Genomic SELEX: Unraveling Transcription Factor Binding Sites for Precision Drug Discovery
This article provides a comprehensive overview of genomic SELEX (Systematic Evolution of Ligands by Exponential Enrichment), a powerful high-throughput method for identifying transcription factor (TF) binding specificities.
Genomic SELEX: Unraveling Transcription Factor Binding Sites for Precision Drug Discovery
Abstract
This article provides a comprehensive overview of genomic SELEX (Systematic Evolution of Ligands by Exponential Enrichment), a powerful high-throughput method for identifying transcription factor (TF) binding specificities. Aimed at researchers and drug development professionals, it explores the foundational principles of SELEX technology, detailing innovative methodological adaptations like HT-SELEX and Capillary Electrophoresis SELEX. The content further addresses critical troubleshooting and optimization strategies to enhance motif discovery, and examines rigorous validation frameworks and comparative analyses against other platforms such as ChIP-seq. By synthesizing recent benchmarking studies and emerging computational models, this guide serves as a vital resource for advancing target-based therapeutic development and understanding gene regulatory networks.
The Foundation of Genomic SELEX: Principles and Technological Evolution
Systematic Evolution of Ligands by EXponential enrichment (SELEX) is a combinatorial chemistry technique in molecular biology designed to produce single-stranded DNA or RNA oligonucleotides, known as aptamers, that specifically bind to a target ligand [1]. First introduced in 1990, the SELEX process enables the in vitro selection of nucleic acid molecules from vast random libraries through an iterative process of binding, selection, and amplification [2] [1]. Unlike biological systems that rely on cellular environments for antibody production, SELEX represents a fully in vitro evolution process that mimics natural selection at the molecular level, yielding affinity reagents with several advantages over traditional antibodies, including lower production costs, longer shelf-life, and reduced batch-to-batch variability [3].
The core principle of SELEX involves starting with an immensely diverse library of oligonucleotides, typically containing up to 10¹ⵠunique sequences, and through repeated rounds of selection pressure, enriching for progressively higher-affinity binders to a target of interest [1]. While the estimated success rate of traditional SELEX is below 30%, this can be significantly improved through specialized techniques, optimized libraries, and quality control procedures [3]. As the technology has matured, SELEX has expanded beyond basic molecular targets to include complex entities such as whole cells, subcellular structures, and has been adapted for high-throughput applications including transcription factor binding site identification [4] [5] [6].
Core Principles and Mechanisms
Fundamental SELEX Workflow
The SELEX process follows a systematic, iterative workflow that enables the evolution of nucleic acid sequences toward increasingly specific target binding. Each round of selection consists of several critical steps that collectively drive the enrichment process.
Molecular Interaction Mechanisms
The binding interactions between aptamers and their targets form the biochemical foundation of SELEX efficacy. These interactions are governed by several key mechanisms that vary depending on the relative size and properties of both the aptamer and target.
When aptamers are larger than their target, they typically integrate the target into their structure through stacking interactions (particularly with flat, aromatic ligands and ions), electrostatic complementarity (with oligosaccharides and charged amino acids), and hydrogen bond formation [3]. This mechanism is commonly observed with small molecule targets. Conversely, when the target is a large protein, the situation is generally reversed, with the aptamer being integrated into the target's structure or attaching to its surface [3].
The structural complexity of proteins enables more varied interaction mechanisms compared to small molecules, often involving combinations of hydrogen bonds, polar interactions, and structural complementarity [3]. Naturally occurring RNA- or DNA-binding motifs frequently exhibit such structural complementarity, including leucine zippers, homeodomains, helix motifs, and beta-sheet motifs [3]. The binding event itself can involve conformational changes in either the target, the aptamer, or both according to the "induced fit" principle, resulting in improved shape complementarity that facilitates short-range interactions including hydrogen bonds and van der Waals contacts [3].
The physicochemical properties of both target and aptamer significantly influence binding efficacy. Negative charges on a target molecule's surface can weaken or prevent aptamer binding due to unfavorable interactions with the electronegative phosphate groups in DNA and RNA backbones [3]. Conversely, positive charges can enhance interaction strength but may also increase nonspecific binding [3]. Largely hydrophobic molecules present particular challenges as aptamers are generally hydrophilic, though this limitation can be addressed through incorporation of modified nucleotides [3].
Key SELEX Variants and Their Applications
As SELEX technology has evolved, numerous specialized variants have been developed to address specific research needs and target types. The table below summarizes the principal SELEX variants and their characteristics.
Table 1: Key SELEX Variants and Their Applications
| SELEX Variant | Target Type | Selection Methodology | Primary Applications | Key Advantages |
|---|---|---|---|---|
| Genomic SELEX [4] | Genomic DNA fragments | Selection using fragmented genomic DNA instead of random oligonucleotides | Transcription factor binding site identification, regulatory network mapping | Identifies biologically relevant binding sites within native genomic context |
| Cell-SELEX [2] [5] | Whole living cells | Iterative selection using intact cells as targets | Cancer cell identification, drug delivery, in vivo diagnostics | Preserves native conformation of cell surface targets; no prior knowledge of membrane proteins required |
| High-Throughput SELEX (HT-SELEX) [6] | Proteins, small molecules | Combines SELEX with next-generation sequencing | Comprehensive binding specificity profiling, transcription factor specificity determination | Enables parallel analysis of multiple targets; provides quantitative binding data |
| Filter-Based SELEX [3] [1] | Proteins, large molecules | Target immobilization on nitrocellulose filters | Protein-aptamer interaction studies | Simple, affordable methodology |
| Bead-Based SELEX [3] | Proteins, small molecules, cells | Target coupling to magnetic or chromatographic beads | Small molecule aptamer selection, clinical diagnostics | Versatile target options; easy separation using magnets or centrifugation |
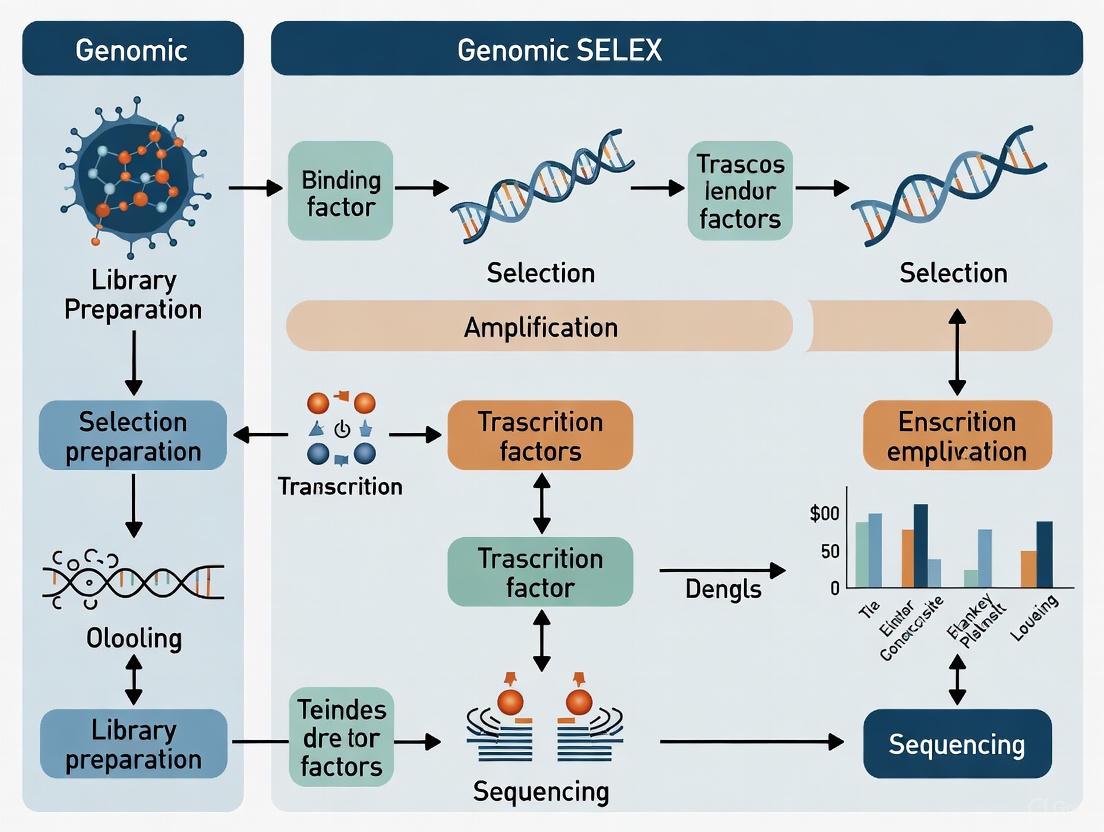
Genomic SELEX for Transcription Factor Binding Site Identification
Genomic SELEX represents a powerful adaptation of the traditional method that replaces synthetic random oligonucleotide libraries with fragments of actual genomic DNA [4]. This approach is particularly valuable for identifying transcription factor binding sites and mapping gene regulatory networks. In vertebrate systems, genomic SELEX has successfully identified transcription factor targets by isolating genomic fragments bound by specific DNA-binding proteins like Fezf2, a conserved zinc finger protein critical for forebrain development [4].
The fundamental advantage of genomic SELEX lies in its ability to identify binding sites within their native genomic context, revealing both known and unexpected regulatory elements. For instance, applications in zebrafish demonstrated that approximately 20% of Fezf2-bound fragments overlapped with well-annotated protein-coding exons, suggesting additional regulatory functions [4]. This approach circumvents limitations of chromatin immunoprecipitation (ChIP)-based methods, which require ChIP-quality antibodies and abundant factor expression in relevant cell types [4].
The genomic SELEX protocol typically involves digesting genomic DNA with restriction enzymes (e.g., Sau3A1), incubating fragments with the transcription factor of interest, and performing multiple rounds of selection and amplification [4]. Computational analysis of bound sequences identifies enriched motifs and consensus binding sites, which can be validated through biochemical assays, reporter constructs, and in vivo models [4].
Experimental Protocols
Standard Genomic SELEX Protocol
This protocol details the application of genomic SELEX for identification of transcription factor binding sites, adapted from established methodologies [4].
Reagent Preparation
- Transcription Factor: Purified DNA-binding domain of transcription factor (e.g., GST-tagged zinc finger domain)
- Genomic DNA: High-molecular-weight genomic DNA (20 μg per selection round)
- Restriction Enzyme: Sau3A1 or appropriate restriction enzyme for genomic fragmentation
- Binding Buffer: 10 mM HEPES (pH 7.9), 50 mM KCl, 1 mM DTT, 5% glycerol, 0.05% NP-40
- PCR Components: Taq polymerase, dNTPs, adapter primers
- Solid Support: Glutathione sepharose beads for GST-tagged proteins
Step-by-Step Procedure
Genomic DNA Fragmentation
- Digest 20 μg genomic DNA with Sau3A1 (or appropriate restriction enzyme) for 2 hours at 37°C
- Verify fragmentation by agarose gel electrophoresis (target range: 100-500 bp)
- Purify fragments using silica membrane columns
Transcription Factor Immobilization
- Incubate purified GST-tagged transcription factor domain with glutathione sepharose beads for 1 hour at 4°C
- Wash beads three times with binding buffer to remove unbound protein
- Quantify bound protein using Bradford assay
First Selection Round
- Incubate immobilized transcription factor with fragmented genomic DNA in binding buffer for 30 minutes at room temperature
- Wash protein-DNA complexes five times with binding buffer to remove non-specifically bound DNA
- Elute specifically bound DNA fragments with elution buffer (10 mM glutathione, 50 mM Tris-HCl pH 8.0)
PCR Amplification
- Ligate eluted DNA fragments with adapter sequences using T4 DNA ligase
- Amplify adapter-ligated fragments using PCR with the following conditions:
- 94°C for 3 minutes (initial denaturation)
- 25 cycles of: 94°C for 30s, 55°C for 30s, 72°C for 30s
- 72°C for 5 minutes (final extension)
- Purify PCR products using silica membrane columns
Subsequent Selection Rounds
- Repeat steps 3-4 for 3-4 additional rounds with increased stringency:
- Increase number of washes in each subsequent round
- Add non-specific competitor DNA (e.g., poly(dI-dC)) in later rounds
- Monitor enrichment by measuring DNA recovery after each round
- Repeat steps 3-4 for 3-4 additional rounds with increased stringency:
Clone and Sequence
- Clone final-round PCR products into sequencing vector
- Sequence 50-100 clones from final round using Sanger sequencing
- Alternatively, subject final pool to high-throughput sequencing
Critical Steps and Troubleshooting
- Protein Quality: Ensure transcription factor preparation is >90% pure and properly folded
- Stringency Control: Increase washing stringency gradually to avoid loss of specific binders early in process
- Amplification Bias: Limit PCR cycles to minimize amplification bias; monitor reaction efficiency
- Background Binding: Include control selections with beads alone to identify background binders
High-Throughput Sequencing SELEX (HT-SELEX) Protocol
HT-SELEX combines traditional selection with next-generation sequencing to comprehensively characterize binding specificities [6].
Procedure Modifications for HT-SELEX
Library Preparation
- Use random oligonucleotide library with fixed flanking sequences for amplification
- Include sequencing adapters compatible with Illumina platforms during initial library construction
Selection Process
- Perform 5-8 rounds of selection with constant protein concentration to maintain fixed stringency
- Retain aliquots from each selection round for sequencing to monitor evolution
Sequencing Library Preparation
- Amplify selected pools from multiple rounds using barcoded primers
- Pool amplified products from different rounds for multiplex sequencing
- Sequence on Illumina platform to obtain >1 million reads per sample
Bioinformatic Analysis
- Use specialized pipelines (e.g., eme_selex) to identify enriched k-mers
- Calculate enrichment ratios for all possible k-mers across selection rounds
- Generate position weight matrices from enriched sequences
Data Analysis and Interpretation
Quantitative Analysis of SELEX Data
The analysis of SELEX data has evolved from simple consensus identification to sophisticated quantitative modeling. Modern approaches enable accurate determination of protein-DNA interaction parameters, providing insights into binding specificity and affinity.
Table 2: Key Parameters in SELEX Data Analysis
| Parameter | Description | Calculation Method | Significance |
|---|---|---|---|
| Enrichment Ratio | Relative abundance of sequence between rounds | (Frequencyroundn)/(Frequencyroundn-1) | Identifies sequences under positive selection |
| K-mer Enrichment | Enrichment of all possible sequences of length k | Normalized count compared to expected frequency | Reveals core binding motifs without assumptions |
| Position Weight Matrix (PWM) | Quantitative representation of binding preferences | Log-likelihood ratios for each base at each position | Enables prediction of binding sites in genomic sequences |
| Dissociation Constant (Kd) | Measure of binding affinity | Determined from fixed-stringency SELEX experiments | Provides quantitative affinity measurements for individual sequences |
Quantitative modeling of SELEX experiments has revealed limitations in traditional approaches for determining protein-DNA interaction parameters [7]. A modified approach maintaining fixed chemical potential (constant free protein concentration) through different selection rounds enables more robust parameter estimation [7]. This fixed-stringency approach generates datasets from which binding energies can be accurately derived, significantly improving the false positive/false negative trade-off compared to traditional methods [7].
For genomic SELEX, computational analysis typically involves multiple motif-finding algorithms (BioProspector, AlignACE, MEME) to identify sequence motifs enriched in selected fragments compared to genomic background [4]. Additional analyses include conservation assessment across species and genomic annotation of selected fragments to identify potential regulatory regions [4].
Validation of Selected Aptamers and Binding Sites
Identification of potential binders through SELEX requires rigorous validation to confirm specificity and biological relevance:
In Vitro Binding Assays
- Fluorescence anisotropy to determine dissociation constants (Kd)
- Electrophoretic mobility shift assays (EMSA) to confirm direct binding
- Competition assays to assess binding specificity
Functional Validation
- Reporter gene assays with selected sequences cloned upstream of minimal promoters
- Site-directed mutagenesis of predicted binding sites to confirm specificity
- Chromatin immunoprecipitation (ChIP) to confirm in vivo binding [4]
Biological Validation
- Loss-of-function studies (e.g., morpholino knockdown) to assess functional consequences
- Gain-of-function experiments to test sufficiency
- Correlation with gene expression changes in relevant biological systems [4]
The Scientist's Toolkit: Essential Research Reagents
Successful SELEX experiments require carefully selected reagents and materials optimized for each selection target and methodology.
Table 3: Essential Research Reagents for SELEX Experiments
| Reagent Category | Specific Examples | Function/Purpose | Selection Considerations |
|---|---|---|---|
| Oligonucleotide Library | Random ssDNA/RNA library with 20-60 nt variable region | Provides sequence diversity for selection | Library complexity should exceed 10¹³ sequences; structural diversity critical |
| Target Molecules | Purified proteins, small molecules, whole cells | Binding target for selection | Purity, conformation, and immobilization method affect selection outcome |
| Immobilization Matrix | Glutathione sepharose, streptavidin beads, nitrocellulose filters | Enables separation of bound and unbound sequences | Matrix should not interfere with target structure or introduce nonspecific binding |
| Amplification Reagents | Taq polymerase, dNTPs, primers with adapter sequences | Amplifies selected sequences for subsequent rounds | Primer design critical to avoid dimerization; polymerase fidelity affects diversity |
| ssDNA Generation | Biotinylated primers, streptavidin beads, lambda exonuclease | Regenerates single-stranded DNA for selection rounds | Efficiency critical; different methods yield 50-70% recovery [1] |
| Buffer Components | Salts, competitors (e.g., poly(dI-dC)), nuclease inhibitors | Creates optimal binding environment | Ionic strength affects electrostatic interactions; competitors reduce background |
| Unesbulin | 5-Fluoro-2-(6-fluoro-2-methyl-1H-benzo[d]imidazol-1-yl)-N4-(4-(trifluoromethyl)phenyl)pyrimidine-4,6-diamine | High-purity 5-Fluoro-2-(6-fluoro-2-methyl-1H-benzo[d]imidazol-1-yl)-N4-(4-(trifluoromethyl)phenyl)pyrimidine-4,6-diamine for research. For Research Use Only. Not for human or veterinary diagnosis or therapeutic use. | Bench Chemicals |
| (R)-Azelastine Hydrochloride | (R)-Azelastine Hydrochloride, CAS:153408-28-7, MF:C22H25Cl2N3O, MW:418.4 g/mol | Chemical Reagent | Bench Chemicals |
Critical Factors and Optimization Strategies
Several factors significantly influence SELEX success and require careful optimization:
Target Considerations
The nature of the target molecule profoundly impacts selection strategy and expected outcomes. Protein targets require consideration of surface charges, as strongly negative surfaces may repel nucleic acids, while positive charges may promote nonspecific binding [3]. Small molecules necessitate careful immobilization to ensure presentation of appropriate epitopes for binding [3]. For transcription factors, using the DNA-binding domain rather than full-length protein often improves selection efficiency [4].
Library Design
Library complexity directly influences selection success. The sequence diversity should significantly exceed the total number of sequences used in the first selection round to ensure adequate structural diversity [3]. Constant domains and primers should be optimized to minimize structural influence on the random region and prevent primer-dimer formation [3]. For specialized applications, chemically modified nucleotides (e.g., 2'-F, 2'-O-methyl RNA) can enhance stability and introduce novel chemical functionalities [1].
Selection Stringency
Appropriate stringency control is essential for successful SELEX. Early rounds should employ lower stringency to avoid losing rare potential binders, while later rounds require progressively increased stringency to eliminate moderate-affinity binders and drive selection toward optimal sequences [3]. Stringency can be modulated through various parameters:
- Wash conditions: Increasing wash volume, duration, or number
- Competitor DNA: Adding nonspecific DNA (e.g., salmon sperm DNA) to compete weak binders
- Salt concentration: Modifying ionic strength to alter binding stringency
- Incubation time: Reducing time to select for faster binding kinetics
Quality Control and Monitoring
Implementing quality control measures throughout the selection process enables informed decisions about progression and termination. Key monitoring approaches include:
- Binding yield: Tracking the percentage of library bound to target each round
- Sequence diversity: Monitoring complexity reduction through clone sequencing or HTS
- Progress assessment: Using specific binding assays to confirm enrichment of target-specific binders
- By-product detection: Identifying amplification artifacts (e.g., primer-dimers) early
SELEX technology represents a powerful platform for generating specific nucleic acid ligands against diverse targets, with genomic SELEX providing particularly valuable insights into transcription factor binding specificities and regulatory networks. The core principles of iterative selection and amplification enable the evolution of high-affinity binders from highly diverse starting libraries. Successful application requires careful consideration of multiple factors, including target presentation, library design, stringency control, and appropriate analytical methods. As SELEX methodologies continue to evolve, particularly with integration of high-throughput sequencing and sophisticated bioinformatic analysis, the applications in basic research, diagnostic development, and therapeutic discovery continue to expand. The protocols and considerations outlined here provide a foundation for designing effective SELEX experiments aimed at identifying specific binding sequences, particularly in the context of transcription factor binding site identification.
HT-SELEX as a High-Throughput Platform for TF Binding Profiling
High-Throughput Systematic Evolution of Ligands by Exponential Enrichment (HT-SELEX) has emerged as a powerful in vitro technique for unbiased determination of DNA binding specificities of transcription factors (TFs). This method enables researchers to characterize preferred target motifs by selecting protein-binding DNA sequences from a vast random oligonucleotide library through iterative cycles of binding, purification, and amplification [8]. Unlike in vivo methods like ChIP-seq, which are constrained by cellular contexts and antibody availability, HT-SELEX provides a controlled environment to explore TF-DNA interactions systematically, making it particularly valuable for profiling poorly studied human transcription factors [9] [10].
The fundamental advantage of HT-SELEX lies in its ability to process thousands to millions of DNA sequences in a single experiment, generating massive datasets that comprehensively capture TF binding preferences [10]. This technological advancement addresses a critical bottleneck in regulatory genomics, as traditional low-throughput methods yielded insufficient data for building accurate models of transcription factor binding sites. Current datasets now encompass hundreds of TF experiments, enabling computational biologists to develop more sophisticated models of DNA recognition beyond simple position weight matrices [11] [10].
Performance and Benchmarking of HT-SELEX
Cross-Platform Performance Assessment
Recent large-scale benchmarking initiatives have evaluated HT-SELEX alongside other prominent technologies for TF binding characterization. The Gene Regulation Consortium Benchmarking Initiative (GRECO-BIT) analyzed an extensive collection of 4,237 experiments for 394 TFs using five different experimental platforms, including HT-SELEX, ChIP-Seq, genomic HT-SELEX (GHT-SELEX), SMiLE-Seq, and PBMs [9].
This systematic comparison revealed that motif consistency across platforms and replicates serves as a key quality metric for successful experiments. Through rigorous human curation, researchers approved a subset of experiments that yielded reliable motifs, with 236 TFs ultimately represented in the high-quality dataset of 1,462 approved experiments [9]. The study demonstrated that motifs with low information content in many cases effectively described binding specificity across different experimental platforms, challenging previous assumptions about motif quality assessment.
Table 1: Comparison of Experimental Platforms for TF Binding Characterization
| Platform | Library Type | Context | Key Advantages | Limitations |
|---|---|---|---|---|
| HT-SELEX | Synthetic random oligonucleotides | In vitro | Unbiased exploration of sequence space; massive throughput | May saturate with strongest binders; lacks cellular context |
| GHT-SELEX | Genomic DNA fragments | In vitro | Natural sequence variation; biochemical environment | Limited to genomic regions in library |
| ChIP-Seq | Genomic regions | In vivo | Native chromatin context; actual binding locations | Antibody-dependent; broad footprints; cellular constraints |
| PBM | Pre-designed probes | In vitro | Highly quantitative; comprehensive probe coverage | Fixed probe sequences; may miss novel motifs |
Quantitative Performance Metrics
The benchmarking effort generated an impressive 219,939 position weight matrices (PWMs), with 164,570 derived from approved experiments [9]. After automatic filtering for common artifact signals (such as simple repeats and widespread ChIP contaminants), 159,063 high-quality PWMs were obtained. The evaluation employed multiple dockerized benchmarking protocols with different scoring methods:
- Sum-occupancy scoring for most sequence types
- Single top-scoring log-odds PWM hit evaluation for ChIP-Seq and GHT-SELEX peaks
- CentriMo motif centrality score accounting for distance to peak summit [9]
Notably, the study found that nucleotide composition and information content did not correlate with motif performance and failed to help in detecting underperformers. This finding challenges conventional wisdom in the field and suggests the need for more sophisticated metrics in assessing motif quality [9].
HT-SELEX Experimental Protocol
The HT-SELEX method follows an iterative selection-amplification process that enriches protein-binding DNA sequences from a random library. The key steps include:
- Library Preparation: Generation of double-stranded DNA oligonucleotides containing random regions flanked by constant sequences for PCR amplification
- Binding Reaction: Incubation of the DNA library with purified, tagged transcription factor
- Partitioning: Separation of protein-DNA complexes from unbound DNA
- Amplification: PCR enrichment of bound sequences for the next selection cycle
- Sequencing: High-throughput sequencing of selected pools, typically after multiple cycles [8] [11]
The process typically requires 3-5 cycles to sufficiently enrich for high-affinity binders, though excessive cycles can reduce library diversity and bias results toward the strongest binding sequences [9] [8].
Detailed Step-by-Step Methodology
Generate Random Library of Double-Stranded DNA Oligonucleotides (Cycle 0)
The initial library consists of synthetic oligonucleotides with a central random region (typically 20-40 bp) flanked by constant sequences that serve as PCR primer binding sites. Technical replicates using three separate random oligonucleotide libraries are recommended to account for distribution variations [8].
Table 2: PCR Mastermix for Library Preparation (24 reactions)
| Reagent | Amount per Mastermix | Final Concentration | Function |
|---|---|---|---|
| Random library DNA template | 12 pmol (0.5 pmol/reaction) | Variable | Provides diverse starting sequences |
| 5× Phusion HF Buffer | 240 μL | 1× | Optimal reaction conditions |
| dNTPs (10 mM) | 24 μL | 200 μM each | DNA synthesis building blocks |
| Library FW Primer (10 μM) | 60 μL | 0.5 μM | Forward amplification primer |
| Library RV Primer (10 μM) | 60 μL | 0.5 μM | Reverse amplification primer |
| Phusion DNA Polymerase | 12 μL | 0.02 U/μL | High-fidelity DNA amplification |
| Nuclease-free water | Up to 1.2 mL | - | Reaction volume adjustment |
PCR Program for Library Amplification:
- Initial Denaturation: 98°C for 1 min (1 cycle)
- Denaturation: 98°C for 20 s
- Annealing: 60°C for 20 s (5 cycles)
- Extension: 72°C for 20 s
- Final Extension: 72°C for 5 min (1 cycle)
- Hold: 4°C indefinitely [8]
After amplification, the double-stranded DNA libraries are purified using commercial PCR purification kits (e.g., Qiagen MinElute), with concentration and integrity assessed via spectrophotometry. A single band at approximately 83 bp should be visible on a 10% polyacrylamide gel without detectable heteroduplexes [8].
Protein Purification and Binding Reactions
The success of HT-SELEX depends critically on the quality and purity of the transcription factor. While protocols vary by specific TF, these general principles apply:
- Affinity Tags: His-tag, GST, or other affinity tags enable both protein purification and isolation of protein-DNA complexes
- Expression Systems: E. coli, wheat germ extracts, or mammalian systems selected based on TF requirements
- Buffer Optimization: Conditions must maintain TF stability and DNA-binding capability [8]
For the binding reaction, recombinant TF (e.g., 0.5 mg/mL concentration) is incubated with the DNA library in appropriate binding buffer. Poly(dI-dC) is often included as non-specific competitor DNA. Complexes are isolated using affinity resin corresponding to the protein tag (e.g., Ni-NTA for His-tagged proteins) [8].
After binding and washing, protein-DNA complexes are eluted, and bound DNA is amplified for subsequent cycles or prepared for sequencing after the final round. Modern implementations typically use 3-4 selection cycles before high-throughput sequencing [8] [11].
Computational Analysis of HT-SELEX Data
Bioinformatics Pipeline
The massive datasets generated by HT-SELEX require sophisticated computational analysis. A typical bioinformatics pipeline includes:
- Sequence Processing: Quality control, demultiplexing, and parsing of sequencing reads
- Enrichment Analysis: Identification of significantly enriched k-mers across selection cycles
- Motif Discovery: Application of algorithms to infer binding motifs from enriched sequences
- Model Building: Construction of quantitative models representing TF binding preferences [8] [10]
The eme_selex pipeline facilitates detection of promiscuous DNA binding by analyzing enrichment of all possible k-mers, providing a comprehensive view of sequence preferences [8].
Advanced Modeling Approaches
While Position Weight Matrices (PWMs) remain the most common representation of binding motifs, their assumption of position independence represents a significant limitation. Recent advances include:
- DCA-Scapes: A global pairwise model that captures interdependencies between nucleotide positions, providing higher-resolution TF recognition specificity landscapes [10]
- Random Forest Models: Combining multiple PWMs to account for multiple modes of TF binding, demonstrating improved performance over single motifs [9]
- Hamiltonian Scoring: A quantitative approach that predicts the likelihood of DNA sequences being TF targets, with more negative scores indicating stronger binding [10]
These advanced models have demonstrated superior performance in predicting in vivo binding sites validated by ChIP-seq data, with Hamiltonian scores showing significant discrimination between bound and unbound genomic regions [10].
Research Reagent Solutions
Table 3: Essential Reagents and Resources for HT-SELEX
| Reagent/Resource | Specifications | Function | Example Sources |
|---|---|---|---|
| Random Oligo Library | 20-40 bp random region with constant flanks | Source of diverse DNA sequences for selection | Integrated DNA Technologies (IDT) |
| High-Fidelity DNA Polymerase | Phusion or equivalent | Error-free amplification of selected sequences | New England Biolabs (NEB) |
| Affinity Purification Resin | Ni-NTA, glutathione, or antibody-conjugated | Isolation of protein-DNA complexes | Cytiva, Qiagen |
| Tagged Recombinant TF | His-tag, GST, or other affinity tag | DNA-binding protein for selection | In-house expression or commercial |
| Poly(dI-dC) | Non-specific competitor DNA | Reduction of non-specific binding | Merck Life Science |
| Next-Generation Sequencing Platform | Illumina or equivalent | High-throughput readout of selected sequences | Various providers |
| Bioinformatics Tools | HOMER, MEME, STREME, RCade, DCA-Scapes | Motif discovery and data analysis | Publicly available packages |
Transcription factor (TF) binding to DNA is a fundamental component of transcriptional regulation, responsible for coordinated gene expression within gene regulatory networks [9]. The accurate identification of DNA sequences recognized by TFs—their binding motifs—is crucial for annotating gene regulatory regions, interpreting regulatory variation, and deciphering the logic of gene regulatory networks [9]. A sequence motif representing the DNA-binding specificity of a TF is commonly modeled with a positional weight matrix (PWM) [9]. However, generating accurate motif models is challenging due to technical biases inherent in different experimental platforms, which influence the types of binding sites detected and the resulting biological interpretations.
The binding specificity of a TF ideally should be studied both in vivo and in vitro with both synthetic and genomic sequences, using multiple experimental platforms to overcome these inherent challenges [9]. This Application Note examines the technical characteristics, advantages, and limitations of major experimental platforms used for TF binding site identification, with a special focus on Genomic SELEX and its variants within the broader context of TF research. We provide detailed protocols and analytical frameworks to help researchers select appropriate methodologies, mitigate technical biases, and integrate complementary data sources for a more comprehensive understanding of TF-DNA interactions.
Experimental Platforms: Comparative Analysis
Multiple experimental platforms have been developed to identify TFBS in random sequences, complete genomes, or their fragments [9]. These can be broadly categorized into in vitro methods using synthetic DNA sequences and in vivo methods examining binding in cellular contexts. Table 1 summarizes the key experimental platforms, their underlying principles, and the types of biases inherent in each approach.
Table 1: Comparison of Experimental Platforms for TF Binding Site Identification
| Platform | Principle | DNA Source | Key Strengths | Technical Biases/Limitations |
|---|---|---|---|---|
| HT-SELEX [9] [12] | Multiple rounds of in vitro selection and amplification | Synthetic random oligos | High-throughput; models for hundreds of TFs; identifies high-affinity sites | Rapid saturation with strongest binders; misses lower-affinity sites; over-representation of high-affinity sites |
| Genomic SELEX (GHT-SELEX) [13] [9] | SELEX with genomic DNA fragments | Natural genomic DNA | Discovers natural genomic aptamers; identifies binding domains in native context | Limited to accessible genomic regions; depends on library representation |
| ChIP-Seq [9] [10] | Chromatin immunoprecipitation with sequencing | Cellular genomic DNA | In vivo binding context; includes chromatin effects | Requires ChIP-grade antibodies; broad footprints; influenced by cellular environment |
| PADIT-seq [12] | In vitro transcription coupled to reporter output | All possible k-mers (e.g., all 10-mers) | Unprecedented sensitivity for low-affinity sites; quantitative affinity measurements | Newer method with less established benchmarks; specialized protocol |
| PBM [9] [12] | Protein binding to microarrayed DNA probes | Pre-defined synthetic sequences | Comprehensive k-mer binding data; high reproducibility | Fixed probe design limits sequence space; potential flanking sequence effects |
| SMiLE-Seq [9] | Microfluidics-based ligand enrichment | Synthetic random sequences | Efficient selection; requires fewer rounds | Platform-specific biases not fully characterized |
Quantitative Performance Metrics Across Platforms
Different platforms exhibit varying capabilities to detect binding sites across affinity ranges. Recent comparative studies, particularly the Gene Regulation Consortium Benchmarking Initiative (GRECO-BIT), have quantitatively evaluated platform performance [9]. Table 2 presents key performance metrics for major platforms based on cross-platform benchmarking studies.
Table 2: Quantitative Performance Metrics of TF Binding Assay Platforms
| Platform | Affinity Range Detected | Sequence Coverage | Sensitivity to Low-Affinity Sites | Correlation with Functional Binding |
|---|---|---|---|---|
| HT-SELEX | High-affinity (Kd < 0.01 μM) [12] | Moderate (107-108 sequences) [9] | Limited (AUROC ~0.7-0.8) [12] | Moderate; biased toward strongest binders |
| PADIT-seq | Broad (Kd ~ 0.1 μM to high affinity) [12] | Comprehensive (all 10-mers) [12] | Excellent (detects hundreds of low-affinity sites) [12] | Strong; correlates with MITOMI Kd (r > 0.9) [12] |
| PBM | Moderate to high affinity [12] | Fixed design (all 8-9mers with flanks) [12] | Moderate (misses lower-affinity sites with E-score < 0.3) [12] | Good for high-affinity sites; variable thresholds by TF |
| ChIP-Seq | In vivo relevant affinities | Genome-wide | Context-dependent | High for in vivo binding but confounded by cellular factors |
| GHT-SELEX | Moderate to high affinity | Depends on genomic library | Better than HT-SELEX for genomic context | Good balance of in vitro and genomic context |
Detailed Methodologies
Genomic SELEX Protocol
Genomic SELEX is a discovery tool for genomic aptamers, which are genomically encoded functional domains in nucleic acid molecules that recognize and bind specific ligands [13]. The major difference between SELEX and Genomic SELEX is the starting pool: while traditional SELEX begins with a library of synthetically derived random DNA molecules, Genomic SELEX starts from libraries derived from genomic DNA [13].
Library Construction and Selection
- Source DNA Preparation: Obtain high-quality genomic DNA from an organism with a fully sequenced genome to allow mapping and analysis of selected sequences [13].
- Primer Design: Design two pairs of primers called "hyb"- and "fix"-primers [13].
- hyb-primers consist of a unique constant sequence region absent in the genome, followed by approximately 9 randomized nucleotides at the 3' terminus
- fix-primers correspond perfectly to the 5' constant regions of respective hyb-primers, with addition of the T7 promoter at the 5' end of the fixFOR primer
- Library Amplification: Perform first- and second-strand Klenow synthesis using hybREV and hybFOR primers, respectively [13].
- Bait Preparation: Use purified RNA-binding proteins as bait. Purity is crucial to avoid enrichment of aptamers binding contaminants [13]. Translational fusion to different tags (e.g., His, Flag or GST) can facilitate purification [13].
- Selection Procedure: Incubate the DNA library with the bait protein. Separate protein-DNA complexes from unbound DNA using methods appropriate for the tag (e.g., glutathione Sepharose for GST-tagged proteins) [14].
- Washing and Elution: Wash thoroughly to remove non-specifically bound DNA. Elute specifically bound DNA [14].
- Amplification and Repeated Selection: Amplify eluted DNA and subject to additional rounds of selection (typically 2-4 rounds) to enrich specific binders [14] [13].
Counter-Selection and Controls
- Counter-Selection: Use modified or inactive baits to strengthen binding specificity of selected sequences [13].
- Neutral SELEX Control: Perform a "Neutral" SELEX experiment in parallel, omitting the selection step, to provide a background signal for comparison [13].
- Sequence Blocking: To prevent flanking primers from becoming part of the structural motif, especially with short libraries, anneal oligonucleotides complementary to flanking regions prior to selection or switch flanking sequences every few SELEX cycles [13].
Genomic SELEX Workflow
Affinity Chromatography-SELEX with Quantitative Binding Assay
Combining affinity chromatography-SELEX with quantitative binding assays provides a streamlined approach to generate accurate models of TF binding specificity [14].
Affinity Chromatography-SELEX Procedure
- Protein Preparation: Express GST-tagged DNA-binding domain (e.g., GST-Zif268) in E. coli and purify using glutathione Sepharose chromatography [14].
- DNA Pool Design: Create double-stranded DNA pool containing random regions flanked by fixed sequences for PCR amplification and cloning [14].
- Binding Reaction: Incubate DNA pool (~10â»â¸ M) with purified GST-tagged protein (~10â»â¸ M) in reaction buffer [30 mM Tris-HCl (pH 8.0), 50 mM NaCl, 0.1 mg/ml BSA, 3 mM DTT, 20 µM ZnSOâ‚„, 25 µg/ml salmon sperm DNA] for 1 hour at room temperature [14].
- Affinity Capture: Add glutathione Sepharose slurry to capture protein-DNA complexes. Wash thoroughly with reaction buffer [14].
- Elution and Amplification: Elute bound DNA with elution buffer [50 mM Tris (pH 8.0), 0.25 M KCl, 10 mM glutathione]. Amplify eluted DNA by PCR for subsequent selection rounds [14].
- Cloning and Sequencing: After 2-4 selection rounds, clone isolated DNA sites into sequencing vector and sequence individual clones [14].
Quantitative Multiplex Fluorescence Relative Affinity (QuMFRA) Assay
- Fluorescent Labeling: Generate double-stranded oligonucleotide binding sites by PCR using fluorophore-labeled primers (FAM, TAMRA, or ROX) [14].
- Competitive Binding: Mix three different fluorophore-labeled DNA binding sites with GST-tagged protein (~10â»â¸ M) in reaction buffer [14].
- Electrophoretic Separation: Separate protein-DNA complexes from free DNA by electrophoresis on 10% polyacrylamide gel [14].
- Fluorescence Detection: Scan gels using a fluorescence scanner (e.g., Typhoon Variable Scanner) to obtain fluorescent intensities at different emission wavelengths [14].
Relative Affinity Calculation: Calculate relative binding constant using the formula:
[ Ka(rel) = \frac{[PD{test}][D{ref}]}{[PD{ref}][D_{test}]} ]
where [PDtest] and [PDref] are concentrations of bound DNA for test and reference sites, and [Dtest] and [Dref] are concentrations of free DNA [14].
PADIT-seq Protocol for Comprehensive Affinity Measurement
PADIT-seq (Protein Affinity to DNA by In Vitro Transcription and RNA sequencing) is a recently developed technology that measures TF-DNA binding preferences at greater sensitivity than prior high-throughput methods, particularly for lower affinity interactions [12].
Reporter Library Construction
- Library Design: Construct a reporter library containing all possible 10-bp DNA sequences (n = 1,048,576) as candidate TF binding sites [12].
- Barcode Association: Randomly associate TF binding sites with barcodes during library construction and determine TFBS-barcode combinations by Illumina sequencing [12].
Binding Assay and Sequencing
- In Vitro Transcription and Translation: Mix PADIT-seq reporter library with either a 'no DBD' control or a constitutive promoter driving expression of the DNA-binding domain of interest [12].
- Reporter RNA Sequencing: Following IVTT, sequence reporter RNAs by Illumina sequencing [12].
- Differential Analysis: Perform differential gene expression analysis of TFBS counts against the 'no DBD' control using DESeq2 [12].
- Activity Calculation: Define logâ‚‚(DBD / 'no-DBD') values as 'PADIT-seq activity,' with 'active' TFBS defined as those significantly increasing reporter gene expression upon TF binding (FDR 5%) [12].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Genomic SELEX and TF Binding Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Expression Vectors | pGEX-4T-1 (GST-tag), pET series | Recombinant protein expression with affinity tags for purification [14] |
| Affinity Matrices | Glutathione Sepharose, Nickel-NTA, Antibody-conjugated beads | Capture and purification of tagged proteins or protein-DNA complexes [14] [13] |
| DNA Library Templates | Random oligo pools, Genomic DNA fragments | Source of potential binding sites for selection experiments [14] [13] |
| Amplification Reagents | High-fidelity DNA polymerases, dNTPs, Fluorophore-labeled primers | Amplification of selected DNA pools; preparation of labeled probes [14] |
| Binding Assay Components | BSA, carrier DNA (salmon sperm), DTT, ZnSOâ‚„ (for zinc fingers) | Reduction of non-specific binding in reaction buffers [14] |
| Sequencing Platforms | Illumina for high-throughput, Sanger for individual clones | Identification of selected sequences; determination of binding motifs [14] [13] |
| Relamorelin | Relamorelin, CAS:661472-41-9, MF:C43H50N8O5S, MW:791.0 g/mol | Chemical Reagent |
| Remodelin hydrobromide | Remodelin hydrobromide, MF:C15H15BrN4S, MW:363.3 g/mol | Chemical Reagent |
Data Analysis and Computational Modeling
Motif Discovery and Benchmarking
With diverse experimental platforms generating TF binding data, appropriate computational analysis is essential for deriving accurate motif models. The GRECO-BIT initiative has systematically evaluated motif discovery tools across multiple experimental platforms [9].
- Data Preprocessing: Uniformly preprocess data, including peak calling for GHT-SELEX and ChIP-Seq data, and normalization for PBMs [9].
- Training-Test Split: Split results of each experiment into training and test sets for validation [9].
- Motif Discovery Tools: Apply multiple motif discovery tools compatible with different data types:
- Benchmarking Protocols: Employ multiple benchmarking protocols using sum-occupancy scoring, HOCOMOCO benchmark (single top-scoring hit), and CentriMo motif centrality score [9].
- Expert Curation: Manually curate results to approve experiments that yield consistent motifs across platforms or similar to known motifs [9].
Advanced Modeling Approaches
Moving beyond standard position weight matrices can improve characterization of TF binding specificities:
- DCA-Scapes Model: A global pairwise model that captures interdependencies between nucleotide positions in TF binding sites, providing higher resolution TF recognition specificity landscapes [10].
- Hamiltonian Scoring: For a given DNA sequence, the Hamiltonian score quantitatively predicts the likelihood of it being a TF target, with more negative scores indicating greater likelihood of favorable TF recognition [10].
- Random Forest Approaches: Combining multiple PWMs into a random forest accounts for multiple modes of TF binding and improves binding prediction [9].
Computational Analysis Workflow
Understanding and addressing technical biases in experimental platforms for TF binding site identification is crucial for generating accurate biological insights. As demonstrated through comparative analyses, each platform possesses distinct strengths and limitations in detecting binding sites across the affinity spectrum [9] [12]. HT-SELEX efficiently identifies high-affinity sites but saturates quickly and misses lower-affinity interactions [12], while newer technologies like PADIT-seq offer unprecedented sensitivity for detecting lower-affinity sites but require specialized expertise [12].
The integration of multiple experimental approaches—combining in vitro and in vivo methods, synthetic and genomic DNA sources—provides the most comprehensive characterization of TF binding specificities [9]. Furthermore, advanced computational models that move beyond simple position weight matrices to account for nucleotide interdependencies and multiple binding modes promise to extract more biological insight from experimental data [9] [10].
As the field advances, researchers should select experimental platforms based on their specific biological questions, employ appropriate controls to address platform-specific biases, and integrate complementary data sources to develop accurate models of TF-DNA interactions that reflect the complexity of gene regulatory systems.
The Role of SELEX in Profiling Poorly Studied and Novel Transcription Factors
The comprehensive characterization of transcription factor (TF) binding specificities is a fundamental challenge in molecular biology, particularly for poorly studied and novel TFs. DNA–transcription factor interactions are essential for gene regulation, and fully characterizing TF recognition specificities is critical to understanding TF function and regulatory networks [10]. Among the various techniques available, the Systematic Evolution of Ligands by Exponential Enrichment (SELEX) method has emerged as a particularly powerful in vitro approach for determining the binding preferences of TFs, even in the absence of prior biological knowledge [14]. Recent advancements have seen the evolution of SELEX into High-Throughput SELEX (HT-SELEX), which combines the biochemical robustness of traditional SELEX with the scale of modern sequencing technology [15]. This protocol outlines detailed methodologies for employing SELEX and HT-SELEX to profile novel TFs, framed within the broader context of genomic SELEX research, and provides the necessary tools for researchers to identify TF binding motifs with high accuracy and reliability.
Key Methodologies and Principles
Core Principles of SELEX and HT-SELEX
SELEX operates on the principle of in vitro selection, where a purified TF is used to isolate high-affinity binding sites through successive rounds of selection and amplification from a vast pool of random oligonucleotide sequences [14]. The power of this method lies in its ability to isolate a small set of specific binding sites from a very large pool of random sequences, typically ranging from thousands to millions of possibilities [14]. HT-SELEX builds upon this foundation by incorporating high-throughput sequencing capabilities, enabling the processing of protein binding measurements for thousands to millions of DNA sequences and providing massive datasets that comprehensively comprise TF binding preferences [10]. This technological advancement has been crucial for addressing the limitations of traditional SELEX, which was often constrained by the limited number of sequences that could be practically analyzed.
Comparative Analysis of SELEX Methodologies
The table below summarizes the key characteristics of different SELEX approaches, highlighting the advantages of HT-SELEX for profiling novel transcription factors.
Table 1: Comparison of SELEX Methodologies for Transcription Factor Profiling
| Method | Throughput | Key Features | Data Output | Primary Applications |
|---|---|---|---|---|
| Traditional SELEX | Low | Gel mobility shift for complex separation; radio-labeled DNA [14] | 20-100 sequences | Initial binding site identification; qualitative specificity assessment |
| Affinity Chromatography-SELEX | Medium | GST-tagged protein purification; glutathione Sepharose for complex isolation [14] | 100-1,000 sequences | Rapid screening; quantitative model refinement with QuMFRA [14] |
| HT-SELEX | High | Illumina sequencing; multiple selection rounds; robust bioinformatic pipelines [10] [15] | 10,000+ sequences [16] | Comprehensive specificity determination; genome-wide binding site prediction; quantitative modeling |
Experimental Protocol: HT-SELEX for Novel Transcription Factors
The following diagram illustrates the comprehensive HT-SELEX workflow for profiling transcription factor binding specificities:
Detailed Procedural Steps
Random DNA Library Design and Preparation
- Library Design: Construct a double-stranded DNA oligonucleotide pool containing a central randomized region (typically 20-40 bp) flanked by fixed sequences for PCR amplification. For a 20-nucleotide random core, the library complexity is 4²Ⱐ(approximately 1×10¹² unique sequences), ensuring comprehensive coverage of potential binding sites [10].
- Library Synthesis: Generate double-stranded DNA molecules by PCR amplification using high-fidelity DNA polymerase. Purify the products using agarose gel electrophoresis and quantify with fluorescence-based methods such as PicoGreen dsDNA quantitation [14].
Recombinant Transcription Factor Production
- Protein Expression: Clone the DNA-binding domain (DBD) of the novel TF into an appropriate expression vector (e.g., pGEX-4T-1 for GST-tagged fusion proteins). Transform into E. coli expression strains such as BL21 [14].
- Protein Purification: Induce expression with IPTG and purify the recombinant TF using affinity chromatography (e.g., glutathione Sepharose for GST-tagged proteins). Dialyze into appropriate storage buffer, concentrate, and quantify using protein assay kits [14]. Verify purity by SDS-PAGE with silver staining.
Selection Rounds (4 Rounds Recommended)
- Binding Reaction: Incubate the DNA library (∼10â»â¸ M) with purified TF (∼10â»â¸ M) in reaction buffer [30 mM Tris-HCl (pH 8.0), 50 mM NaCl, 0.1 mg/ml BSA, 3 mM DTT, 20 µM ZnSOâ‚„, 25 µg/ml salmon sperm DNA] for 1 hour at room temperature [14].
- Complex Separation: For affinity chromatography SELEX, add glutathione Sepharose slurry to capture GST-tagged TF-DNA complexes. Wash extensively with reaction buffer to remove non-specifically bound DNA [14].
- Elution and Amplification: Elute bound DNA with elution buffer [50 mM Tris (pH 8.0), 0.25 M KCl, 10 mM glutathione]. Amplify eluted DNA by PCR using flanking primers. Purify PCR products for subsequent selection rounds [14].
- Round Progression: Typically perform 3-4 rounds of selection, using DNA from the previous round's elution as input for the next round. The increasing stringency of selection enriches for high-affinity binding sites [10].
Sequencing and Data Processing
- Library Preparation: After the final selection round, prepare sequencing libraries from the amplified DNA. For HT-SELEX, this typically involves adapter ligation and PCR amplification compatible with Illumina sequencing platforms [15].
- High-Throughput Sequencing: Sequence the enriched library to obtain millions of reads, providing comprehensive coverage of the selected sequences. Data from the initial non-selected pool should also be sequenced to serve as a background control [10].
Data Analysis and Computational Modeling
Bioinformatic Processing Pipeline
The analysis of HT-SELEX data involves multiple computational steps to transform raw sequencing reads into quantitative models of TF binding specificity. The following diagram illustrates this analytical workflow:
Quantitative Models for Binding Specificity
Position Weight Matrix (PWM) Modeling
The Position Weight Matrix is the most commonly used model to represent DNA-binding preferences of TFs. PWM is a matrix derived from position frequency matrices, with a probability score for each nucleotide at each position. These probabilities can be added to estimate the overall binding affinity of DNA elements [10]. Major databases including JASPAR, TRANSFAC, and CIS-BP collect sequencing data and use PWM-based methods to generate and store binding motif patterns [10]. However, PWM models assume nucleotide positions are independent and may not capture more complex binding specificities.
Advanced Modeling with DCA-Scapes
For more comprehensive modeling, the global pairwise DCA-Scapes model captures the sequence specificity requirements of TF-DNA interactions from HT-SELEX data [10]. This approach involves:
- Parameter Calculation: The model infers the joint probability distribution of DNA sequences using pairwise couplings and local fields parameters, measured as an average of the four-nucleotide gauged state [10].
- Hamiltonian Scores: These parameters are collectively interpreted as a fitness function score (Hamiltonian score), which quantitatively predicts the likelihood of a DNA sequence being a target for the TF. A more negative Hamiltonian score indicates a greater likelihood of favorable TF recognition [10].
- Null Model Construction: To estimate statistical significance, a null model is constructed by calculating Hamiltonian scores for 1 million random 20-mer DNA sequences with similar nucleotide distribution to the human genome [10].
Performance Validation with Genomic Data
To test the accuracy of computational models in predicting in vivo binding sites, ChIP-seq data from the ENCODE project can be used for validation [10]. The performance evaluation involves:
- Peak Sequence Analysis: Extract sequences from the top 500 peaks in each ChIP-seq experiment and compare with control sequences from upstream and downstream regions [10].
- ROC Curve Analysis: Calculate Hamiltonian scores for both peak and control regions using a sliding window approach. Use the three most negative Hamiltonian scores to represent TF recognition specificity and generate receiver operating characteristic (ROC) curves to evaluate prediction performance as the area under the ROC curve (AUC) [10].
Table 2: Quantitative Performance Metrics for SELEX-Based TF Binding Site Prediction
| Analysis Method | Data Input | Key Output | Validation Approach | Performance Metric |
|---|---|---|---|---|
| Position Weight Matrix (PWM) | Enriched sequences from final SELEX round [10] | Nucleotide probability matrix | Prediction of ChIP-seq peaks [10] | Limited accuracy for weak preferences [10] |
| DCA-Scapes Model | HT-SELEX reads from round 4 with initial pool as background [10] | Hamiltonian binding scores | ROC analysis against ChIP-seq data [10] | High AUC (accurate genomic binding prediction) [10] |
| Quantitative Model with QuMFRA | Subset of SELEX sequences with measured affinities [14] | Relative binding constants | Independent dataset binding affinity prediction [14] | Significantly improved prediction performance [14] |
Implementation Guide
Essential Research Reagents and Solutions
The successful implementation of SELEX for novel transcription factors requires carefully selected reagents and materials. The following table details the essential components and their functions:
Table 3: Essential Research Reagent Solutions for SELEX Experiments
| Reagent Category | Specific Examples | Function in Protocol | Technical Notes |
|---|---|---|---|
| DNA Library | Random oligonucleotide pool with fixed flanking sequences [14] | Source of potential binding sites; typically 20-40 bp random core | Ensure high complexity (>10¹² variants); HPLC purification |
| Expression Vector | pGEX-4T-1 (GST-tag) [14] | Recombinant TF production with affinity tag | Enables glutathione Sepharose purification |
| Chromatography Matrix | Glutathione Sepharose [14] | Separation of protein-DNA complexes from free DNA | Alternative to traditional gel shift methods |
| Binding Reaction Buffer | Tris-HCl (pH 8.0), NaCl, BSA, DTT, ZnSOâ‚„, carrier DNA [14] | Optimal binding conditions for TF-DNA interactions | Adjust salt concentration based on TF stability |
| Sequencing Platform | Illumina sequencers [15] | High-throughput analysis of enriched sequences | Enables processing of millions of sequences |
Practical Considerations for Novel Transcription Factors
When applying SELEX to poorly characterized transcription factors, several practical considerations enhance success:
- Protein Purity and Integrity: Ensure recombinant TF domains are properly folded and functional. Use structural bioinformatics to identify appropriate domain boundaries for cloning.
- Selection Stringency: Adjust binding and wash conditions across selection rounds to balance between signal-to-noise ratio and recovery of diverse binding specificities.
- Controls: Include positive control TFs with known specificities when establishing the protocol to verify system performance.
- Bioinformatic Resources: Utilize specialized databases such as HTPSELEX, which provides access to primary and derived data from high-throughput SELEX experiments [16].
SELEX and HT-SELEX provide powerful, unbiased methods for determining the binding specificities of poorly studied and novel transcription factors. By combining robust in vitro selection with advanced computational modeling, researchers can generate high-resolution TF recognition landscapes, predict genomic binding sites, and uncover tissue-specific regulatory mechanisms. The continuous development of both experimental and bioinformatic methodologies ensures that SELEX remains an indispensable tool in the functional annotation of transcription factors and the reconstruction of gene regulatory networks.
Advanced SELEX Methodologies and Applications in Biomedical Research
Systematic Evolution of Ligands by Exponential Enrichment (SELEX) is a powerful in vitro selection process used to identify aptamers—short, single-stranded DNA or RNA sequences—that bind to specific target molecules with high affinity and specificity [17] [18]. Since its development in the early 1990s, SELEX has revolutionized the field of molecular recognition by providing an alternative to antibodies with several distinct advantages, including easier synthetic production, enhanced stability, lower immunogenicity, and the ability to select under non-physiological conditions [18] [19]. The traditional SELEX process involves iterative rounds of selection where a random oligonucleotide library is incubated with a target, bound sequences are separated from unbound ones, and the selected sequences are amplified by PCR to generate an enriched library for subsequent rounds [18] [20]. This process continues until a population of high-affinity binders is obtained, typically requiring 8-15 rounds over several weeks or months [17] [19].
Despite its proven utility, conventional SELEX faces significant challenges, including being time-consuming, labor-intensive, and having a relatively low success rate [20]. In response to these limitations, several innovative SELEX variants have been developed that leverage advanced technologies to improve the efficiency and effectiveness of aptamer selection. Capillary Electrophoresis SELEX (CE-SELEX) utilizes the high resolving power of capillary electrophoresis to separate target-bound sequences based on their mobility shift, dramatically reducing selection time [17] [21]. Microfluidic SELEX employs miniaturized devices to automate the selection process, significantly reducing reagent consumption and enabling precise fluid control [22] [23] [20]. Cell-SELEX uses whole living cells as targets, allowing for the identification of aptamers that recognize proteins in their native conformation and cellular context [24] [25]. These advanced SELEX methodologies have transformed aptamer development, making it possible to isolate high-affinity aptamers in dramatically shorter timeframes—from weeks to days or even hours—while also expanding the range of accessible targets [17] [22] [23].
Capillary Electrophoresis SELEX (CE-SELEX)
Principle and Advantages
CE-SELEX represents a significant advancement in aptamer selection technology by leveraging the exceptional separation capabilities of capillary electrophoresis. In this method, the target molecule is incubated with a random sequence nucleic acid library, and the mixture is injected into a capillary for separation using free zone capillary electrophoresis [17]. The fundamental principle relies on the mobility shift that occurs when oligonucleotides bind to their targets; non-binding oligonucleotides migrate through the capillary with consistent mobility, while target-binding sequences undergo a complexation that alters their size and charge, causing them to migrate as a separate fraction [17] [21]. This distinct fraction of binding sequences is then collected at the capillary outlet for amplification and further enrichment rounds.
The CE-SELEX approach offers numerous advantages over conventional selection methods. Perhaps most significantly, it can isolate high-affinity aptamers in fewer rounds (typically 2-4 rounds) and without tedious negative selection compared to conventional SELEX methods, shortening a several-week process down to as little as a few days [17]. The selection occurs in free solution, eliminating the need for filtration or solid-phase attachment of the target, which increases the number and types of viable targets—including targets smaller than the aptamer itself [17]. CE-SELEX also provides exceptional flexibility to manipulate selection stringency by varying target concentration, separation parameters, and collection window timing [17]. Furthermore, this method is compatible with many non-natural nucleic acid libraries and modifications that cause issues for other SELEX techniques and can work with limited samples, having been successfully used with target concentrations as low as 1 pM [17].
Protocol for CE-SELEX
The CE-SELEX protocol involves several key steps and specialized reagents. Begin by preparing a 5'-FAM labeled ssDNA library consisting of a random region (typically 40 bases) flanked by 20-base constant primer regions, diluted to 400 μM in nuclease-free water [17]. Prepare separation buffer (5x TGK buffer: 125 mM Tris-HCl, 960 mM glycine, 25 mM KH₂PO₄, pH 8.3) and sample buffer that matches anticipated application conditions [17].
Procedure:
- Incubation: Mix the ssDNA library with the target molecule in sample buffer and incubate to allow binding interactions.
- Capillary Electrophoresis: Inject several nanoliters of the incubation mixture onto a bare fused silica capillary (50 μm i.d.) using an automated P/ACE MDQ Plus CE instrument. Apply separation voltage in 1x TGK buffer.
- Collection: Monitor separation by laser-induced fluorescence (LIF) detection. Collect the shifted fraction corresponding to target-ssDNA complexes at the capillary outlet.
- Amplification: PCR amplify the collected sequences using FAM-labeled forward primer and biotin-labeled reverse primer.
- Purification: Purify the amplified product using streptavidin agarose resin to generate single-stranded DNA for subsequent selection rounds.
- Iteration: Repeat steps 1-5 for 2-4 rounds until no further improvement in affinity is observed.
Critical Steps:
- Maintain sample buffer with at least 5 mM K⺠to allow formation of DNA G-quadruplex motifs if needed for binding.
- Precisely optimize collection window timing to balance specificity and recovery.
- Monitor enrichment after each round by analyzing the increasing proportion of shifted complex in capillary electrophoresis.
Applications and Performance
CE-SELEX has demonstrated exceptional performance in generating high-affinity aptamers for various targets. The method has been successfully used to select DNA aptamers with affinities in the nanomolar to picomolar range [17] [21]. For example, researchers have selected aptamers targeting neuropeptide Y using CE-SELEX, achieving high-affinity binders in significantly fewer rounds than conventional methods [17]. The technique has also been adapted in various forms, including Non-SELEX approaches that eliminate PCR amplification between rounds, further accelerating the selection process [21]. Single-step CE-SELEX represents another innovation that integrates mixing, reaction, separation, and detection into a single online step, dramatically shortening experimental time and reducing resource consumption while enhancing sample utilization from 5% to 100% [21].
Table 1: Key Advantages of CE-SELEX Over Conventional SELEX
| Parameter | CE-SELEX | Conventional SELEX |
|---|---|---|
| Selection Rounds | 2-4 rounds [17] | 8-15 rounds [17] |
| Time Required | Few days [17] | Several weeks [17] |
| Selection Environment | Free solution [17] | Solid-phase immobilization [17] |
| Target Limitations | Compatible with targets smaller than aptamer [17] | Size limitations for immobilization |
| Stringency Control | Precise via separation parameters [17] | Limited manipulation options |
| Sequence Motifs | Rare, allowing more optimization flexibility [17] | More common |
Microfluidic SELEX
Principle and Advantages
Microfluidic SELEX leverages the principles of miniaturization and automation to revolutionize the aptamer selection process. This approach utilizes integrated microfluidic chips equipped with micropumps, microvalves, micromixers, and micro nucleic acid amplification modules to perform the entire SELEX process in an automated fashion [23] [20]. The fundamental principle involves the precise manipulation of minute fluid volumes within microchannels and chambers to facilitate the binding, separation, washing, and amplification steps of SELEX in a continuous, automated system [22] [20]. These systems can implement various force fields—including hydrodynamic, electric, magnetic, and acoustic—to enhance the efficiency of aptamer selection [20].
The advantages of microfluidic SELEX are substantial. The most prominent benefit is the dramatic reduction in selection time; where conventional SELEX requires weeks, microfluidic systems can complete the entire process in hours [22] [23]. One reported system completed 7 rounds of SELEX in only 14 hours, while another achieved selection of high-affinity DNA aptamers against immunoglobulin E (IgE) in just 4 rounds requiring approximately 10 hours [22] [23]. Microfluidic systems also offer significantly reduced consumption of samples and reagents, making them cost-effective for working with precious or expensive targets [20]. The automated nature of these systems minimizes manual handling, improving reproducibility and reducing operator-induced variability [22]. Additionally, microfluidic platforms enable precise control over shear forces during washing steps, which is crucial for selecting high-affinity aptamers under physiologically relevant conditions [23]. This precise control allows researchers to optimize selection stringency by adjusting flow rates and shear forces to mimic in vivo conditions, potentially leading to aptamers with better performance in practical applications [23] [20].
Protocol for Microfluidic SELEX
Implementing microfluidic SELEX requires specialized equipment and careful optimization. Begin with an integrated microfluidic device featuring selection and amplification chambers with integrated thin-film resistive heaters and temperature sensors, interconnected by reagent transport channels [22]. The device should include mechanisms for both electrokinetic and pressure-driven transport of oligonucleotides [22].
Procedure:
- Device Preparation: Functionalize microbeads with the target molecule and immobilize them in the selection chamber using a microweir structure [22].
- Negative Selection (Optional): Introduce the random ssDNA library to non-target functionalized beads to remove non-specific binders.
- Positive Selection: Introduce the pre-cleared library to the target-functionalized beads in the selection chamber and allow binding.
- Washing: Apply precisely controlled shear forces using serpentine-shaped micropumps to remove weakly bound sequences [23].
- Elution: Thermally release bound oligonucleotides from the target using integrated heaters.
- Transfer: Electrokinetically migrate eluted oligonucleotides through a gel-filled channel to the amplification chamber [22].
- Amplification: Capture oligonucleotides on reverse primer-functionalized magnetic beads, introduce PCR reagents, and perform on-chip PCR amplification.
- ssDNA Generation: Thermally denature amplified products and transfer the ssDNA via pressure-driven flow back to the selection chamber.
- Iteration: Automatically repeat steps 3-8 for multiple rounds (typically 4-7 rounds).
Critical Steps:
- Precisely characterize and optimize micropump volumes and mixing indices for consistent operation.
- Implement a custom shear force control strategy during washing steps to enhance affinity of selected candidates [23].
- Incorporate negative and competitive selection rounds as needed to enhance specificity [23].
Applications and Performance
Microfluidic SELEX has demonstrated impressive performance in selecting high-affinity aptamers for various targets. In one notable application, researchers used an integrated microfluidic system equipped with a shear force control device to select aptamers targeting folate receptor alpha (FRα), a key biomarker for ovarian cancer diagnosis [23]. The system completed seven SELEX rounds within 14 hours, incorporating five positive selections, one negative selection, and one competitive selection round to enhance specificity [23]. The resulting top candidate aptamer displayed a dissociation constant (Kd) as low as 23 nM, which is superior to aptamers obtained through conventional SELEX [23]. The selected aptamer was successfully applied in a detection assay to quantify FRα in spiked serum samples (1-15 μg/L), demonstrating its potential for early ovarian cancer diagnosis [23].
Another study demonstrated the selection of DNA aptamers against the protein IgE with high affinity (Kd = 12 nM) in a rapid manner (4 rounds in approximately 10 hours) using a microfluidic approach that employed bead-based biochemical reactions and hybrid electrokinetic and pressure-driven transport [22]. These systems have also been adapted for cell-SELEX applications, further expanding their utility in identifying aptamers against complex cellular targets [20].
Table 2: Performance Metrics of Microfluidic SELEX Platforms
| Parameter | Reported Performance | Significance |
|---|---|---|
| Selection Time | 4 rounds in ~10 hours [22]; 7 rounds in 14 hours [23] | Dramatic reduction from weeks to hours |
| Affinity (Kd) | 12 nM for IgE [22]; 23 nM for FRα [23] | High-affinity binders comparable or superior to conventional SELEX |
| Automation Level | Full integration of binding, washing, amplification, and ssDNA generation [22] [20] | Minimal manual intervention, improved reproducibility |
| Reagent Consumption | Nanoliters to microliters per round [20] | Significant cost savings, enables work with precious targets |
| Shear Force Control | Custom serpentine micropumps for optimized washing [23] | More physiologically relevant selection conditions |
Cell-SELEX
Principle and Advantages
Cell-SELEX represents a paradigm shift in aptamer selection by using whole living cells as targets rather than purified molecules. This approach involves incubating the random oligonucleotide library with intact cells, allowing aptamers to bind to native cell surface structures in their physiological conformation and environment [24] [25]. The fundamental principle leverages the complex molecular landscape of the cell surface, enabling the identification of aptamers that recognize naturally folded proteins, protein complexes, and other cell surface components without prior knowledge of specific molecular targets [24]. The process typically involves iterative rounds of selection against target cells, counter-selection against control cells (to remove binders to common surface molecules), and amplification of bound sequences.
The advantages of Cell-SELEX are substantial and complementary to other SELEX variants. Most importantly, it allows for the discovery of aptamers against unknown cell surface biomarkers, making it particularly valuable for cancer research where specific surface profiles may not be fully characterized [24] [25]. The selected aptamers recognize their targets in native conformations with appropriate post-translational modifications, increasing the likelihood that they will function effectively in biological applications [24] [25]. Cell-SELEX can also reveal novel insights into cell surface biology; for example, one study demonstrated that mutant K-Ras expression dynamically alters cell surface composition and can cause abnormal translocation of a mitochondrial matrix protein to the cell surface without detectable changes in mRNA or protein levels [24]. This capability makes Cell-SELEX a powerful tool for investigating cell surface remodeling under different physiological and pathological conditions. Furthermore, aptamers selected through Cell-SELEX often show excellent specificity for their target cell type, able to distinguish between closely related cells based on subtle surface differences [24] [25].
Protocol for Cell-SELEX
Implementing Cell-SELEX requires careful cell culture practices and specific modifications to incorporate enhanced functionality. Begin by preparing a single-stranded DNA library with a central random region (typically 30-60 nucleotides) flanked by constant primer regions, enzymatically synthesized to incorporate modified bases such as tryptamino-dU (trp-dU) instead of dT to enhance DNA aptamer functionality by introducing artificial hydrophobic residues [24].
Procedure:
- Cell Culture: Maintain target cells (e.g., cancer cells) and control cells (e.g., non-malignant counterparts) under standard conditions.
- Negative Selection: Incubate the ssDNA library with control cells to remove sequences binding to common surface molecules. Collect unbound sequences.
- Positive Selection: Incubate the pre-cleared library with target cells. Allow binding under appropriate conditions.
- Washing: Gently wash cells to remove weakly bound or non-specifically bound sequences.
- Elution: Recover bound sequences by heating cell-aptamer complexes or using other elution methods.
- Amplification: Amplify eluted sequences by PCR or RT-PCR (for RNA aptamers).
- ssDNA Generation: Generate single-stranded DNA from amplified products for subsequent selection rounds.
- Iteration: Repeat steps 2-7 for multiple rounds (typically 5-15 rounds), monitoring enrichment of cell-specific binders.
- Aptamer Profiling: Use next-generation sequencing to analyze enriched libraries and identify candidate aptamers.
Critical Steps:
- Incorporate modified bases like trp-dU to enhance library functionality and binding diversity [24].
- Use comparative profiling with different cell types (e.g., normal vs. mutant protein-expressing cells) to identify selective aptamers [24].
- Implement careful washing conditions to maintain cell viability while removing non-specific binders.
- Monitor sequence enrichment through next-generation sequencing to identify candidate aptamers without the need for cloning [24].
Applications and Performance
Cell-SELEX has proven particularly valuable in cancer research, where it enables the identification of aptamers specific to cancer cell surfaces without prior knowledge of molecular targets. In one compelling application, researchers used comparative aptamer profiling to investigate cell surface remodeling in normal versus mutant K-Ras-expressing cells [24]. This approach revealed that mutant K-Ras expression dynamically alters cell surface composition and led to the identification of aptamers that showed specific binding to mutant K-Ras-expressing cells without requiring sequence optimization [24]. Remarkably, target identification of one aptamer revealed abnormal translocation of a mitochondrial matrix protein to the cell surface under oncogenic signaling, highlighting how Cell-SELEX can uncover previously unrecognized cell surface markers and biological phenomena [24].
The field has further evolved from Cell-SELEX to Tissue-SELEX, which selects aptamers using tissue samples to ensure optimal binding properties in more native in vivo environments [25]. These advancements include methods such as tissue slide-based SELEX, morph-X-SELEX, ex vivo-SELEX, and microfluidic tissue-SELEX, applied to various tissues including cornea, breast, ovary, lung, cardiac, and thyroid tissues [25]. Applications of these tissue-SELEX derived aptamers in drug delivery include local administration for ocular diseases and systemic administration for lung cancer, demonstrating the translational potential of this technology [25].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of innovative SELEX variants requires specific reagents and materials optimized for each platform. The following table compiles key research solutions essential for conducting CE-SELEX, Microfluidic SELEX, and Cell-SELEX experiments.
Table 3: Essential Research Reagents and Materials for Innovative SELEX Platforms
| Category | Specific Reagent/Material | Function/Application | SELEX Variant |
|---|---|---|---|
| Oligonucleotide Library | 5'-FAM labeled ssDNA library with 40 random bases flanked by 20-base primer regions [17] | Starting pool for selection | CE-SELEX |
| Modified base-incorporated library (e.g., trp-dU instead of dT) [24] | Enhanced aptamer functionality through artificial hydrophobic residues | Cell-SELEX | |
| Separation Matrix | Bare fused silica eCap Capillary (50 μm i.d., 375 μm o.d.) [17] | Separation channel for mobility-based partitioning | CE-SELEX |
| Functionalized microbeads (NHS-activated) [22] | Target immobilization for binding selection | Microfluidic SELEX | |
| Buffers & Solutions | 5x TGK separation buffer (125 mM Tris-HCl, 960 mM glycine, 25 mM KHâ‚‚POâ‚„, pH 8.3) [17] | Capillary electrophoresis separation buffer | CE-SELEX |
| Streptavidin-binding buffer (50 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA) [17] | Purification of biotinylated PCR products | All variants | |
| Amplification Components | FAM-labeled forward primer, biotin-labeled reverse primer [17] | PCR amplification with labeled primers for detection and purification | All variants |
| GoTaq Flexi DNA polymerase [22] | PCR amplification of selected sequences | All variants | |
| Specialized Equipment | P/ACE MDQ Plus Capillary Electrophoresis system [17] | Automated CE separation with LIF detection | CE-SELEX |
| Integrated microfluidic device with micropumps, microvalves, and micromixers [23] | Automated fluid handling and process integration | Microfluidic SELEX | |
| Target Materials | Purified protein targets [17] | Standard molecular targets | CE-SELEX, Microfluidic SELEX |
| Cultured cells (e.g., mutant K-RasV12-transformed MDCK cells) [24] | Complex cellular targets in native conformation | Cell-SELEX | |
| Rezivertinib | Rezivertinib, CAS:1835667-12-3, MF:C27H30N6O3, MW:486.6 g/mol | Chemical Reagent | Bench Chemicals |
| Rhosin | Rhosin, MF:C20H18N6O, MW:358.4 g/mol | Chemical Reagent | Bench Chemicals |
Workflow Comparison of SELEX Variants
The following diagram illustrates the key procedural differences and unique features of CE-SELEX, Microfluidic SELEX, and Cell-SELEX:
SELEX Workflow Comparison: Key procedural differences between three innovative SELEX variants
The development of innovative SELEX variants—CE-SELEX, Microfluidic SELEX, and Cell-SELEX—represents significant advancements in aptamer selection technology that address critical limitations of conventional SELEX. Each platform offers unique advantages: CE-SELEX provides exceptional resolution and efficiency for purified targets, Microfluidic SELEX enables unprecedented automation and speed, while Cell-SELEX allows discovery of aptamers against complex cellular targets in their native state. These technologies have dramatically reduced selection time from weeks to days or hours while maintaining or even improving the quality of selected aptamers, with reported dissociation constants in the nanomolar range [17] [22] [23]. The integration of these advanced SELEX methodologies with next-generation sequencing and bioinformatic analysis has further enhanced their power, enabling researchers to monitor enrichment in real time and identify optimal aptamer candidates more efficiently [21] [24]. As these technologies continue to evolve and converge, they hold great promise for accelerating the development of aptamers for diverse applications in genomics research, diagnostic assays, targeted drug delivery, and therapeutic interventions, particularly in the context of transcription factor binding site identification and gene regulation studies.
The identification of transcription factor binding sites (TFBS) is a cornerstone of transcriptional regulation research. Positional Weight Matrices (PWMs), also referred to as Position-Specific Scoring Matrices (PSSMs), serve as the quantitative model for representing the binding specificity of transcription factors (TFs) to DNA [26]. Derived from collections of experimentally validated binding sites, PWMs assign weighted scores to nucleotides at each position within a binding site, reflecting their relative importance in protein-DNA interactions [27] [26]. This model operates on the biophysical principle that the PWM score for a given DNA sequence is proportional to the binding free energy between the TF and DNA, with higher scores indicating stronger predicted binding [28] [29]. In the context of genomic SELEX (Systematic Evolution of Ligands by Exponential Enrichment)—a high-throughput method that identifies protein-binding sequences from random oligonucleotide libraries—PWMs provide the computational framework to transform massive sequencing data into interpretable models of TF specificity [30] [31].
Theoretical Foundation: From Binding Sites to Quantitative Models
The Mathematical Basis of PWMs
The construction of a PWM follows a defined mathematical pathway, beginning with a set of aligned DNA sequences known to bind a specific transcription factor. The process involves creating a Position Frequency Matrix (PFM), which tabulates the observed counts of each nucleotide at every position across the binding site [27] [26]. The following example PFM, constructed from 8 binding sites for a hypothetical transcription factor, illustrates this initial step:
Table: Example Position Frequency Matrix (PFM) from 8 Binding Sites
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0 | 4 | 4 | 0 | 3 | 7 | 4 | 3 | 5 | 4 | 2 | 0 | 0 | 4 |
| C | 3 | 0 | 4 | 8 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 2 | 4 |
| G | 2 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 6 | 8 | 5 | 0 |
| T | 3 | 1 | 0 | 0 | 5 | 1 | 4 | 2 | 2 | 4 | 0 | 0 | 1 | 0 |
To convert the PFM to a PWM, frequencies are transformed into log-likelihood scores using the formula:
[ S{\alpha,j} = \log2 \left( \frac{v{\alpha,j}}{q{\alpha}} \right) ]
Where (S{\alpha,j}) is the PWM score for nucleotide (\alpha) at position (j), (v{\alpha,j}) is the corrected frequency of nucleotide (\alpha) at position (j), and (q_{\alpha}) is the background genomic frequency of nucleotide (\alpha) [27] [26]. The corrected frequency is calculated to avoid zeros by incorporating pseudocounts:
[ v{\alpha,j} = \frac{n{\alpha,j} + q{\alpha} \cdot \mu}{\sum{x} n_{x,j} + \mu} ]
where (n_{\alpha,j}) is the observed count of nucleotide (\alpha) at position (j), and (\mu) is the pseudocount parameter, often set to 1 or (\sqrt{N}) where N is the number of binding sites [27] [29]. This transformation produces the final PWM, where each element represents the log-likelihood ratio of observing a particular nucleotide at a specific position compared to random chance.
Scoring DNA Sequences with PWMs
To evaluate any DNA sequence of length L against a PWM of the same length, the position-specific scores for the observed nucleotides are summed:
[ \text{score}{\text{sequence}} = \sum{j=1}^{L} S_{\text{sequence}[j], j} ]
This aggregate score provides a quantitative measure of how well the sequence matches the binding preference of the transcription factor [26]. Higher scores indicate sequences that more closely resemble known binding sites and are thus more likely to be bound by the TF in biological contexts. The score can be interpreted in terms of binding energy, as it is proportional to the free energy of binding between the TF and DNA sequence [29]:
[ E_{\text{binding}} \propto -\frac{S}{\lambda} ]
where (\lambda) is a scaling factor that enables comparison between different TFs [29].
Experimental Workflow: Genomic SELEX for PWM Derivation
Genomic SELEX represents a powerful high-throughput approach for identifying TF binding specificities and deriving accurate PWMs. The method combines iterative in vitro selection of binding sequences with high-throughput sequencing and computational analysis.
Diagram Title: Genomic SELEX Workflow for PWM Derivation
Genomic SELEX Protocol
The genomic SELEX methodology involves the following key experimental steps [30] [31]:
Library Preparation: Create a double-stranded DNA library containing random sequences (typically 20-40 bp) flanked by constant primer binding sites. Alternatively, genomic DNA fragments can be used for a more biologically relevant representation of potential binding sites.
Incubation with Transcription Factor: Mix the purified TF (produced via in vitro transcription/translation systems such as E. coli extracts or wheat germ extracts) with the DNA library in binding buffer. Incubate to allow specific protein-DNA complexes to form [9].
Partitioning of Bound Complexes: Separate protein-bound DNA sequences from unbound sequences using appropriate methods:
- Immunoprecipitation: For TFs with tags (e.g., GST, GFP), use antibody-coated beads to pull down TF-DNA complexes [9].
- Microfluidics-based Systems: In techniques like SMiLE-Seq, TFs are bound to the surface of microfluidic devices, and unbound DNA is washed away [31].
- Membrane Filtration: Use nitrocellulose filters that retain protein-DNA complexes while allowing unbound DNA to pass through.
Elution and Amplification: Recover bound DNA sequences by disrupting protein-DNA interactions (e.g., using proteinase K or high salt buffers). Amplify the eluted DNA using polymerase chain reaction (PCR) with primers complementary to the constant regions.
Iterative Selection: Use the amplified DNA as input for subsequent rounds of selection (typically 2-4 cycles) to enrich for high-affinity binding sequences.
Sequencing and Analysis: After the final selection round, subject the enriched DNA pool to high-throughput sequencing. Process the resulting sequences to identify enriched motifs.
Research Reagent Solutions for Genomic SELEX
Table: Essential Reagents and Materials for Genomic SELEX Experiments
| Reagent/Material | Function in Protocol | Specification Notes |
|---|---|---|
| Purified Transcription Factor | DNA-binding protein for selection | Full-length, preferably with affinity tag (GST, GFP) for pulldown [9] |
| Random Oligonucleotide Library | Source of potential binding sites | 20-40 bp random region with constant primer sites |
| PCR Amplification System | Amplification of selected DNA | High-fidelity DNA polymerase, dNTPs, sequence-specific primers |
| Separation Matrix | Partitioning of bound/unbound DNA | Antibody-coated beads (GST/GFP), nitrocellulose filters, or microfluidic devices [31] |
| High-Throughput Sequencer | Analysis of enriched sequences | Illumina, PacBio, or other NGS platforms |
| Motif Discovery Software | PWM construction from sequences | MEME, HOMER, STREME, ChIPMunk [9] |
Computational Methods: From Sequences to PWM Models
Motif Discovery Algorithms
Following genomic SELEX experiments, computational motif discovery tools identify enriched sequence patterns and convert them into PWMs. Multiple algorithms are available, each with specific strengths and compatibility with different data types [9]:
Table: Comparison of Motif Discovery Tools for PWM Construction
| Tool | Method | Input Data Compatibility | Key Features |
|---|---|---|---|
| MEME | Expectation-Maximization | SELEX, ChIP-seq, PBM | Discovers ungapped motifs using probabilistic modeling [9] |
| HOMER | Hypergeometric Optimization | SELEX, ChIP-seq | De novo motif discovery with known motif comparison [9] |
| STREME | Hypergeometric testing | SELEX, DAP-seq | Finds short, ungapped motifs in large datasets [9] |
| ChIPMunk | Heuristic Gibbs sampling | ChIP-seq, ChIP-chip | Fast, accurate motif discovery with minimal parameters [9] |
| Dimont | Dinucleotide weight matrices | HT-SELEX, ChIP-seq | Accounts for dinucleotide dependencies [9] |
| RCade | Machine learning | Zinc finger TFs | Specialized for zinc finger transcription factors [9] |
PWM Construction Protocol
The computational derivation of PWMs from genomic SELEX data follows this detailed protocol [31] [27]:
Sequence Preprocessing:
- Quality control: Filter raw sequencing reads based on quality scores.
- Demultiplexing: Separate sequences by barcodes if multiple samples were pooled.
- Remove adapter sequences and constant flanking regions.
- For genomic SELEX, align sequences to the reference genome if using genomic DNA fragments.
Enriched Motif Identification:
- Apply motif discovery tools (see Table above) to identify significantly enriched sequence patterns.
- For HT-SELEX data, compare sequence enrichment across multiple selection rounds.
- Set motif length based on known TF family characteristics or optimize using information content measures.
Position Frequency Matrix Construction:
- Extract all occurrences of the identified motif from the enriched sequences.
- Create a multiple sequence alignment of these occurrences.
- Tabulate the frequency of each nucleotide (A, C, G, T) at each position to form the PFM.
PWM Calculation:
- Convert nucleotide frequencies to probabilities using pseudocounts to avoid zeros:
[ v{\alpha,j} = \frac{n{\alpha,j} + q{\alpha} \cdot \mu}{\sum{x} n_{x,j} + \mu} ]
where (n{\alpha,j}) is the count of nucleotide (\alpha) at position (j), (q{\alpha}) is the background frequency of nucleotide (\alpha), and (\mu) is the pseudocount (typically (\mu = 1) or (\sqrt{N}) where N is the number of sites) [27] [29].
- Calculate log-odds scores:
[ S{\alpha,j} = \log2 \left( \frac{v{\alpha,j}}{q{\alpha}} \right) ]
PWM Validation:
- Assess PWM quality using information content:
[ ICj = \sum{\alpha} v{\alpha,j} \log2 \left( \frac{v{\alpha,j}}{q{\alpha}} \right) ]
Advanced Considerations in PWM Applications
PWM Scaling for Binding Strength Comparisons
A significant challenge in PWM applications is that raw scores are not directly comparable between different transcription factors. The scaling parameter λ enables conversion of PWM scores to binding energy estimates, allowing meaningful comparisons across TFs [29]:
[ E_{\text{binding}} = -\frac{S}{\lambda} ]
Two methods for estimating λ have been developed [29]:
Genomic Distribution Approach: Based on the distribution of PWM scores across the genome, where λ is estimated by assuming the top 0.1% of scoring sites represent true binding sites.
Residence Time Conversion: Leverages the relationship between binding energy and residence time, using known λ values for one PWM to estimate appropriate values for different PWMs of the same TF.
These approaches facilitate quantitative studies of TF binding dynamics and enhancer activity modeling, where accurate energy estimations are crucial.
Limitations and Alternative Models
While PWMs remain the standard model for TF binding specificity, they have recognized limitations. The core assumption of position independence fails to capture known dependencies between nucleotides at different positions, particularly adjacent bases [33]. More complex models have been developed to address these limitations:
- Binding Energy Models (BEMs): Incorporate energy parameters for nonindependent contributions, including adjacent dinucleotide interactions [33].
- Machine Learning Approaches: Models like gkmSVM and ExplaiNN can capture complex dependencies but require more training data and computational resources [9].
- Random Forest Models: Combining multiple PWMs into random forest classifiers can account for multiple modes of TF binding [9].
Despite these advancements, PWMs maintain widespread use due to their simplicity, interpretability, and direct connection to biophysical principles of protein-DNA interactions.
Validation and Benchmarking of PWM Models
Performance Metrics for PWM Evaluation
Rigorous validation is essential to ensure PWM quality and predictive power. Standard evaluation metrics include [32] [31]:
- Area Under Precision-Recall Curve (AUPRC): Measures performance across all classification thresholds, particularly suitable for imbalanced datasets where binding sites are sparse.
- Area Under ROC Curve (AUC ROC): Assesses the trade-off between true positive and false positive rates across score thresholds.
- Pearson Correlation: Quantifies how well PWM score differences (ΔPWM) between alternative alleles predict experimental binding differences.
- CentriMo Motif Centrality: Evaluates the enrichment of motif occurrences centrally located in ChIP-seq peaks [9].
Benchmarking Insights
Comprehensive all-against-all benchmarking studies have revealed that the best-performing PWM for a given TF often belongs to another TF, usually from the same family [28]. This underscores the importance of TF family relationships in PWM selection and application. Benchmarking-based selection of family-representative motifs has proven more effective than motif clustering-based approaches [28].
For genomic SELEX-derived PWMs, performance validation against independent in vivo data (e.g., ChIP-seq) is crucial. Recent studies demonstrate that carefully selected PWMs can achieve prediction reliability comparable to more complex models like deltaSVM, particularly for strongly bound sites [32]. When applying PWMs to predict the regulatory impact of single-nucleotide variants, performance is notably higher for SNPs with strong predicted binding (PWM score p-value < 10^-4), with correlation coefficients reaching ~0.828 compared to experimental measurements [32].
Positional Weight Matrices continue to serve as a fundamental tool in computational biology, providing a bridge between high-throughput experimental data like genomic SELEX and quantitative models of transcription factor binding specificity. The derivation of accurate PWMs requires careful execution of both wet-lab protocols—including iterative selection and sequencing—and computational methods for motif discovery and matrix construction. While the PWM model has limitations in its assumption of positional independence, ongoing developments in scaling methods, benchmarking approaches, and alternative models ensure its continued relevance in transcriptional regulation research and drug development applications. As high-throughput technologies evolve, PWMs will remain essential for interpreting regulatory variants, modeling enhancer function, and understanding the complex landscape of gene regulation.
For decades, the position weight matrix (PWM) has served as the fundamental model for representing transcription factor (TF) binding preferences, quantifying nucleotide preferences at each position of a binding site independently [34]. While PWMs form the backbone of major TF binding databases such as JASPAR and HOCOMOCO, they operate under a significant limitation: the assumption that each nucleotide in a binding site contributes independently to binding affinity [35] [36]. This simplification ignores potential dependencies between nucleotide positions, which can be critical for accurately modeling the binding landscape of transcription factors with extended or structured recognition elements.
The emergence of genomic SELEX (Systematic Evolution of Ligands by Exponential Enrichment) technologies has revolutionized our ability to characterize TF binding specificities by enabling the high-throughput screening of random DNA oligonucleotides or actual genomic DNA fragments against target transcription factors [4] [15]. Unlike chromatin immunoprecipitation (ChIP)-based methods that identify both direct and indirect binding events in vivo, SELEX identifies directly bound DNA in vitro, providing a clearer picture of intrinsic binding preferences without confounding factors like chromatin accessibility or cooperative binding [4]. When combined with high-throughput sequencing (HT-SELEX), this approach generates massive datasets of bound DNA sequences that serve as ideal training data for more sophisticated binding models.
Direct-coupling analysis (DCA), initially developed for analyzing coevolution in protein families, has recently been adapted to model TF-DNA interactions through a framework known as DCA-Scapes [35] [37]. This advanced modeling approach captures both individual nucleotide preferences and, crucially, the pairwise dependencies between different nucleotide positions within a binding site. By moving beyond the independent position assumption of PWMs, DCA-Scapes provide a more comprehensive and accurate representation of the complex recognition code between transcription factors and their DNA targets.
The DCA-Scape Framework: Principles and Components
Theoretical Foundation of Direct-Coupling Analysis
The DCA-Scape framework represents a significant paradigm shift in modeling TF-DNA interactions. At its core, the method employs a global probabilistic model that estimates a joint probability distribution over all possible DNA sequences of a fixed length (typically 20mers). This distribution is parameterized by two types of recognition parameters: local biases (hi) that capture individual nucleotide preferences at each position, and pairwise couplings (eij) that quantify interactions between different nucleotide positions [35] [37]. These parameters are formally expressed in the joint probability distribution:
P(x1, ..., xL) = (1/Z) * exp[Σhi(xi) + Σeij(xi, xj)]
where Z is a normalization constant, L is the length of the binding site (20 nucleotides), xi represents the nucleotide at position i, hi(xi) is the local bias for nucleotide xi at position i, and eij(xi, xj) is the pairwise coupling between nucleotides xi and xj at positions i and j [37].
The power of this approach lies in its ability to capture higher-order interactions within binding sites that traditional PWMs would miss. For transcription factors that recognize extended binding elements or depend on specific structural features in DNA, these pairwise couplings can be essential for accurate binding prediction [35].
Key Metrics and Computational Outputs
The DCA-Scape framework generates several key metrics that collectively provide a comprehensive view of TF-DNA interaction landscapes:
- Direct Information (DI) Pairs: DI values provide a quantitative measure of pairwise interactions between individual bases, serving as a proxy to detect coevolving nucleotide positions that may be in direct physical contact or structurally constrained [35].
- Coupling Landscapes: These visual representations depict the strength of nucleotide connectivity across all possible pairs of positions in the binding site, offering finer-scale resolution of interaction strengths compared to DI values alone [35].
- Hamiltonian Scores: This global metric sums all recognition parameters (both local biases and pairwise couplings) for a given DNA sequence, providing a quantitative prediction of how likely a particular protein is to associate with that sequence [37]. More negative Hamiltonian scores indicate more favorable TF recognition.
Table 1: Key Components of the DCA-Scape Framework
| Component | Mathematical Representation | Biological Interpretation | Application in Binding Prediction |
|---|---|---|---|
| Local Biases (hi) | hi(xi) for nucleotide xi at position i | Position-specific nucleotide preferences analogous to PWM scores | Captures independent contributions at each position |
| Pairwise Couplings (eij) | eij(xi, xj) for nucleotides at positions i and j | Interactions between nucleotide positions | Captures dependencies and structural constraints |
| Hamiltonian Score | Σhi(xi) + Σeij(xi, xj) | Overall binding affinity for a sequence | Quantitative prediction of binding likelihood |
| Direct Information | DIij for positions i and j | Strength of direct coupling between positions | Identifies critical base-pair interactions |
Experimental Training Data Requirements
The accuracy of DCA-Scape models depends critically on the quality and quantity of experimental training data. These models are typically trained using TF-bound DNA sequences from the fourth round of HT-SELEX experiments, with the initial non-selected sequence pool (round 0) serving as a background set to establish baseline nucleotide frequencies [37]. This training approach allows the model to distinguish true binding signals from random background sequences.
For robust model training, the Coevolved-TF-DNA web service utilizes a comprehensive set of 184 TFs (147 human and 37 mouse), with each model trained on thousands of bound DNA sequences obtained through HT-SELEX [37]. The resulting models are categorized into "Validated" (demonstrated high accuracy in predicting in vivo ChIP-seq data with average AUC > 0.7) and "Not-validated" groups, providing users with quality metrics for reliable application of these tools.
Quantitative Performance Comparison: DCA-Scapes vs Traditional Methods
Prediction Accuracy in Genomic Contexts
The true test of any binding site prediction method lies in its ability to accurately identify functional binding sites within complex genomic sequences. DCA-Scapes have demonstrated remarkable performance in this regard. When applied to the complete genomes of bacteriophages λ and P22, the DCA-Scape approach successfully identified known binding elements for the N protein with exceptional accuracy [35]. For λ N, the highest-ranking sequence in the entire genome corresponded precisely to the known binding site (AUC = 0.937, FDR ~ 10^-110), while for P22 N, known binding elements ranked in the top 10 out of 41,705 sequences tested (AUC = 0.9923, FDR ~ 10^-12) [35].
The statistical significance of these predictions is striking. When calculating Hamiltonian scores for genomic regions flanking the operator regions of P22 and λ N, the dominant valleys (indicating high-probability binding sites) were situated directly on known binding elements with extremely low p-values (P22 nut-boxB right p-value = 7.3×10^-12, P22 nut-boxB left p-value = 3.2×10^-13, λ nut-boxB right p-value = 3.5×10^-13, and λ nut-boxB left p-value = 6.3×10^-15 under one-tailed z-test) [35]. This level of precision far exceeds what can typically be achieved with traditional PWM-based approaches, particularly for transcription factors with extended binding motifs.
Functional Validation and Cell-Type Specificity
Beyond mere sequence prediction, DCA-Scapes show promise in addressing the challenge of cell-type specific TF binding. Recent research has revealed that a surprisingly large proportion of TFs (approximately two-thirds) show statistically significant cell-type specific DNA binding signatures [36]. While traditional models assume that a TF's inherent DNA-binding preferences are invariant across cell types, the reality is more complex, with factors like cooperative binding with different partners, post-translational modifications, and steric hindrance influencing binding specificity in different cellular contexts.
DCA-Scapes, with their ability to capture more complex binding patterns, are better equipped to model these cell-type specific variations than traditional PWMs. When combined with deep learning approaches like SigTFB (Signatures of TF Binding), these models can detect and quantify cell-type specificity in a TF's genomic binding sites, potentially revealing how the same TF can perform different regulatory roles in different cellular environments [36].
Table 2: Performance Comparison of Binding Site Prediction Methods
| Method | Theoretical Basis | Key Assumptions | Genome-Wide Prediction Accuracy | Limitations |
|---|---|---|---|---|
| Position Weight Matrix (PWM) | Position-independent nucleotide frequencies | Independence between nucleotide positions | Moderate (high false positive rate) | Cannot capture dependent nucleotide effects |
| DCA-Scape | Global pairwise coupling analysis | Pairwise interactions suffice to explain binding specificity | High (AUC >0.9 for validated TFs) | Computationally intensive; requires large training datasets |
| ChIP-seq | Empirical in vivo binding data | Binding reflects direct and indirect interactions | High but context-dependent | Does not distinguish direct from indirect binding |
| Genomic SELEX | Empirical in vitro binding data | Binding reflects direct TF-DNA interactions | High for intrinsic specificity | May miss in vivo contextual factors |
Experimental Protocols: Implementing Genomic SELEX and DCA Analysis
Genomic SELEX Workflow
The genomic SELEX protocol provides the essential experimental foundation for building accurate DCA-Scape models. This method isolates DNA fragments bound by a transcription factor from a pool of genomic DNA fragments, offering the advantage of selecting binding sites from their native genomic context [4]. The step-by-step protocol proceeds as follows:
Protein Preparation: Generate a recombinant DNA-binding domain of the transcription factor (e.g., GST-Fezf2 zinc finger domain) and immobilize it to a solid surface [4].
Genomic DNA Fragmentation: Digest genomic DNA (e.g., zebrafish genomic DNA) with an appropriate restriction enzyme (e.g., Sau3A1) to generate fragments of optimal size for protein binding [4].
Selection Rounds:
- Incubate immobilized TF with genomic DNA fragments
- Wash to remove non-specifically bound DNA
- Elute specifically bound DNA fragments
- Amplify eluted DNA using PCR with appropriate adapters
- Repeat for multiple rounds (typically 3-4 rounds) to enrich for high-affinity binders [4]
Sequencing Preparation: Add sequencing adapters and unique barcodes to amplified products from each round to enable multiplexed high-throughput sequencing [35] [15].
High-Throughput Sequencing: Sequence the selected DNA fragments using platforms such as Illumina to generate comprehensive datasets of bound sequences [15].
Genomic SELEX and DCA Modeling Workflow
DCA Model Construction Protocol
Once genomic SELEX data has been generated, the following computational protocol is used to construct the DCA-Scape model:
Sequence Preprocessing:
- Quality control of sequencing reads
- Demultiplexing of different selection rounds
- Alignment and trimming to isolate the random region
- Elimination of duplicate sequences
Background Model Estimation:
- Use round 0 (initial library) sequences to establish background nucleotide frequencies
- Generate a null model of one million random sequences with similar genomic nucleotide bias [37]
Parameter Estimation:
Model Validation:
- Calculate Hamiltonian scores for known binding sites from independent experiments (e.g., ChIP-seq)
- Compute AUC scores to quantify prediction accuracy
- Classify models as "validated" (AUC > 0.7) or "not-validated" [37]
Application to Genomic Sequences:
- Scan input sequences with a sliding window of 20nt
- Calculate Hamiltonian score for each 20mer
- Compute p-values based on the null model
- Report significant binding sites
Research Reagent Solutions: Essential Tools for Implementation
Table 3: Essential Research Reagents for Genomic SELEX and DCA Analysis
| Reagent/Tool | Function | Example Sources/Implementations |
|---|---|---|
| Recombinant TF DNA-binding domain | Protein for in vitro binding assays | GST-tagged zinc finger domains [4] |
| Restriction endonucleases | Genomic DNA fragmentation | Sau3A1 for partial digestion [4] |
| Magnetic resin | Immobilization of recombinant TF | Magnetic GST-binding beads [35] |
| High-fidelity PCR enzymes | Amplification of selected DNA | Phusion or similar high-fidelity polymerases [15] |
| High-throughput sequencer | Sequencing of selected DNA fragments | Illumina platforms [15] |
| Coevolved-TF-DNA webserver | DCA-Scape model application | dcascapes.org/TOOLS.html [38] [37] |
| eme_selex pipeline | Bioinformatic analysis of HT-SELEX data | GitHub: kashyapchhatbar/eme_selex [15] |
Application Notes: Practical Implementation Guidelines
Using the Coevolved-TF-DNA Web Server
For researchers wishing to apply DCA-Scape models without building them from scratch, the Coevolved-TF-DNA web service offers an accessible interface (dcascapes.org/TOOLS.html) [38]. The practical workflow for using this resource involves:
Transcription Factor Selection: Choose from 184 available TFs (147 human, 37 mouse) from the dropdown list [38] [37].
Sequence Input:
- Select input method (direct paste or FASTA upload)
- Ensure sequences are between 20nt and 1,000,000nt
- For genomic scans, sequences up to 4,000nt are recommended for manageable processing time [38]
Parameter Configuration:
- Input desired number of hits (0 to display none)
- Select display options for most likely (negative Hamiltonian) or unlikely binding results
- Choose between table, graph, or combined output formats [38]
Result Interpretation:
- Hamiltonian scores are displayed with 20mer sequence, position, and sequence origin
- More negative scores indicate more favorable binding
- P-values are calculated based on the null model of random sequences [37]
Troubleshooting and Optimization
Successful implementation of DCA-Scapes requires attention to several potential challenges:
Sequence Length Considerations: For sequences longer than 4,000nt, select "Display only the table(s) for Hamiltonian scores" to avoid visualization delays [38].
Model Selection Priority: Prioritize "Validated TFs" (18 with ChIP-seq validation, AUC>0.7) over "not-validated" models (129 TFs) for more reliable predictions [37].
Handling Promiscuous Binding: For TFs with promiscuous binding patterns, use the eme_selex bioinformatic pipeline to analyze all possible k-mers and detect multiple binding modes [15].
Cell-Type Specific Considerations: When working with TFs known to exhibit cell-type specific binding (approximately two-thirds of TFs), consider complementing DCA-Scape predictions with cell-type specific chromatin accessibility data [36].
The development of DCA-Scapes represents a significant advancement in our ability to model the complex interactions between transcription factors and DNA. By moving beyond the limitations of traditional PWMs and capturing pairwise dependencies between nucleotide positions, these models offer more accurate predictions of binding sites across genomic sequences. When combined with the experimental power of genomic SELEX, DCA-Scapes provide a framework for deciphering the transcriptional regulatory code at unprecedented resolution.
As the field progresses, the integration of these advanced binding models with cell-type specific information and three-dimensional genomic architecture promises to further enhance our understanding of transcriptional regulation. The availability of user-friendly web services like the Coevolved-TF-DNA server makes these powerful tools accessible to a broad range of researchers, accelerating discoveries in gene regulatory networks and their implications for development, disease, and therapeutic intervention.
Applications in Target Discovery and Cell-State Specific Binding Analysis
Transcription factors (TFs) are crucial proteins that regulate gene expression by binding to specific DNA sequences in regulatory regions. Understanding the precise binding specificities of TFs is fundamental to deciphering transcriptional regulatory networks. Genomic SELEX (Systematic Evolution of Ligands by EXponential Enrichment) has emerged as a powerful discovery tool for identifying genomically encoded functional domains in nucleic acid molecules that recognize and bind specific ligands [13]. Unlike traditional SELEX that begins with libraries of synthetically derived random DNA molecules, Genomic SELEX starts from libraries derived from genomic DNA, ensuring that only naturally occurring aptamers encoded in the screened genome will be identified [13]. This approach is particularly valuable for studying TF binding specificities, as it enables researchers to work with biologically relevant DNA sequences while maintaining the controlled conditions of in vitro assays.
The core principle of Genomic SELEX involves iterative cycles of selection and amplification to enrich DNA sequences with high binding affinity for a protein target of interest [39]. When combined with high-throughput sequencing technologies, Genomic SELEX transforms into a powerful discovery tool for identifying genomic aptamers—functional domains within genomically encoded RNA or DNA molecules that recognize and bind ligands such as proteins [13]. This methodology is especially useful for investigating TFs with unknown DNA-binding specificities or for exploring non-canonical binding sites that might be missed by in vivo methods due to chromatin constraints or cellular contexts.
Application in Transcription Factor Target Discovery
Biological Context and Significance
Comprehensive identification of TF binding specificities is essential for understanding gene regulatory networks and their roles in development, cellular differentiation, and disease. Traditional methods for studying protein-DNA interactions often face limitations: computational predictions primarily rely on conservation and structural stability, while in vivo approaches like ChIP-seq require that the searched RNAs be expressed during the conditions used for RNA extraction [13]. Genomic SELEX bypasses these limitations by enabling the discovery of DNA binding specificities independent of cellular context or expression levels, making it particularly valuable for studying TFs that are difficult to assay in vivo or that bind DNA with weak but biologically relevant affinities.
Recent research has revealed that lower affinity DNA binding sites are widespread and play crucial roles in precise spatiotemporal control of gene expression during development [12]. The ability to detect these sites is critical because they can create overlapping binding opportunities that collectively modulate TF genomic occupancy in vivo. Studies have shown that TF binding is not necessarily determined by individual high-affinity binding sites, but rather by the sum of multiple, overlapping binding sites [12]. This paradigm shift underscores the importance of sensitive in vitro methods like Genomic SELEX that can comprehensively map both high and low-affinity interactions.
Methodological Approach
The standard Genomic SELEX protocol for TF binding site identification involves several key stages. First, a genomic DNA library is prepared from the organism of interest, ensuring the DNA is of high quality and reliably represents the genome [13]. Specialized primers are designed for library construction, including "hyb"- and "fix"-primers with constant sequence regions flanking randomized genomic regions [13]. The purified TF (often as a tagged fusion protein for ease of purification) is incubated with the genomic library, and protein-DNA complexes are isolated from unbound DNA using methods such as affinity chromatography, gel shift, or filtration [14] [39].
The bound DNA sequences are subsequently amplified by PCR to create an enriched library for the next selection round. Typically, multiple selection rounds (3-8 cycles) are performed to sufficiently enrich for specific binders [39]. After the final round, the enriched sequences are identified through high-throughput sequencing, and motif discovery tools are applied to identify the consensus binding motif for the TF.
A key methodological consideration is the selection stringency, which can be controlled by adjusting the protein concentration and binding conditions [39]. Lower protein concentrations favor selection of higher affinity sites, while higher concentrations allow detection of lower affinity interactions. Recent advancements have introduced fixed-stringency/high-throughput SELEX approaches that maintain constant selection conditions across rounds, enabling more quantitative modeling of protein-DNA interaction parameters [39].
Experimental Protocol: Genomic SELEX for TF Binding Site Identification
Materials Required:
- High-quality genomic DNA from target organism
- Purified transcription factor (e.g., GST-tagged fusion protein)
- Primers for library construction and amplification
- Affinity matrix (e.g., glutathione sepharose for GST-tagged proteins)
- PCR reagents and equipment
- High-throughput sequencing platform
Procedure:
- Library Preparation:
- Fragment genomic DNA to appropriate size (typically 100-500 bp)
- Ligate with hybrid primers containing constant regions for amplification
- Confirm library diversity and quality by sequencing
Selection Rounds:
- Incubate purified TF (∼10â»â¸ M) with genomic library (∼10â»â¸ M) in binding buffer for 1 hour at room temperature [14]
- Separate protein-DNA complexes from unbound DNA using appropriate method (affinity chromatography for tagged proteins)
- Wash to remove non-specifically bound DNA
- Elute specifically bound DNA sequences
- Amplify eluted DNA by PCR for next selection round
Sequencing and Analysis:
- After 3-8 selection rounds, prepare sequencing library from enriched DNA
- Perform high-throughput sequencing (Illumina platform recommended)
- Analyze sequences using motif discovery tools (MEME, HOMER, STREME)
- Validate binding specificities through independent assays (e.g., EMSA, PBM)
Technical Notes:
- Include control selections without protein to identify background binding
- Monitor enrichment progression by quantifying recovery rates after each round
- Consider using different stringency conditions to capture both high and low-affinity sites
- For TFs with known binding preferences, include positive control sequences to monitor selection efficiency
Performance Data and Applications
Table 1: Comparison of Genomic SELEX with Other High-Throughput TF Binding Assays
| Assay Type | Sensitivity for Low-Affinity Sites | Throughput | Quantitative Accuracy | Primary Applications |
|---|---|---|---|---|
| Genomic SELEX | Moderate | High | Moderate | De novo motif discovery, binding site identification |
| PADIT-seq | High | Very High | High | Comprehensive affinity profiling, low-affinity site detection |
| HT-SELEX | Low | High | Low | Rapid screening of binding preferences |
| Protein Binding Microarrays (PBM) | Moderate | High | High | Specificity quantification, k-mer affinity measurement |
| ChIP-seq | Context-dependent | Moderate | N/A | In vivo binding site identification |
Recent studies have demonstrated the power of sensitive in vitro binding assays to reveal previously unrecognized aspects of TF binding specificity. For example, PADIT-seq—a novel technology to measure protein affinity to DNA by in vitro transcription and RNA sequencing—has enabled researchers to comprehensively assay the binding preferences of TFs to all possible 10-bp DNA sequences, detecting hundreds of novel, lower affinity binding sites [12]. This expanded repertoire revealed that nucleotides flanking high-affinity DNA binding sites create overlapping lower-affinity sites that together modulate TF genomic occupancy in vivo.
In large-scale benchmarking studies, researchers have processed results from 4,237 experiments for 394 TFs using five different experimental platforms, including Genomic SELEX (GHT-SELEX) and HT-SELEX [9]. This work highlighted that motifs with low information content, in many cases, describe well the binding specificity assessed across different experimental platforms, challenging previous assumptions about motif quality assessment [9].
Application in Cell-State Specific Binding Analysis
Biological Context and Significance
The ability to identify cell-state specific binding profiles is crucial for understanding cellular differentiation, response to stimuli, and disease mechanisms. Traditional SELEX approaches against purified proteins cannot capture the complexity of cellular environments where TF binding specificity may be influenced by post-translational modifications, co-factors, or cellular context. Cell-SELEX addresses this limitation by using intact cells as selection targets, enabling identification of aptamers that recognize cell-surface markers or reflect cell-state specific binding profiles.
This approach is particularly valuable for identifying biomarkers for disease states such as cancer, where cell-surface protein expression patterns differ between normal and malignant cells. By performing parallel selections against different cell types, researchers can identify aptamers that specifically recognize target cells while minimizing binding to control cells. This differential binding strategy has proven effective for identifying cell-type specific molecular signatures without prior knowledge of specific membrane biomarkers [40].
Methodological Approach: Differential Binding Cell-SELEX
The differential binding cell-SELEX workflow adapts tools from functional genomics to achieve more informative metrics about the selection process [40]. This method involves parallel selection against target and control cell lines, followed by high-throughput sequencing and statistical analysis to identify sequences with statistically significant binding differences.
The key innovation of this approach is the application of bioinformatics tools like edgeR—commonly used for RNA-seq analysis—to identify differentially abundant aptamers between target and control cells [40]. This statistical framework enables rigorous assessment of binding specificity and helps eliminate sequences that bind nonspecifically to both cell types.
A recent study demonstrated this approach by performing aptamer selection against a clear cell renal cell carcinoma (ccRCC) RCC-MF cell line using the RC-124 cell line from healthy kidney tissue for negative selection [40]. Despite challenges in achieving complete selectivity through conventional monitoring, the differential analysis revealed cell-specific binding patterns that would have been missed by traditional enrichment-based selection criteria.
Experimental Protocol: Differential Binding Cell-SELEX
Materials Required:
- Target and control cell lines
- Initial randomized oligonucleotide library
- Cell culture reagents and equipment
- Flow cytometer (for binding monitoring)
- PCR reagents and equipment
- High-throughput sequencing platform
Procedure:
- Cell Preparation:
- Culture target and control cells under standard conditions
- Harvest cells at appropriate confluence, ensure viability >95%
Selection Rounds:
- Incubate oligonucleotide library with target cells (positive selection)
- Wash to remove unbound sequences
- Elute cell-bound sequences
- Counter-select eluted sequences against control cells (negative selection)
- Collect unbound sequences for amplification
- Amplify sequences for next selection round
Differential Binding Assessment:
- After multiple selection rounds (e.g., 4th and 11th cycles), perform parallel binding to target and control cells
- Retrieve bound sequences from both cell types separately
- Prepare sequencing libraries with sample-specific barcodes
- Perform high-throughput sequencing
Bioinformatic Analysis:
- Process sequencing data (quality filtering, adapter removal)
- Align sequences to reference library
- Use edgeR or similar tools to identify differentially abundant sequences
- Apply multiple testing correction (Benjamini-Hochberg procedure)
- Filter sequences by fold-change and statistical significance
Technical Notes:
- Use consistent cell numbers and viability across selections
- Normalize for sequencing depth between samples
- Include technical replicates to assess variability
- Consider using flow cytometry to monitor overall library binding progression
- Validate identified aptamers using independent methods (e.g., fluorescence microscopy, flow cytometry)
Performance Data and Applications
Table 2: Analysis Metrics from Differential Binding Cell-SELEX Experiment
| Parameter | 4th Selection Cycle | 11th Selection Cycle |
|---|---|---|
| Total Unique Sequences | 3,627,938 | 503,107 |
| Sequences After Filtering | 1,015 | 35,859 |
| Statistical Power | Lower (early selection) | Higher (late selection) |
| Sequence Diversity | Higher | Lower (enrichment) |
| Identification of Differential Binders | Possible but noisier | More reliable |
In a proof-of-concept study, researchers employed the differential binding approach after the 4th and 11th selection cycles of cell-SELEX [40]. After data clean-up, they identified 3,627,938 unique sequences within the 4th selection cycle experiment and 503,107 unique sequences in the 11th selection cycle experiment. After filtering reads by edgeR to remove sequences with lower counts per million (CPM) than two per sample and those present in less than two replicates, they retained 1,015 unique sequences for the 4th cycle aptamers and 35,859 sequences for the 11th cycle aptamers [40].
This approach enabled the identification of statistically significant differential binding despite the failure of conventional methods to achieve complete selectivity. The adaptation of RNA-seq analysis tools for SELEX data demonstrates how methodologies from functional genomics can enhance the analysis of aptamer selection experiments, providing more robust and statistically valid results.
Visualization of Methodologies
Genomic SELEX Workflow
Differential Binding Cell-SELEX Design
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Genomic SELEX Applications
| Reagent/Material | Function/Purpose | Examples/Specifications |
|---|---|---|
| Tagged TF Construct | Enables purification and immobilization | GST-, His-, or Flag-tagged fusion proteins [14] |
| Genomic DNA Library | Source of naturally occurring binding sites | High-quality DNA from target organism; 100-500 bp fragments [13] |
| Specialized Primers | Library construction and amplification | "hyb"- and "fix"-primers with constant regions [13] |
| Affinity Matrix | Separation of protein-DNA complexes | Glutathione sepharose (GST-tags), nickel-NTA (His-tags) [14] |
| High-Fidelity Polymerase | Error-free amplification during selection rounds | Phusion, Q5 polymerases |
| Binding Buffers | Maintain protein activity and proper nucleic acid folding | Near-physiological conditions with additives (Zn²⺠for zinc fingers) [14] |
| Cell Culture Materials | For cell-SELEX applications | Validated cell lines, culture media, supplements |
| HTS Platform | Sequence analysis of enriched pools | Illumina platforms for millions of reads [40] |
| Bioinformatics Tools | Data analysis and motif discovery | MEME, HOMER, STREME, FASTAptamer, edgeR [9] [40] |
| Ribocil-C | Ribocil-C, MF:C21H21N7OS, MW:419.5 g/mol | Chemical Reagent |
Genomic SELEX methodologies provide powerful and versatile approaches for transcription factor binding site identification and cell-state specific binding analysis. The integration of these experimental protocols with advanced bioinformatics tools and high-throughput sequencing technologies enables researchers to obtain comprehensive insights into TF binding specificities under controlled in vitro conditions or in biologically relevant cellular contexts. The continued refinement of these approaches, including the development of more sensitive binding assays and sophisticated analytical frameworks, promises to further enhance our understanding of transcriptional regulation and its role in health and disease.
As these methodologies become more accessible and standardized, they offer tremendous potential for drug development professionals seeking to identify novel therapeutic targets, understand mechanisms of transcriptional dysregulation in disease, and develop targeted interventions that modulate specific transcriptional programs. The applications outlined in this document provide a foundation for researchers to implement these powerful techniques in their own investigations of transcription factor binding specificities.
Optimizing Genomic SELEX: From Bench to Bioinformatics
Addressing Platform-Specific Biases in HT-SELEX and PBM Data
Transcription factor (TF) binding specificity is commonly represented by positional weight matrices (PWMs) derived from various high-throughput experimental platforms [9]. Among these, High-Throughput Systematic Evolution of Ligands by Exponential Enrichment (HT-SELEX) and Protein Binding Microarrays (PBMs) have emerged as powerful in vitro technologies for determining DNA binding specificities of hundreds of TFs [12]. However, each platform exhibits distinct technical biases that significantly impact the reliability and completeness of the resulting binding models. HT-SELEX, which involves iterative selection of protein-binding sequences from a random oligonucleotide library, tends to saturate quickly with the strongest binding sequences, creating a systematic under-representation of lower-affinity interactions [9] [12]. Conversely, PBM assays, while comprehensive in their coverage of possible k-mers, may be confounded by variable flanking sequences surrounding each tested k-mer and struggle to reliably detect lower-affinity binding sites [12]. These platform-specific limitations have profound implications for understanding transcriptional regulation, particularly as lower-affinity binding sites are increasingly recognized as important for precise spatiotemporal control of gene expression [12]. This application note provides detailed methodologies for identifying, quantifying, and addressing these biases through integrated experimental and computational approaches.
Quantitative Comparison of Platform Characteristics and Biases
Table 1: Characteristics and Specific Biases of Major TF Binding Assays
| Platform | Throughput | Affinity Range | Primary Biases | Strengths |
|---|---|---|---|---|
| HT-SELEX | High | Primarily high-affinity | Rapid saturation with strong binders; Under-representation of low-affinity sites [9] [12] | Unbiased exploration of sequence space; No requirement for specific antibodies [15] |
| PBM | High | Broad but limited sensitivity for low-affinity | Variable flanking sequence effects; Fixed threshold challenges across TFs [12] | Comprehensive coverage of k-mer space; Highly reproducible [12] |
| PADIT-seq | High | High to low-affinity | Newer method with less established benchmarks | Detects hundreds of novel lower-affinity sites; Direct functional coupling [12] |
| ChIP-seq | Medium | Context-dependent | Requires specific antibodies; Influenced by cellular context [9] | In vivo binding information; Genome-wide binding sites [41] |
Table 2: Performance Comparison of Motif Discovery Tools Across Platforms
| Tool | Compatible Platforms | Strengths | Limitations |
|---|---|---|---|
| MEME | Multiple | Classic, widely-used [9] | Not all tools compatible with all data types [9] |
| HOMER | Multiple | Popular for high-throughput data [9] | May be technically handicapped when not used by creators [9] |
| RCade | Specialized | Excellent for zinc finger TFs [9] | Exclusively for zinc finger TFs [9] |
| DimontHTS | HT-SELEX | Specialized adaptation for HT-SELEX data [9] | Limited to specific platform [9] |
| ExplaiNN | Multiple | Advanced method [9] | Not specified in search results |
| DeepAptamer | SELEX | Hybrid deep learning model; Predicts binding affinities from early SELEX rounds [42] | Primarily focused on aptamer discovery [42] |
Experimental Protocols for Bias Assessment and Mitigation
HT-SELEX Protocol for Comprehensive Binding Site Identification
Summary: This optimized HT-SELEX protocol enables unbiased determination of preferred target motifs of DNA-binding proteins in vitro through iterative selection from random oligonucleotide libraries [15].
Key Materials:
- Random DNA library (typically 1013 to 1016 sequences)
- Recombinant DNA-binding protein
- PCR amplification reagents
- Illumina sequencing adapters
- Binding and wash buffers
Procedure:
- Library Preparation: Synthesize a random oligonucleotide library with fixed flanking sequences for amplification.
- Incubation: Mix protein with DNA library in binding buffer.
- Partitioning: Separate protein-bound DNA from unbound DNA.
- Elution and Amplification: Recover bound DNA and amplify using PCR.
- Sequencing: Prepare Illumina sequencing libraries from amplified DNA.
- Iteration: Repeat steps 2-5 for multiple rounds (typically 3-8 rounds).
- Bioinformatic Analysis: Use the eme_selex pipeline to analyze enrichment of all possible k-mers and detect promiscuous DNA binding [15].
Critical Considerations:
- Monitor diversity reduction through sequencing of intermediate rounds.
- Avoid excessive rounds that lead to oversaturation with strongest binders.
- Use early-round data for computational recovery of lower-affinity binders.
Cross-Platform Validation Protocol Using PADIT-seq
Summary: Protein Affinity to DNA by in Vitro Transcription and RNA sequencing (PADIT-seq) provides a sensitive method for detecting lower-affinity binding sites missed by HT-SELEX and PBM [12].
Key Materials:
- PADIT-seq reporter library (all possible 10-bp DNA sequences)
- In vitro transcription and translation system
- T7 RNA Polymerase
- ALFA-nbALFA interaction system
- Illumina sequencing platform
Procedure:
- Library Construction: Assemble reporter library with all possible 10-bp DNA sequences as candidate TF binding sites.
- TF Binding: Mix PADIT-seq reporter library with TF of interest.
- Transcriptional Output: Measure reporter gene expression proportional to TF-DNA binding strength.
- Sequencing: Perform Illumina sequencing of reporter RNAs.
- Quantification: Calculate PADIT-seq activity as log2(DBD / 'no-DBD') values using DESeq2 [12].
- Validation: Compare with uPBM and HT-SELEX data to identify sites uniquely detected by PADIT-seq.
Critical Considerations:
- Include 'no DBD' controls for baseline measurement.
- Perform custom PBM validation for selected lower-affinity sites.
- Use AUROC analysis to compare with uPBM E-scores and Z-scores.
Integrated Workflow for Comprehensive TF Binding Characterization
Diagram 1: Integrated workflow for comprehensive TF binding characterization across multiple platforms.
Computational Approaches for Bias Correction
Advanced Motif Discovery with Multi-Tool Integration
Protocol for Robust PWM Derivation:
- Multi-Platform Data Integration: Process results from multiple experimental platforms (ChIP-Seq, GHT-SELEX, HT-SELEX, SMiLE-Seq, PBM) for consistent motif discovery [9].
- Tool Diversification: Apply multiple motif discovery tools (MEME, HOMER, ChIPMunk, Autoseed, STREME, Dimont, ExplaiNN, RCade, gkmSVM) to the same dataset [9].
- Cross-Platform Benchmarking: Evaluate PWM performance across different experimental types using multiple dockerized benchmarking protocols [9].
- Expert Curation: Manually approve experiments that yield consistent motifs across platforms and replicates.
- Advanced Modeling: Combine multiple PWMs into random forest models to account for multiple modes of TF binding [9].
Deep Learning Approaches for Bias Correction
DeepAptamer Implementation Protocol:
- Data Preparation: Collect SELEX data from multiple rounds, including early rounds with greater diversity.
- Model Architecture: Implement hybrid neural network combining convolutional neural networks and bidirectional long short-term memory [42].
- Feature Integration: Incorporate both sequence composition and structural features to predict aptamer binding affinities.
- Training: Train comprehensive model on SELEX data to identify high-affinity sequences from unenriched early rounds [42].
- Validation: Experimentally validate predictions to confirm recovery of genuine binders missed by conventional analysis.
Diagram 2: Computational workflow for deep learning-based correction of platform-specific biases.
Table 3: Key Research Reagent Solutions for Bias-Aware TF Binding Studies
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Random DNA Library | Provides diverse sequence space for in vitro selection | Size typically 1013-1016 sequences; Fixed flanking regions for amplification [43] |
| PADIT-seq Reporter Library | Comprehensive testing of all possible 10-bp sequences | Contains 1,048,576 sequences; Enables detection of low-affinity sites [12] |
| Codebook Motif Explorer | Catalog of motifs and benchmarking results | Interactive resource at https://mex.autosome.org [9] |
| eme_selex Pipeline | Bioinformatic analysis of HT-SELEX data | Quantifies all possible k-mers to detect promiscuous DNA binding [15] |
| Recombinant TFs | Protein for in vitro binding assays | Produced via GST-tagged IVT (E. coli), GFP-tagged IVT (wheat germ), or whole cell lysate [9] |
| DeepAptamer Model | Hybrid deep learning for affinity prediction | Identifies high-affinity sequences from early SELEX rounds [42] |
Addressing platform-specific biases in HT-SELEX and PBM data requires an integrated approach combining multiple experimental platforms, computational tools, and validation strategies. The protocols outlined herein enable researchers to identify technical limitations in their TF binding data and implement appropriate corrective strategies. As the field advances, the development of more sensitive assays like PADIT-seq, coupled with advanced deep learning models, will progressively overcome current limitations in detecting the full spectrum of TF-DNA interactions. Furthermore, resources like the Codebook Motif Explorer provide centralized access to curated motifs and benchmarking results, supporting continued progress in understanding transcriptional regulation. By systematically addressing platform-specific biases, the research community can build more accurate models of TF binding that better reflect the complexity of gene regulatory networks.
The identification of transcription factor binding sites (TFBSs) is fundamental to deciphering the regulatory code that controls gene expression. DNA sequence motifs, representing the binding specificity of transcription factors (TFs), are central to this effort. The position weight matrix (PWM) remains the most widely used model to represent these motifs, despite the development of more complex models [9] [44]. Motif discovery tools that generate PWMs from experimental data are crucial for annotating regulatory regions, interpreting the impact of genetic variation, and understanding regulatory networks.
The performance of these tools, however, is highly dependent on the type of experimental data from which motifs are derived. High-throughput methods like ChIP-Seq, HT-SELEX, and protein binding microarrays (PBMs) each have distinct technical characteristics, biases, and resolutions, which in turn influence the effectiveness of motif discovery algorithms [9] [44]. Until recently, systematic benchmarking of motif discovery tools across a diverse range of experimental platforms was lacking. This application note synthesizes findings from a major consortium effort that addressed this gap by evaluating ten motif discovery tools across five experimental platforms, providing a data-driven guide for researchers seeking to identify optimal tools for their specific experimental context [9].
Key Benchmarking Findings from the GRECO-BIT Initiative
The Gene Regulation Consortium Benchmarking Initiative (GRECO-BIT) conducted a large-scale cross-platform analysis to evaluate the performance of PWM-based motif discovery. The study processed 4,237 experiments for 394 human transcription factors, assayed using five different platforms [9]. After rigorous human curation, a subset of 1,462 high-quality datasets across 236 TFs was approved for detailed benchmarking. The key quantitative results are summarized in the table below.
Table 1: Performance Overview of Motif Discovery Tools Across Experimental Platforms
| Motif Discovery Tool | Underlying Algorithm | Compatible Data Types (from study) | Key Performance Findings |
|---|---|---|---|
| HOMER | Hypergeometric Optimization | ChIP-Seq, GHT-SELEX, HT-SELEX, SMiLE-Seq, PBM | Consistently high-ranking PWMs; widely compatible and reliable [9] |
| MEME | Expectation-Maximization (EM) | ChIP-Seq, GHT-SELEX, HT-SELEX, SMiLE-Seq, PBM | Robust performance across multiple platforms; a established standard [9] |
| STREME | EM-based | ChIP-Seq, GHT-SELEX, HT-SELEX, SMiLE-Seq, PBM | Effective for identifying short, core motifs in high-throughput data [9] |
| ChIPMunk | Greedy Algorithm | ChIP-Seq, GHT-SELEX | Efficient and accurate for in vivo data like ChIP-Seq peaks [9] |
| Dimont/DimontHTS | Thermodynamic Modeling | HT-SELEX (specialized adaptation) | Specialized for HT-SELEX data; models binding energy [9] |
| RCade | Custom Model | Zinc Finger TFs (Platform-agnostic) | Specialized for zinc finger transcription factors [9] |
| ExplaiNN | Deep Learning | ChIP-Seq, GHT-SELEX, HT-SELEX, SMiLE-Seq, PBM | Advanced method capturing complex binding patterns [9] |
| rGADEM | Genetic Algorithm + EM | ChIP-Seq | Identified in other studies as a top performer for ChIP-Seq data [44] |
The benchmarking revealed several critical insights that challenge conventional assumptions in the field. Notably, the nucleotide composition and information content of a motif were not correlated with its performance in cross-platform validation. Motifs with low information content often described binding specificity effectively across different experiments, indicating that weak motifs can be biologically relevant and technically reproducible [9]. Furthermore, the study demonstrated that combining multiple PWMs into a random forest model could account for multiple modes of TF binding, outperforming single PWMs and highlighting the potential of ensemble approaches [9].
Experimental Platforms and Their Impact on Motif Discovery
The choice of experimental platform is a primary determinant of successful motif discovery. Each method captures TF-DNA interactions under different conditions, with inherent strengths and biases.
Table 2: Characteristics of Key Experimental Platforms for TF Binding Profiling
| Experimental Platform | Methodology | Resolution | Key Advantages | Key Limitations / Biases |
|---|---|---|---|---|
| ChIP-Seq (in vivo) | Chromatin Immunoprecipitation with sequencing | 100-1000 bp regions | Captures binding in native chromatin context; genome-wide [9] [44] | Broad peaks; requires high-quality antibody; influenced by chromatin accessibility and co-factors [10] [44] |
| HT-SELEX (in vitro) | High-throughput Systematic Evolution of Ligands by EXponential Enrichment | Single binding site | Explores uniform sequence space; high specificity [9] [10] | Can saturate with strongest binders; may miss weaker functional sites [9] [10] |
| GHT-SELEX (in vitro) | Genomic HT-SELEX | Single binding site | Uses complex genomic DNA library, closer to natural sequence context [9] | Library complexity depends on genomic source [9] |
| PBM (in vitro) | Protein Binding Microarray | Single binding site | High-throughput; quantitative binding data [9] [45] | Limited to pre-designed probe sequences on the array [9] |
| SMiLE-Seq (in vitro) | Selective Microfluidics-based Ligand Enrichment followed by sequencing | Single binding site | Microfluidics enable sensitive screening [9] | Relatively newer method; broader benchmarking data may be limited [9] |
Detailed Protocol for Cross-Platform Motif Discovery and Benchmarking
The following protocol is adapted from the GRECO-BIT consortium's workflow for systematic motif discovery and evaluation [9].
Data Acquisition and Preprocessing
- Obtain Raw Data: Access sequencing data (FASTQ files) from public repositories like the European Nucleotide Archive (ENA) for HT-SELEX data (e.g., study PRJEB14744) or the ENCODE project for ChIP-seq data [10] [44].
- Platform-Specific Processing:
- For ChIP-Seq and GHT-SELEX: Perform peak calling using standard tools (e.g., MACS2) to identify genomic regions or DNA fragments enriched for TF binding. Extract these peak sequences for analysis.
- For HT-SELEX, SMiLE-Seq: Process the sequenced oligonucleotide pools from the final selection round(s). Quality control should include checks for library complexity and enrichment over earlier rounds.
- For PBM Data: Apply appropriate normalization procedures to the probe intensity data [9] [45].
- Dataset Splitting: For robust benchmarking, split the data from each experiment into training and test sets (e.g., 70/30 split). The training set is used for de novo motif discovery, while the test set is held back for final performance evaluation.
De Novo Motif Discovery
- Tool Selection: Choose a set of motif discovery tools based on your experimental data type and the benchmarking results in Table 1. A recommended starting set includes HOMER, MEME, and STREME for their broad compatibility and strong performance.
- Execution: Run the selected tools on the training set of your preprocessed data. Use default parameters initially, unless specific modifications are required for the data type (e.g., using DimontHTS for HT-SELEX data).
- Output Extraction: The primary output will be Position Frequency Matrices (PFMs). Convert these to Position Weight Matrices (PWMs) using standard log-odds scoring methods for subsequent scanning and evaluation [9].
Benchmarking and Validation
- Motif Scanning: Use the generated PWMs to scan the held-out test set sequences. Tools like FIMO (for individual sites) and MCAST (for clusters) have been identified as top performers for this task [44].
- Apply Benchmarking Metrics: Evaluate the performance of each PWM using multiple dockerized benchmarking protocols. Key metrics include:
- Sum-Occupancy Scoring: Evaluates the total binding signal in a sequence based on all PWM hits [9].
- HOCOMOCO Benchmark: Considers only the single top-scoring PWM hit in each ChIP-Seq or GHT-SELEX peak sequence [9].
- CentriMo Score: Assesses the centrality of the motif occurrence relative to the peak summit, which is a strong indicator of true binding [9].
- AUC-ROC (Area Under the Receiver Operating Characteristic Curve): Measures the ability of the motif model to distinguish true bound sequences (peaks) from unbound background sequences [10].
- Cross-Platform Validation (Gold Standard): The most rigorous validation involves testing a PWM discovered from one experimental platform (e.g., HT-SELEX) on test data from a different platform (e.g., ChIP-Seq) for the same TF. Consistent high performance across platforms is the strongest evidence for a motif's quality and biological relevance [9].
Result Curation and Artifact Filtering
- Expert Curation: Manually inspect the discovered motifs and their benchmarking results. Approve experiments where motifs are consistent across replicates, platforms, or are similar to known motifs for related TFs.
- Filter Artifacts: Automatically filter out PWMs that match common artifact signals, such as simple sequence repeats (e.g., poly-A tracts) or widespread ChIP-seq contaminants [9].
The following workflow diagram illustrates the complete protocol:
Table 3: Key Reagents, Data, and Software for Motif Discovery Research
| Category | Item | Function and Application Notes |
|---|---|---|
| Data Resources | ENCODE Project Data | Primary source for high-quality, annotated ChIP-seq data from human cell lines [10]. |
| European Nucleotide Archive (ENA) | Repository for high-throughput sequencing data, including comprehensive HT-SELEX datasets (e.g., PRJEB14744) [10]. | |
| JASPAR / CIS-BP / HOCOMOCO | Public databases of curated, non-redundant transcription factor binding motifs (PWMs) used for validation and comparison [9] [44]. | |
| Software & Algorithms | HOMER Suite | Integrated suite for motif discovery and next-gen sequencing analysis. Particularly strong on ChIP-seq data [9]. |
| MEME Suite | A comprehensive collection of tools for motif discovery (MEME), scanning (FIMO), and enrichment analysis (CentriMo) [9] [44]. | |
| rGADEM | An R/Bioconductor package for de novo motif discovery, effective for ChIP-Seq data [44]. | |
| Experimental Platforms | HT-SELEX | In vitro method for characterizing binding specificities using randomized oligonucleotide libraries [9] [10]. |
| ChIP-Seq | The current 'gold standard' in vivo method for genome-wide mapping of TF binding events in a native chromatin context [9] [44]. | |
| Validation Resources | ChIP-Seq Data for Perturbed TFs | Datasets from genetic knockouts/overexpression of TFs provide a "gold standard" for validating motif-based predictions of TF activity [46]. |
| Codebook Motif Explorer (MEX) | An interactive catalog (mex.autosome.org) providing access to motifs, benchmarking results, and underlying data from the GRECO-BIT study [9]. |
The systematic benchmarking of motif discovery tools reveals that performance is intimately linked to the experimental source data. No single tool is universally superior, but tools like HOMER, MEME, and STREME demonstrate robust performance across multiple platforms. The critical practice of cross-platform validation emerges as the most reliable strategy for identifying high-quality motifs. Researchers are advised to select tools compatible with their experimental data type and to leverage the presented protocols and the public Codebook Motif Explorer resource to guide their discovery of biologically relevant transcription factor binding specificities.
Strategies for Improving Signal-to-Noise and Overcoming Artifact Motifs
In the context of genomic SELEX for transcription factor (TF) binding site identification, a paramount challenge is the distinction of genuine biological signals from experimental noise and artifact motifs. These artifacts can arise from various sources, including technical biases inherent to high-throughput sequencing platforms, non-specific protein-DNA interactions, and the presence of repetitive or structurally peculiar DNA sequences. The consequences of such artifacts are severe, leading to inaccurate motif models, misannotation of regulatory genomes, and flawed biological inferences. This application note details standardized protocols and strategic frameworks designed to enhance signal-to-noise ratio and systematically mitigate the influence of artifact motifs, thereby increasing the reliability and reproducibility of TF binding specificity data for the drug discovery and research communities. The recommendations are framed within the findings of the Codebook/GRECO-BIT initiative, a large-scale benchmarking effort that analyzed 4,237 experiments for 394 human TFs across five experimental platforms [9].
Artifact motifs, or "passenger motifs," are non-functional sequence patterns that are recurrently identified in TF binding datasets but do not represent the true intrinsic binding specificity of the protein under investigation [9]. The Codebook/GRECO-BIT analysis, which processed data from Chromatin Immunoprecipitation followed by sequencing (ChIP-Seq), high-throughput SELEX (HT-SELEX), genomic HT-SELEX (GHT-SELEX), Protein Binding Microarray (PBM), and SMiLE-Seq, found that a significant proportion of initial datasets contained such confounding signals [9] [47]. Through rigorous human curation, they approved only a subset of experiments (1,462 datasets for 236 TFs) that displayed consistent motifs across platforms and replicates, highlighting the pervasive nature of this problem [9].
Table 1: Common Sources of Artifact Motifs in Genomic SELEX
| Source Category | Specific Examples | Impact on Data |
|---|---|---|
| Technical Biases | Platform-specific saturation (e.g., HT-SELEX), probe design in PBM, antibody non-specificity in ChIP-seq [9]. | Skews the representation of binding sequences, over-representing strong binders. |
| Sequence Composition | Simple sequence repeats (e.g., homopolymers), low-complexity regions [9]. | Generates high-scoring but biologically irrelevant motif models. |
| Protein Production | Differences between GST-tagged IVT (E. coli), GFP-tagged IVT (wheat germ), and whole cell lysate methods [9] [47]. | May introduce bacterial or other contaminating DNA-binding proteins. |
| Contextual Factors (in vivo) | Cobinding factors, chromatin accessibility, nucleosome positioning [48] [41]. | Identifies tethered binding events not directly mediated by the TF's DNA-binding domain. |
A critical insight from large-scale benchmarking is that traditional motif quality metrics, such as nucleotide composition and information content, show little to no correlation with actual motif performance and are not reliable indicators for detecting underperformers or artifacts [9] [47]. Consequently, more robust, multi-faceted strategies are required for validation.
Strategic Framework for Signal Enhancement
Multi-Platform Experimental Design
The most effective strategy for verifying a genuine TF binding motif is to assay the same TF using multiple, complementary experimental platforms. The concordance of a discovered motif across different technologies (e.g., in vitro HT-SELEX/PBM and in vivo ChIP-seq) provides powerful evidence for its validity [9] [47]. The Codebook initiative demonstrated that motifs consistently identified across platforms are far more likely to represent true biological specificity. This approach controls for the unique technical biases and limitations of any single method.
Cross-Platform Benchmarking as a Validation Tool
A robust computational pipeline for cross-platform benchmarking is essential for evaluating motif quality and identifying artifacts. The recommended protocol involves:
- Motif Discovery on Training Data: Apply multiple motif discovery tools (e.g., MEME, HOMER, STREME) to a training subset of data from one platform [9].
- Performance Evaluation on Test Data: Test the resulting Position Weight Matrices (PWMs) on held-out test datasets from other experimental platforms [9] [47].
- Metric Aggregation: Use multiple benchmarking metrics, such as:
- Sum-occupancy scoring for genomic peaks (ChIP-Seq, GHT-SELEX) [9] [47].
- HOCOMOCO benchmark, which considers the single top-scoring log-odds PWM hit per sequence [9] [47].
- CentriMo, to assess the centrality of the motif hit relative to the peak summit [9] [47].
- PBM-specific scoring adapted from established methodologies [9].
A motif that performs well in classifying or explaining binding data from a platform different from the one it was derived from is a high-confidence genuine motif.
Diagram 1: Cross-platform motif discovery and validation workflow.
Integration of Contextual Genomic Information
In vivo binding is profoundly influenced by cellular context. Integrating data on chromatin accessibility significantly improves the prediction of true TF binding sites. A modern strategy involves leveraging assays like ATAC-seq to inform motif discovery and validation [48]. Tools like TRAFICA, an open chromatin language model, demonstrate that pre-training on sequences from open chromatin regions before fine-tuning on in vitro binding profiles (e.g., from PBM or HT-SELEX) leads to state-of-the-art performance in predicting both in vitro and in vivo TF-DNA binding affinity [48]. This effectively uses biological context as a filter to prioritize plausible binding sequences.
Advanced Modeling to Capture Binding Complexity
While PWMs are the standard model, they operate on the simplifying assumption of independent nucleotide contributions. For TFs with complex binding modes, such as many zinc finger proteins, more advanced models can provide a better signal-to-noise ratio. The Codebook/GRECO-BIT initiative demonstrated that combining multiple PWMs into a random forest model can account for multiple modes of TF binding and improve performance [9]. Similarly, deep learning models like ExplaiNN or language models like TRAFICA can capture interdependencies within the binding site that are missed by a PWM [9] [48].
Detailed Protocol: A Multi-Tool Motif Discovery and Curation Pipeline
This protocol outlines a standardized workflow for generating high-confidence, artifact-free motifs from genomic SELEX data, based on the GRECO-BIT pipeline [9] [47].
Materials and Reagent Solutions
Table 2: Essential Research Reagents and Computational Tools
| Item Name | Function/Description | Example/Note |
|---|---|---|
| HT-SELEX Library | Synthetic DNA library with random core region. | e.g., 40N random inserts [9]. |
| Motif Discovery Tools | Software to identify overrepresented sequence patterns. | MEME, HOMER, STREME, ChIPMunk, Autoseed [9]. |
| Benchmarking Suite | Dockerized protocols for cross-platform PWM testing. | Protocols from Ambrosini et al. and HOCOMOCO [9] [47]. |
| Codebook Motif Explorer | Online catalog for visualizing approved motifs and benchmarks. | https://mex.autosome.org [9]. |
| Advanced Modeling Tools | Software for complex binding modes. | RCade (for zinc fingers), gkmSVM, ExplaiNN, Random Forest [9]. |
Step-by-Step Procedure
Data Preprocessing and Quality Control.
- For genomic binding data (ChIP-Seq, GHT-SELEX): Perform peak calling using MACS2 or a similar tool. Extend peaks 100 bp on either side of the summit for sequence extraction [49].
- For HT-SELEX data: Handle sequencing reads and account for potential saturation of strong binders in later cycles [9].
- For PBM data: Apply appropriate normalization to fluorescence intensity data [9] [47].
- Critical Step: Split the data from each experiment into training (e.g., 2/3) and test (e.g., 1/3) sets.
Multi-Tool Motif Discovery.
- Apply a diverse set of at least 5-10 motif discovery tools to the training set. This should include:
- Run each tool with default parameters optimized for the respective data type.
Automated Artifact Filtering.
Cross-Platform Benchmarking.
- Convert all motif models to a standardized PWM format.
- Using the held-out test sets, execute benchmarking protocols:
- Use sum-occupancy scoring on ChIP-Seq and GHT-SELEX peak sequences [9] [47].
- Run the HOCOMOCO benchmark (single best hit) on the same genomic data [9].
- Calculate the CentriMo centrality score for motifs relative to peak summits [9] [47].
- Apply PBM-specific evaluation metrics for microarray data [9].
Human Expert Curation and Approval.
- This is a critical, non-automated step for final validation.
- Criterion 1: Approve experiments where motifs discovered from the data are consistent across platforms and biological replicates [9].
- Criterion 2: Approve experiments where the dataset yields high scores for consistent, high-ranking motifs discovered from other approved experiments for the same TF [9].
- Compare motifs to known motifs for related TFs (both positive controls and Codebook TFs from well-studied families) to validate real cases and exclude recurring passenger motifs [9].
Overcoming artifact motifs and improving the signal-to-noise ratio in genomic SELEX experiments requires a systematic, multi-layered strategy that no longer relies on simple motif metrics. The integration of a multi-platform experimental design, rigorous cross-platform computational benchmarking, and final human expert curation, as exemplified by the Codebook/GRECO-BIT initiative, provides a robust framework for establishing high-confidence TF binding specificities. The adoption of these protocols and the utilization of the resulting curated resources, such as the Codebook Motif Explorer, will empower researchers and drug developers to build their work on a more reliable genomic foundation, accelerating the accurate annotation of gene regulatory regions and the interpretation of regulatory variants in health and disease.
The identification of transcription factor (TF) binding sites is a cornerstone of genomic research, critical for deciphering gene regulatory networks and their implications in development and disease. Genomic SELEX (Systematic Evolution of Ligands by EXponential Enrichment) has emerged as a powerful in vitro technique for discovering genomically encoded DNA aptamers that bind with high affinity to TFs of interest [13]. This method screens entire genomes for functional domains within nucleic acid molecules, independent of their native expression levels, thus uncovering novel regulatory sequences [13] [50]. However, the massive, complex datasets generated by high-throughput SELEX technologies present significant analytical challenges. Traditional position weight matrix (PWM) models, while simple and interpretable, often fail to capture the full complexity of TF-DNA interactions due to their assumption of positional independence and fixed binding configurations [51] [9]. To address these limitations, researchers are increasingly turning to multi-model computational approaches, with Random Forest (RF) leading this paradigm shift. RF ensembles effectively model high-dimensional data with correlated variables, making them exceptionally well-suited for capturing the nuanced dependencies in TF binding specificities [51]. This Application Note details how RF and other ensemble methods are revolutionizing TF binding site prediction from genomic SELEX data, providing researchers with robust protocols and frameworks to enhance their discovery pipelines.
Key Applications of Multi-Model Approaches
Ensemble Random Forest for Cross-Tissue TF Binding Prediction
A primary challenge in TF binding site prediction is building models that generalize accurately across diverse cell types and tissues. Schulz et al. tackled this in the ENCODE-DREAM in vivo TF binding site prediction challenge by developing a novel ensemble RF approach [52]. Their method leveraged DNase1-seq data and TF motif information from position-specific energy matrices (PSEMs) to predict binding locations, using ChIP-seq data as the gold standard for training and validation. The ensemble model aggregated predictions from multiple tissue-specific RF classifiers into a final, robust prediction, outperforming both individual tissue-specific models and classifiers built on aggregated data from all tissues [52]. This approach demonstrated superior generalizability, effectively capturing tissue-specific co-factor information that is often lost in simpler models. Analysis of feature importance within the RF models revealed that the algorithm preferentially selected motifs of other TFs known to be close interaction partners in existing protein-protein interaction networks, providing biological validation of the method [52].
Table 1: Performance Comparison of TF Binding Prediction Models from the ENCODE-DREAM Challenge
| Model Type | Key Features | Reported Performance (ROC-AUC/PR-AUC) | Advantages |
|---|---|---|---|
| Ensemble Random Forest | Combines multiple tissue-specific RF models; uses TRAP-computed TF affinities [52] | Superior generalizability across tissues [52] | Reduces false positives; identifies biologically relevant co-factors [52] |
| Tissue-Specific Classifier | Single RF model trained on data from one tissue type [52] | Lower performance on unseen tissues [52] | Captures tissue-specific signals; lacks generalizability [52] |
| Aggregated Classifier | Single RF model trained on data pooled from all tissues [52] | Intermediate generalizability [52] | Simpler architecture; may miss tissue-specific nuances [52] |
| Position Weight Matrix (PWM) | Traditional motif scoring; assumes base independence [51] | AUROC ~0.59 for pairwise binding [51] | Simple, interpretable; fails to capture dependencies [51] |
Random Forest for Modeling TF-TF Cooperative Binding
Transcription factors often bind DNA cooperatively as pairs, forming complexes that recognize novel DNA motifs with distinct spacing and orientation preferences. The limitations of PWMs are particularly pronounced in modeling these flexible pairwise interactions. Lähdesmäki et al. addressed this challenge by developing two RF-based methods—ComBind and JointRF—trained on large-scale CAP-SELEX data, which comprises DNA sequences enriched for binding of specific TF pairs [51]. ComBind, their more advanced approach, utilizes random forests to simultaneously consider multiple orientations and spacings of two TFs without requiring prior knowledge of their precise binding configuration. This method achieved an AUROC of 0.78, significantly outperforming both orientation and spacing-specific pairwise PWMs (AUROC 0.59) and JointRF (AUROC 0.75), which relies on pre-determined PWMs [51]. The RF framework's ability to handle correlated variables and identify class-specific clusters of features makes it uniquely suited for capturing the complex dependencies inherent in cooperative TF-TF-DNA binding.
Integration with Multiple Motif Discovery Tools
The GRECO-BIT (Gene Regulation Consortium Benchmarking Initiative) benchmarking study further demonstrated the power of ensemble approaches by combining PWMs from multiple motif discovery tools into a random forest model [9]. This large-scale analysis processed 4,237 experiments for 394 TFs across five experimental platforms (ChIP-Seq, GHT-SELEX, HT-SELEX, SMiLE-Seq, and PBM) and employed ten different motif discovery tools. By integrating the diverse PWMs generated by these tools into an RF model, researchers could account for multiple modes of TF binding, thereby enhancing prediction accuracy and robustness [9]. This approach highlights a strategic application of ensemble learning where the RF meta-model synthesizes insights from multiple base models, each with different strengths and biases, to create a more comprehensive representation of TF binding specificities.
Table 2: Multi-Model Approaches for TF Binding Site Prediction
| Method | Data Source | Algorithm | Key Innovation | Use Case |
|---|---|---|---|---|
| ComBind [51] | CAP-SELEX [51] | Random Forest | Models multiple TF-TF orientations/spacings simultaneously [51] | Predicting cooperative TF pair binding sites [51] |
| Ensemble RF [52] | DNase1-seq, ChIP-seq [52] | Random Forest Ensemble | Combines tissue-specific classifiers for cross-tissue generalization [52] | Genome-wide TF binding prediction across multiple cell types [52] |
| GRECO-BIT Integration [9] | Multi-platform TF data [9] | RF with multiple PWMs | Combines PWMs from various discovery tools into an RF model [9] | Improving motif discovery robustness and accounting for multiple binding modes [9] |
| DCA-Scapes [10] | HT-SELEX [10] | Global Pairwise Model | Captures nucleotide interdependencies beyond PWM limitations [10] | High-resolution TF recognition specificity landscapes [10] |
Experimental Protocols
Protocol 1: Ensemble RF for Cross-Tissue TF Binding Prediction
This protocol adapts the methodology from Schulz et al. for building an ensemble RF model that generalizes across cell types [52].
Step 1: Data Preparation and Feature Engineering
- Obtain TF ChIP-seq data as positive labels and DNase1-seq data from ENCODE or similar consortia for multiple cell types [52].
- Identify DNaseI hypersensitive sites (DHSs) using a peak caller such as JAMM [52].
- Compute TF binding affinities within DHSs for a comprehensive set of PWMs (e.g., 557 motifs) using the TRAP tool, which employs a biophysical model to capture low-affinity binding sites [52].
- Segment the genome into non-overlapping bins (e.g., 200 bp) and label them as bound or unbound based on ChIP-seq peak overlap [52].
- Compile features including TRAP-computed affinity scores for all TFs, DNA sequence features, and chromatin accessibility metrics.
Step 2: Training Tissue-Specific Base Models
- For each cell type with sufficient ChIP-seq data, train an individual Random Forest classifier using the extracted features to predict TF binding.
- Perform hyperparameter optimization via grid search or randomized search for each base model.
- Validate each tissue-specific model using cross-validation on held-out chromosomes.
Step 3: Building the Ensemble Model
- Aggregate predictions from all tissue-specific RF models using a meta-learner or averaging approach.
- Alternatively, train a final-layer RF classifier on the concatenated prediction probabilities from all base models.
- Validate the ensemble model on completely held-out cell types to assess cross-tissue generalizability.
Step 4: Model Interpretation and Biological Validation
- Analyze feature importance scores from the ensemble model to identify the most predictive TF motifs.
- Validate top features against known protein-protein interaction networks, as co-operative TFs often show up as important features [52].
Protocol 2: ComBind for TF-TF Cooperative Binding Prediction
This protocol implements the ComBind approach for modeling flexible pairwise TF-TF-DNA binding specificities from CAP-SELEX data [51].
Step 1: CAP-SELEX Data and Control Set Preparation
- Obtain CAP-SELEX data for the TF pair of interest, which contains DNA sequences (typically 40 bp) enriched for binding of the specific TF pair [51].
- Generate a negative control set by shuffling each CAP-SELEX sequence once using uShuffle or similar tools that preserve dinucleotide counts [51].
- Include reverse complements of all sequences in the analysis.
Step 2: Sequence Selection with Individual TF PWMs
- Retrieve individual PWMs for each TF in the pair from databases such as JASPAR or CIS-BP.
- Extend each PWM with neutral columns (all nucleotides weighted equally) to create a 25-nucleotide long matrix, accommodating variable spacing between motifs [51].
- For each sequence in the training set, select the highest-scoring 25 bp sub-sequence for two orientations: TF1-TF2 and TF2-TF1, based on the extended individual PWMs.
Step 3: Training Orientation-Specific Random Forests
- Train two separate RF classifiers:
- TF1-TF2 orientation RF: Uses sub-sequences selected based on TF1's PWM, considering potential binding of TF2 in the downstream region.
- TF2-TF1 orientation RF: Uses sub-sequences selected based on TF2's PWM, considering potential binding of TF1 in the downstream region.
- Use nucleic acids at each position as categorical features for the random forests.
Step 4: Sequence Scoring and Prediction
- For a new DNA sequence, extract all 25 bp sub-sequences and compute scores using both orientation-specific RF models.
- Calculate the maximum score across all positions for each orientation.
- The final ComBind score is the maximum of the two orientation scores, representing the most likely binding configuration [51].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Genomic SELEX and Random Forest Modeling
| Reagent/Resource | Function and Application | Technical Considerations |
|---|---|---|
| CAP-SELEX Libraries [51] | Provides DNA sequences enriched for specific TF-TF pair binding; essential for training cooperative binding models [51] | Contains 40 bp sequences; requires negative set generation via shuffling [51] |
| HT-SELEX/GHT-SELEX Data [9] [10] | Delivers comprehensive binding preferences for individual TFs from in vitro selection [10] | Can saturate with strongest binders; multiple rounds provide enrichment data [9] |
| Position Weight Matrices (PWMs) [9] | Foundation for feature generation; represent binding motifs for individual TFs [9] | Assume positional independence; can be extended for flexible spacing [51] |
| TRAP Tool [52] | Computes TF binding affinities from PWMs using biophysical model; captures low-affinity sites [52] | Preferable to binary classification; produces continuous affinity scores [52] |
| JAMM Peak Caller [52] | Identifies DNaseI hypersensitive sites from DNase1-seq data [52] | Provides unified peak calls across samples; critical for feature extraction [52] |
| uShuffle Tool [51] | Generates negative control sequences while preserving dinucleotide frequencies [51] | Maintains sequence composition properties of background [51] |
| Random Forest Implementation (e.g., scikit-learn) | Builds ensemble classifiers; handles high-dimensional correlated features [51] | Requires careful parameter tuning; provides feature importance metrics [51] |
The integration of Random Forest and other multi-model approaches with genomic SELEX data represents a significant advancement in transcription factor binding site identification. By moving beyond the limitations of traditional PWM models, these ensemble methods capture the complexity of TF-DNA interactions, including tissue-specific binding preferences, cooperative TF-TF binding with flexible configurations, and multi-motif binding modes. The protocols outlined in this Application Note provide researchers with robust methodologies to implement these powerful approaches in their own workflows. As genomic SELEX technologies continue to evolve and generate increasingly complex datasets, the role of sophisticated computational approaches like Random Forest will only grow in importance, ultimately accelerating our understanding of gene regulatory networks and their implications in health and disease.
Validating and Contextualizing SELEX Findings in Functional Genomics
Transcription factors (TFs) are regulatory proteins that bind DNA in a sequence-specific manner to control gene expression, with alterations in TF-DNA interactions contributing significantly to gene expression changes due to sequence variants [9] [53]. A rigorous understanding of TF binding specificities requires integration of multiple experimental approaches, as each method carries unique technical biases and provides complementary information [9]. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) generates genome-wide maps of TF binding sites in vivo but offers limited spatial resolution and may include non-functional binding events [53]. High-throughput systematic evolution of ligands by exponential enrichment (HT-SELEX) isolates DNA ligands through iterative selection and amplification in vitro, providing large sequence libraries for motif discovery [11] [15]. Protein binding microarrays (PBMs) enable high-throughput testing of TF binding to thousands of double-stranded DNA molecules in a single experiment [9] [53]. Cross-platform validation integrates these complementary approaches to generate robust, high-confidence models of TF binding specificities.
Experimental Platforms and Methodologies
High-Throughput SELEX (HT-SELEX)
HT-SELEX is an in vitro technique for unbiased determination of preferred target motifs of DNA-binding proteins [15]. The procedure involves iterative selection of DNA binding sites from a random oligonucleotide library by purifying protein-DNA complexes and amplifying bound DNA using PCR.
- Random DNA Library Preparation: A synthetic oligonucleotide library consisting of a random internal segment (typically 20-40 bp) flanked by constant sequences is converted to double-stranded DNA. The constant regions serve as priming sites for PCR amplification [11].
- Selection Cycles: The double-stranded DNA library is incubated with the purified TF of interest. Protein-DNA complexes are isolated using methods such as electrophoretic mobility shift assay or affinity purification. Bound DNA is eluted, purified, and amplified by PCR to create the input for the next selection cycle [11] [15].
- Sequencing and Analysis: After multiple selection cycles (typically 3-5), the enriched DNA pools are sequenced using high-throughput platforms. The random parts of the selected oligonucleotides are excised, concatemerized, and ligated into cloning vectors to increase sequencing throughput [11]. Bioinformatic pipelines such as
eme_selexanalyze the enrichment of all possible k-mers to detect preferred binding motifs, including promiscuous DNA binding [15].
Chromatin Immunoprecipitation Sequencing (ChIP-seq)
ChIP-seq enables the study of protein-DNA interactions within their native genomic context, generating genome-wide maps of TF binding sites in vivo [53].
- Cross-linking and Fragmentation: Cells are treated with formaldehyde to cross-link TFs to their genomic DNA binding sites. Chromatin is then fragmented by sonication or enzymatic digestion.
- Immunoprecipitation: An antibody specific to the TF of interest is used to immunoprecipitate the protein-DNA complexes. After purification and reversal of cross-links, the bound DNA fragments are isolated.
- Sequencing and Peak Calling: The purified DNA fragments are sequenced, and reads are mapped to the reference genome. Peak-calling algorithms identify genomic regions significantly enriched for TF binding, representing putative binding sites [9]. Protocols such as ChIP-exo use an exonuclease to trim TF-bound DNA, improving the signal-to-noise ratio and resolution of binding sites [53].
Protein Binding Microarrays (PBM)
PBMs provide a high-throughput platform for measuring TF-DNA interactions in vitro, testing binding against thousands of predefined DNA sequences simultaneously [9] [53].
- Microarray Design: DNA probes, which can be synthetic pseudo-random sequences or genomic fragments, are printed on microarray slides.
- Protein Binding and Detection: Purified TFs, often tagged with a fluorescent marker like GFP, are incubated with the microarray. After washing, bound TFs are detected through fluorescence measurement [53].
- Data Analysis: Fluorescence intensity at each probe provides a quantitative measure of binding affinity. These data are used to determine dissociation constants (KD values) and derive binding motifs [53].
Cross-Platform Integration and Validation Framework
The Gene Regulation Consortium Benchmarking Initiative (GRECO-BIT) established a systematic workflow for cross-platform motif discovery and benchmarking, analyzing data from 4,237 experiments for 394 TFs assayed across five experimental platforms [9]. This integrated approach enables robust validation of TF binding specificities.
Multi-Platform Data Integration Workflow
The following diagram illustrates the logical workflow for integrating data from SELEX, ChIP-seq, and PBM platforms to validate TF binding specificities:
Computational Analysis Pipeline
The computational workflow for processing and integrating data across platforms involves uniform preprocessing, motif discovery, and systematic benchmarking:
Quantitative Benchmarking Results
Systematic evaluation of motif performance across platforms reveals critical insights into the reliability of different experimental approaches and computational tools.
Table 1: Performance Metrics of Motif Discovery Tools Across Experimental Platforms [9]
| Motif Discovery Tool | Compatible Platforms | Key Strengths | Performance Notes |
|---|---|---|---|
| MEME | All platforms | Classic, widely-used algorithm | Applied uniformly across datasets |
| HOMER | ChIP-seq, SELEX | Popular for high-throughput data | High rankings in benchmarks |
| ChIPMunk | ChIP-seq, GHT-SELEX | De novo motif discovery | Compatible with peak-based data |
| STREME | All platforms | Modern motif discovery | Applied in second discovery round |
| Dimont/DimontHTS | HT-SELEX | Specialized for SELEX data | Platform-specific optimization |
| RCade | Selected platforms | Specialized for zinc finger TFs | Applied to specific TF families |
| ExplaiNN | All platforms | Advanced neural network method | Comprehensive application |
| ProBound | Approved experiments | Advanced probabilistic modeling | Second discovery round only |
Table 2: Experimental Platform Characteristics and Benchmarking Outcomes [9]
| Experimental Platform | Context | Key Advantages | Limitations | Approval Rate |
|---|---|---|---|---|
| ChIP-seq | In vivo | Genomic context, functional sites | Low resolution, non-specific binding | Varies by TF and cell type |
| HT-SELEX | In vitro | Uniform sequence exploration | Saturates with strong binders | High for consistent TFs |
| GHT-SELEX | In vitro | Genomic DNA fragments | Technical biases in selection | Moderate |
| PBM | In vitro | High-throughput, quantitative | Limited commercial availability | High for quantitative data |
| SMiLE-Seq | In vitro | Microfluidics-based | Protocol complexity | Moderate |
The GRECO-BIT analysis generated 219,939 PWMs, with 164,570 derived from approved experiments after rigorous curation. Notably, 159,063 PWMs passed additional automatic filtering for common artifact signals such as simple repeats and widespread ChIP contaminants [9]. The study found that nucleotide composition and information content are not correlated with motif performance and do not help in detecting underperformers. Importantly, motifs with low information content in many cases describe TF binding specificity effectively across different experimental platforms [9].
Table 3: Key Research Reagent Solutions for Cross-Platform TF Binding Studies
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Recombinant TFs | Protein source for in vitro assays | Produced as GST-tagged or GFP-tagged fusions in E. coli or wheat germ extracts [9] |
| Random Oligo Library | Starting pool for SELEX | Contains random internal segment (20-40 bp) with constant flanking regions for PCR [11] |
| Antibodies | TF immunoprecipitation for ChIP-seq | Specific antibodies for target TFs; critical for success of ChIP experiments [53] |
| PBM Microarrays | High-throughput binding measurement | Contain thousands of DNA probes; custom designs often required [53] |
| HTPSELEX Database | Repository for SELEX data | Primary and derived data from HT-SELEX experiments; enables re-analysis [11] |
| Codebook MEX | Motif catalog and exploration | Interactive catalog of motifs for 394 TFs with benchmarking results [9] |
| JASPAR Database | TF binding motif repository | Curated, non-redundant TF binding profiles; reference for comparison [53] |
| IceQream (IQ) | Quantitative accessibility modeling | Physical TF models for predicting chromatin accessibility from sequence [54] |
Application Notes and Technical Considerations
Experimental Design Recommendations
For comprehensive TF characterization, researchers should employ multiple complementary platforms. The GRECO-BIT initiative recommends:
- Platform Selection: Combine in vivo (ChIP-seq) and in vitro (SELEX, PBM) approaches to balance biological relevance with controlled specificity assessment [9].
- Replication and Controls: Include both technical replicates and well-studied TFs as positive controls, particularly when working with poorly characterized TFs [9].
- Protein Production: Consider multiple expression systems (E. coli, wheat germ, human cell lysates) as TF production method can influence binding specificity assessments [9].
Data Analysis and Interpretation Guidelines
- Motif Discovery Tool Selection: Apply multiple motif discovery algorithms with different underlying assumptions, as performance varies across experimental platforms [9].
- Cross-Platform Validation: Use high-confidence motifs from one platform to validate results from another. An experiment should be approved if it yields consistent motifs across platforms or provides high scores for consistent motifs from other experiments [9].
- Advanced Modeling: Consider extending beyond simple position weight matrices (PWMs) to more sophisticated models like random forests or physical TF models (e.g., IceQream) that can account for multiple modes of TF binding and synergistic interactions [9] [54].
Troubleshooting Common Issues
- Artifact Identification: Implement automatic filtering for common artifact signals such as simple repeats and widespread ChIP contaminants [9].
- Low Information Content Motifs: Recognize that low information content motifs can still represent valid binding specificities and should not be automatically dismissed [9].
- Platform-Specific Biases: Account for technical biases inherent to each platform, such as HT-SELEX saturation with strong binders and non-specific binding in ChIP-seq [9].
Integrated cross-platform analysis represents the gold standard for establishing robust, high-confidence models of TF binding specificity. By leveraging the complementary strengths of SELEX, ChIP-seq, and PBM approaches, researchers can overcome the limitations of individual methods and generate biologically meaningful insights into transcriptional regulation. The protocols, benchmarking data, and analytical frameworks presented here provide a foundation for rigorous TF characterization studies that effectively bridge in vitro and in vivo contexts.
Assessing Predictive Power for In Vivo Binding and Functional Genomics
The accurate prediction of transcription factor (TF) binding sites is a cornerstone of functional genomics, directly impacting the interpretation of regulatory variants in drug development and disease research. While genomic SELEX (Systematic Evolution of Ligands by EXponential Enrichment) and other high-throughput in vitro assays provide a foundational understanding of TF-DNA binding preferences, translating these models to accurately predict in vivo binding remains a significant challenge. This application note delineates the performance boundaries of current predictive models and provides a structured protocol for researchers to benchmark tools, leveraging quantitative data from recent large-scale evaluations. The integration of multi-platform data and the selection of context-appropriate computational tools are critical for generating biologically relevant predictions in functional genomics and drug discovery pipelines.
Quantitative Benchmarking of Predictive Models
The predictive accuracy of computational models varies substantially between in vitro and in vivo contexts. A 2024 benchmark evaluation of 14 different models provides a clear hierarchy of tool performance based on the experimental context of the prediction [55].
Table 1: Top-Performing Models for In Vitro vs. In Vivo TF Binding Prediction
| Prediction Context | Best-Performing Model Types | Example Tools | Key Performance Insight |
|---|---|---|---|
| In Vitro Binding(e.g., SNP-SELEX) | Kmer/gkm-based Machine Learning | deltaSVM_HT-SELEX, QBiC-Pred | Trained on in vitro datasets (e.g., HT-SELEX), these exhibit superior performance for predicting variant effects in controlled biochemical experiments [55]. |
| In Vivo Binding(e.g., ASB, ChIP-seq) | Deep Neural Network (DNN)-based Multitask Models | DeepSEA, Sei, Enformer | Models trained on large-scale in vivo datasets (e.g., ChIP-seq) show relatively superior performance for predicting allele-specific binding in cellular environments [55]. |
| General PWM Performance | PWM-based Methods | tRap | Among simpler Position Weight Matrix (PWM) based methods, tRap demonstrates better performance in both in vitro and in vivo evaluations [55]. |
A critical finding is that the accuracy of each model in predicting SNP effects in vitro significantly exceeds that achieved in vivo [55]. This underscores the complex influence of the cellular environment—including chromatin accessibility, co-factors, and epigenetic modifications—on actual TF binding.
Furthermore, the DNA-binding domain (DBD) of a TF influences predictability. For instance, models predict binding for TFs with basic leucine zipper factors more accurately, while predictions for C2H2 zinc finger factors are less accurate, aligning with the evolutionary conservation and binding complexity of these TF classes [55].
Experimental Protocols for Validation and Binding Assessment
Protocol: Validating In Vivo Binding Predictions using ChIP-seq
This protocol is designed to test whether binding sites predicted from in vitro derived models (e.g., from SELEX) correspond to actual in vivo binding regions.
1. Reagents and Equipment
- Crosslinked cells of relevant cell type/tissue
- Antibody specific to the transcription factor of interest
- Protein A/G magnetic beads
- Lysis buffers, wash buffers, and elution buffer
- Protease K
- DNA purification kit (e.g., phenol-chloroform, columns)
- Equipment for sonication (e.g., Bioruptor)
- qPCR machine and next-generation sequencer
2. Procedure
- Crosslinking & Lysis: Harvest cells and crosslink DNA-protein complexes with 1% formaldehyde for 10 minutes at room temperature. Quench with glycine. Pellet cells and lyse with appropriate lysis buffers.
- Chromatin Shearing: Sonicate chromatin to fragment DNA to an average size of 200-500 bp. Confirm fragment size by agarose gel electrophoresis.
- Immunoprecipitation: Incubate chromatin lysate with TF-specific antibody overnight at 4°C. Add Protein A/G beads and incubate for 2 hours. Pellet beads and wash extensively with low-salt, high-salt, and LiCl wash buffers, followed by a final TE buffer wash.
- Elution & Reverse Crosslinking: Elute bound complexes from beads with elution buffer (e.g., 1% SDS, 0.1M NaHCO3). Reverse crosslinks by adding NaCl and incubating at 65°C overnight.
- DNA Purification: Treat with RNase A and Protease K. Purify DNA using a DNA purification kit. Quantify the recovered DNA.
- Sequencing & Analysis: Prepare a sequencing library from the purified DNA and perform high-throughput sequencing (ChIP-seq). Map sequenced reads to the reference genome and call significant peaks using tools like MACS2.
3. Validation of Predictions
- Obtain the top 500 ChIP-seq peaks and extract their genomic sequences [10].
- As negative controls, extract same-length sequences from both upstream and downstream flanking regions of these peaks, ensuring they do not overlap with other positive peaks for the same TF [10].
- Score both positive and negative sequences using the computational model (e.g., a PWM or a DNN model). Use the three most negative Hamiltonian (affinity) scores to represent each region [10].
- Calculate the Area Under the Curve (AUC) of the Receiver Operating Characteristic (ROC) curve to evaluate the model's performance in distinguishing true in vivo binding sites [10].
Method: Quantitative Microplate-Based Fluorescence Relative Affinity (QuMFRA) Assay
The QuMFRA assay is a robust method for quantitatively measuring the relative binding affinities of multiple DNA sequences in parallel, which can be used to refine models based on SELEX data [14].
1. Reagents and Equipment
- Purified TF protein (e.g., GST-Zif268 fusion protein used in the study)
- Double-stranded oligonucleotide binding sites generated by PCR
- Fluorophore-labeled primers (FAM, TAMRA, or ROX)
- Glutathione Sepharose beads (if using GST-tagged protein)
- Polyacrylamide gel electrophoresis system
- Typhoon Variable Scanner or similar fluorescent gel imager
2. Procedure
- Sample Preparation: Generate PCR products for the DNA sites to be tested, using primers labeled with different fluorophores (e.g., FAM, TAMRA, ROX). Include one site as a reference to which all other affinities will be compared [14].
- Competitive Binding Reaction: Mix the three different fluorophore-labeled DNA-binding sites with a defined concentration of the TF protein in reaction buffer. Include poly(dI–dC) as non-specific competitor DNA [14].
- Separation & Imaging: After equilibration, electrophorese the reaction on a non-denaturing polyacrylamide gel. Scan the gel using a fluorescence scanner at the respective emission wavelengths for each fluorophore to obtain the fluorescent intensities of the protein-bound and free DNA bands [14].
- Data Analysis: For a given sequence, the relative binding constant (Ka(rel)) to the reference site is calculated using the formula:
Ka(rel) = [PDtest][Dref] / [PDref][Dtest]
where [PD] is the concentration of protein-bound DNA and [D] is the concentration of free DNA for the test and reference sites [14]. These quantitative affinities can then be used to build a more accurate model.
Workflow Visualization
Diagram 1: A workflow integrating in vitro data and in vivo validation to refine predictive models.
Diagram 2: A computational pipeline for predicting TF binding, highlighting context-specific model selection.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for SELEX and Binding Validation
| Reagent / Material | Function / Application | Examples / Specifications |
|---|---|---|
| GST-Tagged TF Proteins | Purified transcription factors for in vitro binding assays (SELEX, QuMFRA). Allows purification via glutathione affinity. | GST-Zif268 fusion protein expressed in E. coli [14]. |
| Fluorophore-Labeled Primers | Labeling PCR-amplified DNA sequences for quantitative fluorescence-based binding assays. | Primers labeled with FAM, TAMRA, or ROX for QuMFRA [14]. |
| Oligonucleotide Library | Starting pool for SELEX; contains random sequences for TF binding selection. | 53-base template with a 4-base randomized region for finger 1 interaction [14]. |
| TF-Specific Antibodies | Immunoprecipitation of TF-DNA complexes in in vivo ChIP-seq experiments. | ChIP-grade antibody validated for the specific transcription factor. |
| Glutathione Sepharose | Affinity resin for purifying GST-tagged proteins and protein-DNA complexes. | Used in affinity chromatography-SELEX and QuMFRA [14]. |
| ChIP-Seq Kits | Commercial kits providing optimized buffers and beads for Chromatin Immunoprecipitation. | Kits from suppliers like Abcam, Cell Signaling Technology, or Diagenode. |
The journey from a high-throughput in vitro binding experiment to a model that accurately predicts in vivo function requires careful navigation. This note provides a framework for this process, emphasizing that model performance is highly context-dependent. The key to success lies in:
- Selecting the right model for the biological question, guided by benchmarking studies [55].
- Integrating multi-platform data where possible, as motifs derived from and consistent across multiple experimental platforms (ChIP-seq, SELEX, PBM) are more reliable [9].
- Rigorously validating predictions using in vivo data like ChIP-seq, acknowledging that non-sequence factors (TF expression, protein interactions, PTMs) significantly influence in vivo predictive performance [55].
For researchers in drug development, accurately predicting the functional impact of non-coding variants is paramount. The protocols and benchmarks outlined here provide a pathway to build more accurate, biologically grounded models of TF binding, thereby enhancing the interpretation of regulatory genomics in complex diseases.
Understanding where and how transcription factors (TFs) bind to DNA to regulate gene expression is a fundamental challenge in modern genomics. The accurate identification of transcription factor binding sites (TFBS) is crucial for deciphering gene regulatory networks, understanding disease mechanisms, and guiding drug development. Two principal methodological approaches have emerged: in vitro techniques, particularly SELEX (Systematic Evolution of Ligands by Exponential Enrichment) and its high-throughput variants, and in vivo methods, primarily ChIP-seq (Chromatin Immunoprecipitation followed by sequencing). SELEX characterizes the intrinsic DNA-binding preferences of TFs under controlled conditions, while ChIP-seq captures TF binding in its native cellular context, reflecting the complexity of chromatin structure, nucleosome positioning, and co-factors [56]. This analysis provides a structured comparison of these methodologies, their performance characteristics, and detailed protocols to guide researchers in selecting the appropriate approach for their experimental goals.
Comparative Analysis of Performance and Applications
Key Characteristics and Performance Metrics
Table 1: Comparative Analysis of SELEX and In Vivo Methods for TFBS Identification
| Feature | SELEX/HT-SELEX (In Vitro) | ChIP-seq (In Vivo) |
|---|---|---|
| Experimental Principle | Iterative selection and amplification of protein-bound DNA sequences from a random synthetic library [11] | Immunoprecipitation of cross-linked protein-DNA complexes from cells, followed by sequencing [10] |
| Binding Context | Purified, recombinant TFs in a controlled environment [15] | Native chromatin within a cellular context [10] |
| Primary Output | High-resolution binding motifs (PWMs) representing intrinsic specificity [56] | Genomic regions bound by the TF in vivo, with broad signal footprints [10] |
| Pros | Unbiased sampling of all possible k-mers; reveals full binding potential; not affected by chromatin state [56] | Reflects biologically relevant binding; captures effects of chromatin, cooperativity, and cellular environment [57] |
| Cons | May miss biologically relevant context; can exhibit technological biases (e.g., underrepresentation of palindromes) [56] | Binding site resolution is lower; highly dependent on antibody quality; unsuitable for many lesser-studied TFs [10] |
| Predictive Performance | HT-SELEX-derived models predict in vivo binding better than PBM-derived models [56]. In one study, HT-SELEX models achieved an average AUC of 0.76 [57]. | Serves as the "gold standard" for validating in vivo relevance. JASPAR models (largely based on in vivo data) achieved an average AUC of 0.83 [57]. |
Quantitative Performance in TFBS Prediction
Large-scale comparisons have quantified the effectiveness of models derived from different technologies in predicting genuine in vivo binding sites. A systematic evaluation of 179 binding models revealed that manually curated JASPAR matrices (primarily derived from in vivo data) and HT-SELEX-derived models showed superior performance compared to models from other in vitro technologies like Protein Binding Microarrays (PBMs) [57].
When tested on a "high-confidence" dataset of in vivo binding sites:
- 60% of JASPAR models reached an Area Under the Curve (AUC) score of ≥0.7, with an average AUC of 0.83 [57].
- 70% of HT-SELEX models reached an AUC of ≥0.7, with an average AUC of 0.76 [57].
- In contrast, only 26.6% of PBM-derived models from the hPDI database achieved an AUC of ≥0.7, with an average AUC of 0.57 [57].
Another independent study confirmed that while PBM-based 8-mer ranking was more accurate, models derived from HT-SELEX predicted in vivo binding more effectively [56]. For predicting the effects of non-coding genetic variants, models trained on in vitro HT-SELEX data (e.g., deltaSVM_HT-SELEX, QBiC-Pred) excel at predicting in vitro binding impacts, while DNN-based multitask models trained on large-scale in vivo ChIP-seq datasets (e.g., DeepSEA, Sei) show relatively superior performance for predicting in vivo allele-specific binding events [55].
Experimental Protocols
Detailed Protocol: HT-SELEX
HT-SELEX is a powerful, unbiased technique for determining the preferred DNA sequence motifs of TFs in vitro [15].
Key Research Reagent Solutions:
- Random DNA Library: A double-stranded synthetic oligonucleotide pool featuring a central random region (e.g., 20-40 bp) flanked by constant primer sequences for amplification [11].
- Recombinant TF Protein: Purified DNA-binding domain or full-length transcription factor, often with an affinity tag for purification.
- Binding Buffer: Optimized buffer containing salts, pH stabilizers, and carrier proteins (e.g., BSA) to facilitate specific TF-DNA interactions.
- PCR Reagents: High-fidelity polymerase, dNTPs, and primers complementary to the library's constant flanks for amplifying bound sequences.
- Sequencing Kit: Library preparation and high-throughput sequencing kit compatible with platforms like Illumina.
Step-by-Step Workflow:
- Incubation: The recombinant TF protein is incubated with the double-stranded random DNA library in a suitable binding buffer. This allows the formation of protein-DNA complexes [15].
- Partitioning and Purification: TF-bound DNA sequences are separated from unbound DNA. This can be achieved through various methods, such as using an affinity tag on the TF (e.g., GST-tag pulled down by glutathione beads) or native gel electrophoresis [11] [15].
- Amplification: The purified, bound DNA is eluted and amplified by PCR using primers targeting the constant flanking regions. This enriches the pool for high-affinity binders [15].
- Cycle Repetition: The amplified DNA is used as the input for the next round of selection (steps 1-3). Typically, 3-5 cycles are performed to sufficiently enrich for the highest-affinity binding sites [15] [58].
- Sequencing and Analysis: The final enriched DNA pool, and sometimes intermediate cycles, are subjected to high-throughput sequencing. Bioinformatic pipelines (e.g.,
eme_selex) are then used to identify significantly enriched k-mers and generate position weight matrices (PWMs) [15].
Detailed Protocol: ChIP-seq
ChIP-seq identifies the genomic regions bound by a TF in its native cellular environment.
Key Research Reagent Solutions:
- Crosslinking Reagent: Formaldehyde to covalently cross-link TFs to their genomic DNA targets in living cells.
- Cell Lysis & Sonication Buffers: Detergents and salts for cell lysis and chromatin fragmentation.
- Antibody: High-quality, ChIP-grade antibody specific to the TF of interest.
- Protein A/G Beads: Magnetic or sepharose beads that bind the antibody for immunoprecipitation.
- DNA Clean-up Kit: For purifying the immunoprecipitated DNA after reverse cross-linking.
- Library Prep Kit: For preparing sequencing libraries from the purified, fragmented DNA.
Step-by-Step Workflow:
- Cross-linking: Cells are treated with formaldehyde to cross-link proteins to DNA, freezing TF-DNA interactions in place [59].
- Cell Lysis and Chromatin Shearing: Cells are lysed, and chromatin is fragmented into small pieces (~200-600 bp), typically via sonication [59].
- Immunoprecipitation (IP): The sheared chromatin is incubated with a specific antibody against the target TF. Antibody-TF-DNA complexes are then pulled down using Protein A/G beads [10] [59].
- Washing and Reverse Cross-linking: Beads are washed stringently to remove non-specifically bound DNA. The cross-links are then reversed, and proteins are digested, freeing the immunoprecipitated DNA [59].
- Library Preparation and Sequencing: The purified DNA fragments are used to construct a sequencing library, which is then subjected to high-throughput sequencing [59].
- Data Analysis: Sequenced reads are mapped to a reference genome. Regions with significant enrichment of reads (peaks) compared to a control input sample are identified as putative in vivo binding sites [10] [59].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for TFBS Identification Studies
| Reagent / Solution | Function / Application | Key Considerations |
|---|---|---|
| Random DNA Oligo Library | Core input for SELEX; provides diverse sequence space for probing TF specificity [11]. | The length of the random region (e.g., 20-40 bp) balances coverage and selection pressure. |
| Recombinant TF Protein | The DNA-binding protein of interest for in vitro assays like SELEX [15]. | Purity, concentration, and preservation of DNA-binding activity are critical. Affinity tags (e.g., GST, His) facilitate purification. |
| ChIP-grade Antibody | Specifically immunoprecipitates the target TF-DNA complex in ChIP-seq [10] [57]. | Antibody specificity and affinity are paramount; poor antibodies are a major failure point. |
| Protein A/G Magnetic Beads | Solid-phase support for efficient immunoprecipitation in ChIP-seq [59]. | Enable efficient washing and reduction of non-specific background binding. |
| High-Fidelity PCR Mix | Amplifies bound DNA sequences during SELEX cycles and prepares libraries for sequencing [15]. | Minimizes PCR-introduced biases and errors during amplification steps. |
| Crosslinking Reagent (Formaldehyde) | Preserves transient in vivo TF-DNA interactions for ChIP-seq [59]. | Cross-linking time and concentration must be optimized to balance efficiency and epitope masking. |
The choice between SELEX and ChIP-seq is not a matter of which is universally superior, but which is appropriate for the specific research question. HT-SELEX excels at defining the intrinsic, high-resolution DNA-binding specificity of a TF, free from cellular confounding factors, making it ideal for building foundational models of TF-DNA recognition and for studying TFs lacking ChIP-grade antibodies. ChIP-seq remains the gold standard for mapping the biologically relevant, in vivo binding landscape of a TF within a specific cellular context, capturing the effects of chromatin, cooperativity, and other in vivo determinants. A synergistic approach, using SELEX to define the core motif and ChIP-seq to identify its functional genomic locations, provides the most comprehensive understanding of transcription factor binding and gene regulatory networks. This integrated strategy is powerfully enabled by emerging computational models that leverage the strengths of both data types to improve the prediction of variant effects and functional outcomes.
The identification of transcription factor (TF) binding motifs is a cornerstone of molecular biology, enabling researchers to decipher the complex regulatory codes that control gene expression. These binding preferences are most commonly represented as position weight matrices (PWMs), which provide a quantitative model of the nucleotide preferences at each position within a binding site [60]. The challenge for contemporary researchers lies in the sheer volume and redundancy of available PWM data; multiple databases and high-throughput methods often generate numerous motifs for the same TF, creating a significant bottleneck in data interpretation [60] [61]. This application note addresses these challenges by presenting an integrated framework that leverages the comprehensive Codebook Motif Explorer (MEX) alongside robust public databases and advanced clustering tools like abc4pwm. Within the context of genomic Systematic Evolution of Ligands by Exponential Enrichment (SELEX) research, this resource combination enables more accurate identification of TF binding sites and facilitates the transition from motif discovery to meaningful biological insight.
Table 1: Key Resource Comparison for PWM Analysis
| Resource Name | Primary Function | Key Advantage | Data Scale/Context |
|---|---|---|---|
| Codebook Motif Explorer (MEX) | Consolidated catalog of DNA motifs from experimental data | Provides uniformly processed data & motifs for previously uncharacterized TFs | 332 putative human TFs; ~5,000 experiments [62] [61] |
| abc4pwm | Clustering of PWMs from multiple sources | Integrates DNA-binding domain (DBD) information & automatic quality assessment | ~1,770 human TF PWMs [60] |
| Genomic SELEX | Identification of TF binding sites and target genes | Uses genomic DNA fragments, capturing binding in a more native context | Applied to vertebrate transcription factors like Fezf2 [63] |
The Codebook Motif Explorer Initiative
The Codebook Motif Explorer represents a monumental international effort to systematically determine the DNA binding specificities of human transcription factors, with a particular focus on previously uncharacterized proteins. This project generated an unprecedented data structure through nearly 5,000 independent experiments across multiple assays, including HT-SELEX, GHT-SELEX, SMiLE-seq, and Protein Binding Microarrays (PBMs) [61]. A critical achievement of this project was the establishment of "approved" motif datasets for 177 out of 332 putative TFs analyzed. The data are notably dominated by C2H2 zinc finger proteins, for which 67% (121/180) yielded successful experiments, while approximately half (49%) of TFs with other established DNA-binding domains also produced reliable motifs [61]. This resource provides the scientific community with a uniquely uniform and benchmarked dataset, significantly expanding the catalog of human TF motifs and reducing the interpretive challenges posed by method-specific biases.
Public PWM Databases and Clustering Tools
Alongside the specialized Codebook resource, several public databases and analytical tools remain essential for comprehensive TF binding site analysis. General repositories such as JASPAR and TRANSFAC maintain extensive collections of experimentally derived TF binding motifs [60]. However, the exponential increase in available PWMs from multiple sources has created significant redundancy problems, where different high-throughput methods may generate distinct PWMs for the same TF, confusing researchers and complicating result interpretation [60]. To address this challenge, the abc4pwm (Affinity Based Clustering for Position Weight Matrices) Python package was developed. This tool efficiently clusters biologically similar PWMs from multiple sources, optionally using DNA-binding domain information, generates representative motifs for each cluster, and automatically evaluates clustering quality to filter out incorrect assignments [60]. This functionality is particularly valuable when analyzing large sets of putative PWMs predicted from high-throughput sequencing experiments like ChIP-seq or ATAC-seq.
Experimental Protocols for Motif Discovery and Validation
Genomic SELEX for Binding Site Identification
Genomic SELEX represents a powerful alternative to traditional SELEX that uses random oligonucleotides. This method employs fragmented genomic DNA as the selection library, thereby identifying binding sites within a natural genomic context. The following protocol outlines the key steps for implementing genomic SELEX:
- Library Preparation: Fragment vertebrate genomic DNA (e.g., from zebrafish) mechanically or enzymatically to sizes between 100-500 bp. Ligate adapters compatible with subsequent PCR amplification [63].
- Protein Expression and Incubation: Express the recombinant TF of interest (e.g., Fezf2) with an affinity tag. Incubate the TF with the genomic DNA library in a suitable binding buffer to allow complex formation [63].
- Partitioning and Recovery: Isolate protein-DNA complexes using methods that leverage the affinity tag (e.g., pull-down with magnetic beads). Wash beads thoroughly to remove non-specifically bound DNA [63].
- Amplification and ssDNA Generation: Elute bound DNA fragments from the complexes. Amplify the eluted DNA by PCR. Generate single-stranded DNA (ssDNA) for the next selection round. Asymmetric PCR with a primer-blocker has been shown to yield favorable results in terms of specificity and efficiency for ssDNA generation [64] [65].
- Iterative Selection and Analysis: Repeat steps 2-4 for 3-6 rounds to enrich for high-affinity binding sites. After the final round, clone and sequence the enriched DNA fragments, or analyze them by high-throughput sequencing. The resulting sequences can be computationally analyzed to predict a core consensus binding site [63].
HT-SELEX and Motif Discovery Pipeline
For researchers seeking an in vitro method that surveys a vast space of random sequences, High-Throughput SELEX (HT-SELEX) is a robust option. The protocol below is summarized from a detailed established workflow [15]:
- Random Library Design: Utilize a ssDNA library consisting of a central random region (typically 20-30 bp) flanked by fixed primer sequences for amplification.
- Selection Rounds: Incubate the library with the purified DNA-binding protein. Separate protein-bound DNA from unbound DNA (e.g., using nitrocellulose filters or affinity tags). Recover the bound DNA and amplify it by PCR.
- ssDNA Regeneration: Convert the double-stranded PCR products back to ssDNA for subsequent selection rounds. The
eme_selexbioinformatic pipeline can be used to quantify the enrichment of all possible k-mers, facilitating the detection of promiscuous DNA binding [15]. - Sequencing and Motif Discovery: After several rounds of selection, the enriched DNA pool is sequenced using high-throughput platforms (e.g., Illumina). Apply multiple motif discovery tools (e.g., from the MEME suite, ExplaiNN, or ProBound) to the sequenced fragments to derive candidate PWMs [61].
Integrated Data Analysis Workflow
The journey from raw sequencing data to biological insight requires a structured analytical workflow. HINT-ATAC provides an example of a specialized tool for identifying TF binding sites from ATAC-seq data, which uses a position dependency model to correct for the specific cleavage biases of the Tn5 transposase, significantly improving the prediction of TF binding sites with footprints [66]. Following footprint detection or motif discovery from genomic SELEX/HT-SELEX, the integration with the Codebook database and subsequent clustering is crucial.
A critical step in this workflow is the use of abc4pwm for clustering PWMs. This tool performs a pairwise comparison of PWMs using a similarity score and a dynamical forward-backward PWM alignment method [60]. It can cluster PWMs within specific DNA-binding domain families, and its integrated quality assessment metrics help filter out poorly clustered motifs, ensuring that downstream analyses such as TF motif searching are based on high-quality, non-redundant representative motifs [60].
Research Reagent Solutions
The successful execution of genomic SELEX and subsequent motif analysis depends on key reagents and resources.
Table 2: Essential Research Reagents and Resources
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Codebook MEX Dataset | Provides pre-processed motifs and benchmarking data for human TFs. | Ideal for benchmarking newly discovered motifs or finding motifs for uncharacterized TFs [62] [61]. |
| abc4pwm Software | Clusters PWMs from multiple sources and assesses quality. | Crucial for reducing redundancy and generating robust representative motifs before scanning sequences [60]. |
| Genomic DNA Library | Provides a native genomic context for SELEX. | Uses fragmented genomic DNA instead of random oligonucleotides, capturing evolutionarily conserved sites [63]. |
| Asymmetric PCR with Primer-Blocker | Generates single-stranded DNA (ssDNA) for subsequent SELEX rounds. | Recommended for its specificity, efficiency, and reproducibility [64] [65]. |
| HINT-ATAC Software | Identifies transcription factor binding sites from ATAC-seq data. | Corrects for Tn5 transposase cleavage bias, improving footprinting accuracy [66]. |
Conclusion
Genomic SELEX has firmly established itself as an indispensable, high-throughput method for defining the DNA binding landscape of transcription factors, including many previously uncharacterized proteins. The integration of sophisticated experimental platforms with advanced computational models like DCA-Scapes and random forests is moving the field beyond simple PWMs towards a more nuanced understanding of binding specificity, including multi-modal binding and cell-state specificities. Future directions point towards the seamless integration of in vitro derived motifs with in vivo functional data to build predictive models of gene regulation in specific cellular environments. For drug discovery professionals, this progress translates into an enhanced ability to identify novel therapeutic targets, understand disease mechanisms at the transcriptional level, and develop more precise interventions. The ongoing benchmarking efforts and resource development, such as the Codebook Motif Explorer, will continue to be critical for validating and applying these powerful tools in biomedical research.
References
- 1. Systematic evolution of ligands by exponential enrichment [https://en.wikipedia.org/wiki/Systematic_evolution_of_ligands_by_exponential_enrichment]
- 2. Cell-SELEX: in vitro selection of synthetic small specific ... [https://pubmed.ncbi.nlm.nih.gov/25791604/]
- 3. SELEX: Critical factors and optimization strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9788027/]
- 4. Genomic Selection Identifies Vertebrate Transcription ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3099680/]
- 5. A Comprehensive Guide to Screening Cell-Specific Aptamers [https://www.alpha-lifetech.com/news/blog-cell-selex-a-comprehensive-guide-to-screening-cell-specific-aptamers/]
- 6. High-throughput sequencing SELEX for the determination ... [https://pubmed.ncbi.nlm.nih.gov/35776646/]
- 7. Quantitative modeling and data analysis of SELEX experiments [https://pubmed.ncbi.nlm.nih.gov/16582458/]
- 8. High-throughput sequencing SELEX for the determination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9243297/]
- 9. Cross-platform motif discovery and benchmarking to ... [https://www.nature.com/articles/s42003-025-08909-9]
- 10. Characterizing DNA recognition preferences of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12214022/]
- 11. HTPSELEX—a database of high-throughput SELEX ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1347412/]
- 12. Multiple overlapping binding sites determine transcription ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12447533/]
- 13. Genomic SELEX: A discovery tool for genomic aptamers [https://pmc.ncbi.nlm.nih.gov/articles/PMC2954320/]
- 14. Combining SELEX with quantitative assays to rapidly obtain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1236725/]
- 15. High-throughput sequencing SELEX for the determination ... [https://www.sciencedirect.com/science/article/pii/S2666166722003707]
- 16. HTPSELEX - Database Commons [https://ngdc.cncb.ac.cn/databasecommons/database/id/1279]
- 17. CE-SELEX: Rapid Aptamer Selection Using Capillary ... [https://sciex.com/tech-notes/ce/ce-selex-rapid-aptamer-selection-using-capillary-electrophoresi]
- 18. A beginners guide to SELEX and DNA aptamers [https://www.sciencedirect.com/science/article/abs/pii/S0003269725001289]
- 19. Recent Advances in the Optimization of Nucleic Acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12564743/]
- 20. https://public-pages-files-2025.frontiersin.org/journals/cell-and- ... [https://public-pages-files-2025.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.730035/xml]
- 21. Advancements in SELEX Technology for Aptamers and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190505/]
- 22. AN INTEGRATED MICROFLUIDIC SELEX APPROACH ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5417355/]
- 23. An integrated microfluidic system equipped with a shear ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267025012218]
- 24. Comparative aptamer profiling reveals cell surface remodeling ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d5cb00110b]
- 25. From cell-SELEX to tissue-SELEX for targeted drug ... [https://pubmed.ncbi.nlm.nih.gov/40618926/]
- 26. Position Weight Matrix - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/position-weight-matrix]
- 27. Position weight matrix - Dave Tang's blog [https://davetang.org/muse/2013/10/01/position-weight-matrix/]
- 28. Insights gained from a comprehensive all-against-all ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-01996-3]
- 29. Reliable scaling of position weight matrices for binding ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-015-0666-1]
- 30. The genomic SELEX-based method identifies 350 SigA- ... [https://pubmed.ncbi.nlm.nih.gov/40903908/]
- 31. Position Weight Matrix (PWM) model generation and evaluation [https://epd.expasy.org/pwmtools/pwmeval_selex.php]
- 32. Positional weight matrices have sufficient prediction power ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9237556/]
- 33. Improved Models for Transcription Factor Binding Site ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3389974/]
- 34. Functional analysis of transcription factor binding sites in ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2012-13-9-r50]
- 35. Global pairwise RNA interaction landscapes reveal core ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6023938/]
- 36. Identifying transcription factors with cell-type specific DNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10859-1]
- 37. Introduction [https://dcascapes.org/Introduction.html]
- 38. TOOLS [https://dcascapes.org/TOOLS.html]
- 39. SELEX experiments: New prospects, applications and data ... [https://www.sciencedirect.com/science/article/abs/pii/S1389034407000299]
- 40. Differential binding cell-SELEX method to identify ... [https://www.nature.com/articles/s41598-019-44654-w]
- 41. Predicting transcription factor binding sites [https://bio-protocol.org/exchange/minidetail?id=790617&type=30]
- 42. DeepAptamer: Advancing high-affinity aptamer discovery ... [https://www.sciencedirect.com/science/article/pii/S2162253124003238]
- 43. New Strategies for Evaluation and Analysis of SELEX ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3977542/]
- 44. Evaluating tools for transcription factor binding site prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC6889335/]
- 45. DNA-Binding Specificities of Human Transcription Factors [https://www.sciencedirect.com/science/article/pii/S0092867412014961]
- 46. Benchmarking tools for transcription factor prioritization [https://www.sciencedirect.com/science/article/pii/S2001037024001648]
- 47. Cross-platform DNA motif discovery and benchmarking to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11601219/]
- 48. TRAFICA: an open chromatin language model to improve ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582366/]
- 49. Positional distribution of transcription factor binding sites in the ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0329226]
- 50. Monitoring Genomic Sequences during SELEX Using High ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0009169]
- 51. Modeling binding specificities of transcription factor pairs with ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04734-7]
- 52. Predicting transcription factor binding using ensemble ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6823902/]
- 53. Technologies for profiling the impact of genomic variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11006259/]
- 54. IceQream: Quantitative chromosome accessibility analysis ... [https://www.nature.com/articles/s41467-025-63925-x]
- 55. Comparative analysis of models in predicting the effects ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10959158/]
- 56. A comparative analysis of transcription factor binding models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4005680/]
- 57. A systematic, large-scale comparison of transcription factor ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-016-2729-8]
- 58. DNA-guided transcription factor interactions extend human ... [https://www.nature.com/articles/s41586-025-08844-z]
- 59. Explainable AI in Genomics: Transcription Factor Binding ... [https://arxiv.org/html/2507.09754v1]
- 60. abc4pwm: affinity based clustering for position weight ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8896320/]
- 61. Perspectives on Codebook: sequence specificity of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11601247/]
- 62. Codebook Motif Explorer Supplementary Dataset [https://zenodo.org/records/10182957]
- 63. Genomic selection identifies vertebrate transcription factor ... [https://pubmed.ncbi.nlm.nih.gov/21471212/]
- 64. Specific Aspects of SELEX Protocol: Different Approaches ... [https://www.mdpi.com/2409-9279/8/2/36]
- 65. Specific Aspects of SELEX Protocol: Different Approaches ... [https://pubmed.ncbi.nlm.nih.gov/40278510/]
- 66. Identification of transcription factor binding sites using ATAC-seq [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1642-2]