can be sourced from reference materials, proficiency test samples, standardized controls, or de-identified clinical samples that have previously been tested with a validated method [29].
- Testing Procedure: Each sample is tested in parallel using the new method (the test method) and the established comparative method.
- Calculation: Accuracy is calculated as the percentage of results where the new method agrees with the comparative method.
Accuracy (%) = (Number of results in agreement / Total number of results) × 100[29]. - Acceptance Criteria: The calculated percentage of agreement should meet or exceed the performance claims made by the method's manufacturer or a threshold determined by the laboratory director [29].
Precision
Precision, or reliability, evaluates the degree of agreement among repeated measurements from the same homogeneous sample. It assesses the random error of a method and is typically broken down into within-run (repeatability) and between-run (reproducibility) variance [1] [29].
Experimental Protocol for Verifying Precision:
- Sample Number and Type: A minimum of two positive and two negative samples are tested. For semi-quantitative assays, samples should cover a range from high to low values [29].
- Testing Procedure: The selected samples are tested in triplicate over the course of five days by two different operators. If the test system is fully automated, testing for operator variance may not be necessary [29].
- Calculation: Precision is calculated for each level of sample as the percentage of agreement between the replicate results.
Precision (%) = (Number of results in agreement / Total number of results) × 100[29]. - Acceptance Criteria: As with accuracy, the results should conform to the manufacturer's stated claims or the laboratory's predefined acceptance criteria [29].
Detection Limit
The Limit of Detection (LOD) is the lowest quantity or concentration of an analyte that can be reliably distinguished from its absence. For a qualitative method, this is the smallest amount of target microorganism that yields a positive result [1].
Experimental Protocol for Verifying the LOD:
- Objective: To confirm that the LOD stated by the method's manufacturer can be achieved under the laboratory's specific conditions.
- Sample Preparation: Prepare samples spiked with the target microorganism at a concentration near the claimed LOD. Serial dilutions may be used to bracket the manufacturer's stated detection limit.
- Testing Procedure: The spiked samples are tested repeatedly (e.g., in multiple replicates) using the new method.
- Evaluation: The LOD is verified as the lowest concentration at which ≥95% of the replicates test positive [1]. The laboratory confirms that its results are consistent with the validation data provided by the manufacturer.
Reportable Range
The reportable range defines the upper and lower limits of the analyte that the method can directly measure without modification, such as dilution or concentration. It establishes the bounds of what constitutes a valid, reportable result [29].
Experimental Protocol for Verifying the Reportable Range:
- Sample Number and Type: A minimum of three samples is recommended. For qualitative methods, use known positive samples. For semi-quantitative assays, use samples with values near the upper and lower cutoffs established by the manufacturer [29].
- Testing Procedure: Test the selected samples using the new method.
- Evaluation: The reportable range is verified by confirming that the results for all tested samples fall within the limits defined by the manufacturer and that they are reported appropriately (e.g., "Detected," "Not detected," or a specific cycle threshold value) [29].
Table 1: Summary of Core Verification Parameters and Protocols
| Parameter | Definition | Experimental Protocol Summary | Key Calculations/Acceptance |
|---|---|---|---|
| Accuracy | Agreement with a reference method [29] | - 20 samples (pos/neg mix)- Test vs. comparative method | % Agreement = (Agreements/Total) x 100Meet mfr. claims or lab criteria [29] |
| Precision | Closeness of repeated measurements [29] | - 2 pos & 2 neg samples- Triplicate, 5 days, 2 operators | % Agreement between replicatesMeet mfr. claims or lab criteria [29] |
| Detection Limit | Lowest detectable analyte level [1] | - Samples spiked near claimed LOD- Multiple replicate tests | Lowest concentration with ≥95% positive rateConfirm mfr.-stated LOD [1] |
| Reportable Range | Range of reportable results [29] | - 3 samples across the range- Test with new method | Results fall within mfr.-defined limits and are reported correctly [29] |
The Experimental Workflow for Method Verification
Executing a method verification study requires careful planning. The following workflow diagram and accompanying explanation outline the key stages, from initial planning to final implementation for routine use.
Diagram 1: Method Verification Workflow
1. Define Purpose and Assay Type: The first step is to confirm that the study is a verification (for an unmodified, previously validated method) and not a validation (required for laboratory-developed tests or modified methods) [29]. The assay type—qualitative, quantitative, or semi-quantitative—must be defined, as this determines the specific verification protocols [29].
2. Create a Written Verification Plan: Before beginning experimental work, a detailed plan must be drafted and signed off by the laboratory director. This plan is a blueprint for the entire study and should include [29]:
- The purpose of the test and a description of the method.
- The study design, specifying the number and types of samples, quality controls, number of replicates, analysts, and the performance characteristics to be evaluated (e.g., Accuracy, Precision) along with their acceptance criteria.
- A list of all required materials, equipment, and resources.
- Safety considerations and an expected timeline for completion.
3. Conduct Verification Experiments: The laboratory executes the experiments as outlined in the verification plan. This involves testing the specified samples for accuracy, precision, reportable range, and other relevant parameters according to the predefined methodology [29].
4. Analyze Data and Compile Report: The data collected from the experiments are analyzed and compared against the acceptance criteria. The results, along with a detailed description of the process, are compiled into a verification report [14].
5. Director Review and Approval: The final verification report is submitted to the laboratory director for formal review and approval. Once approved, the method can be released for routine diagnostic use [29].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successfully conducting a verification study requires access to well-characterized biological and chemical materials. The following table details key reagents and their critical functions in the process.
Table 2: Essential Reagents for Verification Studies
| Reagent / Material | Function in Verification |
|---|---|
| Reference Strains (QC Strains) | Well-characterized microbial strains used as positive controls, for spiking studies to assess accuracy and LOD, and for precision testing [29]. |
| Clinical Isolates / De-identified Samples | Provide real-world, complex matrices to confirm method performance correlates with the validated comparative method during accuracy testing [29]. |
| Proficiency Test (PT) Samples | Blinded samples of known value from an external provider; provide an objective assessment of accuracy and help identify potential systematic errors [29]. |
| Quality Controls (QC) | Includes positive, negative, and internal controls. They are run with each batch to monitor the test system's performance and ensure day-to-day reliability [29]. |
| Selective Agar Media | Used for the isolation and confirmation of target microorganisms, forming part of the comparative method against which a new method is verified [2]. |
| Molecular Detection Kits (e.g., PCR) | Commercial kits (e.g., for Salmonella or Listeria detection) provide the core reagents for the test method itself and must be used as specified by the manufacturer [30]. |
| (2E,9Z)-octadecadienoyl-CoA | (2E,9Z)-octadecadienoyl-CoA, MF:C39H66N7O17P3S, MW:1030.0 g/mol |
| (2R)-2-methyltetradecanoyl-CoA | (2R)-2-methyltetradecanoyl-CoA, MF:C36H64N7O17P3S, MW:991.9 g/mol |
The rigorous assessment of accuracy, precision, detection limit, and reportable range forms the bedrock of a reliable microbiological method verification. This process, distinct from the broader scope of method validation, provides documented evidence that a laboratory can successfully implement a standardized method within its own operational environment. For researchers and professionals in drug development, mastering these concepts and protocols is not merely a regulatory hurdle but a fundamental component of quality assurance. It ensures that the data generated in the laboratory is scientifically sound, reproducible, and ultimately fit for making critical decisions regarding product safety and public health.
Within the framework of microbiological method verification and validation research, a foundational distinction dictates all subsequent study design: verification is for unmodified, FDA-approved tests to confirm that a test performs as established in your laboratory, while validation is a more extensive process to establish that a new or modified test works as intended for its specific purpose [15]. International standards like ISO 15189:2022 and the European In Vitro Diagnostic Regulation (IVDR) have intensified the need for robust, well-documented procedures for both verification and validation [31]. The choice between a qualitative and quantitative assay is the primary determinant of the statistical approach, sample size, and experimental protocols required to satisfy these regulatory and scientific demands. This guide provides a detailed examination of the minimum requirements for study design and sample size, contextualized for researchers and drug development professionals establishing reliability in microbiological testing.
Core Differences Between Qualitative and Quantitative Assays
Understanding the fundamental distinctions between qualitative and quantitative methods is essential for selecting the correct validation pathway. The table below summarizes the key characteristics.
Table 1: Core Characteristics of Qualitative and Quantitative Microbiological Assays
| Aspect | Qualitative Assays | Quantitative Assays |
|---|---|---|
| Data Type | Descriptive, categorical (e.g., Present/Absent, Detected/Not Detected) [32] | Numerical, measurable (e.g., CFU/g, CFU/mL) [32] |
| Primary Objective | Detection or identification of a specific microorganism or attribute [33] | Enumeration or precise measurement of microorganism population [33] |
| Reportable Result | "Positive/25g" or "Not Detected/375g" [33] | Numerical value, such as "1.5 x 10³ CFU/g" [33] |
| Limit of Detection (LOD) | Very low, theoretically 1 CFU per test portion [33] | Higher, typically 10-100 CFU/g for plate counts [33] |
| Common Examples | Tests for Salmonella, Listeria monocytogenes, STEC [33] | Aerobic Plate Count (APC), Staphylococcus aureus enumeration, yeast and mold counts [33] |
| Statistical Analysis | Percent agreement, sensitivity, specificity, positive/negative predictive value [34] | Standard deviation, coefficient of variation, confidence intervals, regression analysis [32] [34] |
These core differences directly influence the parameters assessed during verification/validation and the sample sizes required to demonstrate that the method is fit for purpose.
Minimum Sample Size Requirements
Sample size determination is a critical step that balances statistical rigor with practical laboratory constraints. An inadequately small sample size makes it challenging to reproduce results and may produce false negatives, while an excessively large sample size can be ethically problematic and waste resources [35] [36].
Sample Size for Verification of Qualitative and Semi-Quantitative Assays
For the verification of unmodified, FDA-cleared qualitative and semi-quantitative assays in a clinical microbiology laboratory, CLIA standards provide a practical framework. The following table summarizes the minimum sample size requirements for key performance characteristics.
Table 2: Minimum Sample Size for Verification of Qualitative/Semi-Quantitative Assays (per CLIA) [15]
| Performance Characteristic | Minimum Sample Number & Type | Experimental Design & Acceptance Criteria |
|---|---|---|
| Accuracy | A minimum of 20 clinically relevant isolates or samples [15] | Use a combination of positive and negative samples. Calculate as: (Number of results in agreement / Total number of results) x 100. The percentage must meet manufacturer's claims or lab director's criteria [15]. |
| Precision | A minimum of 2 positive and 2 negative samples [15] | Test samples in triplicate for 5 days by 2 operators. For fully automated systems, operator variance is not needed. Calculate percent agreement; must meet manufacturer's claims [15]. |
| Reportable Range | A minimum of 3 samples [15] | Use known positive samples. For semi-quantitative assays, use samples near the upper and lower manufacturer cutoffs. Verify that results are reportable as defined by the lab (e.g., "Detected," Ct value cutoff) [15]. |
| Reference Range | A minimum of 20 isolates [15] | Use de-identified clinical or reference samples representing the laboratory's typical patient population. Verify the manufacturer's reference range is appropriate for your population [15]. |
Sample Size for Quantitative Assay Validation and Descriptive Studies
For quantitative assays and descriptive studies (e.g., estimating prevalence), sample size calculation relies on statistical principles rather than fixed numbers. The required size depends on several factors [35]:
- Acceptable Precision Level (Margin of Error): The smaller the allowed margin of error, the larger the required sample size.
- Study Power: The probability of correctly rejecting a false null hypothesis; typically set at 80% or 90% [36].
- Confidence Level: The probability that the confidence interval contains the true population parameter; typically 95%.
- Effect Size (ES): The magnitude of the difference or relationship the study is designed to detect. This is often the most challenging parameter to determine [35] [36].
For a study estimating a proportion (e.g., prevalence), the sample size can be calculated using the formula for a survey study [35] [36]. For a study comparing two means (e.g., comparing a new quantitative method to a reference method), the sample size for each group can be calculated using the formula for two means [36]. Free software tools such as OpenEpi and G*Power can greatly simplify these calculations [35].
Experimental Protocols for Key Validation Parameters
A robust verification or validation study requires carefully planned experiments to assess critical method parameters. The protocols below are aligned with international standards.
Accuracy and Specificity
Objective: To confirm the acceptable agreement of results between the new and a comparative method, and to demonstrate the method can resolve the target microorganism in the presence of interferents [34].
Protocol:
- Sample Preparation: Select a minimum of 20 clinically relevant isolates or samples [15]. For qualitative tests, use a combination of positive and negative samples. For quantitative tests, use samples spanning the reportable range.
- Challenge Strains: Include a panel of target microorganisms and related species/strains to test specificity. For growth-based methods, use a low challenge level of <100 CFU [34].
- Interference Testing: Spike the target microorganism into samples containing potential interferents (e.g., excipients, active pharmaceutical ingredients, other microorganisms).
- Testing Procedure: Test all samples in parallel using the new method and the reference/comparative method.
- Analysis: Calculate the percent agreement for qualitative tests. For quantitative tests, use statistical tests like the Student's t-test to compare means. All challenge microorganisms should be recovered, and no interference should be observed [34].
Precision: Repeatability and Intermediate Precision
Objective: To confirm acceptable variance within a single run and between different runs, operators, and equipment [15] [34].
Protocol:
- Sample Preparation: Select a minimum of 2 positive and 2 negative samples (for qualitative) or samples with high and low values (for quantitative) [15].
- Repeatability (Within-run): A single operator tests each sample in triplicate in a single run using the same reagents and equipment.
- Intermediate Precision (Between-run): Two different operators test each sample in triplicate over 5 separate days, using different lots of reagents and different equipment where possible [15].
- Analysis: Express precision as standard deviation, coefficient of variation (relative standard deviation), or percent agreement. The results should meet the manufacturer's stated claims or pre-defined acceptance criteria [15] [34].
Limit of Detection (LOD)
Objective: To determine the lowest number of microorganisms that can be detected by the method under stated conditions [34].
Protocol:
- Sample Preparation: Perform serial dilutions of a microbial suspension to create a low-level challenge range (e.g., from <100 CFU to <10 CFU) [34].
- Testing Procedure: Test each dilution level in multiple replicates (e.g., 3 or more).
- Analysis: The LOD is the lowest concentration at which the microorganism is consistently detected (e.g., in 95% of replicates). It is recognized that for microbiological methods, the LOD can be theoretical due to the Poisson distribution of microorganisms in liquid [34].
The following diagram illustrates the logical workflow and decision points for establishing a verification or validation study, integrating the concepts of sample size and experimental protocols.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key materials required for conducting the experiments described in this guide.
Table 3: Essential Research Reagents and Materials for Method Assessment
| Item | Function / Application |
|---|---|
| Certified Reference Materials | Commercially available strains with known characteristics; used for accuracy and specificity challenges to provide a "true" value for comparison [34]. |
| Clinical Isolates | Well-characterized, de-identified microbial strains from clinical samples; used to ensure the method works with relevant, wild-type organisms [15]. |
| Quality Control (QC) Strains | Strains with defined positive, negative, and borderline reactions; used for daily monitoring of test performance and during precision studies [15]. |
| Selective and Enrichment Media | Culture media designed to promote the growth of target organisms while inhibiting others; critical for specificity testing and traditional cultural methods [34] [33]. |
| Statistical Software Packages | Tools like G*Power or OpenEpi; used for a priori sample size calculation and subsequent data analysis to ensure statistical validity [35]. |
| 23-Methylpentacosanoyl-CoA | 23-Methylpentacosanoyl-CoA, MF:C47H86N7O17P3S, MW:1146.2 g/mol |
| trans-23-methyltetracos-2-enoyl-CoA | trans-23-methyltetracos-2-enoyl-CoA, MF:C46H82N7O17P3S, MW:1130.2 g/mol |
The journey from method selection to routine use is governed by a clear understanding of verification and validation requirements, which are fundamentally shaped by the qualitative or quantitative nature of the assay. Adherence to structured experimental protocols and evidence-based sample size determination—whether following the fixed minima for qualitative assay verification or the statistically-powered calculations for quantitative studies—is paramount. This rigorous approach ensures that microbiological methods are not only compliant with international standards but also reliably safeguard public health and product quality in pharmaceutical development and clinical diagnostics.
Navigating Challenges: Solutions for Common Verification and Validation Pitfalls
The accurate assessment of microbial distribution and viability is a cornerstone of pharmaceutical development, food safety, and clinical diagnostics. However, the complexity of microbiological methods often presents significant challenges for researchers and quality control professionals. Method complexity is not merely a technical issue; it is a multifaceted problem impacting time, resources, regulatory compliance, and the fundamental reliability of viability data. This complexity is compounded by the diverse physiological states of microorganisms, including the viable but non-culturable (VBNC) state, where bacteria are metabolically active but resistant to cultivation on standard media, leading to potential underestimation of viable populations [37] [38].
Within this technical landscape, a clear understanding of the regulatory framework governing method validation and verification is paramount. These are distinct but complementary processes. Method validation is a comprehensive process that proves an analytical method is acceptable for its intended use and is required when developing new methods or substantially modifying existing ones [1] [15]. It answers the question, "Is this method scientifically sound and fit-for-purpose?" In contrast, method verification is the process of confirming that a previously validated method performs as expected in a specific laboratory setting [2] [1]. It answers the question, "Can my laboratory perform this method correctly?" The ISO 16140 series provides a standardized framework for both the validation and verification of microbiological methods in the food and feed chain, outlining specific protocols for different scenarios [2]. Adhering to this structured approach provides a strategic pathway for managing method complexity, ensuring data integrity, and maintaining regulatory compliance.
Method Validation vs. Verification: A Strategic Framework
A clear grasp of the distinction between method validation and verification is critical for selecting the appropriate strategy to address method complexity. The choice between them is not arbitrary but is dictated by the origin and status of the analytical method in question.
Method Validation is an extensive exercise conducted to prove that a newly developed or significantly modified method is fit for its intended purpose. It is a one-time, in-depth assessment performed before a method is put into routine use. Key parameters assessed during validation include accuracy, precision, specificity, detection limit, quantitation limit, linearity, and robustness [1]. For microbiological methods, this often involves a method comparison study against a reference method, followed by an interlaboratory study to ensure reliability across different environments [2]. Validation is typically required for Laboratory Developed Tests (LDTs) or when applying an established method to a new sample matrix not covered by the original validation [15].
Method Verification, on the other hand, is a more focused process. It is conducted when a laboratory adopts a pre-validated, standard method (e.g., from a pharmacopoeia like USP or an FDA-cleared kit) without modification. The goal is not to re-establish the method's performance from scratch but to provide objective evidence that the method performs as claimed under the specific conditions of the user's laboratory, including its personnel, equipment, and reagents [1] [15]. According to ISO 16140-3, verification involves two stages: implementation verification (demonstrating the lab can perform the method correctly using a known item) and item verification (demonstrating performance with challenging items specific to the lab's scope) [2].
The following table provides a comparative summary of these two processes:
Table 1: Comparative Analysis: Method Validation vs. Verification
| Comparison Factor | Method Validation | Method Verification |
|---|---|---|
| Definition | Process of proving a method is fit for its intended purpose [1]. | Process of confirming a validated method performs as expected in a user's lab [1]. |
| When Performed | For new methods, method transfers, or significant modifications [1] [15]. | When implementing a pre-validated, standard method without modification [15]. |
| Scope & Complexity | Comprehensive, assessing all performance parameters [1]. | Limited, focusing on key parameters like accuracy and precision [1]. |
| Resource Intensity | High (time, cost, personnel) [1]. | Moderate to low [1]. |
| Regulatory Driver | Required for regulatory submissions of new methods [1]. | Required by CLIA for non-waived systems and for ISO/IEC 17025 accreditation [1] [15]. |
| Primary Goal | Establish performance characteristics of the method itself [1]. | Demonstrate laboratory competency with an established method [1]. |
The relationship between these processes and the management of method complexity can be visualized as a strategic pathway. Validation builds the foundational knowledge of a method's capabilities and limitations, while verification translates that knowledge into a specific, operational context. By correctly applying this framework, laboratories can avoid the common pitfall of over-verification (unnecessarily expending resources on established methods) or under-validation (risking compliance and data quality with new methods).
Advanced and Emerging Viability Assessment Technologies
Overcoming method complexity requires a toolkit of sophisticated methods. The "gold standard" for viability assessment remains the Colony Forming Unit (CFU) assay, which measures culturability [39] [38]. However, its limitations—including the inability to detect VBNC cells and being time- and resource-intensive—have driven the development of alternative and complementary technologies [37] [39]. These methods are broadly categorized by the viability criterion they assess: membrane integrity, metabolic activity, and nucleic acid integrity.
Membrane Integrity-Based Assays
These assays operate on the principle that a viable cell has an intact cytoplasmic membrane. Dyes like propidium monoazide (PMA) selectively penetrate compromised membranes of dead cells, intercalate with DNA, and inhibit PCR amplification upon photoactivation. This allows for the differentiation of DNA from viable and dead cells in molecular analyses like qPCR, a technique known as viability PCR (vPCR) [40] [41]. Flow cytometry can also be used with dye combinations (e.g., SYTO green and PI red) to count and characterize cells based on membrane integrity [38] [41].
Metabolic Activity-Based Assays
These methods assess cellular functions as a proxy for viability. A common approach uses fluorogenic enzyme substrates. For example, fluorescein diacetate (FDA), a non-polar, non-fluorescent compound, passively diffuses into cells. Intracellular esterases in viable cells hydrolyze FDA into fluorescein, which is polar and emits a measurable fluorescent signal that accumulates inside cells with intact membranes [37]. Another strategy is to monitor the uptake and utilization of specific metabolites. The dye 2-NBDG, a fluorescent glucose analog, is consumed by metabolically active cells, and its decomposition can be correlated with viability, though its applicability varies by bacterial species [37].
Nucleic Acid-Based and Novel Assays
The frontier of viability testing includes innovative molecular and technological solutions. Nanopore sequencing empowered by Artificial Intelligence represents a breakthrough. This method leverages the raw electrical signal data ("squiggles") from nanopore sequencers. Deep learning models can be trained on this data to distinguish between DNA derived from viable and dead microorganisms, potentially due to differences in DNA modification or damage, offering a fully computational viability assessment [40]. Geometric Viability Assay (GVA) addresses the resource complexity of the traditional CFU assay. GVA mixes a microbial sample with melted agarose inside a standard pipette tip. After solidification and incubation, colonies grow embedded within the cone. The assay computationally calculates the original viable concentration based on the geometric distribution of colonies along the tip's length, leveraging the fact that the probability of a colony forming is proportional to the cross-sectional area at that point. This method can reduce time and consumables by over 10-fold while maintaining a dynamic range of over 6 orders of magnitude [39].
Table 2: Technical Comparison of Microbial Viability Assessment Methods
| Method | Principle | Key Advantages | Key Limitations | Throughput |
|---|---|---|---|---|
| CFU Assay | Culturability [39] | Gold standard, simple, wide dynamic range [39] | Laborious, slow, misses VBNC cells [37] [39] | Low |
| Membrane Integrity (e.g., PMA-qPCR) | Membrane integrity [40] | Specific for membrane-compromised cells, culture-independent [38] | May overestimate viability if membrane remains intact post-death [38] | Medium |
| Metabolic Activity (e.g., FDA Staining) | Enzyme activity & membrane integrity [37] | Rapid, indicates metabolic potential | Sensitive to pH, potential dye efflux [37] | Medium |
| Geometric Viability Assay (GVA) | Embedded culturability & probability [39] | High-throughput, low waste, ~30x faster than drop CFU [39] | Requires specialized imaging and analysis | High (1200/day) [39] |
| AI-Nanopore Viability Inference | AI analysis of raw DNA sequencing signals [40] | Culture-independent, can be applied to metagenomes, high information content | Emerging technology, may require stressor-specific model training [40] | High |
The experimental workflow for implementing these techniques, from sample to answer, involves critical branching decisions that align with the specific viability question being asked.
Detailed Experimental Protocols
Geometric Viability Assay (GVA) Protocol [39]:
- Sample Preparation: Grow microbial culture to the desired phase (e.g., stationary phase for E. coli).
- Agarose Mixing: Serially dilute the sample if necessary. Mix the sample 1:1 with melted LB agarose (cooled to ≤55°C) to a final agarose concentration of 0.5%. Include a viability stain like Triphenyl tetrazolium chloride (TTC) for better colony contrast.
- Loading and Solidification: Aspirate the sample-agarose mixture into a standard pipette tip. Eject the tip into a sterile rack and allow the agarose to solidify completely at room temperature.
- Incubation: Place the entire rack of tips in a humidified incubator at the appropriate temperature and duration for the microbe (e.g., 37°C overnight for E. coli).
- Imaging and Analysis: Image the tips using a custom optical setup with a high-resolution camera. Analyze the images to detect the positions (distance from the tip) of all visible colonies. Compute the original CFU/mL concentration using the formula: CFUs/mL = N / (V * ∫ PDF(x) dx) where N is the number of colonies counted in a sub-volume, V is the volume of the cone, and PDF(x) is the probability density function proportional to x² (the squared distance from the tip).
Viability PCR (vPCR) with PMA Protocol [40] [38]:
- Sample Treatment: Add PMA dye to the microbial suspension to a final concentration of 10-100 µM. Incubate in the dark for 5-10 minutes to allow dye penetration into dead cells.
- Photoactivation: Expose the sample to bright light (e.g., a 500-W halogen lamp) for 5-15 minutes on ice. This crosslinks PMA to the DNA from dead cells, rendering it unamplifiable.
- DNA Extraction: Proceed with standard genomic DNA extraction protocols.
- qPCR Amplification: Perform qPCR using primers targeting a conserved gene (e.g., 16S rRNA). The resulting Cq values will be higher for PMA-treated samples compared to untreated controls, as DNA from dead cells is suppressed. The reduction in signal quantifies the non-viable population.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the methods described relies on a suite of key reagents and materials. The following table details these essential components and their functions.
Table 3: Research Reagent Solutions for Microbial Viability Assessment
| Reagent/Material | Function/Application | Technical Notes |
|---|---|---|
| Propidium Monoazide (PMA) | DNA intercalating dye for viability PCR; penetrates dead cells with compromised membranes [40] [38]. | Requires photoactivation with bright light. Critical for differentiating DNA sources in complex samples. |
| Fluorescein Diacetate (FDA) | Fluorogenic substrate for esterase activity; measures metabolic activity as a viability marker [37]. | Hydrolyzed to fluorescent fluorescein inside viable cells. Signal can be pH-sensitive and subject to efflux. |
| Triphenyl Tetrazolium Chloride (TTC) | Colorimetric viability stain; reduced to red formazan by metabolically active cells [39]. | Used in solid media (like in GVA) to enhance colony contrast for automated imaging and counting. |
| 2-NBDG | Fluorescent D-glucose analog; tracks glucose uptake as a measure of metabolic activity [37]. | Not universally taken up by all bacteria; applicability must be verified for the target organism. |
| Low-Melt Agarose | Polymer for embedding microbes in GVA and other soft-agar assays [39]. | Allows mixing with samples at low temperatures (37°C) to preserve microbial viability. |
| Nanopore Sequencing Kit | Preparation and sequencing of native DNA for AI-based viability inference [40]. | Enables sequencing of DNA without prior amplification, preserving modification signals used for viability prediction. |
| SYTO Dyes / PI Stain | Nucleic acid stains for flow cytometry; SYTO penetrates all cells, PI only dead cells [38] [41]. | A combination allows for differentiation of intact (SYTO+/PI-) and membrane-compromised (SYTO+/PI+) populations. |
| Fucosyl-lacto-N-sialylpentaose c | Fucosyl-lacto-N-sialylpentaose c, MF:C43H72N2O33, MW:1145.0 g/mol | Chemical Reagent |
| 13-Methyltetracosanoyl-CoA | 13-Methyltetracosanoyl-CoA, MF:C46H84N7O17P3S, MW:1132.2 g/mol | Chemical Reagent |
Addressing method complexity in microbial distribution and viability studies is not achieved by seeking a single perfect method, but through the strategic integration of a structured regulatory framework and a diversified technological toolkit. The critical first step is the correct application of method validation for novel approaches and method verification for established procedures, ensuring scientific and regulatory rigor from the outset. Subsequently, researchers can strategically select from a spectrum of viability methods—from the classic CFU assay and emerging high-throughput platforms like GVA to sophisticated culture-independent techniques like PMA-qPCR and AI-driven nanopore analysis. The choice of method must be guided by the specific research question, the need for throughput, the requirement for culture-independent data, and the physiological state of the target microbes. By embracing this multi-faceted strategy, researchers and drug development professionals can effectively navigate complexity, enhance the reliability of their data, and accelerate development cycles with greater confidence.
In the highly regulated fields of clinical diagnostics, pharmaceutical development, and food safety, the reliability of microbiological testing is paramount. Ensuring this reliability requires two distinct but complementary processes: method validation and method verification. For researchers, scientists, and drug development professionals, effectively managing the resources of time, cost, and personnel while maintaining stringent regulatory compliance presents a significant challenge. Method validation is a comprehensive process that establishes and documents that an analytical method is capable of producing accurate, precise, and reliable results for its intended purpose, typically required for newly developed or significantly modified methods [19]. In contrast, method verification is the process of confirming that a previously validated method performs as expected under the specific conditions of a given laboratory, including its instruments, personnel, and sample matrices [1] [19]. Understanding the strategic application of each process is fundamental to efficient resource management without compromising data integrity or regulatory standing. This guide provides a detailed framework for navigating these processes, complete with experimental protocols, resource comparisons, and practical tools for implementation.
The decision to validate or verify a method is not a matter of choice but is determined by the method's origin and history. A clear grasp of this distinction prevents both unnecessary expenditure of resources and critical compliance missteps.
Method Validation is performed when a method is new, developed in-house, or has undergone significant modification beyond the manufacturer's specifications or acceptable compendial allowances [15] [19]. It is a foundational process that proves a method is fit-for-purpose. Scenarios demanding validation include developing a new HPLC method to quantify a novel impurity, introducing a new sample matrix that may cause interference, or significantly adjusting the parameters of a compendial method [19]. Under regulatory frameworks like the Clinical Laboratory Improvement Amendments (CLIA), validation is required for non-FDA cleared tests, such as laboratory-developed tests (LDTs) or modified FDA-approved tests [15].
Method Verification, however, is applicable when adopting a method that has already been fully validated elsewhere. This includes using unmodified FDA-cleared or approved tests, compendial methods from the USP or Ph. Eur., or methods transferred from a validated marketing authorization dossier [15] [19]. The goal is not to re-establish all performance characteristics, but to provide documented evidence that the method performs reliably in the hands of the user's laboratory, with its specific operators, equipment, and reagents [1]. For example, a water testing lab adopting a standard EPA method for pesticide residues would perform verification to confirm the method works with their local instruments and water matrices [1].
The following workflow diagram illustrates the decision-making process for determining whether a method requires validation or verification.
Quantitative Resource Comparison
The strategic choice between validation and verification has profound implications for a project's budget, timeline, and personnel requirements. The table below summarizes the key performance characteristics assessed and the typical resource investments for each process, based on regulatory guidelines and industry standards.
Table 1: Resource and Scope Comparison: Validation vs. Verification
| Comparison Factor | Method Validation | Method Verification |
|---|---|---|
| Purpose & Scope | Establishes method performance for a new or significantly modified method [19]. | Confirms performance of a pre-validated method in a user's lab [1] [19]. |
| Typical Duration | Weeks to months [1]. | Days to a few weeks [1]. |
| Personnel Effort | High (requires extensive planning, execution, and data analysis) [1]. | Moderate (focused, limited testing) [1]. |
| Financial Cost | High (significant investment in reference standards, reagents, and labor) [1]. | Low to Moderate (economical due to narrower scope) [1]. |
| Key Performance Characteristics Assessed | Accuracy, Precision, Specificity, Limit of Detection (LOD), Limit of Quantitation (LOQ), Linearity, Range, Robustness [1] [19]. | Accuracy, Precision, and sometimes LOD/LOQ confirmed under local conditions [1] [19]. |
| Regulatory Driver | Required for new drug applications, clinical trials, and novel assays [1]. | Acceptable for standard methods in established workflows [1]. |
| Flexibility | Highly adaptable to new matrices, analytes, or workflows [1]. | Limited to the conditions defined by the validated method [1]. |
Detailed Experimental Protocols
To effectively plan and resource these studies, laboratory managers need detailed, actionable protocols. The following section outlines standard methodologies for key experiments in the verification and validation of qualitative microbiological methods, incorporating sample size guidance from CLIA and other regulatory bodies.
Protocol for Verifying Accuracy
Accuracy verification confirms the acceptable agreement of results between the new method and a comparative method [15].
- Objective: To demonstrate that the new test method produces results that correlate with a reference or comparative method.
- Sample Requirements: A minimum of 20 clinically relevant isolates or samples. Use a combination of positive and negative samples for qualitative assays. For semi-quantitative assays, use a range of samples with high to low values [15].
- Sample Sources: Acceptable specimens can be obtained from accredited standards or controls, reference materials, proficiency test samples, or de-identified clinical samples previously tested with a validated method [15].
- Testing Procedure: Test all samples using both the new method and the established comparative method. Ensure testing is performed in parallel or from previously characterized samples to minimize variability.
- Data Analysis: Calculate the percentage agreement. Formula: (Number of results in agreement / Total number of results) × 100 [15].
- Acceptance Criteria: The percentage of accuracy should meet the performance claims stated by the test manufacturer or criteria determined by the laboratory director under CLIA regulations [15].
Protocol for Verifying Precision
Precision verification confirms acceptable method variance within-run, between-run, and between different operators [15].
- Objective: To establish the reproducibility of the test results under normal operating conditions.
- Sample Requirements: A minimum of two positive and two negative samples [15].
- Testing Procedure: Test the selected samples in triplicate, over five different days, and by two different operators. If the test system is fully automated, testing for user variance may not be required [15].
- Data Analysis: Calculate the percentage of agreement for the results across all replicates and conditions. Formula: (Number of results in agreement / Total number of results) × 100 [15].
- Acceptance Criteria: The calculated precision should meet the manufacturer's stated claims or the laboratory director's determined criteria [15].
Protocol for Establishing Reportable and Reference Ranges
These protocols verify the limits of what the test can report and the "normal" expected result for a patient population.
Reportable Range Verification:
- Objective: To confirm the acceptable upper and lower limits of the test system.
- Sample Requirements: Verify using a minimum of three known positive samples. For semi-quantitative assays, include samples near the manufacturer's cutoff values [15].
- Evaluation: The reportable range is defined as what the laboratory establishes as a reportable result (e.g., "Detected," "Not detected," Ct value cutoff), verified by testing samples within that range [15].
Reference Range Verification:
- Objective: To confirm the normal expected result for the tested patient population.
- Sample Requirements: A minimum of 20 isolates. Use de-identified clinical samples or reference samples with a result known to be standard for the laboratory’s patient population (e.g., samples negative for MRSA for an MRSA detection assay) [15].
- Evaluation: The reference range is verified by testing samples representative of the laboratory's patient population. If the manufacturer's range does not represent the local population, the laboratory must screen additional samples and may need to redefine the range [15].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful method validation and verification rely on well-characterized reagents and materials. The following table details key components required for these studies.
Table 2: Essential Materials for Verification and Validation Studies
| Reagent/Material | Function in Validation/Verification | Specific Examples & Applications |
|---|---|---|
| Quality Control (QC) Organisms | To validate testing methodologies, monitor test performance, and verify instrument, operator, and reagent quality [3]. | Well-characterized microorganisms from type culture collections (e.g., ATCC strains) with defined biochemical profiles used as positive/negative controls in pharmaceutical, food, and clinical testing [3]. |
| Reference Materials & Standards | To provide a verified baseline for assessing the accuracy of a new method during comparison studies [15]. | Accredited reference materials, proficiency test samples, or commercially available certified reference materials (CRMs) used to spike samples for recovery studies in accuracy and LOD/LOQ experiments [15] [3]. |
| De-identified Clinical Samples | To assess method performance with real-world sample matrices in a controlled manner [15]. | Previously characterized patient samples used to verify accuracy, precision, and reference ranges in a clinical laboratory setting [15]. |
| Culture Media | To support the growth of QC organisms and clinical isolates during testing; requires validation through growth promotion testing [3]. | Selective and non-selective agars used for the recovery and isolation of microorganisms specified in the test method [3]. |
Navigating Regulatory Frameworks and Standards
Adherence to recognized standards is non-negotiable. The following key documents provide structured frameworks for validation and verification:
- ISO 16140 Series (Microbiology of the food chain - Method validation): This multi-part standard is dedicated to the validation and verification of microbiological methods in food and feed testing. Part 2 covers the validation of alternative methods, while Part 3 provides the protocol for the verification of reference and validated alternative methods in a single laboratory [2].
- CLSI Guidelines: Essential documents for clinical laboratories include:
- EP12-A2: User Protocol for Evaluation of Qualitative Test Performance.
- M52: Verification of Commercial Microbial Identification and AST Systems.
- MM03-A2: Molecular Diagnostic Methods for Infectious Diseases.
- Cumitech 31A: Verification and Validation of Procedures in the Clinical Microbiology Laboratory [15].
- ICH Q2(R2): Provides guidance on the validation of analytical procedures for the pharmaceutical industry [19].
- USP <1225> & <1226>: Define requirements for the validation and verification of compendial procedures, respectively [19].
In the demanding environment of microbiological research and diagnostics, the strategic application of method validation and verification is a cornerstone of effective resource management. Validation is a necessary, resource-intensive investment for pioneering new methods, while verification is a cost-effective, focused confirmation for implementing established techniques. By understanding the distinct requirements of each process, leveraging structured experimental protocols, and utilizing well-characterized reagents, professionals can confidently balance the competing demands of time, cost, and regulatory compliance. This ensures not only the integrity of laboratory data but also the safety of products and patients that rely on these critical analytical results.
In the highly regulated fields of pharmaceutical microbiology and clinical diagnostics, the erroneous substitution of method verification for method validation represents a critical compliance risk that can compromise product quality, patient safety, and data integrity. This persistent confusion stems from a misunderstanding of the fundamental purposes and regulatory triggers for each process. Framed within a broader thesis on microbiological method lifecycle management, this guide provides researchers and drug development professionals with the experimental protocols and decision frameworks necessary to correctly apply these distinct principles.
Definitions and Regulatory Significance
The terms "validation" and "verification" have specific, non-interchangeable definitions in regulatory language.
- Method Validation is a comprehensive process of establishing and documenting that an analytical method is capable of producing results that are accurate, precise, and reliable for its intended purpose [19]. It is required for new methods developed in-house, significantly altered compendial methods, or methods used for new products or formulations [19]. Validation proves that a method is fit-for-purpose from its inception.
- Method Verification, in contrast, is the process of confirming that a previously validated method performs as expected in a specific laboratory setting [1]. It assesses whether a method, already proven to work, is suitable for the actual conditions of use in a particular lab with its specific personnel, equipment, and reagents [25]. The U.S. Food and Drug Administration (FDA) regulation 21 CFR 211.194 states that users of compendial methods must verify their suitability but are not required to re-validate them [25].
The core misapplication occurs when a laboratory attempts to "verify" a method that has never been formally validated, using a limited verification protocol as a shortcut. This creates a fundamental gap in the evidence demonstrating that the method is actually capable of meeting its analytical requirements.
Method Validation: Establishing Foundational Performance
Method validation is a foundational study that provides the initial evidence of a method's robustness. It is typically required in scenarios such as developing a new HPLC method for quantifying a new impurity or adapting a method for a new sample matrix that may cause interference [19].
Experimental Protocol for Method Validation
A full validation requires a multi-parameter experimental approach. The International Council for Harmonisation (ICH) Q2(R1) and USP <1225> provide the key validation parameters and their experimental methodologies [1] [19].
Table 1: Key Performance Characteristics and Experimental Protocols for Method Validation
| Performance Characteristic | Experimental Protocol & Methodology | Objective & Data Analysis |
|---|---|---|
| Accuracy [1] [19] | Analyze a minimum of 3 replicates at 3 different concentration levels (e.g., 80%, 100%, 120% of target) using a reference standard or a spiked placebo. | Establish agreement between the measured value and the true value. Reported as percent recovery of the known amount of analyte. |
| Precision [1] [19] | Repeatability: Analyze at least 6 replicates at 100% of the test concentration.Intermediate Precision: Incorporate variations like different days, different analysts, or different equipment. | Determine the degree of scatter in the results. Expressed as percent relative standard deviation (%RSD). |
| Specificity [1] [19] | Inject and analyze blank, placebo, standard, and sample to demonstrate resolution from potential interferents (e.g., degradants or matrix components). | Demonstrate that the method can unequivocally assess the analyte in the presence of other components. |
| Linearity [1] [19] | Prepare and analyze a minimum of 5 concentrations across a specified range (e.g., 50-150% of the target concentration). | Establish a directly proportional relationship between the response and analyte concentration. Assessed by the correlation coefficient and y-intercept. |
| Range [1] [19] | Defined by the interval between the upper and lower concentration levels for which linearity, accuracy, and precision have been demonstrated. | Confirm the method is suitable for the entire range of intended use. |
| Limit of Detection (LOD) & Quantitation (LOQ) [1] | LOD: Signal-to-Noise ratio of 3:1, or based on standard deviation of the response.LOQ: Signal-to-Noise ratio of 10:1, or based on standard deviation of the response and the slope. | LOD: The lowest amount of analyte that can be detected.LOQ: The lowest amount of analyte that can be quantified with acceptable accuracy and precision. |
| Robustness [1] | Deliberately introduce small, intentional variations in method parameters (e.g., flow rate, temperature, pH). | Measure the method's capacity to remain unaffected by small changes in operational parameters. |
Regulatory and Standardization Frameworks
Validation is mandated by international regulatory bodies for new drug submissions and novel assay development [1]. For microbiological methods, the ISO 16140 series provides a standardized protocol for the validation of alternative (proprietary) methods against a reference method [2].
Method Verification: Confirming Local Suitability
Method verification is not a repetition of the full validation; it is a targeted assessment to confirm that the performance characteristics already established during validation are maintained in the receiving laboratory's environment [19] [25].
Experimental Protocol for Method Verification
The scope of verification is narrower and focuses on critical parameters. USP General Chapter <1226> guides laboratories on how to verify compendial procedures [25].
Table 2: Typical Verification Parameters for a Compendial Method
| Verification Parameter | Typical Experimental Approach | Acceptance Criteria |
|---|---|---|
| Specificity [19] | Demonstrate that the method can resolve the analyte from the specific sample matrix used in the lab. | No interference from the matrix at the retention time of the analyte. |
| Precision (Repeatability) [25] | Perform a minimum of 6 replicate determinations of a homogeneous sample. | The %RSD meets pre-defined criteria based on the method's validated performance. |
| Accuracy [19] [25] | Analyze a sample of known concentration (e.g., a reference standard or spiked sample). | The mean result falls within the acceptance range, typically 98.0-102.0% for an assay. |
The extent of verification is risk-based and depends on factors such as the complexity of the method, the experience of the analysts, and the nature of the article being tested [25]. For a standard compendial method, verification may only require an assessment of precision and specificity, whereas a more complex method transfer might warrant a broader verification [25].
In microbiological testing, the ISO 16140-3 standard outlines a two-stage verification process: implementation verification (demonstrating the lab can perform the method correctly using a known item) and (food) item verification (demonstrating the method works for the specific items within the lab's scope) [2].
A Framework for Correct Application
The decision to validate or verify a method is dictated by the method's origin and history. The following diagram illustrates the logical decision pathway to prevent misapplication.
Consequences of Misapplication
Substituting verification for validation creates significant risks:
- Regulatory Non-Compliance: Failing a regulatory inspection (FDA, EMA) due to insufficient data proving a method is fit-for-purpose [1] [42].
- Compromised Data Integrity: Generating unreliable results that can lead to incorrect decisions in drug development or product release, potentially affecting patient safety [1].
- Product Quality Failures: In pharmaceutical microbiology, an invalidated sterility test or microbial identification method can fail to detect contaminants, leading to product recalls [43].
Essential Research Reagent Solutions for Microbiological Method Assessment
The following reagents and materials are critical for executing the experimental protocols for both validation and verification.
Table 3: Key Research Reagent Solutions for Microbiological Methods
| Reagent / Material | Function in Validation/Verification |
|---|---|
| Reference Microbial Strains [15] [2] | Used as positive controls and for accuracy studies. Essential for challenging the method with known organisms. |
| Certified Reference Materials [15] | Provide a traceable standard for quantifying analytes and establishing method accuracy and linearity. |
| Selective Culture Media & Agars [2] | Used in specificity protocols to demonstrate the method's ability to distinguish the target microorganism from others. |
| Inhibitors & Interfering Substances | Used in robustness and specificity studies to challenge the method's resilience to matrix effects and potential interferents. |
| Standardized Suspension Media | Ensures consistency in microbial inoculum preparation for precision and limit of detection studies. |
The distinction between method validation and verification is a cornerstone of quality assurance in research and drug development. Validation is a creative, comprehensive process to establish performance, while verification is a confirmatory process to transfer proven performance. Adhering to this distinction, supported by robust experimental protocols and a clear decision framework, is non-negotiable for ensuring data integrity, regulatory compliance, and ultimately, the safety and efficacy of pharmaceutical products and diagnostic results.
The implementation of a new microbiological method, whether through a full method validation for novel procedures or a method verification for established ones, marks a critical beginning rather than an end point [15] [19]. In the broader context of microbiological method management, distinguishing between validation and verification is fundamental. Method validation is a comprehensive process establishing that an analytical method is acceptable for its intended use, typically required for new methods, significantly modified compendial methods, or methods used for new products [1] [19]. Conversely, method verification confirms that a previously validated method performs as expected under specific laboratory conditions, applicable when adopting standard methods in a new lab or with different instruments [1] [19]. This distinction, clearly outlined in standards such as the ISO 16140 series for food chain microbiology, defines the initial implementation stage [2].
However, the ultimate goal of ensuring patient safety, product quality, and reliable data extends far beyond this initial phase. Quality Control Integration represents the systematic, ongoing processes that ensure the method continues to perform reliably throughout its operational life. This continuous monitoring and assessment form an essential part of the method's lifecycle, confirming that the performance characteristics established during validation or verification remain stable during routine use [15]. Without robust post-implementation quality control, laboratories risk the gradual degradation of method performance, potentially leading to inaccurate results, compromised decisions, and significant financial impacts from erroneous data [3]. This guide details the protocols and strategies for integrating these essential quality control measures to sustain method reliability.
Core Components of an Integrated QC Strategy
Ongoing Quality Control Monitoring
Routine quality control (QC) monitoring provides the day-to-day assurance that the microbiological method is functioning within established parameters. This involves several key activities:
Quality Control Organisms: The regular use of QC strains is foundational to reliable testing [3]. These are well-characterized microorganisms with defined profiles that serve as verified standards with predictable biochemical reactions. Laboratories should utilize QC organisms relevant to the method's targets, typically sourced from type culture collections or qualified in-house isolates. Their function is to validate testing methodologies, monitor instrument and reagent performance, and serve as positive controls for diagnostic procedures [3]. The frequency of testing should be risk-based, with higher-risk methods requiring more frequent QC testing.
Control Charts: Implementing statistical control charts for quantitative methods enables trend analysis and early detection of method drift. Plotting QC results over time against established control limits allows laboratories to differentiate between random variation and systematic shifts in method performance. Control limits should be derived from initial method performance data established during validation or verification studies.
Response Plans: Establishing predefined corrective action protocols for QC failures is critical. When QC results fall outside acceptance criteria, immediate actions should include stopping patient or product testing, investigating root causes, implementing corrections, and documenting all activities. Preventive actions might include reagent lot testing, equipment maintenance verification, or analyst re-training.
Proficiency Testing (PT) and External Quality Assessment (EQA)
Proficiency Testing provides an external benchmark for method performance by comparing results with other laboratories using the same or different methods [3]. This external verification is crucial for:
- Identifying Methodological Limitations: PT can reveal method-specific issues not apparent through internal QC, particularly with challenging matrices or low analyte concentrations.
- Benchmarking Performance: Comparing results with peer laboratories provides context for internal performance metrics.
- Meeting Accreditation Requirements: Most laboratory accreditation standards, including ISO/IEC 17025, require regular participation in PT programs [1].
- Documentation and Response: All PT results should be formally reviewed, with investigations conducted for unsatisfactory performance and corrective actions implemented and documented.
Continuous Data Review and Trend Analysis
Ongoing reliability requires moving beyond point-in-time assessments to continuous monitoring of method performance trends:
- Data Mining: Regular analysis of patient or sample results can reveal subtle shifts in population characteristics that might indicate method changes.
- Westgard Rules Application: Implementing multi-rule QC procedures helps detect both random and systematic errors.
- Seasonal Variation Monitoring: For certain microbiological targets, recognizing and accounting for seasonal prevalence patterns helps distinguish true method changes from epidemiological variations.
- Reference Range Monitoring: Periodic assessment of reference ranges ensures they remain appropriate for the tested patient population, as recommended by CLIA standards [15].
Experimental Protocols for Ongoing Verification
Protocol for Quarterly Method Performance Assessment
Purpose: To comprehensively evaluate whether the method maintains the performance characteristics established during the initial verification/validation.
Materials:
- QC strains representing method targets (positive and negative controls)
- Clinical or product samples with known status (blinded)
- All standard reagents, media, and equipment as specified in the method
Procedure:
- Sample Preparation: Include a minimum of 20 positive samples and 20 negative samples for qualitative methods [15]. For quantitative methods, include samples across the reportable range.
- Testing Sequence: Perform testing over five separate runs with two different operators [15].
- Control Integration: Include routine QC organisms in each run [3].
- Data Collection: Record all results, including any atypical signals or borderline findings.
Calculation and Acceptance Criteria:
- Calculate accuracy as (Number of correct results/Total number of results) × 100 [15]
- Compare to initial verification/validation performance
- Acceptance criterion: Performance must not deviate significantly from established baselines (p > 0.05 using appropriate statistical tests)
Protocol for Environmental Isolate Challenge Testing
Purpose: To verify that the method continues to detect organisms relevant to the specific testing environment.
Materials:
- Environmental isolates collected from the testing facility (minimum of 5-10 unique strains)
- Reference strains for comparison
- Standard culture media and reagents
Procedure:
- Strain Selection: Select environmental isolates representative of the facility's microflora, particularly those with atypical characteristics.
- Preparation: Prepare suspensions of each isolate to specified concentrations.
- Testing: Test each isolate alongside reference strains using the validated method.
- Comparison: Compare detection capabilities, growth characteristics, and identification results.
Interpretation:
- Environmental isolates should be detected/identified with performance comparable to reference strains
- Significant discrepancies trigger investigation and potential method re-validation
Table 1: Quarterly QC Testing Schedule for Microbiological Methods
| QC Component | Frequency | Acceptance Criteria | Corrective Action |
|---|---|---|---|
| Control Organisms [3] | Each testing session | Expected reactivity pattern | Repeat test; evaluate reagents; check equipment |
| Quantitative Accuracy | Quarterly | Within established control limits | Calibration verification; standard preparation review |
| Precision Evaluation | Quarterly | CV ≤ established threshold | Equipment performance check; technique review |
| Proficiency Testing [3] | As per program schedule | Satisfactory performance | Root cause analysis; corrective action implementation |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagent Solutions for Quality Control Integration
| Reagent/Material | Function in QC Integration | Implementation Considerations |
|---|---|---|
| Quality Control Organisms [3] | Verify test validity; monitor method performance | Select strains relevant to method targets; include fastidious organisms |
| Reference Materials | Calibration; trueness verification | Use certified reference materials when available; document traceability |
| Proficiency Test Samples [3] | External performance assessment | Handle as patient samples; incorporate into routine workflow |
| Environmental Isolates [3] | Challenge method with relevant strains | Maintain collection; characterize fully; monitor stability |
| Culture Media [44] | Support microbial growth | Performance testing; growth promotion testing; qualify each lot |
Visualization of Quality Control Integration Workflow
The following diagram illustrates the continuous cycle of quality control integration in the post-implementation phase:
Quality Control Integration Cycle depicts the continuous process of monitoring, assessment, and action that ensures ongoing method reliability after implementation.
Documentation and Regulatory Compliance
Essential Documentation Practices
Comprehensive documentation provides the evidence trail demonstrating ongoing method control:
- QC Result Records: Maintain detailed records of all QC testing, including organism identities, preparation methods, results, and any deviations.
- Trend Analysis Reports: Document quarterly and annual reviews of method performance with statistical analysis.
- Proficiency Testing Reports: File all PT results with accompanying investigations for unsatisfactory performance.
- Change Control Records: Document any deviations from established procedures, however minor, with impact assessments.
Meeting Regulatory Requirements
Regulatory bodies expect laboratories to monitor method performance continuously, not just at implementation:
- CLIA Requirements: For clinical laboratories, CLIA mandates verification of manufacturer performance specifications before implementing unmodified FDA-approved tests, but ongoing monitoring is equally critical [15].
- ISO/IEC 17025: Requires laboratories to monitor validity of results through quality control activities and participate in interlaboratory comparisons [1].
- Pharmacopeial Standards: USP chapters provide guidance on ongoing verification of compendial methods [19].
In the context of microbiological method management, where the distinction between validation and verification sets the initial implementation approach, Quality Control Integration forms the essential framework for sustaining reliability throughout the method's operational life. This ongoing process, incorporating systematic monitoring, regular performance assessment, and responsive corrective actions, ensures that the investment in proper method establishment continues to yield reliable results. For researchers and drug development professionals, mastering this integration is not merely regulatory compliance—it is fundamental to scientific integrity and public health protection.
Strategic Selection: A Comparative Framework for Method Verification and Validation
In the highly regulated field of drug development, the concepts of method validation and verification are critical pillars of quality assurance. These processes ensure that microbiological methods are both reliable (verified) and fit for their intended purpose (validated). For researchers and scientists, understanding the distinction and knowing when to apply each process is not merely an academic exercise—it is a fundamental requirement for regulatory compliance, data integrity, and ultimately, patient safety. Method validation is the process of proving that a method is fit for its purpose, providing assurance that it will consistently yield accurate and reproducible results for a defined scope [13]. Method verification, in contrast, is the process whereby a laboratory demonstrates that it can successfully execute a method that has already been validated [2] [13]. This guide provides a decisive framework for selecting the appropriate path for your microbiological testing projects.
Core Concepts and Regulatory Framework
What is Method Validation?
Method validation confirms a method's performance characteristics, testing its ability to detect target organisms under a particular range of conditions [13]. It is performed for a specific matrix category and provides objective evidence that the method meets predefined acceptance criteria for parameters such as specificity, accuracy, and precision. In essence, validation answers the question, "Is this method scientifically sound and suitable for its intended use?" [13]. For novel methods or those applied to new sample matrices, validation is a mandatory first step.
What is Method Verification?
Method verification demonstrates that a specific laboratory is capable of performing a previously validated method correctly [2] [13]. It is a demonstration of competency, showing that the laboratory's implementation of the method—with its specific personnel, equipment, and environment—can achieve the performance standards established during the initial validation. Verification answers the question, "Can our laboratory execute this validated method proficiently?" [13].
The International Standard: ISO 16140 Series
The ISO 16140 series, "Microbiology of the food chain - Method validation," provides the definitive international protocol for these processes [2]. This series outlines distinct pathways:
- ISO 16140-2: Governs the validation of alternative (proprietary) methods against a reference method, involving a method comparison study and an interlaboratory study [2].
- ISO 16140-3: Specifies the protocol for the verification of reference methods and validated alternative methods in a single laboratory [2]. The standard explicitly notes that verification is only applicable to methods that have undergone a full interlaboratory validation study [2].
The Decision Matrix: Validation or Verification?
The following matrix provides a clear, actionable pathway for determining whether your project requires method validation or verification.
When Validation is Mandatory
- Developing a new microbiological test method. Any novel assay, whether proprietary or not, must undergo a full validation to establish its baseline performance characteristics [2].
- Applying an existing method to a new category of sample matrix. If a method validated for dairy products is to be used for meat products, a new validation is required, as the matrix can interfere with detection [13].
- There is no pre-existing validated reference method. In this case, the method must be validated within a single laboratory according to protocols like ISO 16140-4 [2].
When Verification is Sufficient
- Implementing a commercially purchased, pre-validated test kit (e.g., from AOAC or ISO) into your laboratory's workflow [13].
- The method is a validated standard (e.g., an ISO or USP method) and your laboratory is using it for the exact matrices and test portion sizes covered in its validation scope [2] [13].
- The core performance characteristics of the method are already established and your goal is to demonstrate laboratory-specific competency.
Experimental Protocols for Validation and Verification
Detailed Protocol for Method Validation (Based on ISO 16140-2)
Method validation is a comprehensive process designed to generate robust evidence of a method's reliability.
Objective: To demonstrate that an alternative (proprietary) microbiological method is equivalent to or better than a reference method for a defined scope [2].
Workflow: The validation process is a two-phase endeavor, as outlined in the following workflow:
Key Experiments & Methodologies:
- Specificity/Selectivity: Challenge the method with a panel of target and non-target strains. For a Listeria test, this includes relevant Listeria species and closely related non-Listeria organisms to confirm accurate detection and absence of cross-reactivity [44].
- Accuracy/Quantitative Bias: For quantitative methods (e.g., bioburden), compare the results of the alternative method against a reference method using artificially contaminated samples across the dynamic range. Recovery rates should typically be ≥80% [44].
- Precision: Repeat the testing multiple times (repeatability) and across different days, analysts, or equipment (intermediate precision) to determine the method's consistency. The coefficient of variation is a key metric here.
- Detection Limit (LOD): Determine the lowest number of microorganisms that can be reliably detected by the method. This is critical for sterility tests or pathogen detection.
- Robustness: Deliberately introduce small, intentional variations in protocol parameters (e.g., incubation temperature ±1°C, media pH) to evaluate the method's resilience [44].
Detailed Protocol for Method Verification (Based on ISO 16140-3)
Verification is a more targeted process focused on proving laboratory proficiency.
Objective: To demonstrate that a user laboratory can satisfactorily perform a validated method [2].
Two-Stage Process:
- Implementation Verification: The laboratory tests one of the exact same food items used in the original validation study. Successful replication of the expected result demonstrates fundamental competency with the method [2].
- Item Verification: The laboratory tests several challenging food items that fall within its specific scope of accreditation but may differ from those in the original validation. This proves the method performs well for the laboratory's unique application [2].
Key Experiments & Methodologies:
- Comparison to Reference Results: For quantitative methods, analyze a certified reference material or a sample with a known value. The laboratory's result should fall within an acceptable range of the reference value.
- Precision Study: Perform replicate testing (e.g., n=6) on a homogeneous sample to establish that the laboratory's repeatability meets the performance standards set during validation.
- LOD Confirmation: Verify that the method can detect the target organism at the validated detection limit by testing low-level inoculated samples.
- Inclusivity/Exclusivity Check: Test a small, representative subset of the inclusivity and exclusivity strains used in the full validation to confirm the method's specificity is maintained in the laboratory's hands.
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below details key reagents and their critical functions in microbiological method assessment.
| Reagent/Material | Function in Validation/Verification | Key Considerations |
|---|---|---|
| Indicator Organisms | Representative strains used to challenge the method's ability to detect and/or quantify targets [44]. | Must include type strains and relevant environmental isolates; fastidious organisms (e.g., mycoplasmas) require special attention [44]. |
| Culture Media | Supports the growth and recovery of microorganisms for detection [44]. | Validation must cover nutrient composition, pH, and ionic strength. Growth promotion testing must show ≥80% recovery of inoculum [44]. |
| Reference Materials | Samples with known properties (e.g., certified microbial count) used as benchmarks [2]. | Critical for verifying quantitative method accuracy and precision in a single laboratory. |
| Inhibitor Neutralizers | Agents that counteract antimicrobial properties in the sample matrix [44]. | Essential for validating methods for testing products with preservatives or inherent antimicrobial activity. |
| Selective Agars | Media used for the confirmation and typing of microorganisms [2]. | The validation scope is tied to the specific agars used; changing agars may require re-validation [2]. |
Comparative Analysis: Validation vs. Verification
This table summarizes the core quantitative and qualitative differences between the two processes, providing a clear, side-by-side comparison.
| Aspect | Method Validation | Method Verification |
|---|---|---|
| Fundamental Question | "Is the method fit-for-purpose?" [13] | "Can our lab perform the validated method?" [13] |
| Primary Goal | Establish method performance characteristics [13]. | Demonstrate laboratory competency [13]. |
| Regulatory Basis | ISO 16140-2, -4, -5 [2]. | ISO 16140-3 [2]. |
| Typical Scope | 5+ food categories/matrices [2]. | Laboratory's specific scope of application. |
| Key Parameters | Specificity, Accuracy, Precision, LOD, Robustness [44]. | Comparison to reference result, Precision, LOD confirmation. |
| Statistical Power | High (involves interlaboratory study) [2]. | Moderate (focused on single-lab performance). |
| Resource Intensity | High (time, cost, samples, labs) [2]. | Moderate (manageable for a single lab). |
| Output | Validation report; method deemed suitable for broad use [2]. | Verification report; lab is qualified to use the method. |
Choosing between validation and verification is a critical, definitive decision in the drug development pipeline. The decision matrix provided offers a straightforward, actionable tool for project leaders. By rigorously applying these principles and adhering to international standards like the ISO 16140 series, research scientists can ensure the integrity of their microbiological data, streamline regulatory approvals, and uphold the highest standards of product quality and patient safety.
In regulated laboratory environments, the choice between method validation and method verification is a critical strategic decision that directly impacts data integrity, regulatory compliance, and operational efficiency. For researchers, scientists, and drug development professionals working in microbiology, understanding the distinction between these two processes is fundamental to ensuring product safety and meeting regulatory obligations. While both processes aim to confirm that analytical methods are suitable for their intended purpose, they apply to different circumstances and require different levels of investment [1].
This technical guide provides an in-depth comparison of method validation versus verification within the context of microbiological testing. The distinction is particularly crucial in microbiology due to the unique challenges posed by biological test systems, including the variability of living organisms, complex matrix effects, and the need for specialized growth conditions [44]. We examine the scope, regulatory requirements, and resource commitments for each approach, providing a structured framework for decision-making in pharmaceutical development, clinical diagnostics, and food safety testing environments.
Definitions and Core Concepts
Method Validation
Method validation is defined as a comprehensive, documented process that establishes, through extensive laboratory studies, that the performance characteristics of a method meet the requirements for its intended analytical applications [45] [46]. It is performed when developing new methods, substantially modifying existing methods, or transferring methods between laboratories or instruments [1]. The United States Pharmacopeia (USP) General Chapter <1225> outlines key validation parameters, including accuracy, precision, specificity, detection limit, quantitation limit, linearity, range, and robustness [45] [46].
In microbiology, validation presents unique challenges compared to chemical analysis due to the biological nature of analytes, which are often variable, difficult to culture, and may exist in complex matrices that affect their recovery and detection [44]. The purpose of a test also influences validation requirements; for instance, a sterility test for a product destined for immunocompromised patients demands greater sensitivity than one for orally administered drugs [44].
Method Verification
Method verification is the process of confirming that a previously validated method performs as expected in a specific laboratory setting [1]. It demonstrates that a laboratory can successfully execute a compendial or previously validated method and correctly detect target organisms using its own analysts, equipment, and environmental conditions [13]. Verification applies when adopting standard methods from recognized sources such as USP, European Pharmacopoeia (EP), AOAC INTERNATIONAL, or ISO [1] [45].
For USP compendial methods, verification is required to determine the "suitability of the test method" under actual conditions of use [46]. This often includes suitability testing, which confirms that the testing method can recover microorganisms in the presence of a specific product or formulation without inhibitory effects [47] [48].
Key Conceptual Differences
The fundamental distinction lies in their objectives: validation creates the evidence that a method works for its intended purpose, while verification provides evidence that a laboratory can competently perform an already-validated method [1] [13]. Validation answers the question "Does this method work scientifically for its stated purpose?" while verification answers "Can our laboratory perform this validated method correctly?" [13].
Scope of Application
Table 1: Scope Comparison for Method Validation vs. Verification
| Aspect | Method Validation | Method Verification |
|---|---|---|
| When Applied | New method development; method transfer between labs; major modifications [1] [45] | Implementing compendial methods; introducing previously validated methods to new lab [1] [13] |
| Method Type | Laboratory-developed methods; non-compendial methods; modern microbial methods [45] [49] | USP, EP, AOAC, ISO standard methods; FDA-cleared tests used without modification [1] [15] |
| Microbiological Examples | New HPLC method for drug quantification; modern rapid microbial methods; novel ELISA development [1] [45] | USP <61> microbial enumeration; USP <62> specified microorganisms; EPA water methods [1] [47] |
| Lifecycle Considerations | Required for significant changes: formulation updates, manufacturing process changes, reagent source changes [45] | Required when introducing products with new formulations; periodic QC checks; supplier changes [47] [48] |
Regulatory Suitability and Requirements
Table 2: Regulatory Landscape for Validation and Verification
| Regulatory Aspect | Method Validation | Method Verification |
|---|---|---|
| Primary Regulatory Drivers | FDA requirements for new drug applications; ICH Q2(R2) guidelines; USP <1225> [1] [45] | CLIA requirements for non-waived systems; ISO/IEC 17025 accreditation; FDA 21 CFR 111 [1] [15] |
| Key Guidance Documents | USP <1225>; ICH Q2(R2); USP <1223> (Alternative Methods); EP 5.1.6 [45] [49] | USP <1226>; CLSI EP12; CLSI M52; FDA 21 CFR 493.1253 [46] [15] |
| Industry-Specific Requirements | Required for pharmaceutical submissions; clinical trial methods; novel assay development [1] [45] | Required for compendial methods in GMP labs; FDA-cleared tests in clinical labs [1] [15] |
| Documentation Expectations | Comprehensive validation protocol and report; statistical analysis; full parameter assessment [1] [45] | Limited verification report; demonstration of competency; suitability testing results [1] [48] |
Resource Investment and Operational Considerations
Table 3: Resource Comparison for Validation vs. Verification
| Resource Factor | Method Validation | Method Verification |
|---|---|---|
| Time Investment | Weeks to months depending on method complexity [1] | Typically days to complete [1] |
| Personnel Requirements | Multidisciplinary team: method development scientists, statisticians, QA [1] | Laboratory analysts with technical competency in the method [13] |
| Financial Impact | Significant investment: reference standards, reagents, instrumentation, training [1] | Moderate costs: primarily analyst time and quality controls [1] |
| Instrumentation & Materials | May require specialized equipment; extensive reference materials; multiple reagent lots [1] [44] | Standard laboratory equipment; quality control materials [13] [15] |
| Ongoing Maintenance | Periodic revalidation for major changes; continuous monitoring [45] | Annual competency assessment; proficiency testing [15] |
Experimental Protocols and Methodologies
Validation Protocol Design
A comprehensive microbiological method validation evaluates multiple performance parameters, which vary based on whether the method is quantitative or qualitative [45] [44]. The following core parameters are typically assessed:
- Accuracy: Determined by comparing test results to a known reference value or by spiking studies with known concentrations of target microorganisms. Recovery of at least 70-80% is generally required, with higher expectations for critical assays [44].
- Precision: Evaluated through repeatability (within-run) and intermediate precision (between-day, between-analyst) studies. Expressed as standard deviation, coefficient of variation, or confidence interval [45].
- Specificity: Demonstrates the method's ability to detect the target analyte in the presence of potentially interfering substances. For microbiological methods, this includes testing against closely related non-target organisms and assessing matrix effects [45] [46].
- Limit of Detection (LOD) and Quantitation (LOQ): For qualitative methods, LOD represents the lowest number of microorganisms detectable. For quantitative methods, LOQ is the lowest level measurable with acceptable precision and accuracy [45] [44].
- Linearity and Range: Established by testing samples across the claimed analytical measurement range. In microbiology, this may be limited by the Poisson distribution at low microbial counts [44].
- Robustness: Evaluates method reliability when operational parameters are deliberately varied (e.g., incubation temperature ±1°C, media pH variations, analyst technique) [45] [44].
Verification Protocol Design
Method verification for microbiological tests follows a more targeted approach, focusing on key performance characteristics:
- Accuracy Assessment: For qualitative microbiological assays, test a minimum of 20 clinically relevant isolates comprising both positive and negative samples. Calculate percentage agreement between the new method and a comparative method [15].
- Precision Evaluation: Test minimum 2 positive and 2 negative samples in triplicate over 5 days with 2 different operators. For fully automated systems, operator variance testing may not be required [15].
- Reportable Range Verification: Confirm the upper and lower limits of detection using at least 3 samples with known results near the manufacturer's established cutoff values [15].
- Reference Range Confirmation: Verify normal expected results for the laboratory's patient population using at least 20 samples representative of typical specimens [15].
Suitability Testing for Compendial Methods
Suitability testing (also called preparatory testing) is a critical component of verification for USP microbiological methods [47] [48]. This process confirms that the product itself does not inhibit microbial growth, which could lead to false negatives. The protocol involves:
- Inoculating the product with a low level (typically <100 CFU) of specified microorganisms
- Demonstrating that the method can recover the inoculated organisms
- Comparing recovery to controls without product
- Implementing neutralizers or modified sample preparation if inhibition occurs [48]
A suitability test "failure" indicates inherent antimicrobial properties in the product, which must be documented with a neutralizing statement per USP <2021> [48].
Decision Framework and Implementation Strategy
Selection Criteria
The decision between validation and verification depends on multiple factors, which can be systematically evaluated using the following framework:
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Essential Materials for Microbiological Method Validation and Verification
| Reagent/Material | Function in Validation/Verification | Key Considerations |
|---|---|---|
| Reference Microorganism Strains | Accuracy, specificity, and LOD/LOQ determination | Use ATCC or recognized culture collection strains; include environmental isolates relevant to manufacturing setting [44] |
| Growth Media | Microbial cultivation and recovery assessment | Validate nutrient composition, pH, ionic strength; test multiple lots; consider fastidious organism requirements [44] |
| Neutralizing Agents | Counteract antimicrobial product properties in suitability testing | Validate neutralizer effectiveness without toxicity; examples include lectihins, polysorbates, histidine [48] |
| Quality Control Organisms | Ongoing method performance monitoring | Maintain traceable stocks; define acceptance criteria for growth characteristics [44] [15] |
| Standardized Reference Materials | Method comparison and trueness assessment | Use certified reference materials when available; document source and characterization data [15] |
Implementation Planning
Successful implementation requires careful planning and resource allocation:
- For Method Validation: Develop a comprehensive validation master plan specifying acceptance criteria before study initiation. Include predefined criteria for each parameter based on regulatory guidance and product requirements. Avoid "specification creep" where acceptance criteria gradually become less rigorous [44].
- For Method Verification: Create a verification plan signed by the laboratory director detailing sample types, number of replicates, operators, and acceptance criteria. Incorporate relevant CLSI guidelines such as EP12 for qualitative tests or M52 for identification systems [15].
- For Modern Microbial Methods: Follow a structured approach including instrument qualification, validation of the alternative technology, and method suitability testing. Utilize available resources such as the Modern Microbial Methods (M3) Collaboration's User Requirements Specification template [49].
The distinction between method validation and verification represents a fundamental concept in quality assurance for microbiological testing. Validation is a comprehensive process establishing that a method is fit-for-purpose, while verification confirms that a laboratory can successfully perform an already-validated method. The choice between these approaches has significant implications for regulatory compliance, resource allocation, and operational workflow.
For drug development professionals and researchers, understanding this distinction enables appropriate strategic planning and resource management. Validation demands substantial investment but is essential for novel methods and regulatory submissions. Verification offers a more efficient pathway for implementing standardized methods while still demonstrating methodological control. In both cases, careful experimental design, documentation, and adherence to regulatory guidelines are essential for generating reliable, defensible microbiological data that ensures product quality and patient safety.
In the regulated pharmaceutical industry, ensuring the reliability of analytical methods is paramount to product quality and patient safety. This whitepaper delineates the critical distinctions between method validation for novel pharmaceutical development and method verification for compendial method adoption. Through detailed technical exploration and case studies, we examine the experimental protocols, regulatory frameworks, and practical applications that define these separate but complementary processes. Framed within broader research on microbiological method verification versus validation differences, this guidance equips researchers, scientists, and drug development professionals with the strategic understanding necessary to implement these approaches effectively, maintain regulatory compliance, and optimize resource allocation throughout the product lifecycle.
The foundation of reliable pharmaceutical testing lies in appropriately establishing the scientific validity of analytical methods. Two distinct processes—method validation and method verification—serve this purpose, each with specific applications in pharmaceutical development and quality control.
Method validation constitutes a comprehensive, documented process that proves an analytical method is suitable for its intended purpose through extensive laboratory studies [1] [46]. It is performed when developing new analytical procedures, significantly modifying existing methods, or transferring methods between laboratories or instruments [1]. Validation demonstrates through rigorous assessment that the method's performance characteristics meet predefined acceptance criteria aligned with its analytical application [50].
Method verification, in contrast, represents a confirmation process undertaken when a laboratory adopts a previously validated compendial method—such as those published in the United States Pharmacopeia (USP), European Pharmacopoeia (EP), or other recognized standards—and must demonstrate that the method performs as expected under actual conditions of use within that specific laboratory [51] [46]. Verification provides evidence that the adopting laboratory can successfully execute the standardized method with its specific personnel, equipment, and reagents [1].
The fundamental distinction is succinctly summarized by regulatory guidance: "compendial methods are verified while in house developed methods are validated" [51]. This paradigm ensures that laboratories avoid redundant validation of already-qualified methods while still confirming local suitability.
Regulatory Framework and Guidelines
Pharmaceutical analytical methods operate within a well-defined regulatory ecosystem with specific requirements governing validation and verification activities. The International Council for Harmonisation (ICH) guideline Q2(R2), "Validation of Analytical Procedures," provides the primary framework for method validation, defining criteria and methodologies for establishing method suitability [52]. This guideline addresses validation requirements for registration applications supporting commercial drug substances and products.
Complementing ICH guidelines, USP general chapters <1225> "Validation of Compendial Procedures" and <1226> "Verification of Compendial Procedures" offer practical implementation guidance [46]. These standards recognize that compendial methods have undergone prior validation; consequently, laboratories need only verify their capability to perform these methods successfully [46].
Regulatory agencies including the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) require that methods supporting product release and stability testing undergo appropriate validation or verification based on method origin [26]. For compendial methods, the FDA stipulates that "the suitability of all testing methods used shall be verified under actual conditions of use" [50].
Table 1: Regulatory Guidelines Governing Method Validation and Verification
| Guideline | Focus Area | Key Requirements | Applicable Scope |
|---|---|---|---|
| ICH Q2(R2) | Method Validation | Defines validation parameters: accuracy, precision, specificity, LOD, LOQ, linearity, range, robustness | New or revised analytical procedures for drug substances and products |
| USP <1225> | Validation of Compendial Procedures | Categorizes analytical methods based on required validation level | Compendial methods during development and prior to publication |
| USP <1226> | Verification of Compendial Procedures | Guidance for demonstrating suitability of compendial methods under actual conditions of use | Laboratories implementing compendial methods |
| FDA Guidance on Analytical Procedures | Validation for Submissions | Details validation package expectations for IND, NDA, and BLA submissions | Methods supporting regulatory applications |
Method Validation in Pharmaceutical Development: A Novel HPLC Method Case Study
Background and Objective
Pharmaceutical development frequently requires creation of novel analytical methods tailored to specific drug formulations. This case study examines the validation of a new High-Performance Liquid Chromatography (HPLC) method developed for quantifying the active pharmaceutical ingredient (API) in a novel drug formulation undergoing regulatory submission [1].
The method required validation to demonstrate suitability for its intended purpose—release testing and stability studies of the commercial product—in accordance with ICH Q2(R2) and USP <1225> requirements [52] [46].
Experimental Protocol and Acceptance Criteria
The validation protocol employed a systematic approach assessing multiple performance parameters with predefined acceptance criteria derived from regulatory guidelines and product specifications.
Table 2: Validation Parameters and Acceptance Criteria for HPLC Method
| Validation Parameter | Experimental Protocol | Acceptance Criteria |
|---|---|---|
| Accuracy | Spiked recovery with placebo: 50%, 100%, 150% of target concentration (n=9) | Mean recovery: 98.0–102.0%; RSD ≤2.0% |
| Precision | Repeatability: Six independent preparations at 100% target concentration | RSD ≤2.0% |
| Intermediate Precision | Different analyst, different day, different instrument (n=12) | Overall RSD ≤3.0%; No significant difference between analysts (p>0.05) |
| Specificity | Forced degradation studies: acid, base, oxidation, thermal, photolytic stress | No interference from degradation products; Peak purity >99.0% |
| Linearity | Five concentrations: 50–150% of target concentration (n=3 each) | Correlation coefficient (r) ≥0.999; y-intercept not significantly different from zero |
| Range | Established from linearity data | 50–150% of target concentration |
| Robustness | Deliberate variations: flow rate (±0.1 mL/min), column temperature (±2°C), mobile phase pH (±0.1) | System suitability criteria met under all conditions; RSD ≤2.0% |
Implementation Workflow
The validation process followed a structured workflow encompassing planning, execution, and documentation phases to ensure regulatory compliance and scientific rigor.
Results and Regulatory Impact
The comprehensive validation provided definitive evidence of method reliability, with all parameters meeting predefined acceptance criteria. The validation package successfully supported regulatory submissions, including an Investigational New Drug (IND) application and subsequent New Drug Application (NDA) [50]. The documented robustness study established system suitability parameters for routine method transfer to quality control laboratories.
Method Verification in Compendial Method Adoption: A Sterility Testing Case Study
Background and Objective
This case study examines the verification of a compendial sterility testing method according to USP <71> adopted by a pharmaceutical quality control laboratory for sterile injectable products [53]. While the method was previously validated and published in the pharmacopeia, verification remained necessary to demonstrate it performed as expected under the laboratory's specific conditions—including environmental factors, personnel technique, and equipment [51].
Experimental Protocol and Acceptance Criteria
Unlike full validation, verification focuses on demonstrating the laboratory's proficiency with the compendial method rather than re-establishing all validation parameters.
Table 3: Verification Parameters for Compendial Sterility Testing Method
| Verification Parameter | Experimental Protocol | Acceptance Criteria |
|---|---|---|
| Accuracy (Method Suitability) | Inoculation with <100 CFU of S. aureus, B. subtilis, P. aeruginosa, C. albicans, A. brasiliensis | Growth promotion comparable to positive controls; No inhibition from product |
| Precision (Repeatability) | Multiple analysts testing identical samples on different days (n=6) | Consistent negative results for product; 100% recovery for positive controls |
| Specificity | Testing with product and product spiked with low levels of microorganisms | No false positives or negatives; Clear distinction between sterile and contaminated samples |
| Robustness | Minor variations in incubation temperature and media lots | All variations support growth of compendial test strains |
Implementation Workflow
The verification process for compendial methods is more streamlined than full validation but requires careful attention to conditions specific to the implementing laboratory.
Results and Implementation Impact
The verification study confirmed that the compendial sterility testing method performed adequately within the laboratory's specific environment. Growth promotion tests demonstrated appropriate media performance, while method suitability testing confirmed the product itself did not inhibit microbial growth [53]. The successful verification enabled the laboratory to implement USP <71> for routine sterility testing with demonstrated capability, meeting regulatory expectations for compendial method adoption [51] [46].
Comparative Analysis: Validation Versus Verification
The distinction between method validation and verification extends beyond regulatory definitions to practical implementation differences across multiple dimensions.
Table 4: Comprehensive Comparison of Method Validation vs. Verification
| Comparison Factor | Method Validation | Method Verification |
|---|---|---|
| Objective | Establish method suitability for intended use | Confirm laboratory capability with compendial method |
| Regulatory Basis | ICH Q2(R2), USP <1225> | USP <1226> |
| Scope of Testing | Comprehensive: All relevant validation parameters | Limited: Key parameters confirming local suitability |
| Resource Requirements | High: Significant time, expertise, and material investment | Moderate: Focused testing with efficient resource utilization |
| Timeline | Weeks to months | Days to weeks |
| Documentation | Extensive validation protocol and report | Streamlined verification report |
| Applicable Methods | Novel methods, significantly modified methods | Compendial methods (USP, EP, etc.) |
| Risk Assessment | Identifies and characterizes method limitations | Confirms acceptable performance under local conditions |
The Scientist's Toolkit: Essential Research Reagent Solutions
Implementing robust method validation and verification requires specialized reagents and materials designed to ensure accuracy, precision, and regulatory compliance.
Table 5: Essential Research Reagent Solutions for Method Validation and Verification
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Well-Characterized Reference Standards | Provide known response for accuracy and linearity determination | API quantification, impurity method validation |
| Certified Microbial Strains | Growth promotion testing and method suitability | Sterility testing verification, microbial enumeration validation |
| Quality Control Organisms | Monitor test method performance and reagent quality | Media quality testing, method transfer verification |
| Matrix-Matched Materials | Evaluate specificity and matrix effects | Biologics method validation, complex formulation analysis |
| Stability-Indicating Materials | Demonstrate method stability-indicating capabilities | Forced degradation studies, specificity assessment |
The pharmaceutical industry's evolving landscape, with increasingly complex molecules and advanced manufacturing technologies, continues to emphasize the critical importance of proper method validation and verification. While these processes serve distinct purposes—validation establishing method suitability and verification confirming local implementation capability—both are essential components of a robust pharmaceutical quality system.
Future developments in analytical science, including the adoption of rapid microbiological methods [53] and advanced spectroscopic techniques, will necessitate ongoing evolution of validation approaches. Nevertheless, the fundamental principle remains unchanged: analytical methods must demonstrate reliability for their intended use through scientifically sound, thoroughly documented processes that ensure product quality and patient safety.
By understanding and implementing the appropriate approach—validation for novel methods and verification for compendial methods—pharmaceutical professionals can navigate regulatory requirements while advancing drug development and manufacturing excellence.
In the rigorous world of pharmaceutical development and microbiological analysis, the reliability of analytical methods is paramount. Two cornerstone processes—method validation and method verification—serve as the foundation for ensuring data integrity and regulatory compliance. Method validation is a comprehensive process that proves an analytical method is acceptable for its intended use, typically required during the development of new methods [1]. Conversely, method verification is the process of confirming that a previously validated method performs as expected under specific laboratory conditions [1]. Within the context of microbiological method selection, a fundamental tension exists between the comprehensiveness of validation and the efficiency of verification. This whitepaper provides an in-depth technical analysis of this critical trade-off, equipping researchers, scientists, and drug development professionals with the knowledge to make strategically sound decisions that align with both scientific rigor and project constraints.
Core Conceptual Frameworks
Defining Method Validation
Method validation is a documented process that proves an analytical method is acceptable for its intended use. It is a comprehensive exercise involving rigorous testing and statistical evaluation, typically required when developing new methods or transferring methods between labs or instruments [1]. According to USP general information chapter <1225>, "VALIDATION OF COMPENDIAL PROCEDURES", method validation is an evaluation process on the performance characteristics of an established analytical procedure through laboratory studies to ensure all performance characteristics meet the intended analytical applications [54]. The process is designed to provide exhaustive evidence that a method is scientifically sound and reproducible across its entire operational range.
Defining Method Verification
Method verification is the process of confirming that a previously validated method performs as expected in a specific laboratory setting [1]. As defined in USP general chapter <1226>, "Verification of Compendial Procedures", verification focuses on how the analytical test procedure is suitable for its intended use under actual experimental conditions, including specific drug substances/products, environment, personnel, equipment, and reagents [54]. It is typically employed when adopting standard methods (e.g., compendial or published methods) in a new lab or with different instruments [1]. Verification builds upon existing validation data rather than generating entirely new evidence, making it a more targeted assessment of method performance under local conditions.
Table 1: Fundamental Definitions and Applications
| Aspect | Method Validation | Method Verification |
|---|---|---|
| Primary Objective | Prove method fitness for intended use [1] | Confirm performance in a specific lab [1] |
| Regulatory Basis | ICH Q2(R1), USP <1225> [1] [54] | USP <1226> [54] |
| Typical Scenario | New method development [1] | Implementing compendial method [54] |
| Technology Transfer Role | Establishes method for transfer [1] | Confirms transfer success at receiving unit [42] |
The Comprehensiveness-Efficiency Spectrum
The relationship between method validation and verification represents a classic trade-off between comprehensiveness and efficiency. Validation offers comprehensiveness through its exhaustive assessment of all possible performance characteristics, while verification provides efficiency by leveraging existing validation data to execute a focused assessment. This spectrum dictates strategic decision-making in laboratory operations. The comprehensive nature of validation delivers higher confidence in data quality and universal applicability but consumes significant time and resources [1]. Verification, while more efficient and faster to implement, offers a narrower scope of assessment and is entirely dependent on the quality of prior validation work [1]. A proper understanding of this spectrum enables laboratories to optimize resource allocation without compromising data integrity.
Comparative Analysis: Advantages and Limitations
Advantages of Method Validation (Comprehensiveness)
Method validation provides several significant advantages rooted in its comprehensive nature:
High Confidence in Data Quality: By systematically assessing parameters like precision, accuracy, and specificity, validation ensures the data produced is scientifically robust and reproducible [1]. This comprehensive assessment leaves minimal uncertainty about method performance.
Universal Applicability: Once validated, a method can be confidently used across various instruments, analysts, and locations, enhancing consistency and scalability [1]. The comprehensive data set supports method transfer and implementation across global laboratory networks.
Comprehensive Risk Mitigation: Through extensive evaluation, validation uncovers methodological weaknesses early in the development process, reducing the risk of costly errors in regulated workflows [1]. This proactive approach prevents future analytical failures.
Regulatory Compliance for Submissions: Validation is essential for new drug applications, clinical trials, and novel assay development [1]. Regulatory bodies like the FDA and EMA require complete validation data for submissions, making it non-negotiable for new product development.
Limitations of Method Validation (Comprehensiveness)
Despite its robust nature, method validation presents several limitations:
Resource Intensity: Validation requires significant investment in training, instrumentation, reference standards, and statistical analysis tools [1]. The comprehensive nature demands substantial financial and personnel resources that may strain laboratory budgets.
Time-Consuming Process: Designing, executing, and documenting a full validation protocol can extend project timelines significantly, especially in complex analytical methods [1]. The process may take weeks or months depending on method complexity.
Potential Over-Engineering: For standardized, well-established tests, full validation may exceed the necessary scope for quality assurance [1]. The comprehensive approach can introduce unnecessary financial and operational burden in routine applications.
Advantages of Method Verification (Efficiency)
Method verification offers distinct advantages based on its efficient approach:
Time and Cost Efficiency: Because it involves a narrower set of performance characteristics, method verification is faster to execute and more economical [1]. This efficiency is particularly valuable in fast-paced or budget-conscious laboratories.
Rapid Deployment: Verification can typically be completed in days, enabling rapid method implementation [1]. This speed supports quicker response to changing analytical needs and faster turnaround for critical projects.
Ideal for Compendial Methods: When applying established methods from regulatory compendia like USP, EP, or AOAC, verification ensures compliance without the need for full revalidation [1]. This efficient approach leverages existing validated methods.
Focus on Real-World Conditions: Verification confirms that the method works effectively under the lab's actual operational environment, instruments, and sample matrices [1]. This practical focus ensures relevance to specific laboratory conditions.
Limitations of Method Verification (Efficiency)
The efficiency of verification comes with inherent limitations:
Dependency on Prior Validation: Verification is only applicable when the method has already undergone complete validation, limiting its use to existing, pre-approved methodologies [1]. The quality of verification is entirely dependent on the quality of the original validation.
Limited Scope Assessment: Since verification does not assess every parameter, it might overlook subtle weaknesses in the method that could impact data integrity in the long run [1]. The efficient approach may miss method vulnerabilities.
Regulatory Constraints: Certain regulatory contexts or novel applications might still demand full validation, especially when dealing with new analytes or critical patient data [1]. Verification cannot always substitute for validation in regulated environments.
Potential for Misapplication: Inappropriately substituting verification for validation could result in non-compliance, erroneous results, or failed audits, particularly in highly regulated industries [1]. The efficient approach carries implementation risks if applied incorrectly.
Table 2: Comprehensive Comparison of Performance Characteristics
| Performance Characteristic | Method Validation Approach | Method Verification Approach |
|---|---|---|
| Accuracy | Full assessment comparing results to true value [54] | Limited confirmation using known samples [54] |
| Precision | Complete evaluation of repeatability and reproducibility [54] | Verification of repeatability only [54] |
| Specificity | Comprehensive assessment of interference effects [54] | Limited confirmation with expected matrix [42] |
| Linearity & Range | Full characterization across specified range [54] | Spot-checking at critical levels [42] |
| Detection/Quantitation Limits | Established through rigorous statistical methods [54] | Confirmed using pre-established limits [42] |
| Robustness | Deliberate variation of method parameters [54] | Typically not assessed [42] |
| Implementation Timeline | Weeks to months [1] | Days to weeks [1] |
Experimental Protocols and Methodologies
Comprehensive Validation Protocol (ICH Q2/R1)
The validation protocol for a new microbiological assay requires meticulous planning and execution. The following detailed methodology outlines the key experiments for a quantitative procedure:
Accuracy Assessment: Prepare a minimum of 9 determinations across 3 concentration levels covering the specified range (e.g., 80%, 100%, 120% of target). For drug substance analysis, compare results against a reference standard; for drug product, use placebo spiking with known analyte quantities. Calculate percent recovery or difference between found and accepted true values. Document all sample preparations, including matrix composition and spike volumes [54].
Precision Evaluation:
- Repeatability: Perform 9 determinations at 100% concentration or 6 determinations at 3 concentrations. Prepare independent sample preparations from homogeneous sample batch. Calculate relative standard deviation (RSD), which should not exceed predefined criteria based on method stage and analyte [54].
- Intermediate Precision: Incorporate variations representing within-laborature differences (different days, analysts, equipment). Use experimental design approaches to efficiently evaluate multiple variables. Perform statistical analysis (F-test) to compare data sets [54].
Specificity Demonstration: For identity tests, analyze samples containing analytes of interest versus those without. For assay and impurity tests, demonstrate separation from closely related compounds, matrix components, or degradation products. Use chromatographic or spectroscopic techniques to show resolution and selectivity. For forced degradation studies, subject samples to stress conditions (acid, base, oxidation, thermal, photolytic) [54].
Linearity and Range Establishment: Prepare analyte in minimum of 5 concentrations from below to above expected range. Use linear least-squares regression to analyze data. Report correlation coefficient, y-intercept, slope, and residual sum of squares. The range is established as the interval between upper and lower concentration levels where linearity, accuracy, and precision are acceptable [54].
Robustness Testing: Deliberately vary method parameters (pH, mobile phase composition, column temperature, flow rate) using experimental design (e.g., Plackett-Burman). Evaluate effects on system suitability criteria. Establish system suitability tests to ensure method validity during routine use [54].
Efficient Verification Protocol (USP <1226>)
The verification protocol for a compendial microbiological method focuses on demonstrating suitability under actual conditions of use:
Verification of Precision and Accuracy: Conduct a minimum of 6 determinations at 100% concentration using qualified reference standards. For microbiological assays, this may include parallel line analysis or diffusion assays. Compare results against acceptance criteria derived from validation data. Use statistical tests (t-test) to show no significant difference from accepted value [54].
Specificity Confirmation: Test the method with the specific product formulation to demonstrate absence of interference from excipients or related substances. For microbiological assays, demonstrate that the recovery of microorganisms is not adversely affected by the product matrix [42].
Determination of System Suitability: Establish that the verification system meets all compendial requirements before and during analysis. For chromatographic methods, this includes resolution, tailing factor, and theoretical plates. For microbiological methods, include positive and negative controls, reference standard responses, and zone diameter or turbidity measurements [42].
Diagram 1: Method Selection: Verification vs Validation
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of method validation and verification studies requires carefully selected, high-quality materials. The following table details essential research reagent solutions and their specific functions in microbiological and analytical methods.
Table 3: Essential Research Reagent Solutions for Method Validation and Verification
| Reagent/Material | Function in Validation/Verification | Key Specifications |
|---|---|---|
| Reference Standards | Establish accuracy and method calibration; serve as known value comparator [54] | Certified purity, stability, proper storage conditions |
| Culture Media | Support microbial growth in microbiological assays; medium composition affects results [42] | Growth promotion testing, sterility, pH, batch-to-batch consistency |
| Qualified Microorganisms | Challenge method specificity and accuracy in microbiological assays [42] | Certified strains, known characteristics, proper viability |
| Matrix Placebos | Evaluate specificity and detect interference in product testing [54] | Composition matching final product without active ingredient |
| System Suitability Standards | Verify system performance before and during analysis [54] | Resolution mixtures, tailing factor standards, reference cultures |
Strategic Implementation and Decision Framework
Decision Matrix for Method Selection
Choosing between validation and verification requires systematic evaluation of multiple factors. The following decision framework supports consistent, defensible choices:
Regulatory Requirements: Method validation is mandatory for new drug applications, clinical trials, and novel assay development [1]. Verification is acceptable for standard methods in established workflows but cannot replace validation in regulatory submissions [1].
Method Provenance: Laboratory-developed methods or significantly modified existing methods require full validation [54]. Compendial methods (USP, EP) or previously validated methods transferred between sites typically require verification [54].
Resource Constraints: When time and budget are limited, and a validated method exists, verification offers a scientifically sound, efficient alternative [1]. For critical quality attributes or novel mechanisms, comprehensive validation remains necessary despite resource demands [1].
Risk Management Considerations: Higher risk applications (sterility testing, endotoxin detection, novel therapies) warrant comprehensive validation regardless of method provenance [42]. Lower risk applications (raw material identification, in-process checks) may be suitable for verification approaches [42].
Diagram 2: Strategic Decision Framework
Hybrid Approaches and Practical Considerations
Strategic laboratories often implement hybrid approaches that balance comprehensiveness and efficiency:
Tiered Validation Approach: Conduct full validation for critical methods while implementing verification for compendial methods. This risk-based approach optimizes resource allocation while maintaining data integrity [42].
Leveraging Prior Knowledge: Utilize existing validation data from method development or technology transfer to design focused verification protocols. This approach maximizes efficiency while maintaining scientific rigor [54].
Lifecycle Management: Implement ongoing performance monitoring to determine when verification suffices versus when revalidation becomes necessary. Changes in instrumentation, sample matrix, or regulatory requirements may trigger revalidation [55].
The choice between method validation and verification represents a critical decision point in pharmaceutical and microbiological analysis, balancing the competing demands of comprehensiveness and efficiency. Validation offers exhaustive assessment and regulatory robustness but requires substantial resources, while verification provides practical efficiency with narrower scope. Through the strategic frameworks, experimental protocols, and decision matrices presented in this whitepaper, researchers and drug development professionals can make informed choices that align with their specific regulatory, scientific, and operational requirements. The optimal approach often involves a hybrid strategy that applies validation where innovation and regulatory submission demand comprehensiveness, while employing verification where established methods can be efficiently implemented. This balanced perspective enables laboratories to maintain both scientific excellence and operational effectiveness in an increasingly complex regulatory landscape.
Conclusion
Understanding the distinction between microbiological method verification and validation is not merely a regulatory formality but a fundamental component of scientific rigor and product quality in drug development and clinical research. A strategic approach, where validation establishes the foundational fitness of a new method and verification confirms its performance in a specific laboratory setting, ensures both innovation and reliability. As microbiological technologies continue to evolve, embracing standardized frameworks from ISO 16140, CLSI, and global pharmacopoeias will be crucial. Future directions will involve adapting these principles to advanced molecular techniques, AI-driven diagnostics, and personalized medicine, further solidifying the role of robust method establishment in safeguarding public health and accelerating biomedical discovery.
References
- 1. Method Validation vs Method Verification: Which is Better ... [https://www.labmanager.com/method-validation-vs-method-verification-which-is-better-for-use-in-laboratories-33982]
- 2. Method validation and method verification - Introduction - ISO [https://committee.iso.org/sites/tc34sc9/home/essential-information/content-left-area/validation-of-methods/method-validation-and-method-ver.html]
- 3. Mastering Quality: New Method Validation, QC Standards, PT [https://www.rapidmicrobiology.com/news/mastering-quality-new-method-validation-qc-standards-and-pt-a-rapidmicrobiology-special-focus]
- 4. AMM/RMM – Pharmacopoeial Developments and More [https://www.rapidmicrobiology.com/news/amm-rmm-pharmacopoeial-developments-and-more]
- 5. Review and comparison of quality standards, guidelines and regulations for laboratories - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5644525/]
- 6. CAP 15189 Accreditation Program… | College of American Pathologists [https://www.cap.org/laboratory-improvement/accreditation/cap-15189-accreditation-program/cap-15189-accreditation-program-frequently-asked-questions]
- 7. CMS's CLIA Rule Now Fully in Effect [https://www.ascp.org/news/news-details/2025/01/09/cms-s-clia-rule-now-fully-in-effect?srsltid=AfmBOooYdw2omamO_DeZPxuORfKdv38koMkcRKeks6vKSM8BwhBMl-SC]
- 8. ISO - ISO/IEC 17025 — Testing and calibration laboratories [https://www.iso.org/ISO-IEC-17025-testing-and-calibration-laboratories.html]
- 9. ISO/IEC 17025:2017 [https://www.iso.org/standard/66912.html]
- 10. ICH Q1 guideline on stability testing of drug substances and ... [https://www.ema.europa.eu/en/ich-q1-guideline-stability-testing-drug-substances-drug-products]
- 11. Microbiological quality control testing [https://www.usp.org/microbiology]
- 12. ISO 17025 Compliance Guide 2026: Requirements, ... [https://www.scispot.com/blog/iso-17025-compliance-guide-requirements-software-best-practices]
- 13. Food Safety Testing: Understanding Microbiological ... [https://www.eurofinsus.com/food-testing/resources/food-safety-testing-understanding-microbiological-method-validation-verification-and-fitness-for-purpose/]
- 14. Validation vs. Verification: What's the Difference? [https://www.idexxcurrents.com/en/latest/validation-vs-verification-what-s-the-difference/]
- 15. Planning a Method Verification Study in Clinical ... [https://asm.org/articles/2022/january/planning-a-method-verification-study-in-clinical-m]
- 16. How to Verify and Validate Requirements [https://specinnovations.com/blog/how-to-verify-and-validate-requirements]
- 17. Verification vs Validation in Software: Overview & Key ... [https://www.bplogix.com/blog/verification-vs-validation-in-software]
- 18. Verification Vs Validation [https://www.geeksforgeeks.org/software-engineering/differences-between-verification-and-validation/]
- 19. Difference Between Validation And Verification Of ... [https://gmpinsiders.com/method-validation-vs-verification/]
- 20. Analytical Method Validation, Verification and Transfer Right [https://www.complianceonline.com/resources/getting-the-analytical-method-validation-verification-and-transfer-right.html]
- 21. Points to Consider in Quality Control Method Validation ... [https://www.bioprocessintl.com/qa-qc/points-to-consider-in-quality-control-method-validation-and-transfer]
- 22. Explanation of sterility testing of drugs and its method suitabitity test . [https://mpl.loesungsfabrik.de/en/english-blog/method-validation/sterility]
- 23. | CPT Labs Method Transfer [https://cptclabs.com/method-transfer/]
- 24. Analytical method and processes for bacterial endotoxin... transfer [https://www.europeanpharmaceuticalreview.com/article/103929/analytical-method-transfer-and-the-supplier-change-processes-for-bacterial-endotoxin-testing/]
- 25. Method Verification Versus Method Validation [https://www.linkedin.com/pulse/method-verification-versus-validation-pharcell-9xlzf]
- 26. Analytical Methodology: Validation, Verification, or ... [https://vicihealthsciences.com/analytical-methodology-validation-verification-qualification/]
- 27. The Comparison of Methods Experiment [https://www.westgard.com/lessons/basic-method-validation/lesson23.html]
- 28. Quantitative Method Comparison Guide | PDF [https://www.scribd.com/document/735097424/VAL-2002-Quantitative-Accuracy-Pack]
- 29. Planning a Method Verification Study in Clinical ... [https://asm.org/articles/2022/january/planning-a-method-verification-study-in-clinical-m?sr_id=23df675c-421d-4816-a54e-4d751ba62574&sr_pos=3]
- 30. AOAC & MicroVal Method Validation Training [https://microval.org/2025/09/02/aoac-microval-method-validation-training-microbial-method-validation-detailed-insights/]
- 31. How to verify and validate a clinical microbiology test ... [https://www.sciencedirect.com/science/article/pii/S1198743X24003094]
- 32. Difference Between Qualitative and Quantitative Test Results [https://labwale.co/blog/30-differences-between-qualitative-and-quantitative-test-results/?srsltid=AfmBOoq-J1l21WigO5-dCbTVmjpx7UnN4hyr_if2vPzS23BQfzTAeWgJ]
- 33. Quantitative and Qualitative Microbiological Testing [https://www.eurofinsus.com/food-testing/resources/small-world-it-is-quality-and-quantity-that-matter/]
- 34. 10 Critical Validation Parameters For Microbiological ... [https://www.outsourcedpharma.com/doc/critical-validation-parameters-for-microbiological-experiments-0001]
- 35. Sample size determination: A practical guide for health ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10000262/]
- 36. Sample size, power and effect size revisited: simplified and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7745163/]
- 37. Recent Methods for the Viability Assessment of Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9500772/]
- 38. Schrödinger's microbes: Tools for distinguishing the living ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-017-0285-3]
- 39. A high-throughput and low-waste viability assay for microbes [https://www.nature.com/articles/s41564-023-01513-9?error=cookies_not_supported&code=96544415-f4a4-4dfb-8a9e-c7880af922c9]
- 40. Nanopore- and AI-empowered microbial viability inference [https://pmc.ncbi.nlm.nih.gov/articles/PMC12405693/]
- 41. sciencedirect.com/topics/immunology-and-microbiology/ microbial ... [https://www.sciencedirect.com/topics/immunology-and-microbiology/microbial-viability]
- 42. Difference Between Method Verification Vs Validation in ... [https://altabrisagroup.com/method-verification-vs-validation-in-gmp-labs/]
- 43. All you need to know about equipment validation for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12421869/]
- 44. Validation of Microbiological Tests [https://www.biopharminternational.com/view/validation-microbiological-tests]
- 45. Method Validation/Verification: How Important is it in Your ... [https://www.americanpharmaceuticalreview.com/Featured-Articles/597641-Method-Validation-Verification-How-Important-is-it-in-Your-QC-Microbiology-Laboratory/]
- 46. Method Validation Vs. Verification - A Simple Breakdown [https://cptclabs.com/method-validation-vs-verification/]
- 47. What Is Suitability Testing and Why It Matters [https://infinitylaboratories.com/what-is-suitability-testing-and-why-it-matters-a-guide-from-eurofins-north-brunswick/]
- 48. USP Testing | Frequently Asked Questions [https://www.eurofinsus.com/food-testing/resources/usp-microbiology-services-frequently-asked-questions/]
- 49. Facilitating Adoption of Modern Microbial Methods [https://www.pda.org/pda-letter-portal/home/full-article/facilitating-adoption-of-modern-microbial-methods]
- 50. Method Validation Guidelines [https://www.biopharminternational.com/view/method-validation-guidelines]
- 51. What's the difference between method verification and method ... [https://mpl.loesungsfabrik.de/en/english-blog/method-validation/difference-validation-verification]
- 52. ICH Q2(R2) Validation of analytical procedures [https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline]
- 53. Challenges of growth-based microbiological methods in ... [https://link.springer.com/article/10.1007/s44395-025-00020-6]
- 54. What Is The Difference Between Verification And Validation of ... [https://boracdmo.com/what-is-the-difference-between-verification-and-validation-of-analytical-methods-in-the-pharmaceutical-industry/]
- 55. Method Validation, Verification, and (now) Confirmation [https://www.westgard.com/essays/method-validation/validate-verify-confirm.html]