From Relative to Absolute: A Comprehensive Guide to Quantifying Microbial Abundance for Robust Biomedical Research
This article provides a definitive resource for researchers and drug development professionals on measuring absolute abundance in microbial communities.
From Relative to Absolute: A Comprehensive Guide to Quantifying Microbial Abundance for Robust Biomedical Research
Abstract
This article provides a definitive resource for researchers and drug development professionals on measuring absolute abundance in microbial communities. Moving beyond the limitations of standard relative abundance analysis, we explore the critical importance of quantifying actual microbial loads for accurate biological interpretation. The content covers foundational principles, compares established and emerging quantification methods like spike-in sequencing, flow cytometry, and digital PCR, and offers practical troubleshooting guidance. Furthermore, it presents a rigorous framework for validating methodological choices and demonstrates, through case studies in nutrition and pharmacology, how absolute abundance data transforms our understanding of host-microbiome-drug interactions, ultimately leading to more reproducible and clinically relevant findings.
Why Absolute Abundance Matters: Moving Beyond Compositional Illusions in Microbiome Science
Frequently Asked Questions (FAQs)
1. What does it mean that microbiome data is "compositional," and why is this a problem? Microbiome data generated by high-throughput sequencing is compositional because the data you get from the sequencer represents proportions, not absolute counts. The total number of reads per sample is fixed by the instrument's capacity, meaning an increase in the relative abundance of one microbe must be accompanied by a decrease in the relative abundance of others [1]. This is a fundamental problem because it means you lose information about the true, absolute quantity of microbes in the original sample. Consequently, the data you analyze is closed, or sum-constrained, which can lead to spurious correlations and misleading conclusions about which taxa are truly changing between experimental groups [2] [1] [3].
2. How can compositional data lead to incorrect conclusions in my differential abundance analysis? Compositional data can create both false positives and false negatives. A common error occurs when the absolute abundance of a single microbe changes dramatically. This change can cause the relative abundances of all other microbes to shift, even if their absolute counts remain constant, making them appear differentially abundant when they are not [3]. For example, if a drug treatment drastically reduces a dominant bacterium, the relative proportions of all other bacteria will artificially increase, potentially leading you to falsely believe the treatment benefited those other taxa [1].
3. My analysis shows a strong negative correlation between two microbial taxa. Could this be an artifact? Yes, it very likely could be. Compositional data have a known negative correlation bias [1]. The spurious correlation arises because the data exists in a "simplex" space (where all parts sum to a constant), which violates the assumptions of standard correlation methods designed for unconstrained data. This issue was identified by Karl Pearson over a century ago and is a well-known pathology of compositional data analysis. Therefore, any correlation analysis performed on raw relative abundances or read counts should be treated with extreme caution.
4. Are heritability estimates for microbiome taxa affected by compositionality? Yes, significantly. Estimating heritability (the proportion of variance in a taxon's abundance attributable to host genetics) from relative abundance data can be highly misleading [2]. The interdependency between taxa means that a heritable signal from one microbe can create a spurious heritable signal for a non-heritable microbe, and vice versa. This problem is most acute for dominant taxa. With large sample sizes, these effects can lead to a strong overestimation of the number of heritable taxa in a community [2].
5. What are the main methods available for measuring absolute abundance? Researchers have developed several "quantitative microbiome profiling" (QMP) methods to move beyond relative abundances. The main approaches are summarized in the table below.
Table 1: Core Methods for Absolute Microbial Abundance Measurement
| Method | Brief Description | Key Considerations |
|---|---|---|
| Digital PCR (dPCR) / qPCR [4] [5] | Uses universal primers to quantify the absolute number of 16S rRNA gene copies in a sample. Acts as an "anchor" to convert relative sequencing data to absolute counts. | Provides a precise count of gene copies; requires specific instrumentation. dPCR is highly accurate and does not require a standard curve [4]. |
| Spike-in Standards [4] | A known quantity of an exogenous DNA sequence (not found in the sample) is added prior to DNA extraction. | Controls for variations in DNA extraction and sequencing efficiency; requires careful calibration [4]. |
| Flow Cytometry [5] | Directly counts the number of microbial cells in a sample. | Provides a direct cell count; requires specialized equipment and can be challenging for complex samples like mucosa [4]. |
| Machine Learning Prediction [5] | Uses models trained on datasets with known absolute abundance (e.g., from ddPCR) to predict abundance in new samples from easy-to-measure features like DNA concentration. | A promising, low-cost approach for existing datasets; prediction accuracy depends on the training data and may not be as precise as direct measurement [5]. |
6. I already have a dataset with only relative abundances. What are my options for analysis? For existing relative abundance data, you should use statistical methods designed specifically for compositional data. These methods typically use log-ratios of abundances to avoid the pitfalls of the constant sum constraint. Techniques such as Aitchison's log-ratio analysis, ALDEx2, and Ancom are examples of approaches that acknowledge and adjust for the compositional nature of the data [4] [1] [3]. It is critical to avoid standard statistical tests that assume data independence, as they will likely produce inflated false discovery rates.
Troubleshooting Guides
Problem 1: Inconsistent or Misleading Differential Abundance Results
Symptoms:
- You identify many differentially abundant taxa between two groups, but the results are difficult to interpret biologically.
- The direction of change for a taxon (increase/decrease) does not align with other biological evidence.
- Results change dramatically when you add or remove a single, highly abundant taxon from the dataset.
Solutions:
- Implement Absolute Quantification: Transition from relative to absolute abundance measurement. Incorporate a method like dPCR to quantify total 16S rRNA gene copies in your samples. This allows you to transform your relative sequencing data into estimated absolute counts, providing a true picture of microbial load changes [4] [5].
- Use Compositionally Aware Tools: Re-analyze your relative data with a differential abundance tool that accounts for compositionality, such as ANCOM or ALDEx2 [3]. These tools use log-ratio transformations or other strategies to minimize compositional bias.
- Validate with an External Standard: In future experiments, include a spike-in standard. Adding a known amount of DNA from a microbe not expected to be in your samples during the DNA extraction step allows you to calibrate your entire workflow and calculate absolute abundances for all your native taxa [4].
Problem 2: Unreliable Correlation and Network Analysis
Symptoms:
- You observe a high number of strong negative correlations in your microbial co-occurrence network.
- The network structure is unstable and changes significantly when you rarefy your data to a different depth or filter out low-abundance taxa.
Solutions:
- Apply Log-Ratio Transformations: Replace raw abundance or relative abundance with log-ratios before performing correlation analysis. Because a log-ratio between two taxa is not constrained by the composition, it reduces the risk of spurious correlations [1].
- Be Cautious with Interpretation: Understand that any correlation network built from compositional data is likely to contain false edges. Treat the network as a hypothesis-generating tool rather than a definitive map of microbial interactions. Experimental validation is key.
Problem 3: Low Microbial Biomass and High Contamination Background
Symptoms:
- Samples with low microbial loads (e.g., mucosal tissues, small intestine) show high variability and inconsistent results.
- Your sequencing data reveals a high proportion of taxa commonly identified as laboratory or reagent contaminants (e.g., Pseudomonas, Acinetobacter).
Solutions:
- Quantity and Establish Limits: Use dPCR to establish the lower limit of quantification (LLOQ) for your specific sample type. For example, one study established an LLOQ of 4.2 × 10ⵠ16S rRNA gene copies per gram for stool and 1 × 10ⷠcopies per gram for mucosa [4]. Samples below this threshold are not reliable for profiling.
- Monitor Contaminants: Always include negative control extractions (no sample added) in your sequencing batch. This allows you to identify contaminant taxa present in your reagents and laboratory environment, which can then be filtered out from your biological samples during analysis [4].
- Optimize Input Mass: Ensure you are using the maximum amount of sample material possible without exceeding the binding capacity of your DNA extraction column, especially for host-rich samples like mucosa [4].
The Scientist's Toolkit: Key Reagents & Materials
Table 2: Essential Reagents and Kits for Absolute Quantification Workflows
| Item | Function in Experiment |
|---|---|
| Digital PCR (dPCR) System & Assays | Ultrasensitive and absolute quantification of 16S rRNA gene copies without a standard curve; used to "anchor" sequencing data [4] [5]. |
| Exogenous DNA Spike-in Standard | A known quantity of DNA from an organism not in your samples (e.g., Pseudoaheromonas). Added to sample lysate to control for technical variation from extraction through sequencing [4]. |
| Universal 16S rRNA Primers | Primer sets targeting conserved regions of the 16S rRNA gene; used for both dPCR quantification and amplicon sequencing library preparation [4] [5]. |
| Validated DNA Extraction Kit | A kit demonstrated to have consistent and high efficiency across both Gram-positive and Gram-negative bacteria and your specific sample types (e.g., stool, mucosa) [4]. |
| Mycoplasma Detection/Removal Kits | Critical for maintaining pure microbial cultures and preventing contamination of host-cell cultures used in integrated host-microbe studies [6]. |
| Albenatide | Albenatide|GLP-1 Receptor Agonist|For Research |
| Amino-PEG8-Amine | Amino-PEG8-Amine, MF:C18H40N2O8, MW:412.5 g/mol |
Experimental Protocol: Absolute Abundance Measurement via dPCR Anchoring
This protocol outlines the key steps for quantifying absolute microbial abundance using digital PCR (dPCR) to anchor your 16S rRNA gene sequencing data [4] [5].
1. Sample Preparation and DNA Extraction:
- Homogenize your samples (e.g., stool, mucosal scrapings) in a standardized buffer.
- Critical Step: If using a spike-in standard, add a known, consistent quantity to each sample lysate immediately before DNA extraction.
- Extract DNA using a protocol validated for efficiency and evenness across different bacterial taxa and sample matrices. Monitor and record the total DNA concentration.
2. Absolute Quantification with dPCR:
- Design or obtain a dPCR assay targeting a conserved region of the 16S rRNA gene.
- Run the dPCR reaction according to the manufacturer's instructions for your specific system (e.g., droplet-based or chip-based).
- Use the dPCR software to determine the absolute concentration of 16S rRNA gene copies in each sample (e.g., copies per µL of DNA eluate).
3. 16S rRNA Gene Amplicon Sequencing:
- Prepare amplicon sequencing libraries from the same DNA extracts using standard protocols.
- Recommendation: Monitor amplification with real-time qPCR and stop reactions in the late exponential phase to limit chimera formation [4].
- Sequence the libraries to obtain standard relative abundance profiles.
4. Data Integration and Calculation of Absolute Abundance:
- Process sequencing data to get a feature table (counts per ASV/OTU per sample).
- Use the dPCR measurements to convert relative abundances to absolute abundances with the following calculation for each taxon i in sample j:
Absolute Abundance_ij = (Relative Abundance_ij) * (Total 16S rRNA copies per gram from dPCR)
This workflow transforms your data from a closed composition to an open, absolute scale, enabling biologically accurate comparisons.
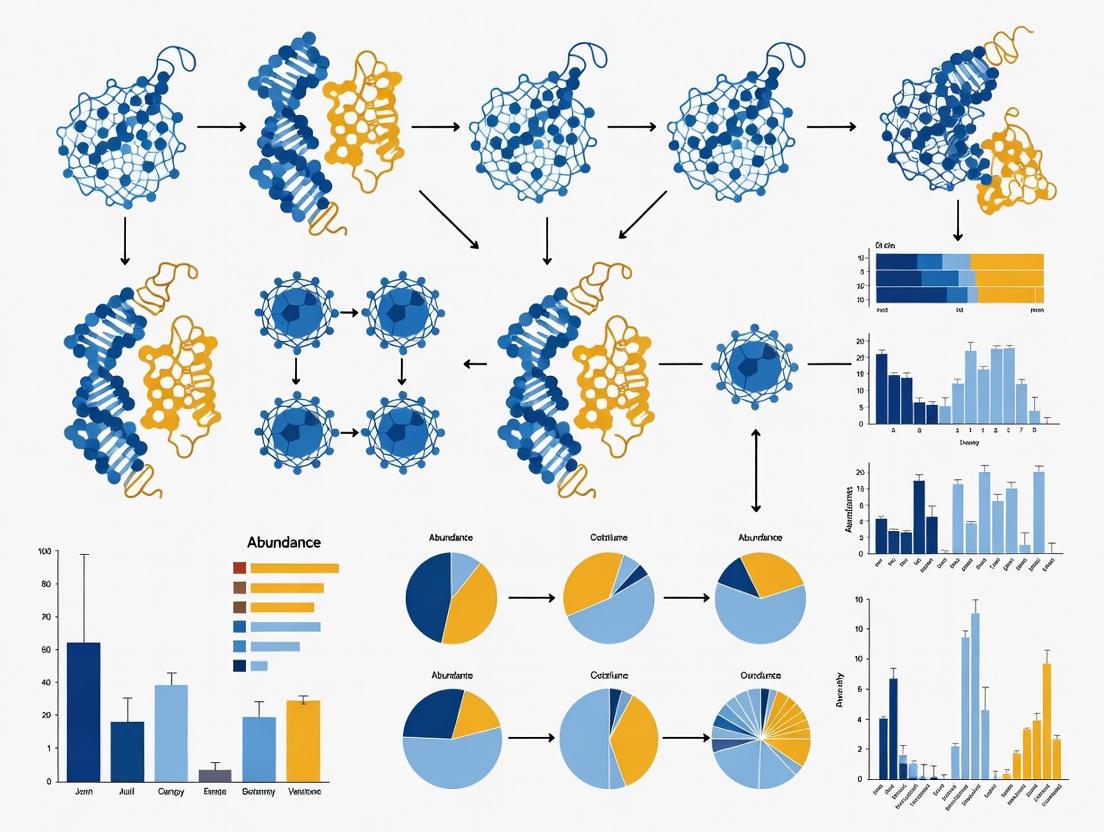
Workflow Visualization: From Relative to Absolute Abundance
The following diagram illustrates the core problem of compositional data and the solution offered by absolute quantification methods.
In microbiome research, a fundamental distinction exists between relative abundance (the proportion of a microorganism within a community) and absolute abundance (the actual quantity of that microorganism in a sample) [7]. Standard sequencing methods, like 16S rRNA gene amplicon sequencing, typically provide only relative abundance data. This case study examines how reliance on relative abundance analysis can produce misleading conclusions in antibiotic intervention studies, and provides troubleshooting guidance for obtaining more accurate, quantitative results.
The Pitfall: How Relative Abundance Misleads
Relative abundance data normalizes all measurements to a constant total, meaning that an increase in one taxon's relative abundance can artificially appear to cause a decrease in all others, even when their actual quantities remain unchanged [8]. In antibiotic studies, this can severely distort the interpretation of a treatment's effect.
Consider this common scenario: An antibiotic eliminates a significant portion of susceptible bacteria. The resistant bacteria, which may not have increased in actual number, now constitute a larger percentage of the surviving community. Relative abundance analysis would incorrectly interpret this as an "increase" or "bloom" of the resistant taxa [9] [10].
Table 1: Comparative Interpretation of a Theoretical Antibiotic Effect
| Metric | Susceptible Taxon A | Resistant Taxon B | Interpretation |
|---|---|---|---|
| Absolute Abundance | Decrease from 60 to 15 million cells | No change (40 million cells) | Antibiotic reduced Taxon A; no effect on Taxon B. |
| Relative Abundance | Decrease from 60% to 27% | Increase from 40% to 73% | Misleadingly suggests Taxon B increased. |
Evidence from veterinary studies demonstrates this pitfall clearly. In a study on piglets treated with tylosin, flow cytometry-based absolute abundance analysis revealed significant decreases in five bacterial families and ten genera that were completely undetectable by standard relative abundance analysis [10]. The relative data showed only a re-shuffling of proportions, obscuring the true, destructive impact of the antibiotic on the community.
Troubleshooting Guide: Resolving Ambiguity in Microbiome Data
Problem: Inconsistent Differential Abundance Results
- Symptoms: Different statistical tools (e.g., LEfSe, DESeq2, ANCOM-II) applied to the same dataset identify drastically different sets of "significant" taxa [8].
- Root Cause: Many differential abundance methods are highly sensitive to data pre-processing steps (e.g., rarefaction, filtering) and the compositional nature of relative data [8].
- Solution:
- Use a consensus approach by running multiple differential abundance methods (e.g., ALDEx2 and ANCOM-II have shown higher consistency) and focus on taxa identified by several tools [8].
- Transition to absolute quantification methods to bypass compositional data constraints. Quantitative Microbiome Profiling (QMP) provides a more reliable foundation for statistical tests [11] [10].
Problem: Unable to Distinguish Ecological Drivers from Direct Effects
- Symptoms: Observing significant changes in taxa that are known to be intrinsically resistant to the administered antibiotic, suggesting complex, indirect ecological impacts [9].
- Root Cause: Relative abundance data cannot differentiate between a taxon's direct susceptibility to a drug and the indirect effects caused by the depletion of its competitors or neighbors.
- Solution:
- Employ a framework like the Microbiome Response Index (MiRIx), which annotates taxa in a sample as susceptible or resistant based on phenotypic and drug-specific databases. A negative MiRIx shift post-antibiotic confirms the treatment enriched resistant organisms [9].
- Correlate shifts in absolute abundance with known antibiotic susceptibility profiles to separate direct inhibition from secondary ecological succession.
Problem: Unclear Impact on Total Microbial Load
- Symptoms: A study can report complete taxonomic recovery post-antibiotic based on relative data, but the host (human or animal) continues to show physiological signs of dysbiosis.
- Root Cause: Relative abundance normalizes to 100%, masking a potentially large and persistent reduction in the total number of microbes (the total microbial load) [11].
- Solution: Quantify the total microbial load using qPCR or flow cytometry. This reveals if the community has truly recovered in size or if a state of depletion persists, which is a crucial clinical insight [11] [10].
Frequently Asked Questions (FAQs)
Q1: My budget only allows for 16S rRNA sequencing. Can I approximate absolute abundance? A: While not a direct measurement, you can approximate absolute abundance if you obtain a single, external measurement of total bacterial load for your sample (e.g., via qPCR targeting the 16S gene). You can then multiply the relative abundances from your 16S sequencing data by this total load to estimate absolute counts [7]. This is most reliable when comparing samples with similar extraction efficiencies.
Q2: What is the most accessible method for transitioning to absolute quantification? A: Spike-in controls are highly accessible and integrate seamlessly with standard sequencing workflows. Adding a known quantity of synthetic DNA or an exotic microbe to your sample before DNA extraction accounts for biases in both extraction and sequencing, allowing for precise back-calculation of absolute abundances for all taxa [12] [11].
Q3: Why can't I just use the raw read counts from my sequencer as a proxy for absolute abundance? A: Raw read counts are heavily influenced by technical variables like sequencing depth and PCR amplification bias. A sample with a higher sequencing depth will have more reads for a taxon, even if its actual abundance is the same as in another sample. Furthermore, organisms with larger genomes can produce more reads, artificially inflating their perceived abundance [7]. Therefore, read counts are only suitable for calculating relative abundance within a sample.
Q4: How do 16S rRNA gene copy number variations affect my analysis, and how can I correct for them? A: The 16S gene exists in multiple copies in a single bacterial genome. This means a bacterium with 10 copies will be overrepresented in sequencing data compared to a bacterium with 1 copy, even if their cell counts are identical [10]. This biases diversity metrics and abundance estimates. To correct for this, you can use databases like rrnDB to normalize your abundance data (both relative and absolute) by the expected 16S copy number for each taxon [10].
Experimental Protocols for Absolute Quantification
Protocol 1: Absolute Abundance via Spike-In Controls
This method uses an internal standard to calibrate sequencing data [12] [11].
- Standard Selection: Choose a synthetic DNA sequence or a non-native, immobilized microbial strain that is absent from your samples.
- Spike-In: Add a precise, known quantity of the standard to your sample lysate before performing DNA extraction.
- Library Preparation & Sequencing: Proceed with standard library prep and sequencing (16S or shotgun).
- Data Calculation:
- Calculate the relative abundance of the spike-in standard in the sequencing data.
- Use the known absolute amount of the spike-in to calculate the total microbial load of the sample:
Total Microbial Load = (Known Spike-in Amount / Relative Abundance of Spike-in). - Calculate the absolute abundance of each taxon:
Absolute Abundance of Taxon = Relative Abundance of Taxon × Total Microbial Load.
Protocol 2: Absolute Abundance via qPCR and Sequencing
This method combines a separate quantitative assay with sequencing data [7] [11].
- qPCR Standard Curve: Run qPCR targeting the 16S rRNA gene on all samples alongside a standard curve of known DNA copy numbers.
- Determine Total Load: From the standard curve, calculate the absolute quantity of 16S gene copies in each sample, which represents the total bacterial load.
- Sequencing & Relative Abundance: Perform 16S rRNA gene sequencing on the same samples to determine the relative abundance of each taxon.
- Data Integration: Calculate the absolute abundance of each taxon:
Absolute Abundance of Taxon = Relative Abundance of Taxon × Total 16S Gene Copies from qPCR.
Table 2: Documented Discrepancies Between Relative and Absolute Abundance in Antibiotic Studies
| Study Context | Finding from Relative Abundance | Finding from Absolute Abundance | Reference |
|---|---|---|---|
| Piglets treated with Tylosin | Missed significant decreases in many taxa. | Revealed decreases in 5 families and 10 genera. | [10] |
| Piglets treated with Tulathromycin | Showed a decrease in only 2 taxa. | Uncovered 8 significantly reduced genera. | [10] |
| Murine Ketogenic Diet Study | Unable to determine direction/magnitude of taxon changes. | Confirmed total microbial load decreased and quantified each taxon's change. | [12] |
| Human Gut Microbiome (General) | Can suggest a taxon increases when it is simply persistent. | Distinguishes between true growth and passive enrichment due to loss of neighbors. | [11] |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Absolute Quantification in Microbiome Research
| Item | Function | Example Application |
|---|---|---|
| Synthetic DNA Spike-Ins | Exogenous internal standard for quantifying absolute abundance from sequencing data. | Added to sample pre-extraction to calibrate for technical biases [12] [11]. |
| qPCR Kits (16S rRNA target) | To quantify total bacterial load via amplification of a universal gene. | Determining total 16S gene copies per gram of sample to convert relative data to absolute [7] [11]. |
| Flow Cytometer | To directly count total bacterial cells in a sample, independent of DNA-based methods. | Providing a direct measurement of total microbial load for fecal or liquid samples [10]. |
| Phenotype & Drug-Susceptibility Databases | To annotate the expected susceptibility of taxa to specific antibiotics. | Calculating a Microbiome Response Index (MiRIx) to contextualize antibiotic intervention data [9]. |
| 16S rRNA Gene Copy Number Database (rrnDB) | To correct for overrepresentation of taxa with multiple 16S gene copies in their genome. | Normalizing sequence counts to more accurately reflect true cellular abundance [10]. |
| Apimostinel | Apimostinel | Apimostinel is an investigational NMDA receptor PAM for neuroscience research. This product is for Research Use Only (RUO), not for human or veterinary use. |
| APN-C3-PEG4-alkyne | APN-C3-PEG4-alkyne, MF:C25H31N3O6, MW:469.5 g/mol | Chemical Reagent |
Workflow Visualization
Comparing Microbiome Analysis Pathways
Antibiotic Effect: Relative vs Absolute Interpretation
Core Concepts: Absolute vs. Relative Abundance
What is the fundamental difference between absolute and relative abundance in microbiome analysis?
- Relative Abundance refers to the proportion of a specific microorganism within the entire microbial community. It is expressed as a percentage, where the sum of all relative abundances in a sample equals 100%. It describes the compositional structure but not the actual quantity of microbes [7].
- Absolute Abundance refers to the actual, quantifiable number of a specific microorganism present in a sample. It is typically reported as the number of microbial cells per unit (e.g., per gram of stool) and reflects the true microbial load [7] [11].
Table 1: Key Differences Between Absolute and Relative Abundance
| Feature | Absolute Abundance | Relative Abundance |
|---|---|---|
| What it measures | Actual number of microbial cells | Proportion of a microbe within the community |
| Data output | Cell count per gram/milliliter | Percentage (%) or fraction |
| Dependence on other taxa | Independent; a change in one taxon does not affect others | Dependent; an increase in one taxon causes an apparent decrease in others [13] |
| Primary data from sequencing | No, requires additional quantification | Yes, directly from sequence read counts |
| Impact of total microbial load | Reveals true changes in population size | Can mask true changes if total load varies [7] |
Why is measuring absolute abundance considered crucial for advanced microbiome research?
Relying solely on relative data can lead to spurious conclusions and mask true biological changes. Absolute abundance is critical because it [13] [11] [10]:
- Prevents Misinterpretation: In relative data, an increase in one taxon can create the false appearance of a decrease in others, even if their actual numbers are unchanged. Absolute quantification distinguishes true increases or decreases in microbial populations.
- Reveals Microbial Load as a Confounder: A 2025 study demonstrated that fecal microbial load is a major determinant of gut microbiome variation and a key confounder in disease association studies. Many disease-linked microbial signatures are more strongly explained by changes in overall microbial load than by the disease itself [14].
- Enables Accurate Cross-Sample Comparison: It allows for reliable comparisons of microbial quantities across different samples, studies, and time points, which is essential for longitudinal and interventional studies.
- Supports Clinical and Diagnostic Applications: It provides the quantitative, robust data required for diagnostics, monitoring treatment efficacy (e.g., antibiotic impact), and developing therapeutics [11] [10].
Methodologies for Quantitative Profiling
Several established and emerging methods enable researchers to move beyond relative abundance to quantitative microbiome profiling (QMP).
Table 2: Methods for Determining Absolute Microbial Abundance
| Method | Underlying Principle | Key Advantages | Key Limitations / Considerations |
|---|---|---|---|
| Spike-in Controls | Adding a known quantity of exogenous microbial cells or DNA to the sample before DNA extraction [13] [11]. | Accounts for technical biases throughout the entire workflow (extraction, amplification) [11]. Highly accurate [13]. | Requires careful selection of non-native spike-in organisms [13]. |
| Flow Cytometry | Directly counting microbial cells in a sample using fluorescent dyes and a flow cytometer [13] [10]. | Direct cell count, not inferred from DNA. Can differentiate between live and dead cells [13]. | Laborious protocol; requires sample dissociation into single cells; can be challenging for low-biomass samples [13] [10]. |
| Quantitative PCR (qPCR) | Amplifying and quantifying a universal marker gene (e.g., 16S rRNA) to estimate total bacterial load [7] [13] [11]. | Cost-effective; feasible for large studies; provides taxonomic specificity with targeted primers [13] [10]. | Subject to primer-dependent amplification bias; does not account for DNA extraction efficiency variations [13] [11]. |
| Total DNA Quantification | Measuring the total DNA concentration of the sample. | Simple and straightforward. | Confounded by the presence of host DNA, especially in low-biomass samples [13]. |
| Machine Learning Prediction | Predicting microbial load from relative abundance profiles using trained models [14]. | Can be applied to existing relative abundance datasets (e.g., large biobanks) without new experiments. | A predictive estimate rather than a direct measurement; accuracy depends on the training data. |
Detailed Experimental Protocol: DNA Spike-in for Absolute Quantification
The following protocol, adapted from a 2025 pilot study on mother-infant gut microbiomes, details the use of marine-sourced bacterial DNA for spike-in quantification [13].
1. Principle: Known amounts of DNA from exogenous microbes not found in the sample of interest are added to the sample prior to DNA extraction. By comparing the sequencing reads of the spike-in to the reads of endogenous microbes, the absolute abundance of the endogenous microbes can be calculated.
2. Reagent Solutions:
- Spike-in Strains: Pseudoalteromonas sp. APC 3896 (Phylum: Pseudomonadota) and Planococcus sp. APC 3900 (Phylum: Bacillota). These marine bacteria are phylogenetically distinct and absent from mammalian gut microbiomes [13].
- Culture Medium: Difco 2216 marine broth.
- DNA Quantification Kit: Qubit 1X dsDNA High Sensitivity (HS) assay kit.
3. Step-by-Step Workflow:
- Step 1: Prepare Spike-in Material. Culture the marine bacterial strains aerobically in marine broth at 30°C for 24 hours. Extract genomic DNA and measure its concentration accurately using the Qubit kit [13].
- Step 2: Calculate Spike-in DNA Copy Number. Determine the number of 16S rRNA gene copies in the spike-in DNA using the formula:
Number of copies (molecules) = (amount of DNA [ng] × 6.022 × 10²³) / (length of dsDNA amplicon × 660 g/mole × 1 × 10⹠ng/g)Obtain the 16S rRNA gene copy number per genome from databases like rrnDB [13]. - Step 3: Add Spike-in to Sample. Add a known, precise volume of the spike-in DNA to the patient sample (e.g., stool) before commencing the DNA extraction process. This ensures the spike-in accounts for biases introduced during extraction [13] [11].
- Step 4: Proceed with Standard Sequencing. Perform DNA extraction, library preparation, and 16S rRNA gene sequencing (e.g., targeting the V3-V4 region) using standard protocols [13].
- Step 5: Bioinformatic and Absolute Abundance Calculation.
- Bioinformatic Processing: Process sequencing reads (e.g., using DADA2 [15]) to generate an Amplicon Sequence Variant (ASV) table.
- Identify Spike-in Reads: The spike-in genera (Pseudoalteromonas and Planococcus) are easily identifiable in the ASV table due to their absence in host samples.
- Calculate Absolute Abundance: For each taxon
iin the sample, its absolute abundance is calculated as:Absolute Abundanceᵢ = (Readsᵢ / Reads_spike-in) × Known_Spike-in_Cells_Added[13] [11].
Troubleshooting Common Issues (FAQs)
FAQ 1: Our lab has already collected a large dataset of 16S rRNA sequencing data with only relative abundance. Can we still derive any absolute quantitative insights?
Answer: Yes, a novel machine-learning approach now allows for the prediction of fecal microbial load directly from relative abundance data. This method can be applied to existing datasets to identify associations between microbial load and host factors, and to statistically adjust for microbial load as a confounder in association studies. However, it is a prediction and not a direct measurement, so its accuracy is dependent on the model and training data [14].
FAQ 2: When we correct for 16S rRNA gene copy number (GCN), some taxa like Lactobacillus show significant changes that were not apparent before. Why does this happen?
Answer: This is a known and important source of bias. The 16S rRNA gene is present in multiple copies in a single bacterial genome. Taxa with a higher GCN (common in Bacillota and Gammaproteobacteria) are overrepresented in sequencing data because a single cell can produce multiple 16S sequences. GCN correction adjusts for this bias, revealing the true per-cell abundance. A 2025 study on antibiotic-treated pigs found that GCN correction was essential to uncover significant decreases in Lactobacillus and Faecalibacterium that were masked by standard relative abundance analysis [10].
FAQ 3: In our antibiotic treatment study, flow cytometry revealed decreases in several genera that were not detected by a spike-in method. Which method is more reliable?
Answer: A 2025 comparative study found that while spike-in methods are highly accurate, flow cytometry can sometimes detect a broader range of significant changes, particularly for certain genera. The study suggested that flow cytometry might be superior for capturing the full effect of strong perturbations like antibiotic treatment, despite being more laborious. The choice of method may depend on your specific research question, sample type, and resources [10].
FAQ 4: Are the raw read counts from a metagenomic sequencing alignment considered a measure of absolute abundance?
Answer: No. Raw read counts from alignment cannot be directly equated to absolute abundance. These counts are influenced by several technical factors, including sequencing depth, PCR amplification bias, and the genome size of different microorganisms. A microorganism with a larger genome will naturally yield more sequencing fragments than one with a smaller genome, even if their cell counts are identical. Therefore, read counts are generally considered an approximation of relative abundance [7].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Resources for Quantitative Microbiome Profiling
| Item / Resource | Function / Purpose | Example & Notes |
|---|---|---|
| Exogenous Spike-in Strains | Provides a known internal standard for calculating absolute abundance. | Marine bacteria (e.g., Pseudoalteromonas sp., Planococcus sp.) [13] or commercially available synthetic cells/DNA. Should be absent from the studied ecosystem. |
| Flow Cytometer with Viability Stains | Directly counts total bacterial cells and can assess cell viability. | Instruments like BD FACSCelesta paired with kits like LIVE/DEAD BacLight [13]. Requires calibration microspheres. |
| qPCR Reagents & Primers | Quantifies total 16S rRNA gene copies or specific taxonomic groups. | PowerUp SYBR Green Master Mix; universal 16S primers (e.g., U16SRT-F/R) or specific primers (e.g., for Bifidobacterium) [13]. |
| DNA Quantification Kits | Precisely measures DNA concentration for spike-in preparation and quality control. | Fluorescence-based kits like Qubit dsDNA HS Assay are preferred over spectrophotometry for accuracy [13]. |
| Bioinformatic Pipelines & Databases | Processes raw sequencing data, performs taxonomy assignment, and facilitates QMP calculations. | 16S processing: DADA2 [16] [15]. Shotgun metagenomics: MetaPhlAn2, Kraken [15]. GCN database: rrnDB [13]. Integrated platform: MicrobiomeAnalyst [16]. |
| Standardized DNA Extraction Kits | Ensures consistent and efficient lysis of microbial cells, which is critical for any quantification method. | Kits such as the QIAamp Mini Stool DNA Kit, often used with bead-beating for mechanical lysis [13]. |
| AQX-435 | AQX-435|Potent SHIP1 Activator for Cancer Research | |
| Arformoterol maleate | Arformoterol | Arformoterol is a selective long-acting beta-2 adrenergic receptor agonist (LABA) for chronic obstructive pulmonary disease (COPD) research. This product is For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
Frequently Asked Questions
FAQ 1: What is the fundamental difference between absolute and relative abundance, and why does it matter for disease association studies?
Relative abundance describes the proportion of a specific microbe within the total microbial community, where all proportions must sum to 100%. In contrast, absolute abundance measures the actual quantity of a microbe, such as the number of cells per gram of sample [7]. This distinction is critical because a change in relative abundance does not reveal whether a microbe is genuinely increasing or if other community members are decreasing. Relying solely on relative data can lead to incorrect conclusions about which taxa are truly associated with a disease state or therapeutic response [17] [12].
FAQ 2: My 16S rRNA sequencing data shows a relative increase in a beneficial taxon after treatment. How can I confirm if this is a true biological effect?
A relative increase could mean the beneficial taxon is growing, or that other taxa are dying off, making the beneficial one appear more prominent. To confirm a true biological effect, you need to measure its absolute abundance. This can be done by quantifying the total microbial load in your sample using methods like digital PCR (dPCR) or flow cytometry and then multiplying the total load by the relative abundance for your taxon of interest [17] [7] [18]. This absolute measurement will reveal if the microbe's actual population size has increased.
FAQ 3: We are studying a drug's mechanism of action on the gut microbiome. Our relative abundance data is inconsistent. What could be the issue?
A common issue is that the total microbial load itself may be changing due to the drug's effect. For instance, if a drug reduces the overall number of gut microbes (total load), a taxon that is actually stable in absolute terms will appear to increase in relative abundance. This can create misleading patterns [17] [12]. Integrating a method for total load quantification (like qPCR or flow cytometry) to calculate absolute abundances will provide a more accurate and reliable picture of the drug's true impact on each microbial population [7].
FAQ 4: How can I differentiate between viable and dead microbes when quantifying absolute abundance?
You can integrate a viability dye, such as propidium monoazide (PMA), into your workflow. PMA selectively enters membrane-compromised (dead) cells and binds to their DNA, preventing its amplification during PCR [19] [18]. When you extract and sequence DNA from a PMA-treated sample, you primarily profile the intact, viable community. Combining PMA treatment with absolute quantification methods (like dPCR) allows you to measure the absolute abundance of viable microbes specifically, which is often more relevant for understanding host-microbe interactions [18].
Troubleshooting Guides
Problem 1: Inconsistent or No Signal in Low-Biomass Samples (e.g., mucosal biopsies, small intestine contents)
Table 1: Troubleshooting Low-Biomass Sample Analysis
| Problem | Potential Cause | Solution |
|---|---|---|
| High levels of host DNA interfering with microbial analysis and DNA extraction. | Host DNA saturates extraction columns, limiting the sample mass that can be processed and reducing microbial DNA yield [17]. | Use a sample input mass that does not exceed the column's binding capacity (e.g., ≤8 mg for mucosal samples). Employ methods to deplete host DNA prior to extraction. |
| Microbial load below the method's limit of detection. | The absolute quantity of microbial 16S rRNA gene copies is too low for accurate quantification or sequencing [17]. | Concentrate samples during DNA extraction if possible. Use an ultrasensitive quantification method like digital PCR (dPCR). Increase sequencing depth to detect low-abundance taxa. |
| High contamination from reagents or the environment. | Contaminating DNA from sources other than the sample becomes significant when microbial biomass is very low [17]. | Include negative control extractions (no sample) in every batch to identify contaminating sequences. Use specialized low-biomass reagent kits. |
Problem 2: Discrepancy Between Viability and Total DNA-Based Absolute Abundance
Table 2: Resolving Viability Discrepancies
| Observation | Interpretation | Resolution |
|---|---|---|
| High absolute abundance of a taxon based on total DNA, but it cannot be cultured. | The taxon may be non-viable (dead) but its DNA is still present and detectable [18]. | Integrate a viability dye like PMA into the workflow. Re-quantify absolute abundance after PMA treatment to measure only intact cells [19] [18]. |
| PMA treatment shows no reduction in signal for a specific taxon. | The cells of this taxon are likely viable and membrane-intact, or the PMA concentration/conditions were not optimized for the sample type [18]. | Validate and optimize PMA concentration for your specific sample matrix (e.g., 2.5–15 µM for seawater) to ensure effective suppression of DNA from dead cells [18]. |
Problem 3: Disagreement Between Molecular and Cell-Counting Anchoring Methods
Table 3: Comparing Anchoring Methods for Absolute Quantification
| Method | Principle | Advantages | Limitations & Pitfalls |
|---|---|---|---|
| Flow Cytometry (FC) | Directly counts intact microbial cells in a sample [18]. | Direct physical count; can distinguish between live/dead cells with specific stains [18]. | Requires a dissociated single-cell suspension, which can be challenging for complex samples like mucosa; does not work well with samples containing high debris [17]. |
| Digital PCR (dPCR) | Quantifies absolute copies of a target gene (e.g., 16S rRNA) per sample volume via endpoint PCR in thousands of droplets [17] [18]. | High precision; resistant to PCR inhibitors; does not require a standard curve [17]. | Quantifies gene copies, not necessarily cell numbers (due to varying copy numbers per genome); requires specific equipment [17]. |
| Spike-in Standards | Adding a known quantity of an exogenous DNA sequence to the sample before DNA extraction [17] [12]. | Can control for technical variations during DNA extraction and library preparation. | Requires accurate initial sample concentration estimate; potential for amplification biases [17] [12]. |
Experimental Protocols for Absolute Abundance Measurement
Protocol 1: Absolute Quantification of Microbial Taxa Using dPCR Anchoring and 16S rRNA Gene Sequencing
This protocol, adapted from a established quantitative framework, is designed for diverse sample types, including stool and mucosal samples [17].
Sample Preparation and DNA Extraction:
- Weigh Sample: Use an amount that does not exceed the DNA extraction column's capacity (e.g., ≤200 mg for stool, ≤8 mg for mucosa) to avoid saturation by host DNA [17].
- Extract DNA: Use a standardized DNA extraction kit with demonstrated efficiency across Gram-positive and Gram-negative bacteria. Include extraction controls.
Quantify Total Microbial Load via dPCR:
- Setup: Prepare a dPCR reaction mixture targeting the 16S rRNA gene using the extracted DNA.
- Partition and Amplify: The reaction is partitioned into thousands of nanoliter-sized droplets, and PCR amplification is carried to endpoint.
- Read and Analyze: Count the number of positive (fluorescent) droplets. Using Poisson statistics, calculate the absolute concentration of 16S rRNA gene copies per microliter of DNA extract, and then per gram of original sample [17].
16S rRNA Gene Amplicon Sequencing:
- Library Prep: Generate 16S rRNA gene amplicon libraries from the same DNA extract. Monitor amplification with real-time qPCR and stop reactions in the late exponential phase to limit chimera formation [17].
- Sequence: Perform high-throughput sequencing on an Illumina or similar platform.
Data Integration and Calculation of Absolute Abundance:
- Bioinformatic Analysis: Process sequencing reads to obtain a table of relative abundances for each taxon in each sample.
- Calculate Absolute Abundance: For each taxon in a sample, multiply its relative abundance (as a proportion) by the total 16S rRNA gene copies/gram obtained from dPCR [17] [7].
- Formula: Absolute Abundanceâ‚œâ‚â‚“â‚’â‚™ = Relative Abundanceâ‚œâ‚â‚“â‚’â‚™ × Total Microbial Load (from dPCR)
Protocol 2: Assessing Absolute Abundance of Viable Microbes with PMA Treatment
This workflow enhances the previous protocol by differentiating viable cells, which is crucial for drug mechanism studies [18].
Sample Treatment:
- PMA Addition: Add PMA dye to the homogenized sample (before DNA extraction) to a final optimized concentration (e.g., 2.5–15 µM for seawater; requires validation for other sample types) [18].
- Incubate and Cross-link: Incubate in the dark for 10 minutes, then expose to a strong light source (e.g., a 464 nm LED transilluminator) for 30 minutes with constant mixing. Light activates PMA, which covalently cross-links to DNA in membrane-compromised cells, rendering it unamplifiable [18].
DNA Extraction and Quantification:
- Proceed with DNA extraction as in Protocol 1.
- Quantify the total 16S rRNA gene copies from viable (intact) cells using dPCR. This value represents your viable microbial load.
Sequencing and Profiling:
- Perform 16S rRNA gene amplicon sequencing on the PMA-treated DNA.
- Calculate the absolute abundance of each viable taxon by multiplying its relative abundance from the PMA-treated sequencing data by the viable microbial load from the PMA-dPCR assay.
Experimental Workflow for Absolute Abundance Analysis
The following diagram illustrates the integrated workflow for obtaining absolute abundance data, incorporating viability assessment.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for Absolute Abundance Studies
| Item | Function/Benefit |
|---|---|
| Digital PCR (dPCR) System | Provides an absolute count of 16S rRNA gene copy numbers without a standard curve, offering high precision for quantifying total microbial load [17] [18]. |
| Propidium Monoazide (PMA) | A viability dye that selectively inhibits PCR amplification of DNA from dead, membrane-compromised cells, allowing for the specific profiling of intact, viable microbes [19] [18]. |
| Validated DNA Extraction Kits | Kits with demonstrated efficiency and evenness in lysing both Gram-positive and Gram-negative bacteria are crucial for unbiased representation and accurate quantification [17]. |
| Flow Cytometer | Enables direct enumeration of total and intact microbial cells in a sample, serving as an independent method to anchor and normalize sequencing data for absolute quantification [18]. |
| Standardized 16S rRNA Gene Primers | "Universal" primer sets with minimized amplification bias are essential for obtaining accurate relative abundance profiles that can be confidently converted to absolute values [17]. |
| Arylquin 1 | Arylquin 1|Par-4 Secretagogue|For Research |
| Asapiprant | Asapiprant, CAS:932372-01-5, MF:C24H27N3O7S, MW:501.6 g/mol |
The Quantitative Toolbox: From Lab Bench to Bioinformatics for Absolute Microbial Quantification
In the study of microbial communities, determining the absolute abundance of microorganisms is a fundamental objective. Flow Cytometry (FCM) has emerged as a powerful, cultivation-independent technique for the rapid enumeration of total microbial cells. Unlike traditional methods like heterotrophic plate counts (HPC), which can significantly underestimate cell numbers by failing to detect viable but non-culturable (VBNC) organisms, FCM provides a direct and sensitive quantification of total cell counts (TCC) within hours [20] [21]. This guide addresses the application of FCM for total microbial load enumeration, providing troubleshooting support and detailed protocols to integrate this method effectively into microbial ecology research.
Troubleshooting Common Flow Cytometry Issues
Researchers often encounter specific challenges when adapting flow cytometry for microbial enumeration, particularly with complex samples. The following table addresses common problems and their solutions.
| Problem Scenario | Possible Causes | Expert Recommendations & Solutions |
|---|---|---|
| High Background/Non-specific Staining | Non-cellular particles (proteins, lipids) binding fluorescent dyes [21] [22]. | Use enzymatic clearing (e.g., proteinase K, savinase) and centrifugation to remove interfering particles [21] [22]. For intracellular targets, ensure proper fixation/permeabilization [23]. |
| Weak or No Fluorescence Signal | Low dye concentration; incorrect instrument settings; poorly expressed target [24] [23]. | Optimize dye concentration and staining incubation time [24]. Verify laser and PMT settings match the fluorochrome. Use bright fluorochromes (e.g., PE) for low-density targets [23]. |
| Loss of Signal or Low Cell Counts | Lysis of delicate cells due to harsh sample preparation; incorrect gating [24]. | For difficult-to-lyse bacteria (e.g., in rich media), reduce glucose concentration, freeze cells before extraction, or add lysozyme [24]. Re-evaluate gating strategy using controls. |
| Variability in Results Day-to-Day | Inconsistent sample preparation; instrument drift; antibody aggregates [23] [25]. | Follow standardized protocols. Centrifuge antibodies before use to remove aggregates [25]. Use internal controls and perform daily instrument quality control. |
| Compensation/Spillover Errors | Incorrectly set spillover matrix; use of inappropriate controls (e.g., beads instead of cells); autofluorescence [26] [25]. | Use well-stained single-color cell controls, not beads. Acquire enough positive events. For spectral flow, check autofluorescence subtraction [26]. |
| Suboptimal Scatter Properties | Incorrect FSC/SSC voltages; cell debris; clogged flow cell [25]. | Adjust FSC/SSC voltages so all cells of interest are on-scale. Filter samples to remove large debris. Run bleach and water to unclog the flow cell [23] [25]. |
Frequently Asked Questions (FAQs)
Q1: How does FCM compare to traditional plate counts for microbial enumeration? FCM is a cultivation-independent method that quantifies total cells, including those that are viable but non-culturable (VBNC), providing a more accurate assessment of total microbial load. HPCs typically detect less than 1% of the total microbial community and require days for results, whereas FCM is quantitative, rapid (results within hours), and demonstrates low operator dependency [20] [21].
Q2: What do the "LNA" and "HNA" bacteria classifications mean in FCM? Based on fluorescence intensity from nucleic acid stains, bacteria in a sample can be broadly classified into two groups: Low Nucleic Acid (LNA) and High Nucleic Acid (HNA) bacteria. The fluorescence intensity serves as an indicator of apparent cellular nucleic acid content. Some studies have found that the HNA subgroup can show a better correlation with active biomass parameters like ATP than the total cell count [20].
Q3: My sample is a complex matrix (e.g., food, milk). How can I prepare it for FCM? Complex matrices require clearing to remove interfering particles. An effective protocol involves a sequence of steps:
- Enzymatic Treatment: Use proteases (e.g., proteinase K, savinase) to digest proteins [21] [22].
- Centrifugation: Pellet cells and remove digested proteins and lipids [21].
- Filtration: A final filtration step can further remove debris [22]. The resulting pellet is then resuspended and stained for FCM analysis.
Q4: How can I distinguish between live and dead microbial cells? A common method is dual-staining with fluorescent dyes that have different membrane permeabilities. A cell-permeant green dye (e.g., SYTO) labels all cells, while a red, non-cell-permeant dye (e.g., Propidium Iodide, PI) only enters cells with compromised membranes. Therefore, cells stained with both green and red are considered dead [24] [21]. It's important to optimize dye concentrations and use filter sets to minimize bleed-through between channels [24].
Quantitative Data from FCM Enumeration Studies
The table below summarizes key findings from field studies that utilized FCM for total cell count (TCC) analysis, illustrating its application and the factors affecting microbial load.
| Study Context / Sample Type | Total Cell Count (TCC) Range | Correlation with Other Parameters | Key Influencing Factor Identified |
|---|---|---|---|
| Drinking Water Distribution Systems [20] | ~120,000 - 220,000 cells/mL (at treatment plant exit) | No consistent relationship found between TCC and HPC or Aeromonas. Some correlation between HNA bacteria and ATP (R² = 0.63). | Water temperature: TCC values were higher at temperatures above 15°C. |
| Raw Milk Analysis [21] | N/A (Detection limit: ≤10ⴠbacteria/mL) | Good correlation (r ≥ 0.98) with plating and microscopic counts in spiked UHT milk; good agreement (r = 0.91) with SPC in raw milk. | Sample preparation: Critical enzymatic clearing required to distinguish bacteria from milk proteins and lipids. |
| General Drinking Water [20] | Can accurately count microbial cells at concentrations as low as 1,000 cells mLâ»Â¹. | Good relationship found between TCC and ATP in some studies [20]. | Treatment processes: Biomass changes are effectively tracked through water treatment steps. |
Essential Experimental Protocols
Protocol 1: Total Cell Count and Viability Assessment for Bacteria
This protocol is adapted for general bacterial suspensions in simple buffers or cleared samples [24] [21].
- Staining Solution Preparation:
- Prepare a working solution of SYBR Green I (SG) by making a 100x dilution in anhydrous DMSO.
- For viability staining, create a SYBR Green I/Propidium Iodide (SGPI) solution by mixing the SG working solution with a 30 mM PI stock at a 50:1 ratio.
- Staining:
- Prepare two 1 mL subsamples of your bacterial suspension.
- Add SG stain to one subsample and SGPI to the other at a recommended volume of 10 μL per mL of sample.
- Vortex gently and incubate in the dark for 10 minutes (SG) or 15 minutes (SGPI).
- Flow Cytometry Analysis:
- Analyze the samples on a flow cytometer equipped with a 488 nm laser.
- Collect green fluorescence (FL1) at ~520 nm and red fluorescence (FL3) at >630 nm.
- Use the SG-stained sample to establish the gate for the total cell population.
- Use the SGPI-stained sample to identify subpopulations: SG-positive/PI-negative (intact/live) and SG-positive/PI-positive (membrane-damaged/dead).
Protocol 2: Enzymatic Clearing of Complex Samples (e.g., Milk, Juices)
This protocol is crucial for analyzing microbial load in samples with high background interference [21] [22].
- Sample Treatment:
- Take a 100 μL sample (e.g., UHT milk, carrot juice).
- Add 0.05 mg of proteinase K or 10 μL of savinase. For raw milk, use 50 μL of savinase plus 50 μL of 0.1% Triton X-100.
- Incubation:
- Mix thoroughly and incubate at 37°C for 30-45 minutes.
- Centrifugation and Washing:
- Add 900 μL of 150 mM NaCl and mix by inversion.
- Centrifuge at 14,000 × g for 10 minutes.
- Carefully remove the top lipid layer and the supernatant containing digested proteins with a micropipette without disturbing the pellet.
- Resuspension:
- Resuspend the pellet (which contains the bacteria) in 100 μL of 150 mM NaCl.
- Staining and Analysis:
- Proceed with staining and FCM analysis as described in Protocol 1.
Workflow and Signaling Pathways
FCM Microbial Enumeration Workflow
The following diagram illustrates the core workflow for total microbial load enumeration using flow cytometry, from sample preparation to data analysis.
The Scientist's Toolkit: Research Reagent Solutions
The table below lists key reagents and their critical functions in flow cytometry-based microbial enumeration.
| Reagent / Material | Function in the Experiment |
|---|---|
| SYBR Green I / SYTO BC | Cell-permeant nucleic acid stain that labels all bacterial cells, enabling total cell count [20] [21]. |
| Propidium Iodide (PI) | Non-cell-permeant nucleic acid stain that only enters cells with damaged membranes, used for viability/dead cell assessment [20] [21]. |
| Proteinase K / Savinase | Protease enzymes used to digest proteinaceous particles in complex samples (e.g., milk, juice) to reduce background noise [21] [22]. |
| Dimethylsulfoxide (DMSO) | A solvent used for preparing stock and working solutions of certain fluorescent dyes [20]. |
| Fixatives (e.g., Formaldehyde) | Used to cross-link and preserve cells, stabilizing the sample for later analysis. Methanol-free formaldehyde is recommended to prevent unwanted permeabilization [23]. |
| Permeabilization Agents (e.g., Saponin, Triton X-100, Methanol) | Used to create holes in the cell membrane, allowing antibodies or dyes to access intracellular targets [23]. |
| ASP6432 | ASP6432, CAS:1282549-08-9, MF:C25H29KN4O7S2, MW:600.75 |
| Asudemotide | Asudemotide, CAS:1018833-53-8, MF:C58H80N10O17, MW:1189.3 g/mol |
In microbiome research, the standard output from high-throughput sequencing is relative abundance—the proportion of each microbe within the total sequenced community. A fundamental limitation of this data is that an increase in the relative abundance of one taxon necessitates an artificial decrease in all others, even if their actual cell counts remain unchanged. This compositional nature of sequencing data can obscure true biological changes, making it impossible to determine from relative data alone whether a microbe has genuinely increased in absolute number or is simply appearing more prevalent because other community members have decreased [27] [7].
Absolute abundance quantification overcomes this by measuring the actual number of microbial cells or gene copies in a sample. Spike-in standards are a powerful method to achieve this, where a known quantity of an exogenous control is added to a sample prior to DNA extraction. This control then serves as an internal calibrator, allowing researchers to convert relative sequencing reads into absolute counts [11] [28]. This technical support center provides a comprehensive guide to implementing these critical controls in your microbiome research.
FAQs on Spike-in Standards
1. What are spike-in standards and why are they crucial for absolute abundance measurement?
Spike-in standards are known quantities of exogenous biological materials—such as synthetic DNA, recombinant bacteria, or engineered cells—added to a sample at the start of an experiment. They undergo the entire wet-lab workflow alongside the native sample, accounting for technical biases introduced during DNA extraction, PCR amplification, and library preparation. By measuring the recovery of the spike-in sequences after sequencing, researchers can create a calibration curve to convert the relative proportions of native microbes into absolute abundances [27] [11] [28].
2. How do I choose between different types of spike-in controls?
The choice of spike-in depends on your experimental goals, sample type, and desired level of control. The table below compares the main categories:
Table 1: Comparison of Major Spike-in Control Types
| Control Type | Description | Key Advantages | Potential Limitations |
|---|---|---|---|
| Synthetic DNA (synDNA) [27] | Chemically synthesized DNA sequences with negligible identity to natural genomes. | - High precision for absolute quantification.- Minimizes nonspecific alignment.- Can be designed to cover a range of GC contents. | Requires accurate initial quantification. |
| Recombinant Bacteria [28] | Genetically engineered bacteria with unique synthetic DNA tags (e.g., in the 16S rRNA gene). | - Controls for cell lysis and DNA extraction efficiency.- Mimics the behavior of natural communities. | - May interact with or influence the native microbiome.- Requires careful selection of host strains. |
| Whole-Cell Standards [28] | Intact, fixed cells of recombinant bacteria. | - Benchmarks the entire process from cell handling to sequencing. | - Cell counting and DNA extraction efficiency can vary between species. |
3. My spike-in recovery is lower than expected. What could be the cause?
Low recovery of spike-in materials can stem from several issues in the experimental workflow:
- Inefficient DNA Extraction: If using whole-cell standards, the cell lysis efficiency may vary between microbial species due to differences in cell wall structures [28].
- PCR Amplification Bias: Spike-in sequences with very high or low GC content can be amplified less efficiently, leading to under-representation in the final library [27]. Using a pool of spike-ins with variable GC content can mitigate this.
- Degradation of Standards: Improper storage or handling of the spike-in reagents can lead to degradation, reducing the effective quantity added [28].
- Binding Losses: In protocols like CUT&RUN, ensure that bead-based binding steps are optimized and that clumping is avoided to maximize recovery [29].
4. Can I use spike-ins from one manufacturer for a different protocol (e.g., using a ChIP-seq spike-in for CUT&RUN)?
Many core spike-in technologies, particularly those based on recombinant nucleosomes with barcoded DNA, are designed for cross-protocol compatibility (e.g., CUT&RUN, CUT&Tag, and ChIP-seq) [30]. However, it is critical to consult the manufacturer's specifications. Always verify that the conserved elements (e.g., antibody recognition sites, barcode locations, and adapter sequences) are compatible with your specific library preparation kit and sequencing platform.
5. How do I normalize my sequencing data using spike-in controls?
The following workflow outlines the general process for data normalization using spike-in controls:
Figure 1: Data normalization workflow using spike-in controls.
After sequencing and read mapping, the absolute abundance of a native microbial taxon can be calculated using the formula:
Absolute Abundance (Taxon A) = (Relative Abundance of Taxon A) × (Total Spike-in Cells Added) / (Spike-in Read Count) [28] [7]
This calculation hinges on knowing the exact number of spike-in cells or genome copies added to your sample, which is provided by the manufacturer or determined through precise quantification methods like digital PCR [28].
Troubleshooting Guide
Table 2: Common Spike-in Experimental Issues and Solutions
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| High Variability in Spike-in Reads | - Inconsistent pipetting of spike-in volume.- Improper mixing of spike-in reagent. | - Use calibrated pipettes and reverse pipetting for viscous solutions.- Vortex and spin down spike-in reagents before use.- Create a master mix of spike-in for multiple samples. |
| Spike-in Reads Dominating Library | - Too high a spike-in-to-sample ratio.- Low microbial load in the native sample. | - Titrate the spike-in amount in a pilot experiment.- Use methods like qPCR or flow cytometry to estimate native microbial load beforehand to inform spike-in dosing [12]. |
| False-Positive Alignment of Native Reads to Spike-ins | - Spike-in sequence shares high similarity with natural genomes. | - Use computationally designed synthetic DNA (synDNA) with verified negligible identity to NCBI databases [27]. |
| Inaccurate Absolute Quantification | - Incorrect initial concentration of the spike-in stock.- DNA extraction bias not fully accounted for. | - Use digital PCR (dPCR) to accurately quantify the spike-in stock solution [12] [28].- Employ whole-cell spike-in standards to control for extraction bias [28]. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials and Reagents for Spike-in Experiments
| Item | Function / Description | Example Context |
|---|---|---|
| synDNA Spike-in Pools [27] | A set of synthetic DNA molecules (e.g., 2,000-bp length) with variable GC content, cloned into plasmids for distribution. | Absolute quantification in shotgun metagenomic sequencing of complex microbial communities. |
| ATCC Spike-in Standards (MSA-1014, MSA-2014) [28] | Defined mixtures of genomic DNA or whole cells from three recombinant bacteria (E. coli, S. aureus, C. perfringens), each containing a unique synthetic 16S rRNA tag. | Quantitative normalization in both 16S rRNA gene amplicon and shotgun metagenomic sequencing. |
| SNAP Spike-in Controls [30] | Panels of defined recombinant nucleosomes with specific histone modifications, each wrapped with a unique barcoded DNA template. | Normalization and antibody validation in epigenomics protocols like CUT&RUN, CUT&Tag, and ChIP-seq. |
| Digital PCR (dPCR) [12] | An ultrasensitive method for absolute nucleic acid quantification without a standard curve by partitioning a sample into thousands of nanoliter reactions. | Precisely quantifying the concentration of spike-in stock solutions and total microbial load in a sample. |
| Quantitative Microbiome Profiling (QMP) Services [11] | Commercial services that integrate spike-in controls or qPCR with sequencing to provide absolute abundance data. | For research groups seeking to outsource the wet-lab and bioinformatic steps of absolute quantification. |
| AT-076 | AT-076, CAS:1657028-64-2, MF:C26H35N3O3, MW:437.6 g/mol | Chemical Reagent |
| Avn-322 | Avn-322, CAS:1194574-68-9, MF:C17H20ClN5O2S, MW:393.9 g/mol | Chemical Reagent |
Experimental Protocol: Implementing synDNA Spike-ins for Shotgun Metagenomics
This protocol is adapted from the synDNA method, which utilizes a pool of 10 synthetic DNA sequences with a range of GC contents (26% to 66%) to minimize amplification bias [27].
Workflow Overview:
Figure 2: synDNA spike-in protocol workflow.
Step-by-Step Methodology:
synDNA Pool Preparation:
- Obtain the synDNA plasmids (e.g., from Addgene) [27].
- Propagate the plasmids in E. coli and purify them using a standard plasmid midi-prep kit.
- Precisely quantify each purified synDNA plasmid using a high-accuracy method like dPCR or fluorometry.
- Mix the 10 synDNAs at different concentrations to create a dilution pool. The concentration of each synDNA in the pool should be known.
Sample Spiking:
- Add a fixed volume of the synDNA pool to your microbial sample (e.g., stool, soil, saliva) immediately before DNA extraction. The volume added should be determined in a pilot experiment to ensure spike-in reads are detectable without dominating the sequencing library.
DNA Extraction and Library Preparation:
- Proceed with your standard DNA extraction protocol. The spike-in DNA will co-extract with the sample's native DNA.
- Use the extracted DNA (sample + spike-in) for shotgun metagenomic library preparation, following standard protocols.
Sequencing and Bioinformatic Analysis:
- Sequence the libraries on your preferred platform (e.g., Illumina).
- Process the raw sequencing reads using a standard metagenomic pipeline for quality control and host read removal.
- Map the cleaned reads to a combined reference database that includes the genomes of expected microbes and the 10 synDNA sequences. The synDNA sequences should be made available by the protocol authors [27].
- Count the number of reads that map uniquely to each synDNA and to each microbial taxon.
Absolute Quantification and Data Normalization:
- For the spike-ins, verify that the measured read counts show a high linear correlation (R² ≥ 0.94) with their known input concentrations [27].
- Use the known number of synDNA molecules added to the sample and the resulting synDNA read counts to generate a linear calibration model.
- Apply this model to the read counts of the native microbial taxa to estimate their absolute abundance in the original sample (e.g., in units of cell equivalents or genome copies per gram of sample).
Frequently Asked Questions (FAQs)
Q1: My Qubit Fluorometer is showing an "out of range" error. What should I check?
- A: An "out of range" error typically means the sample concentration lies outside the instrument's accurate detection limits. Please check the following [31]:
- Dilute concentrated samples: If the concentration is too high, dilute the sample and re-measure.
- Concentrate low-abundance samples: For low concentrations, use a larger sample volume (up to 20 µL) or switch to a more sensitive assay kit (e.g., from BR (Broad Range) to HS (High Sensitivity)).
- Verify standard values: Check the raw fluorescence values under "Check Standards." The value for Standard 2 should be at least 10-50 times higher than Standard 1. If not, your assay kit may be expired or the standards may have degraded.
Q2: Why are my nucleic acid quantification results from a Qubit Fluorometer and a NanoDrop spectrophotometer significantly different?
- A: This is a common occurrence. The Qubit assay uses fluorescent dyes that are specific to the target molecule (e.g., dsDNA), whereas the NanoDrop measures UV absorbance at 260 nm, which can be influenced by contaminants like proteins, single-stranded nucleic acids, or free nucleotides [31]. The discrepancy usually indicates that your sample is contaminated with other molecules that absorb at 260 nm. For accurate quantification of the specific nucleic acid, the Qubit reading is more reliable. To resolve this, further purify your sample to remove contaminants [31].
Q3: The amplification curve from my qPCR run has an unusual shape. What does this indicate?
- A: Abnormal amplification curves can reveal specific issues with your qPCR reaction [32] [33]:
- Exponential amplification in the No Template Control (NTC): Indicates contamination of your reagents or workspace with the target sequence. Decontaminate your work area and prepare fresh reagents [32].
- Jagged or noisy curves: Can be caused by poor probe signal, mechanical errors, or unstable reagents. Ensure you are using a sufficient amount of probe and mix all solutions thoroughly [32].
- Low plateau phase: Suggests that reaction reagents are limiting, degraded, or that the probe concentration is incorrect. Prepare a fresh master mix and check your calculations [32].
- Unexpectedly early Cq (Quantification Cycle): May be due to genomic DNA contamination in RNA samples, high primer-dimer formation, or non-specific amplification. Treat samples with DNase, redesign primers, or optimize annealing temperatures [32].
Q4: When should I use digital PCR (dPCR) over quantitative PCR (qPCR) for my microbial quantification study?
- A: dPCR provides absolute quantification without the need for a standard curve and is superior for applications requiring high sensitivity and precision, especially at low target concentrations [34] [35]. You should consider dPCR when:
- Absolute quantification is required: dPCR directly counts the number of DNA molecules, making it ideal for measuring absolute abundance in microbial communities [4] [5].
- Detecting rare targets: dPCR is more sensitive and can detect rare mutations or low-abundance pathogens that might be missed by qPCR [34].
- Sample contains inhibitors: dPCR is more tolerant to PCR inhibitors present in complex samples like soil or stool because it is an end-point measurement [34] [35].
- For routine quantification of higher concentration samples, qPCR remains a cost-effective and efficient option [35].
Q5: Why is measuring absolute abundance, rather than relative abundance, crucial in microbiome studies?
- A: Standard sequencing techniques provide relative abundance data, where the proportion of one microbe depends on the abundances of all others. This can be misleading [4]. For example, an observed increase in the relative abundance of a taxon could be due to a true increase in its numbers or a decrease in other taxa [4]. Absolute abundance measurements reveal the actual number of microorganisms, providing a true picture of microbial load and enabling accurate comparisons across samples and over time [4] [5]. This is essential for understanding real changes in the ecosystem.
Troubleshooting Guides
Qubit Fluorometer Troubleshooting
| Problem | Potential Cause | Solution |
|---|---|---|
| "Out of Range" Error | Sample concentration is too high or too low. | Dilute the sample or use a more/less sensitive assay. Check sample volume [31]. |
| Inaccurate Readings | Contaminants absorbing light; old reagents; temperature fluctuation. | Purify sample; use fresh kit reagents; ensure all reagents are at room temperature before use [31]. |
| "Error with Standard" Message | Incorrect standard preparation; degraded RNA standard; expired kit. | Prepare fresh standards from new stock; use an unopened RNA standard tube; check kit expiration date [31]. |
| Fluorescence Signal Decreases During Reading | Tube is heating up inside the instrument. | For multiple readings, remove the tube and let it equilibrate to room temperature for 30 seconds before rereading [31]. |
qPCR Amplification Curve Analysis
The table below outlines common qPCR curve anomalies, their causes, and corrective actions [32].
| Observation | Potential Cause | Corrective Steps |
|---|---|---|
| Amplification in No Template Control (NTC) | Contamination from target sequence. | Decontaminate workspace with 10% bleach; prepare reagents in a clean area; use new reagent stocks [32]. |
| High Noise in Early Cycles | Baseline set too early; too much template. | Adjust baseline start/end cycles; dilute input sample [32]. |
| Low-Plateau Phase | Limiting or degraded reagents; inefficient reaction. | Check master mix calculations; use fresh stock solutions; optimize reaction conditions [32]. |
| Jagged Signal | Poor probe signal; mechanical error; bubble in well. | Increase probe amount; use fresh probe; contact equipment technician [32]. |
| Variable Technical Replicates (Cq >0.5 cycles difference) | Pipetting error; insufficient mixing; low template. | Calibrate pipettes; mix solutions thoroughly; use filtered tips; increase sample input [32]. |
| Standard curve slope ≠-3.34, R² < 0.98 | Inaccurate standard dilutions; extremes of curve are variable. | Remake standard dilutions accurately; eliminate extreme concentrations from the curve [32]. |
Digital PCR (dPCR) Troubleshooting
| Problem | Potential Cause | Solution |
|---|---|---|
| Poor Partitioning | Inefficient droplet generation; chip defects. | Check droplet generator or chip for proper function; ensure correct oil and surfactant are used [36]. |
| Rain (Intermediate Fluorescence) | Non-specific amplification; probe degradation; suboptimal annealing temperature. | Optimize primer/probe specificity and concentration; optimize annealing temperature [36]. |
| Low Positive Counts | Sample concentration too low; inhibitors; poor amplification efficiency. | Concentrate the sample; re-purify nucleic acids to remove inhibitors; check primer efficiency [34]. |
Experimental Protocols
Protocol: Absolute Quantification of Bacterial Strains in Fecal Samples using Strain-Specific qPCR
This protocol is adapted from a systematic comparison of qPCR and ddPCR for quantifying Limosilactobacillus reuteri [35].
1. Strain-Specific Primer Design
- Identify unique genomic regions for your target bacterial strain by comparing its genome to closely related strains using tools like BLAST.
- Design primers that are 18-22 nucleotides long, with a GC content of 30-50%, and melting temperatures (Tm) between 50-65°C, ideally within 2-5°C of each other [35].
- Check primer specificity in silico against a comprehensive database.
2. Bacterial Culture and Standard Curve Preparation
- Grow the target bacterial strain under optimal conditions. Harvest cells during the late exponential or early stationary phase [35].
- Determine the cell concentration of the culture using quantitative plating on agar (CFU/mL).
- Perform a 10-fold serial dilution of the culture in phosphate-buffered saline (PBS) to create a standard curve ranging from ~10^2 to 10^7 CFU/mL. Use these to spike control (negative) fecal samples [35].
3. DNA Extraction from Fecal Samples
- Use a kit-based DNA extraction method (e.g., QIAamp Fast DNA Stool Mini Kit) for better reproducibility and purity compared to phenol-chloroform methods [35].
- Include a sample washing step with ice-cold PBS to remove PCR inhibitors.
- Elute DNA in a buffer compatible with downstream PCR and measure DNA concentration and purity spectrophotometrically.
4. qPCR Setup and Execution
- Reaction Mix: Prepare a master mix containing a DNA intercalating dye (e.g., SYBR Green) or a hydrolysis probe (e.g., TaqMan), primers, and polymerase.
- Standards and Samples: Load the standard curve samples (spiked feces with known CFU) and unknown test samples in duplicate or triplicate.
- Cycling Conditions: Use a standard cycling protocol: initial denaturation (95°C for 2-5 min), followed by 40 cycles of denaturation (95°C for 15-30 sec), annealing (primer-specific Tm for 30-60 sec), and extension (72°C for 30 sec).
- Data Analysis: Generate a standard curve by plotting the log of the known CFU values against the Cq values. Use the linear equation from the standard curve to calculate the absolute abundance (CFU/g of feces) of the target strain in your unknown samples [35].
Workflow: Integrating dPCR with 16S rRNA Sequencing for Absolute Microbial Abundance
This workflow describes a framework for converting relative 16S rRNA sequencing data into absolute abundance using dPCR as an anchor [4]. The following diagram illustrates the key steps:
Key Steps:
- Sample Collection & DNA Extraction: Collect the microbial sample (e.g., stool, mucosa) and extract total DNA [4].
- Sample Splitting: Split the extracted DNA into two aliquots for parallel processing.
- Parallel Processing:
- 16S rRNA Gene Amplicon Sequencing: One aliquot is used for standard 16S rRNA gene sequencing to determine the relative abundance of each taxon in the community [4].
- Digital PCR (dPCR): The other aliquot is used with dPCR and universal primers targeting the 16S rRNA gene to obtain an absolute count of the total number of 16S rRNA gene copies in the sample [4] [5].
- Data Integration: The absolute abundance of each individual taxon is calculated by multiplying its relative abundance (from sequencing) by the total bacterial load (from dPCR) [4]. This converts the compositional data into quantitative data.
Research Reagent Solutions
The following table lists key reagents and their functions for nucleic acid quantification and analysis protocols.
| Item | Function/Application |
|---|---|
| Qubit Assay Kits (HS & BR) | Fluorometric quantification of specific nucleic acids (dsDNA, RNA, etc.) with high specificity, ignoring contaminants [31]. |
| Universal 16S rRNA Primers | Used in dPCR or qPCR to amplify a conserved region of the 16S rRNA gene, allowing estimation of total prokaryotic load in a sample [4] [5]. |
| Strain-Specific PCR Primers | Designed to uniquely amplify a genomic region of a specific bacterial strain, enabling its detection and quantification within a complex community [35]. |
| DNA Intercalating Dyes (e.g., SYBR Green) | Binds double-stranded DNA and emits fluorescence, used for detection in qPCR and dPCR [35]. |
| Hydrolysis Probes (e.g., TaqMan) | Fluorescently labeled probes that increase specificity in qPCR/dPCR by only emitting fluorescence upon cleavage during amplification [34] [35]. |
| Kit-Based DNA Extraction Kits | Standardized methods for isolating high-quality DNA from complex matrices like stool, improving reproducibility and yield while removing inhibitors [35] [37]. |
Frequently Asked Questions (FAQs)
Q1: Why can't I use standard relative abundance data from sequencing to get absolute microbial counts? Standard high-throughput sequencing techniques lose information about the total microbial load in a sample during library preparation, as samples are typically normalized to a standard amount of genetic material prior to sequencing. The resulting data is compositional, meaning you only get the proportion of each microbe relative to others in the same sample. An increase in the relative abundance of one taxon can be caused either by its actual growth or by a decrease in the abundance of other taxa, which can lead to misleading interpretations [5] [38] [4].
Q2: My model's predictions of absolute abundance are inaccurate. What could be wrong? Inaccurate predictions can stem from several sources:
- Insufficient Metadata: Models often rely on more than just DNA concentration. Key predictors can include the fraction of host reads, prokaryotic alpha diversity, and sample storage conditions. Ensure you are using a comprehensive set of metadata [5].
- Data Range and Context: Machine learning models trained on data from one cohort (e.g., healthy individuals) may not generalize well to another with a different dynamic range of absolute abundance (e.g., individuals undergoing heavy antibiotic treatment). Always validate your model on an external cohort that matches your experimental context [5].
- Technical Bias: Unexplained technical variation from DNA extraction, PCR amplification, and sequencing can introduce heteroscedasticity and bias. It is crucial to use standardized protocols and account for batch effects [39] [4].
Q3: What are my main options for experimentally measuring absolute abundance to validate my models? The most common and reliable methods are:
- Digital Droplet PCR (dPCR): Used with universal primers for the 16S rRNA gene to absolutely quantify the number of prokaryotic 16S copies in a DNA extraction. This is considered highly precise [5] [4].
- Flow Cytometry: Used to count the total number of microbial cells in a sample [5] [38].
- Spike-in Standards: Adding a known quantity of DNA from an organism not expected to be in the sample (exogenous spike-in) during sample processing. The ratio of spike-in reads to sample reads in sequencing data allows for the calculation of absolute abundances [40] [38] [4].
- Quantitative PCR (qPCR): Similar to dPCR but relies on a standard curve for quantification [4].
Q4: Are there specific machine learning algorithms that work best for predicting absolute abundance? Research shows that even simple models can be highly effective. For example, a random forest model using DNA concentration as its primary input has demonstrated high prediction accuracy (Spearman correlation >0.9) for absolute prokaryotic load. More complex models like XGBoost have also been applied. The choice of algorithm may be less critical than ensuring you have the right, high-quality input features and sufficient training data [5] [41].
Troubleshooting Guides
Issue 1: Handling Compositional Effects in Sequencing Data
Problem: You suspect that observed changes in relative abundance data are misleading and do not reflect true changes in absolute microbial counts.
Solution: Adopt a quantitative microbiome profiling (QMP) framework or use compositionally aware tools.
Anchor with an Absolute Measurement: The most robust solution is to combine your relative sequencing data with a parallel absolute measurement.
- Protocol: Digital PCR for 16S rRNA Gene Quantification
- Step 1: Perform DNA extraction from your samples (e.g., stool, mucosal scrapings) using a standardized protocol. Record the input mass [4].
- Step 2: Using the same DNA extraction, run a digital PCR (dPCR) reaction with universal primers targeting the V4 region of the 16S rRNA gene. dPCR partitions the sample into thousands of nanoliter droplets, allowing for absolute quantification of the 16S gene copies without a standard curve [5] [4].
- Step 3: Calculate the absolute abundance of 16S copies per gram of original sample using the formula:
(16S copies/µL from dPCR) * (DNA elution volume in µL) / (mass of sample input in grams). - Step 4: Use this total absolute abundance value to convert your relative abundances from 16S amplicon sequencing into absolute abundances for each taxon [4].
- Protocol: Digital PCR for 16S rRNA Gene Quantification
Use Computational Normalization Methods: If absolute measurement is not possible, use statistical methods designed for compositional data.
Issue 2: Integrating Metadata for Improved Machine Learning Predictions
Problem: Your model, based only on sequencing read data, has poor performance in predicting absolute abundance.
Solution: Enrich your model with carefully selected metadata features.
Identify Key Predictive Features: Research indicates that the following features are highly predictive:
- DNA Concentration: This is often the strongest single predictor, as the majority of DNA in a stool sample is typically prokaryotic [5].
- Host DNA Fraction: The proportion of sequencing reads that map to the host genome is a critical indicator of microbial load, especially in mucosal samples [5] [4].
- Alpha Diversity: Metrics like Shannon diversity can provide additional signal [5].
- Sample Storage and Processing: Information such as same-day vs. next-day freezing should be recorded and included as a potential feature [5].
Implementation Workflow:
- Step 1: Extract DNA and record its concentration using a method like Qubit or NanoDrop.
- Step 2: Perform shotgun metagenomic sequencing. A portion of this data will be used for taxonomic profiling, while the rest can be used to calculate the fraction of host reads by aligning sequences to the host genome (e.g., human, mouse).
- Step 3: Calculate prokaryotic alpha diversity from the non-host metagenomic reads.
- Step 4: Train a random forest or XGBoost model using the DNA concentration, host read fraction, alpha diversity, and other metadata as input features to predict the absolute abundance (e.g., 16S copies from dPCR). Always use a repeated cross-validation strategy to robustly estimate performance [5].
The logical relationship between inputs, models, and outputs in this workflow is summarized below.
Issue 3: Validating Model Performance and Generalizability
Problem: Your model performs well on your initial dataset but fails when applied to new data from a different study or population.
Solution: Implement rigorous validation protocols.
Cross-Study Validation:
- Action: Never rely solely on internal cross-validation. If possible, train your model on one cohort (e.g., data from five 96-well plates) and test its performance on a completely external cohort (e.g., a sixth plate or a dataset from a different disease population) [5].
- Metrics: Report correlation coefficients (Pearson, Spearman) and error metrics (Mean-Squared Error) on the external validation set. A strong correlation (>0.9) on external data is a key indicator of a robust model [5].
Benchmark Against a Baseline:
- Action: Compare your full model against a simple baseline, such as a "DNA-only" model that uses only DNA concentration for prediction. This helps demonstrate the added value of your more complex metadata integration [5].
Quantitative Data & Experimental Protocols
The following table summarizes the performance of a machine learning model (random forest) using different sets of features to predict absolute prokaryotic abundance, as measured by digital droplet PCR [5].
| Model Type | Key Input Features | Spearman's rho (Ï) | R² | Key Takeaway |
|---|---|---|---|---|
| DNA-Only Model | DNA Concentration | 0.89 | 0.82 | DNA concentration alone is a powerful predictor. |
| Full Model | DNA Concentration, Host Read Fraction, Alpha Diversity, Storage Type | 0.91 | 0.86 | Integrating multiple metadata features provides a statistically significant improvement in accuracy. |
Detailed Experimental Protocol: dPCR for Absolute Microbial Quantification
This protocol is adapted from methods used to validate machine learning predictions and establish a quantitative sequencing framework [5] [4].
Objective: To absolutely quantify the number of 16S ribosomal RNA (rRNA) gene copies in a DNA extraction from a microbial sample (e.g., stool, mucosa).
Principle: Digital PCR partitions a PCR reaction into thousands of nanoliter-sized droplets. A positive droplet (containing at least one target DNA molecule) will fluoresce, allowing for absolute counting without a standard curve.
Materials:
- Extracted DNA sample
- dPCR supermix for probes or EvaGreen
- Universal 16S rRNA gene primers (e.g., targeting the V4 region)
- dPCR droplet generator and reader
- Pipettes, tips, and PCR tubes
Procedure:
- Sample Preparation: Dilute the extracted DNA to a concentration within the dynamic range of the dPCR system (e.g., 1-100 ng/µL). Include a no-template control (NTC) to check for contamination.
- Reaction Mix Setup: Prepare the dPCR reaction mix on ice. A typical 20 µL reaction might contain:
- 10 µL of 2x dPCR supermix
- 1 µL of forward primer (18 µM)
- 1 µL of reverse primer (18 µM)
- X µL of DNA template (e.g., 1-5 µL)
- Nuclease-free water to 20 µL
- Droplet Generation: Transfer the reaction mix to the droplet generator cartridge. Following the manufacturer's instructions, generate thousands of droplets for each sample.
- PCR Amplification: Carefully transfer the droplets to a 96-well PCR plate. Seal the plate and run the PCR protocol on a thermal cycler. A standard 16S amplification protocol can be used.
- Droplet Reading: After amplification, place the plate in the dPCR droplet reader. The instrument will count the number of positive and negative droplets for each sample.
- Data Analysis:
- The dPCR software will calculate the concentration of the target (16S gene) in copies/µL of the reaction mix.
- Calculate the total 16S copies in your original DNA extraction:
(16S copies/µL from dPCR) * (Total DNA elution volume in µL). - To normalize by sample input mass:
(Total 16S copies) / (Mass of sample input in grams) = 16S copies/gram.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Absolute Abundance Research |
|---|---|
| Digital Droplet PCR (dPCR) System | Provides gold-standard absolute quantification of target genes (e.g., 16S rRNA) without a standard curve, used for model validation [5] [4]. |
| Universal 16S rRNA Primers | Primer sets (e.g., 515F/806R for V4 region) used in both dPCR and amplicon sequencing to target a conserved gene across prokaryotes [4]. |
| Exogenous Spike-in Standards | Purified DNA from a non-native organism (e.g., Lycopodium spores, synthetic genes) added in known quantities to samples before DNA extraction to act as an internal standard for calculating absolute abundances from sequencing data [40] [38]. |
| Flow Cytometer | Enables direct counting of total microbial cells in a sample, providing an alternative absolute measurement to validate against [5] [38]. |
| Standardized DNA Extraction Kits | Kits with validated protocols (e.g., ISO 11063) ensure consistent and efficient lysis of both Gram-positive and Gram-negative bacteria, which is critical for accurate quantification [4]. |
| Axitinib sulfoxide | Axitinib Sulfoxide|CAS 1347304-18-0|Research Chemical |
| AZ3451 | AZ3451 PAR2 Antagonist For Research |
Understanding the true dynamics of microbial communities requires moving beyond relative proportions to measure absolute abundance. While high-throughput sequencing reveals microbial composition, the inherent compositional nature of this data means an increase in one taxon's relative abundance necessarily decreases others, regardless of actual population changes [18]. This limitation is particularly problematic in microbial ecotoxicology and drug development research, where establishing quantitative sensitivity thresholds demands knowledge of absolute cell abundances [18].
This guide provides a comprehensive technical framework for integrating absolute abundance measurements into microbial research workflows, enabling researchers to accurately quantify the magnitude and direction of microbial responses to environmental stressors or therapeutic interventions.
Complete Workflow for Absolute Abundance Measurement
The diagram below outlines the integrated workflow from sample collection to absolute abundance data generation, incorporating key decision points and troubleshooting checkpoints.
Frequently Asked Questions & Troubleshooting Guides
Sample Preparation & PMA Treatment
Q1: What is the optimal PMA concentration for low-biomass environmental samples like seawater?
Based on recent optimization studies, PMA concentrations of 2.5-15 μM effectively inhibit PCR amplification of DNA from membrane-compromised cells in natural seawater samples. At these concentrations, researchers observed 24-44% reduction in 16S rRNA gene copies compared to untreated samples, indicating effective exclusion of compromised cells [18].
Troubleshooting Tip: If PMA treatment shows less than 20% reduction in gene copies, check:
- PMA stock solution integrity and storage conditions
- Light exposure consistency during cross-linking
- Sample turbidity interfering with PMA penetration
Q2: How do I validate PMA treatment efficiency in my specific sample type?
Create controlled mixtures of intact and heat-killed (85°C for 5 minutes) cells from your sample matrix. Treat these mixtures with optimized PMA concentrations and quantify 16S rRNA gene copies by ddPCR. Effective treatment should show dose-dependent reduction in signal from heat-killed cell mixtures [18].
Absolute Quantification Methods
Q3: Should I use flow cytometry or ddPCR for microbial load estimation?
Both methods show strong correlation for total and intact cell counts in seawater microbiomes [18]. Consider your specific requirements:
Table: Comparison of Microbial Load Quantification Methods
| Method | Advantages | Limitations | Best For |
|---|---|---|---|
| Flow Cytometry | Direct cell counting, visual validation, high throughput | Requires specialized instrument, staining optimization | High biomass samples, rapid processing |
| Droplet Digital PCR | Absolute quantification without standards, high precision | DNA extraction efficiency dependency, cost | Low biomass samples, highest precision requirements |
Q4: How do I handle discrepancies between flow cytometry and ddPCR results?
Consistent discrepancies may indicate:
- DNA extraction efficiency issues affecting ddPCR
- Staining variability affecting flow cytometry counts
- Presence of extracellular DNA not addressed by PMA treatment
Resolution protocol:
- Replicate measurements with technical controls
- Verify PMA treatment efficiency with heat-killed controls
- Cross-validate with a third method if available
Data Processing & Analysis
Q5: What are the critical steps for converting relative to absolute abundance?
The Quantitative Microbiome Profiling (QMP) approach requires these key steps [18]:
- Normalization: Divide relative abundance values (from sequencing) by the total microbial load (from ddPCR or flow cytometry)
- Validation: Check that sum of absolute abundances matches independent cell counts
- Quality Control: Ensure PMA treatment effectively excluded compromised cells
Q6: Why does absolute abundance reveal different biological patterns than relative abundance?
Unlike relative abundance data (inherently compositional), absolute abundance captures true population dynamics. In stress-response studies, QMP revealed consistent abundance declines in specific taxa that RMP failed to detect because compositional effects masked these changes [18].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table: Key Reagents and Materials for Absolute Abundance Workflows
| Item | Specifications | Function | Technical Notes |
|---|---|---|---|
| PMAxx Dye | 20 mM stock in H₂O, light-protected | Selective binding to DNA from membrane-compromised cells | Test concentration range 2.5-15 μM for optimization [18] |
| SYBR Green I | 100X concentrate in DMSO | Total cell staining for flow cytometry | Use with proper controls for nucleic acid binding |
| Propidium Iodide | 1.0 mg/mL solution | Membrane-impermeant dye for dead cell discrimination | Compatible with SYBR Green for Live/Dead staining [18] |
| Sterivex Filters | 0.22 μm pore size, PES membrane | Sample concentration and PMA treatment | Enables processing of large volume samples (500mL) [18] |
| ddPCR Supermix | Probe-based or EvaGreen | Absolute quantification of 16S rRNA gene copies | Provides digital counting without standard curves [18] |
| Artificial Seawater | 33 ppt salinity, 0.22 μm filtered | Control preparation and sample dilution | Match salinity and pH (8.0) to natural samples [18] |
| Mizacorat | AZD9567|Selective Glucocorticoid Receptor Modulator | AZD9567 is a potent, non-steroidal glucocorticoid receptor modulator for inflammation research. For Research Use Only. Not for human use. | Bench Chemicals |
| (S)-Azelnidipine | (S)-Azelnidipine|L-type Calcium Channel Blocker | (S)-Azelnidipine is a high-purity, long-acting L-type calcium channel antagonist for hypertension research. For Research Use Only. Not for human or veterinary use. | Bench Chemicals |
Data Processing Workflow: From Raw Sequences to Absolute Abundance
The diagram below details the computational workflow for transforming sequencing data and cell counts into validated absolute abundance measurements.
Advanced Applications & Integration with Existing Platforms
Integrating with Analysis Platforms: Tools like MicrobiomeAnalyst provide comprehensive statistical analysis capabilities for processed absolute abundance data. The platform supports 19 different statistical and visualization methods, though it requires pre-processing of raw sequencing data into feature tables [42].
Temporal Dynamics Prediction: For longitudinal studies, graph neural network models can predict microbial community dynamics using historical abundance data. Recent studies demonstrate accurate prediction of species dynamics up to 2-4 months ahead using only relative abundance data [43].
Data Integration Considerations: When incorporating absolute abundance data into larger analyses, ensure consistent:
- Units throughout datasets (cells/volume preferred)
- Normalization approaches across sample types
- Metadata standards for experimental conditions
This integrated approach to absolute abundance measurement addresses critical limitations of relative microbiome profiling, enabling more accurate assessment of microbial community dynamics in response to environmental stressors, therapeutic interventions, and ecological changes.
Navigating Technical Challenges: A Practical Guide to Reliable Absolute Quantification
In the pursuit of measuring absolute abundance in microbial communities, accounting for technical variability is not merely a best practice—it is a fundamental requirement for obtaining biologically accurate data. Sample-to-sample variability, particularly from differences in extraction efficiency and the presence of PCR inhibitors, can significantly skew quantitative measurements, leading to false conclusions about microbial dynamics. This guide provides troubleshooting protocols and FAQs to help researchers identify, quantify, and correct for these sources of error, ensuring that observed changes reflect true biological differences rather than technical artifacts.
Troubleshooting Guide: Common Scenarios & Solutions
FAQ 1: How can I determine if my DNA extraction efficiency is consistent across samples?
The Problem: Inconsistent DNA extraction efficiency, especially with different sample matrices (e.g., stool vs. mucosa) or fragment sizes, can make absolute abundance measurements unreliable.
The Solution: Use a synthetic DNA spike-in control to directly measure and correct for extraction efficiency.
- Recommended Reagent: The CEREBIS (Construct to Evaluate the Recovery Efficiency of cfDNA extraction and BISulphite modification) spike-in is a synthetic, non-human DNA fragment designed to mimic the size of naturally occurring cfDNA (e.g., 180 bp or 89 bp) [44].
- Experimental Protocol:
- Spike Early: Add a known quantity of the CEREBIS construct to your sample lysis buffer before the DNA extraction process begins [44].
- Co-process: Carry the spike-in through the entire DNA extraction and purification workflow alongside the native microbial DNA.
- Quantify Recovery: Use droplet digital PCR (ddPCR) to precisely quantify the amount of CEREBIS DNA recovered in the final eluate [44] [45].
- Calculate Efficiency: The extraction efficiency is calculated as: (Recovered CEREBIS copies / Initial CEREBIS copies added) × 100%.
- Normalize Data: Use the calculated efficiency to adjust the absolute abundance measurements of your target microbes.
Table 1: Example Extraction Efficiencies from Different Methods and Matrices
| Extraction Method | Sample Matrix | Target Spike-in | Mean Extraction Efficiency | Variability (±SD) |
|---|---|---|---|---|
| QIAamp Circulating Nucleic Acid Kit | Plasma | 180 bp CEREBIS | 84.1% | ± 8.17% [44] |
| Zymo Quick-DNA Urine Kit | Urine | 180 bp CEREBIS | 58.7% | ± 11.1% [44] |
| Q Sepharose (Qseph) protocol | Urine | 180 bp CEREBIS | 30.2% | ± 13.2% [44] |
FAQ 2: My samples contain PCR inhibitors. How can I mitigate their effects?
The Problem: Substances like sodium polyanetholsulfonate (SPS) in blood culture media, heme from blood, or humic acids from environmental samples can co-purify with DNA and inhibit downstream PCR or ddPCR, leading to underestimation of microbial load [46].
The Solution: The optimal strategy involves selecting an extraction method that effectively removes inhibitors and using a quantification platform resistant to their effects.
- Experimental Protocol:
- Choose the Right Extraction Chemistry:
- Avoid Silica-Based Columns for Inhibitor-Rich Samples: Studies show silica-based columns can copurify SPS, requiring 100 to 1000-fold sample dilution to mitigate inhibition, which drastically reduces sensitivity [46].
- Consider Alternative Methods: Phenol-chloroform extraction or automated systems using hydrophilic polymer-embedded magnetic particles (e.g., the Automate Express instrument) have proven far more effective at removing SPS, allowing for minimal dilution and significantly improved assay sensitivity [46].
- Use ddPCR for Quantification: Droplet digital PCR (ddPCR) is generally more tolerant of PCR inhibitors than quantitative PCR (qPCR) because it partitions the reaction into thousands of nanoliter-sized droplets, effectively diluting the inhibitor and increasing the chance of a successful amplification in inhibitor-free droplets [47].
- Spike to Monitor Inhibition: Include an internal control, such as a known quantity of exogenous DNA (e.g., from Bovine Coronavirus), in the PCR/ddPCR reaction. A significant drop in the recovery of this control signal indicates the presence of inhibition in the sample [47].
- Choose the Right Extraction Chemistry:
FAQ 3: How does technical variability compare to true biological variation?
The Problem: It can be difficult to discern whether variability in microbial load data stems from pre-analytical technical steps or from genuine biological differences between samples or individuals.
The Solution: Conduct a variance component analysis to partition the total variability in your data.
- Experimental Protocol:
- Experimental Design: Process a set of samples with multiple technical replicates. For example, have different operators extract multiple aliquots from the same biological sample on different days, and perform multiple ddPCR measurements per extract [44] [47].
- Quantification: Use absolute quantification methods like ddPCR to measure target gene copies.
- Statistical Analysis: Perform a variance component analysis (using statistical software) to attribute the proportion of total variance to specific sources:
- Inter-individual (biological) variability
- Inter-operator variability
- Inter-extraction variability
- Intra-assay (ddPCR measurement) variability
Table 2: Example Variance Contributions in a Plasma cfDNA Study
| Source of Variability | Contribution to Total Variance | Context / Setup |
|---|---|---|
| Biological (Inter-individual) | Largest Proportion | Setup with biologically different samples [44] |
| Technical (Inter-extraction) | Substantially Lower | Setup with biologically different samples [44] |
| ddPCR Measurement (Within-triplicate) | Largest Proportion | Technical setup with a pooled plasma sample [44] |
| Analyst Experience Level | Significant (p<0.001) | Impact on accepted droplet generation in ddPCR [47] |
Essential Methodologies for Absolute Quantification
The following experimental workflows are critical for robust absolute abundance measurement. The diagram below illustrates the integrated process for obtaining absolute abundances while controlling for variability.
Protocol 1: Digital PCR (dPCR) Anchoring for Absolute Abundance
This framework uses dPCR to obtain an absolute count of 16S rRNA gene copies, which is then used to transform relative sequencing data into absolute abundances [4].
- Total Microbial DNA Quantification:
- Perform ddPCR targeting the 16S rRNA gene on your extracted DNA samples. This provides an absolute count of 16S rRNA gene copies per unit of sample (e.g., per gram of stool) without relying on a standard curve [4].
- 16S rRNA Gene Amplicon Sequencing:
- Using the same DNA extract, perform standard 16S rRNA gene amplicon sequencing to determine the relative abundance of each taxon in the community.
- Data Integration (Quantitative Microbiome Profiling - QMP):
Protocol 2: Flow Cytometry for Total Cell Counts
This method directly counts intact bacterial cells to provide a total microbial load for normalization [10].
- Sample Staining: Suspend a known weight of sample (e.g., stool) in a suitable buffer. Use a DNA-binding fluorescent dye (e.g., SYTO BC) to stain microbial cells [10].
- Cell Counting: Analyze the stained suspension using a flow cytometer to obtain the total number of intact bacterial cells per gram of sample [10].
- Data Integration: Similar to the dPCR method, multiply the relative abundance from 16S sequencing by the total cell count from flow cytometry to obtain the absolute abundance of each taxon in units of cells per gram [10].
The Scientist's Toolkit: Key Research Reagents & Materials
Table 3: Essential Reagents for Addressing Variability in Absolute Abundance Studies
| Reagent / Tool | Function | Key Consideration |
|---|---|---|
| CEREBIS Spike-in | Synthetic DNA spike-in to measure extraction efficiency and bisulfite conversion recovery [44]. | Choose a fragment length that matches your target DNA (e.g., 180 bp for mononucleosomal cfDNA). |
| Droplet Digital PCR (ddPCR) | Provides absolute quantification of DNA targets without a standard curve; highly resistant to PCR inhibitors [44] [45] [4]. | Ideal for low-abundance targets and complex samples prone to inhibition. |
| Flow Cytometer | Directly counts total intact bacterial cells in a sample for total microbial load measurement [45] [10]. | Requires a well-optimized staining protocol and dissociation of cells from sample matrix. |
| Polymer-Based Magnetic Bead Kits | DNA extraction kits that use a mechanism different from silica, often providing superior removal of common PCR inhibitors like SPS [46]. | Crucial for processing inhibitor-rich samples like blood, soil, or wastewater. |
| 16S rRNA Copy Number Database | Bioinformatics resource (e.g., rrnDB) to correct for gene copy number variation among bacterial taxa when interpreting 16S data [10]. | Essential for moving from 16S rRNA gene copy abundance to approximate cell counts. |
Frequently Asked Questions (FAQs)
Q1: Why is correcting for 16S rRNA gene copy number (GCN) important in microbiome studies? The 16S rRNA gene is the most widely used marker for profiling microbial communities. However, different microorganisms contain different numbers of copies of this gene in their genomes—ranging from 1 to over 15 in bacteria and 1 to 5 in archaea [48]. This variation introduces a significant bias: taxa with higher GCN are over-represented in sequencing read counts compared to their actual cellular abundance. Without correction, this can lead to skewed community profiles and misleading biological interpretations [49] [50].
Q2: What is the range of 16S rRNA gene copy numbers in prokaryotes? Analysis of complete prokaryotic genomes shows that 16S GCN varies substantially across taxa:
- Bacteria: 1 to 37 copies [48]
- Archaea: 1 to 5 copies [48] The copy number is not uniform within phyla. For example, Actinobacteria averages 3.2±1.9 copies, while Bacteroidetes averages 4.1±2.3 copies [48].
Q3: How predictable are 16S rRNA gene copy numbers from phylogeny? 16S GCNs are moderately phylogenetically conserved. Prediction accuracy is highly dependent on how closely related an organism is to sequenced genomes with known GCN. The autocorrelation function of 16S GCNs drops below 0.5 at a phylogenetic distance of approximately 15% (nucleotide substitutions per site) and decays to zero at around 30% distance [49]. This means predictions are generally accurate only for taxa with closely to moderately related representatives (≤15% divergence) [49].
Q4: What methods are available for predicting 16S GCN? Several computational tools have been developed, each with different approaches:
| Method | Prediction Approach | Key Features |
|---|---|---|
| PICRUSt [49] | Phylogenetic Independent Contrasts (PIC) | Predicts GCN and metagenomic content; accuracy decreases with increasing NSTI |
| CopyRighter [49] | Phylogenetic Independent Contrasts (PIC) | Focuses on GCN correction for community profiles |
| PAPRICA [49] | Subtree averaging | Uses arithmetic average of GCNs across descending tips in phylogeny |
| RasperGade16S [50] [51] | Heterogeneous Pulsed Evolution (PE) model | Accounts for intraspecific variation and evolutionary rate heterogeneity; provides confidence estimates |
| 16Stimator [52] | Read-depth analysis | Estimates GCN from draft genomes using coverage depth of 16S vs. single-copy genes |
Q5: When should I avoid correcting for 16S GCN? Correction is not recommended by default [49]. Consider avoiding it when:
- Your microbial community has a high Nearest Sequenced Taxon Index (NSTI >0.15-0.30) [49]
- You are working with little-characterized environments with poor representation in genome databases
- The primary analysis focuses on beta-diversity (PCoA, NMDS, PERMANOVA), where GCN variation has limited impact [50]
- The additional noise introduced by inaccurate GCN predictions outweighs the benefit of correction [49]
Q6: What is the relationship between absolute abundance and relative abundance data? There's a critical mathematical relationship: Absolute abundance = Relative abundance × Total microbial abundance [53]. This means that the absolute abundance of a taxon is linearly correlated with its relative abundance, scaled by the total microbial load in the sample. This relationship forms the foundation for methods that convert relative to absolute abundance.
Troubleshooting Guides
Issue: High Uncertainty in GCN Predictions
Problem: Your GCN predictions have low confidence or different tools give conflicting results.
Solutions:
- Check community NSTI: Calculate the Nearest Sequenced Taxon Index for your community. If NSTI >0.15, predictions will have high uncertainty [49].
- Use tools with confidence estimates: Employ methods like RasperGade16S that provide confidence estimates for predictions [50].
- Consider alternative approaches: For high-NSTI communities, consider:
Issue: Converting Relative to Absolute Abundance
Problem: You have relative abundance data but need absolute microbial counts.
Solutions:
- Digital PCR (dPCR) anchoring:
Spike-in standards:
Computational estimation:
Issue: Primer Selection and Amplification Bias
Problem: Your 16S rRNA primers don't adequately capture the diversity in your samples.
Solutions:
- Evaluate primer coverage: Use in silico tools like TestPrime to assess primer coverage against relevant databases [55].
- Multi-primer strategy: Consider using multiple primer sets for different taxa, as no single "universal" primer perfectly amplifies all bacteria [55].
- Target appropriate variable regions: The V4-V5 region introduces the lowest overestimation rate (4.4%) due to intragenomic variation [48].
- Database consideration: Be aware that primer performance can vary between reference databases (NCBI vs. SILVA) due to different curation methods [55].
Research Reagent Solutions
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| Digital PCR (dPCR) [4] | Absolute quantification of 16S gene copies | Higher precision than qPCR; enables absolute abundance calculations; LLOQ: 4.2×10ⵠcopies/gram for stool |
| ZymoBIOMICS Gut Microbiome Standard [55] | Mock community for validation | Contains 19 bacterial and archaeal strains; useful for primer validation and protocol optimization |
| Halomonas elongata DNA [54] | Spike-in standard for absolute quantification | Strict aerobe not found in gut samples; enables absolute abundance estimation in anaerobic communities |
| SILVA database [49] [56] | Curated 16S rRNA reference | Includes Bacteria, Archaea, and Eukaryota; phylogenetically curated; recommended for primer evaluation |
| GTDB (Genome Taxonomy Database) [56] | Genome-based taxonomy | Modern taxonomy based on whole genomes; useful for linking 16S data to functional potential |
Workflow Diagrams
GCN Correction Decision Framework
Absolute Abundance Quantification Workflow
Quantitative Data Reference
16S rRNA Gene Copy Number Variation Across Phyla
| Phylum | Number of Genomes | Average GCN (Mean ± SD) | Key Notes |
|---|---|---|---|
| Actinobacteria | 2,372 | 3.2 ± 1.9 | Common in human microbiome |
| Bacteroidetes | 879 | 4.1 ± 2.3 | High variation between species |
| Firmicutes | Not specified | Not specified | Diverse GCN patterns |
| Proteobacteria | Not specified | Not specified | Includes E. coli with variable copies |
| Euryarchaeota | 263 | 2.0 ± 0.9 | Archaeal domain |
| Crenarchaeota | 92 | 1.0 ± 0.0 | Single copy dominant |
Method Performance Comparison
| Method | Optimal NSTI Range | Key Advantages | Limitations |
|---|---|---|---|
| PICRUSt | <0.15 | Integrated functional prediction | Accuracy drops with NSTI >0.15 |
| RasperGade16S | <0.30 | Confidence estimates; handles rate heterogeneity | Complex model implementation |
| 16Stimator | N/A (direct measurement) | Direct from sequencing reads; not phylogeny-limited | Requires genomic sequencing data |
| dPCR anchoring | Universal application | Direct absolute quantification; high precision | Additional experimental work required |
Determining Limits of Detection and Quantification Across Diverse Sample Types
Fundamental Concepts: LoB, LoD, and LoQ
What are the key differences between Limit of Blank (LoB), Limit of Detection (LoD), and Limit of Quantitation (LoQ)?
In analytical chemistry and microbiology, these three parameters define the lowest levels at which an analyte can be reliably detected and measured [57].
- Limit of Blank (LoB): The highest apparent analyte concentration expected to be found when replicates of a blank sample containing no analyte are tested. It represents the background noise of the assay [57].
- Limit of Detection (LoD): The lowest analyte concentration likely to be reliably distinguished from the LoB and at which detection is feasible. It is the point where detection is possible but without guaranteed precision or accuracy [57].
- Limit of Quantitation (LoQ): The lowest concentration at which the analyte can not only be reliably detected but at which some predefined goals for bias and imprecision are met. Also called the Lower Limit of Quantification (LLOQ) [57] [4].
Table 1: Key Characteristics of LoB, LoD, and LoQ
| Parameter | Sample Type | Key Question Answered | Statistical Definition |
|---|---|---|---|
| LoB | Sample containing no analyte | What is the background signal of my assay? | LoB = meanblank + 1.645(SDblank) [57] |
| LoD | Sample containing low concentration of analyte | What is the lowest concentration I can detect? | LoD = LoB + 1.645(SDlow concentration sample) [57] |
| LoQ | Sample containing low concentration at expected LoQ | What is the lowest concentration I can accurately measure? | LoQ ≥ LoD; meets predefined bias/imprecision goals [57] |
Why is determining LoD and LoQ particularly important in absolute microbial quantification?
In microbiome research, absolute quantification moves beyond relative abundance to measure the actual number of microbial cells or genes, providing critical insights that relative abundance alone cannot reveal [4] [58].
- Prevents misleading interpretations: Relative abundance data can show an increase in one taxon either because it actually grew or because other taxa decreased—absolute quantification distinguishes these scenarios [4].
- Reveals true microbial dynamics: In soil and gut microbiome studies, absolute quantification has shown that 33-40% of microbial taxa can appear to increase in relative abundance while actually decreasing in absolute abundance [58].
- Enables accurate interaction studies: Absolute bacterial loads are essential for understanding true microbial community interactions including parasitism, competition, and mutualism [58].
Calculation Methods and Experimental Protocols
What are the standard calculation methods for determining LoD and LoQ?
The appropriate calculation method depends on your analytical technique and regulatory requirements. The following table summarizes the most common approaches.
Table 2: Standard Calculation Methods for LoD and LoQ
| Method | Formulas | Application Context | Key Considerations |
|---|---|---|---|
| CLSI EP17 Protocol | LoB = meanblank + 1.645(SDblank)LoD = LoB + 1.645(SDlow concentration sample) [57] | Clinical laboratory testing, immunoassays, microbial quantification | Uses 95% confidence intervals (1.645 SD); requires 60 replicates for establishment, 20 for verification [57] |
| Calibration Curve Approach | LOD = 3.3 × S0/bLOQ = 10 × S0/bWhere S0 = standard deviation of y-intercept, b = slope [59] | HPLC, qPCR, analytical chromatography | S0 represents random error at zero concentration; requires linear response [59] |
| Precision-Based Approach | Based on precision and accuracy at low concentrations rather than signal-to-noise [60] | Research applications where clinical relevance is prioritized | Considered more scientifically relevant by some experts; not always accepted by regulatory authorities [60] |
What is the detailed experimental workflow for establishing LoB, LoD, and LoQ?
The following diagram illustrates the complete workflow for determining these fundamental method validation parameters:
Experimental Workflow for LoB, LoD, and LoQ Determination
What are the specific considerations for establishing LoQ?
The LoQ is determined through iterative testing at and above the LoD concentration until predefined performance goals are met [57]:
- Test samples at the estimated LoD concentration
- Evaluate precision and bias using standards with known concentrations
- If goals aren't met, test at a slightly higher concentration
- Establish LoQ as the lowest concentration where bias and imprecision meet specifications
- "Functional sensitivity" is often defined as the concentration yielding CV=20% [57]
Troubleshooting Common Experimental Issues
How do I address high background signals or non-specific binding in sensitive assays?
High background noise can significantly impact your ability to accurately determine LoD and LoQ, particularly in ELISA and molecular assays [61].
- Incomplete washing: Ensure thorough washing of wells between steps in ELISA procedures. Follow manufacturer's washing techniques exactly—do not overwash (more than 4 times) or allow wash solution to soak in wells [61].
- Contamination of reagents: Avoid performing assays in areas where concentrated forms of analytes are handled. Clean all work surfaces and equipment before assays. Use aerosol barrier pipette tips [61].
- Environmental contamination: Human dander or mucosal aerosols can contaminate assays for human proteins. Don't talk or breathe over uncovered plates; consider using laminar flow hoods for reagent pipetting [61].
- Substrate contamination: For alkaline phosphatase-based assays using PNPP substrate, only withdraw needed amount and recap immediately. Don't return unused substrate to the bottle [61].
What curve fitting methods are recommended for accurate interpolation of low concentration data?
Proper curve fitting is essential for accurate determination of values near the limits of detection and quantification [61].
- Avoid linear regression: Most immunoassays are not linear, and forcing linear fit introduces inaccuracy, particularly at the extremes of the standard curve [61].
- Recommended methods: Use Point-to-Point, Cubic Spline, or 4-Parameter curve fitting routines for most immunoassays [61].
- Validation: Back-fit your standards as unknowns—if they don't report back their nominal values, your curve fit algorithm may be inappropriate [61].
How do I handle samples requiring dilution to fall within the analytical range?
Many samples, particularly from upstream in purification processes, may contain analyte concentrations above the assay's range [61].
- Use matrix-matched diluents: Employ assay-specific diluents that match the standard matrix to minimize dilutional artifacts [61].
- Validate alternative diluents: If using other diluents, ensure they don't yield absorbance values significantly above or below the kit zero standard [61].
- Perform spike recovery: Test recovery at 95-105% across the analytical range using your proposed diluent [61].
- Avoid adsorption issues: Use diluents with carrier protein (not just PBS or TBS) to prevent adsorptive losses of analytes at ng/mL concentrations [61].
Application in Microbial Absolute Quantification
What methods are available for absolute bacterial quantification in microbiome studies?
Multiple approaches exist for determining absolute microbial abundances, each with distinct advantages and limitations for different sample types and research questions [58].
Table 3: Absolute Bacterial Quantification Methods for Microbiome Research
| Method | Major Applications | Advantages | Limitations/Concerns |
|---|---|---|---|
| Flow Cytometry | Feces, aquatic, soil samples | Rapid; single cell enumeration; differentiates live/dead cells | Requires cell dissociation; gating strategy challenges; not ideal for heterogeneous samples [58] |
| 16S qPCR | Feces, clinical samples, soil, plant, air | Directly quantifies specific taxa; cost-effective; high sensitivity | Requires standard curves; 16S rRNA copy number variation; PCR biases [58] |
| ddPCR | Clinical infections, air, feces, soil | No standard curve needed; high precision at low concentrations; absolute quantification | Requires dilution for high-concentration templates; may need many replicates [58] |
| Spike-in Internal Reference | Soil, sludge, feces | Easy incorporation into high throughput sequencing; high sensitivity | Spiking amount and timing affect accuracy; requires 16S copy number calibration [58] |
| Digital PCR (dPCR) Framework | Mucosal and lumenal communities throughout GI tract | Absolute quantification without standard curves; precise counting of DNA molecules | Requires optimization for different sample types; higher cost [4] |
What are the specific LoQ considerations for microbial quantification in different gastrointestinal locations?
The diagram below illustrates the quantitative microbial analysis framework for different GI locations and the factors affecting LoQ:
LoQ Considerations Across GI Sample Types
Key considerations for microbial LoQ determination [4]:
- Sample type dictates LoQ: Stool samples (high microbial load) have much lower LoQ than mucosal samples (low microbial load, high host DNA)
- Extraction efficiency: Must be validated across microbial loads and sample types
- Input requirements: For dPCR-based absolute quantification, >8.3×10ⴠ16S rRNA gene copies needed for ~2x accuracy
- Gram-stain bias: Validate extraction performance for both Gram-positive and Gram-negative microbes
- Low-input artifacts: Samples with <1×10ⴠ16S rRNA gene copies show increased contaminants and dropouts
Research Reagent Solutions for Method Validation
What key reagents and materials are essential for proper LoD/LoQ determination?
Table 4: Essential Research Reagents for LoD/LoQ Studies
| Reagent/Material | Function in LoD/LoQ Studies | Key Quality Requirements |
|---|---|---|
| Blank Matrix | Determines LoB; should be identical to sample matrix without analyte | Commutable with patient specimens; proven analyte-free [57] |
| Low Concentration Standards | Establishes LoD and LoQ; should cover expected low concentration range | Known concentration; matrix-matched; stable over time [57] |
| Assay-Specific Diluent | Diluting samples to fall within analytical range | Matrix-matched to standards; proper pH; contains carrier protein [61] |
| Internal Reference Standards (spike-in) | Converts relative to absolute quantification in microbiome studies | Non-native to sample; known concentration; extraction-resistant [58] |
| Quality Control Materials | Verifies continued assay performance at low concentrations | Stable; characterize precision at LoD/LoQ levels [57] |
Method-Specific Guidance
Are there special considerations for determining LoD and LoQ in qPCR/dPCR methods?
Quantitative PCR methods require specific approaches for LoD/LoQ determination [62]:
- Precision-based LoQ: For qPCR/dPCR in microbial quantification, LoQ is typically determined as the lowest concentration where %CV ≤ 20-25% [4] [58]
- Dynamic range constraints: LoD may reside below the linear range of an assay where the calibration curve is no longer valid [57]
- Sample-specific validation: In dPCR microbial quantification, LoQ must be established for each sample type (stool vs. mucosa) due to differing host DNA content and microbial loads [4]
- Copy number considerations: For 16S rRNA-based quantification, account for variations in gene copy numbers between bacterial taxa when determining LoQ [58]
This technical support guide provides troubleshooting and best practices for using spike-in controls to measure absolute microbial abundance in your research.
FAQs and Troubleshooting Guides
How do I determine the correct quantity of spike-in to use?
The optimal spike-in quantity must bracket the expected abundance range of your endogenous target. Using too much can dominate your sequencing library, while too little may fall below the detection threshold.
- Problem: Poor recovery of spike-in controls or skewed data.
- Solution:
- Pilot Experiment: Conduct a dilution series of your spike-in control to identify the concentration that yields mid-range read counts representative of your target organisms [63].
- General Guideline: A typical approach uses a dilution series spanning 10² to 10⸠molecules per reaction [63].
- Sample-Specific Adjustments: The ideal quantity depends on your sample's microbial load. The table below summarizes key considerations.
| Sample Type | Recommended Spike-in Quantity | Rationale & Considerations |
|---|---|---|
| High Microbial Load (e.g., feces, cell culture) | Use a spike-in control optimized for high biomass [64]. | Prevents the spike-in from being overwhelmed by sample DNA, ensuring accurate quantification. |
| Low Microbial Load (e.g., swabs, water, mucosa) | Use a spike-in control optimized for low biomass and higher input DNA amount [64] [4]. | Ensures the spike-in is detectable above the background noise and helps identify contaminants [4]. |
| Broad-Range Quantification | A pre-optimized commercial mix with a validated concentration range [63]. | Simplifies the process and ensures performance across diverse sample types. |
What is the ideal composition for a spike-in control?
An effective spike-in should mimic your endogenous sample as closely as possible to capture technical biases throughout the workflow.
- Problem: The spike-in control does not accurately reflect the behavior of native microbes, leading to uncorrected biases.
- Solution:
- Diverse Panel: Use a panel of spike-ins with varied GC content, lengths, and predicted secondary structures [63]. This helps correct for biases in steps like adapter ligation and amplification [63] [65].
- Community Composition: For microbiome studies, a defined community of two or more distinct microbial strains (e.g., combining Gram-positive and Gram-negative bacteria) can expose potential biases during DNA extraction [64].
- Key Limitation: Be aware that synthetic spike-ins may lack natural modifications (e.g., 2'-O-methylation on small RNAs), which means they might not fully capture the behavior of every endogenous molecule [63].
When during the experimental protocol should I add the spike-in?
The timing of spike-in addition determines which technical biases it can monitor and correct for.
- Problem: Incorrect timing of addition leads to normalization that does not account for all sources of technical variation.
- Solution: Add the spike-in after sample extraction but before library preparation [63]. This isolates and allows for correction of biases originating from library preparation steps, such as:
- Adapter ligation
- Reverse transcription
- PCR amplification [63]
For the most comprehensive bias assessment from extraction onward, add the spike-in directly to the sample lysis buffer at the very start of DNA/RNA extraction.
How do I troubleshoot poor spike-in recovery?
Poor recovery indicates that the spike-in is not behaving as expected in your sample matrix.
- Problem: Low or inconsistent recovery of the spiked material.
- Solution:
- Check for Matrix Effects: Components in your biological sample (e.g., inhibitors, high host DNA) can affect the efficiency of downstream enzymatic reactions. Perform a spike-and-recovery experiment to assess this [66].
- Adjust the Matrix: If recovery is poor in a neat sample, try diluting the sample in your standard diluent or adjusting its pH [66].
- Validate Extraction Efficiency: Spike a known community into a similar, but sterile, matrix (e.g., from germ-free mice) and perform a dilution series to ensure your extraction protocol yields near-complete and even recovery across taxa [4].
How do I normalize sequencing data using spike-ins?
Using spike-ins for normalization allows you to move beyond relative abundance to absolute quantification.
- Problem: How to convert relative sequencing read counts into absolute counts.
- Solution: Use the known input amount of spike-ins to create a standard curve and calculate scaling factors.
- Standard Curve: Plot the observed read counts against the known input amount for each spike-in control. This relationship allows you to estimate the absolute copy numbers for endogenous targets [63] [65].
- Computational Tools: Use specialized software like the
DspikeInR package, which provides a reproducible workflow for absolute microbial quantification using spike-in controls. It supports scaling factor estimation, abundance conversion, and is compatible with common data structures likephyloseq[67].
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function | Example & Application Notes |
|---|---|---|
| ERCC RNA Controls | A complex set of synthetic RNAs used to assess sensitivity, accuracy, and bias in RNA-seq experiments [65]. | Enables construction of standard curves for absolute transcript quantification and measures protocol-dependent biases [65]. |
| ZymoBIOMICS Spike-in Controls | Defined communities of fully inactivated microbes for absolute quantification in microbiome sequencing [64]. | Spike-in I (High Load): For feces, cell culture. Spike-in II (Low Load): For swabs, water filters. Comprised of Gram-negative and Gram-positive bacteria to expose DNA extraction bias [64]. |
| Custom Synthetic Oligos | RNA or DNA oligonucleotides designed to match specific sequence attributes (length, GC%) of your targets [63]. | Can be tailored to create a diverse normalization panel that corrects for sequence-specific biases in ligation and amplification [63]. |
| PhiX Control v3 Library | A well-characterized library used to improve sequencing performance on Illumina platforms [68]. | Primarily used to balance nucleotide diversity for low-diversity libraries (e.g., 16S rRNA gene amplicons), which improves base calling and data quality. It is spiked in immediately before sequencing [68]. |
| Digital PCR (dPCR) | A method for absolute nucleic acid quantification without a standard curve [4]. | Used as an orthogonal method to validate spike-in recovery and total microbial load measurements by partitioning a sample into thousands of nanoliter reactions for precise counting [4]. |
DspikeIn R Package |
A bioinformatics tool for analyzing absolute abundance data derived from spike-in experiments [67]. | Provides functions for spike-in validation, scaling factor estimation, conversion to absolute counts, and integration with other Bioconductor packages for downstream analysis [67]. |
Experimental Workflow and Protocol Diagrams
Spike-in Experimental Workflow
Troubleshooting Poor Recovery Logic Tree
Mitigating Host DNA Contamination in Mucosal and Tissue Samples
FAQs: Addressing Common Experimental Issues
Q1: Why is host DNA depletion particularly critical for mucosal and tissue samples in absolute abundance studies?
In absolute abundance studies, the goal is to measure the true, countable number of microbial cells or genomes. Mucosal and tissue samples are often low microbial biomass environments, meaning the microbial "signal" is low compared to the host "noise" [69]. An overwhelming amount of host DNA (e.g., >90% of total DNA) severely reduces the sequencing depth available for microbial genomes, impair the detection of low-abundance microorganisms and skews quantitative assessments [70] [71]. Without depletion, results may not reflect the true microbial absolute abundance but rather the variable and often high levels of host contamination.
Q2: What are the primary sources of contamination I should control for in these samples?
You need to consider two main contamination sources:
- Host DNA: The inherent host DNA from the tissue or mucosal sample itself is the primary concern for depletion.
- Exogenous DNA Contamination: This includes DNA from human operators (skin, hair, breath), sampling equipment, laboratory reagents, and kits, or cross-contamination between samples [69]. In low-biomass samples, even trace amounts of this exogenous DNA can be misinterpreted as a true microbial signal.
Q3: We use qPCR for DNA quantification. Our negative controls sometimes show amplification. How can we prevent this?
PCR contamination is a common issue. Key preventive measures include:
- Physical Separation: Designate and use distinct areas for sample preparation, PCR setup, and post-PCR analysis. Do not bring reagents or equipment from the post-PCR area back to the pre-PCR area [72].
- Dedicated Equipment: Use separate sets of pipettes, filter tips, lab coats, and waste containers for pre- and post-PCR work [72].
- Decontamination: Thoroughly clean workspaces, equipment, and pipettes with a diluted bleach solution (10%) to degrade any contaminating DNA [73] [72].
- Always Include Controls: Always run a negative control (e.g., ultrapure water instead of template DNA) to monitor for contamination in every experiment [72].
Q4: Are there computational methods to identify and remove contaminants after sequencing?
Yes, bioinformatic tools are a crucial final step for decontamination. Tools like Decontam can identify and remove contaminant sequences based on their prevalence in negative controls or their inverse correlation with total DNA concentration [71]. Another tool, CLEAN, is a pipeline designed to remove unwanted sequences, including host DNA and common spike-in controls, from both short- and long-read sequencing data [74]. These tools help refine your dataset but should complement, not replace, rigorous wet-lab contamination controls.
Troubleshooting Guides
Problem: Low Microbial Sequencing Depth After Host Depletion
Potential Causes and Solutions:
- Cause: Inefficient Host DNA Removal
- Solution: Benchmark different host depletion methods for your specific sample type. Pre-extraction methods like saponin lysis + nuclease digestion are often very effective for samples with intact microbial cells [70].
- Cause: Method Introduced Bias or Loss of Microbial Cells
- Solution: Some harsh depletion methods can lyse fragile microbes. If studying a community with a mix of robust and fragile cells, consider gentler methods or use a mock community to assess bias. The table below summarizes the performance of various methods from a benchmarking study.
- Cause: High Abundance of Cell-Free Microbial DNA
- Solution: Be aware that pre-extraction methods that target intact cells will miss cell-free microbial DNA. One study found that ~69% of microbial DNA in bronchoalveolar lavage fluid was cell-free [70]. The choice of method should align with your research question (intact cells vs. total microbial DNA).
Problem: Inconsistent Host Depletion Efficiency Across Samples
Potential Causes and Solutions:
- Cause: Variable Sample Quality and Starting Conditions
- Cause: Incomplete Digestion or Reaction Conditions
- Solution: For enzymatic methods (e.g., nucleases, methylation-dependent restriction enzymes), optimize reaction conditions such as enzyme concentration, incubation time, and the use of activators [76]. Include a positive control with known amounts of host and microbial DNA to validate each run.
Experimental Protocols & Data Presentation
This protocol is designed to lyse mammalian cells while leaving bacterial cells intact, followed by degradation of the released host DNA.
Workflow Diagram:
Steps:
- Homogenization: Homogenize the mucosal or tissue sample in a suitable buffer (e.g., PBS).
- Saponin Lysis: Add a pre-optimized volume of saponin stock solution to the sample homogenate to achieve a final concentration of 0.025%. Vortex to mix thoroughly.
- Incubation: Incubate the mixture at room temperature for 15-30 minutes with gentle agitation to allow for lysis of host cells.
- Nuclease Digestion: Add a commercial nuclease enzyme (e.g., Benzonase, Micrococcal Nuclease) according to the manufacturer's instructions. Include the recommended reaction buffer and co-factors (e.g., Mg²âº).
- Incubation: Incubate at 37°C for 30-60 minutes to digest all free DNA, including the released host genomic DNA.
- Termination & Washing: Stop the nuclease reaction by adding a stop solution (e.g., EGTA or EDTA to chelate Mg²âº). Centrifuge the sample to pellet the intact microbial cells. Carefully remove the supernatant containing digested host DNA and wash the pellet with a nuclease-free buffer. Repeat centrifugation.
- DNA Extraction: Proceed with your standard microbial DNA extraction protocol on the final pellet.
This method exploits the differential methylation patterns between host DNA (highly methylated) and most prokaryotic DNA.
Workflow Diagram:
Steps:
- DNA Shearing: Use a sonicator (e.g., Covaris) to shear the total DNA extract to an average fragment size of ~350 bp.
- End-Repair: Perform a standard end-repair step on the sheared DNA fragments to create blunt ends.
- MD-RE Digestion: Set up a 30 µL reaction containing:
- 1x manufacturer's buffer (e.g., NEBuffer 4)
- 10 µg Bovine Serum Albumin (BSA)
- 0.05 µM activator oligonucleotide (enhances digestion)
- 6 units of MD-RE (e.g., MspJI, LpnPI, FspEI)
- 0.1-2 µg of end-repaired DNA Incubate in a thermocycler at 37°C for 16 hours.
- Enzyme Inactivation: Heat the reaction to 65°C for 20 minutes to inactivate the enzyme.
- Size Selection: Purify the digested DNA using solid-phase reversible immobilization (SPRI) beads (e.g., Agencourt Ampure XP). Use a 1:1 sample-to-bead ratio to capture larger, undigested fragments (which are enriched for microbial DNA). Perform two rounds of purification to ensure complete removal of small, digested host DNA fragments.
- Library Preparation: The purified DNA is now enriched for microbial sequences and can be used for standard NGS library preparation.
Performance Comparison of Host Depletion Methods
The following table summarizes key metrics from a benchmarking study on respiratory samples (BALF and oropharyngeal swabs), which are relevant to mucosal sampling [70].
Table 1: Benchmarking Host DNA Depletion Methods for Respiratory Samples
| Method (Abbreviation) | Principle | Median Host DNA Removed (BALF) | Microbial Read Increase (BALF, vs. Raw) | Key Trade-offs / Notes |
|---|---|---|---|---|
| Saponin + Nuclease (S_ase) | Lyses host cells; digests DNA | ~99.99% (to 493.82 pg/mL) | 55.8-fold | High host removal, but can significantly reduce bacterial biomass. |
| HostZERO Kit (K_zym) | Commercial kit (similar principle) | ~99.99% (to 396.60 pg/mL) | 100.3-fold | Highest read increase, but cost and potential bias need evaluation. |
| Filter + Nuclease (F_ase) | Filter to enrich microbes; digest DNA | Data not specified | 65.6-fold | Developed in the study; showed balanced performance. |
| QIAamp Microbiome Kit (K_qia) | Commercial kit | Data not specified | 55.3-fold | Good bacterial retention rate in OP samples. |
| Nuclease only (R_ase) | Digests free DNA (less selective) | Data not specified | 16.2-fold | Highest bacterial retention rate, but lower host depletion. |
| Osmotic Lysis + PMA (O_pma) | Osmotic shock; PMA degrades DNA | Data not specified | 2.5-fold | Least effective in increasing microbial reads. |
The Scientist's Toolkit: Essential Reagents & Materials
Table 2: Key Research Reagent Solutions for Host DNA Depletion
| Item | Function / Principle | Example Use Case |
|---|---|---|
| Saponin | A detergent that selectively lyses mammalian cell membranes but does not disrupt bacterial cell walls. | Pre-extraction depletion of host cells from tissue homogenates [70]. |
| Benzonase / Micrococcal Nuclease | Enzymes that degrade all forms of DNA and RNA. Used after host cell lysis to destroy released host nucleic acids. | Destroying host DNA after saponin lysis or osmotic lysis protocols [70]. |
| Methylation-Dependent Restriction Endonucleases (MD-REs) | Enzymes (e.g., MspJI) that cleave DNA at specific sequences containing methylated cytosine. Targets methylated host DNA. | Post-extraction depletion of host DNA from total DNA extracts; effective for human DNA [76]. |
| Propidium Monoazide (PMA) | A DNA-intercalating dye that penetrates only membrane-compromised cells. Upon light exposure, it cross-links DNA, making it unavailable for PCR. | Differentiating between intact and dead cells; can be used to selectively cross-link DNA from lysed host cells [70]. |
| HostZERO Microbial DNA Kit (Zymo) | Commercial kit designed to selectively remove host cells and digest host DNA. | Standardized protocol for host depletion from various sample types [70]. |
| QIAamp DNA Microbiome Kit (Qiagen) | Commercial kit that enzymatically removes non-microbial DNA and digests common contaminants. | An alternative commercial solution for enriching microbial DNA from complex samples [70]. |
| Droplet Digital PCR (ddPCR) | Provides absolute quantification of nucleic acid targets without a standard curve. Extremely sensitive and precise. | Quantifying trace levels of residual host DNA post-depletion to ensure it meets regulatory limits (e.g., <10 ng/dose) [75]. |
Benchmarking Performance: Validating Methods and Comparing Biological Interpretations
In microbial ecology and related fields, determining the absolute abundance of microorganisms—their exact cell count or nucleic acid copy number per unit of sample—is crucial for understanding true biological changes. Unlike relative abundance, which only shows proportions and can be misleading, absolute abundance reveals whether a microbe is genuinely increasing or decreasing in number [7]. This technical guide compares three core methods for achieving this: flow cytometry, spike-in standards, and droplet digital PCR (ddPCR). Each technique operates on a different principle, leading to distinct strengths, limitations, and optimal use cases, which are detailed in the following troubleshooting and comparison sections.
The table below summarizes the core principles and key technical aspects of the three methods.
Table 1: Core Methodology and Output Specifications
| Method | Fundamental Principle | Primary Output Unit | Throughput |
|---|---|---|---|
| Flow Cytometry | Physical counting of individual stained cells by laser interrogation. | Cells/volume (e.g., cells/μL) [77] [78] | High (>35,000 events/second) [79] |
| Spike-in | Addition of a known quantity of exogenous reference DNA to correct sequencing data. | Calculated cells or copies/sample [58] [4] | High (integrated into sequencing pipeline) |
| Droplet Digital PCR (ddPCR) | Partitioning of sample for end-point PCR and absolute counting via Poisson statistics. | Copies/μL of input [80] [81] [82] | Medium |
Troubleshooting Common Experimental Issues
FAQ 1: My flow cytometry and ddPCR results for the same sample are inconsistent. What could be the cause?
This common issue often stems from the fundamental difference in what each method measures.
- Likely Cause 1: Difference in Target Molecule. Flow cytometry counts intact microbial cells [78], while ddPCR quantifies target DNA molecules. If a sample contains many dead cells (which have DNA but are not counted by flow cytometry) or extracellular DNA (from lysed cells), the results will diverge.
- Troubleshooting Steps:
- Investigate Cell Viability: Use a viability dye, such as Propidium Monoazide (PMA), in your flow cytometry protocol. PMA penetrates only membrane-compromised (dead) cells and binds their DNA, preventing its amplification in subsequent PCR steps. Comparing samples with and without PMA treatment can indicate the level of interference from dead cells/extracellular DNA [78].
- Validate with a Mock Community: Use a defined mix of bacteria with known cell counts. Process this mock community with both techniques to establish a baseline correlation and identify inherent technical biases in your workflow.
- Best Practice: These methods are often complementary. Use flow cytometry for total intact cell load and ddPCR for specific, sensitive quantification of a genetic target (e.g., a taxon-specific gene or CAR-T transgene) [81] [78].
FAQ 2: When using a spike-in standard, what is the critical factor for achieving accurate absolute quantification?
The single most critical factor is the timing and method of the spike's addition.
- Likely Cause: Adding the spike-in standard after DNA extraction and purification only controls for technical variations in downstream steps (like PCR and sequencing). It does not account for the major biases introduced during cell lysis and DNA extraction, which can vary significantly between different microbial taxa and sample types [4].
- Troubleshooting Steps:
- Add Standard Early: Add the known quantity of reference DNA (e.g., from an organism not expected in your sample) to the sample before starting the DNA extraction process [4]. This controls for both extraction efficiency and downstream analysis biases.
- Match the Standard to the Sample: Ensure the spike-in DNA has similar extraction properties (e.g., cell wall hardness for a whole-cell spike, fragment length for purified DNA) to your target microbes to ensure comparable recovery.
- Best Practice: Always report the point of standard addition (pre-extraction vs. post-extraction) in your methods, as this drastically affects data interpretation.
FAQ 3: My ddPCR results show a high coefficient of variation (CV) between replicates. How can I improve precision?
ddPCR is known for high precision, but poor technique can introduce variability.
- Likely Causes:
- Inadequate Sample Homogenization: Nucleic acids are not evenly distributed in the master mix.
- Overloaded or Underloaded Partitioning: Too many target molecules lead to multiple copies per droplet, violating the Poisson assumption; too few lead to high statistical uncertainty.
- Impurities in DNA Sample: Inhibitors can cause failed or delayed amplification in a subset of droplets.
- Troubleshooting Steps:
- Optimize Template Concentration: Serially dilute your DNA sample and re-run the assay. The ideal concentration range for ddPCR is typically 1-100,000 copies per reaction to ensure most droplets contain either 0 or 1 target molecule [82].
- Purify DNA: Use a high-quality DNA cleanup kit to remove PCR inhibitors.
- Improve Pipetting Technique: Ensure the reaction mix is thoroughly mixed before droplet generation. Use calibrated pipettes and tips to ensure accurate and consistent partitioning.
- Best Practice: A well-optimized ddPCR assay can achieve a CV of less than 5% for repeated measures, significantly outperforming qPCR which may have a CV of 25% or more [80].
Detailed Experimental Protocols
Protocol 1: Absolute Quantification of T Cells via ddPCR of Demethylated CD3Z Locus
This protocol is adapted from a study comparing ddPCR to flow cytometry for T cell quantification [80].
- DNA Extraction & Bisulfite Conversion: Extract genomic DNA from whole blood or tissue. Treat the DNA with sodium bisulfite, which converts unmethylated cytosines to uracils, while methylated cytosines remain unchanged.
- ddPCR Reaction Setup:
- Prepare a 20µL reaction mix containing:
- ddPCR Supermix for Probes (no dUTP)
- Methylation-Specific (MS) Primer/Probe Set: Targeting the demethylated (and thus bisulfite-converted) promoter region of the CD3Z gene. This set will not amplify the methylated version from non-T cells.
- Reference Assay: A primer/probe set for a reference gene insensitive to methylation status (a "C-less" reaction) to control for total input DNA [80].
- Restriction Enzyme (e.g., XbaI, EcoRI-HF) to digest genomic DNA and reduce viscosity.
- Bisulfite-converted DNA template (~4µL).
- Prepare a 20µL reaction mix containing:
- Droplet Generation: Transfer the reaction mix to a DG8 cartridge and generate approximately 20,000 nanoliter-sized droplets using a droplet generator.
- PCR Amplification: Transfer the emulsified droplets to a 96-well plate, seal, and run endpoint PCR on a thermal cycler.
- Droplet Reading & Analysis: Place the plate in a droplet reader. It will count the number of fluorescence-positive (containing the demethylated CD3Z target) and negative droplets for each sample.
- Absolute Quantification: The reader's software uses Poisson statistics to calculate the absolute concentration of demethylated CD3Z copies/µL. This value is used to estimate T cell numbers, with two demethylated CD3Z copies corresponding to one T cell [80].
Protocol 2: Quantitative Microbiome Profiling (QMP) via Flow Cytometry
This protocol, used for determining absolute microbial loads in stool, allows for the conversion of 16S rRNA sequencing data from relative to absolute abundance [78].
- Sample Homogenization: Dilute a fixed weight of fecal sample (e.g., 200 mg) in a buffer such as PBS to create a homogeneous suspension.
- Staining: Stain the diluted sample with a fluorescent DNA dye like SYBR Green I. This dye binds to nucleic acids, allowing the microbial cells to be detected by the instrument's laser.
- Flow Cytometry Analysis:
- Run the stained sample through a flow cytometer (e.g., BD FACSCanto II).
- Set a threshold on the fluorescence channel (e.g., FL1) to distinguish stained cells from background debris and noise.
- Use volumetric counting to determine the absolute cell concentration in events/μL.
- Data Integration with Sequencing:
- Perform standard 16S rRNA gene amplicon sequencing on a parallel aliquot of the same sample.
- The absolute abundance of each taxonomic unit is calculated as:
(Relative abundance from sequencing) × (Total bacterial cell count from flow cytometry)
Diagram 1: Flow Cytometry QMP Workflow
Protocol 3: Absolute Quantification in Metagenomics Using Pre-Extraction Spike-in
This protocol uses an external DNA standard added prior to extraction to calibrate metagenomic sequencing data [4].
- Standard Selection & Calibration: Select a purified DNA standard from an organism absent from your sample (e.g., yeast HIS3 or LEU2 genes). Precisely quantify this DNA using fluorometry to know its exact concentration (copies/µL).
- Spike-in Addition: Add a known volume and concentration of the standard DNA to a measured amount of your sample (e.g., stool, soil) before any DNA extraction steps begin. This controls for DNA loss during extraction.
- DNA Extraction & Sequencing: Proceed with your standard metagenomic DNA extraction protocol. Prepare sequencing libraries and perform high-throughput sequencing.
- Bioinformatic Analysis & Quantification:
- Map the sequencing reads to a reference genome for your spike-in standard.
- Calculate the ratio of observed spike-in reads to expected spike-in reads based on the known added amount.
- Use this ratio as a scaling factor to convert the relative abundances of all microbial taxa in the sample to absolute abundances.
Diagram 2: Pre-Extraction Spike-in Workflow
Performance Comparison & Decision Framework
The choice between methods depends heavily on your experimental goals, sample type, and resources. The following table provides a direct, quantitative comparison of key performance metrics.
Table 2: Head-to-Head Performance and Application Matrix
| Metric | Flow Cytometry | Spike-in Standards | ddPCR |
|---|---|---|---|
| What is Quantified? | Intact cells [78] | Total community DNA [4] | Specific DNA target sequences [80] |
| Sensitivity | Limited for rare taxa (<1% may be hard to detect) [77] | High (depends on sequencing depth) | Excellent (can detect rare alleles and low copy numbers) [58] [82] |
| Precision (CV) | High with good technique | High with proper standardization | Very High (e.g., 3.5% vs 25% for qPCR) [80] |
| Ease of Use | Requires expertise in staining and gating | Simple to incorporate into workflow | High; no standard curve needed [58] |
| Throughput | Very High [79] | High (parallelized with sequencing) | Medium |
| Cost | High (instrument, reagents) | Low (after initial standard purchase) | Medium |
| Ideal Use Case | Total microbial load in stool [78], immunophenotyping [79] | Converting existing relative sequencing data to absolute abundance [4] | Rare target detection [82], transgene quantification [81], low biomass samples [58] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Their Functions
| Reagent / Kit | Function | Example Application |
|---|---|---|
| SYBR Green I | Fluorescent nucleic acid stain for detecting cells in flow cytometry. | Distinguishing bacterial cells from debris in a stool sample for total cell counting [77] [78]. |
| PMA/PMAxx | Viability dye that penetrates dead cells and binds DNA, inhibiting its amplification. | Differentiating between intact and membrane-compromised cells to quantify only the viable fraction [78]. |
| ddPCR Supermix for Probes | Optimized PCR mix for droplet-based digital PCR, often without dUTP to accommodate uracil-DNA glycosylase (UDG) clean-up. | Absolute quantification of a CAR-T transgene or a methylation-specific target like demethylated CD3Z [80] [81]. |
| MagMAX DNA Multi-Sample Kit | Magnetic-bead based kit for automated, high-throughput genomic DNA extraction. | Preparing high-quality gDNA from large sets of blood or tissue samples for ddPCR analysis [81]. |
| Exogenous Spike-in DNA | Purified DNA from an organism not found in the sample (e.g., yeast genes). | Adding a known quantity to a microbiome sample pre-extraction to control for technical variability and calculate absolute abundance from sequencing data [4]. |
Troubleshooting Guide: Absolute Abundance Quantification
This guide addresses common issues encountered when transitioning from relative to absolute abundance analysis in microbial ecology studies.
Table 1: Troubleshooting Common Experimental Problems
| Error | Cause | Solution |
|---|---|---|
| Low or inconsistent microbial DNA yield from mucosal samples | High host DNA content saturates extraction columns, limiting the mass of sample that can be processed and reducing microbial DNA recovery [12]. | For mucosal samples, do not exceed 8 mg input mass. The Lower Limit of Quantification (LLOQ) is approximately 1×107 16S rRNA gene copies per gram [12]. |
| Dropouts (low-abundance taxa missing from sequencing data) | Starting with low total microbial DNA input for 16S rRNA library preparation leads to the loss of the least abundant taxa [12]. | Ensure 16S rRNA gene input is greater than 1×104 copies for library preparation. For low-biomass samples, use digital PCR (dPCR) to confirm sufficient template before sequencing [12]. |
| Inaccurate absolute abundance estimates | Amplification biases in standard qPCR or from non-specific amplification of host DNA skews quantification [12]. | Adopt a microfluidic digital PCR (dPCR) anchoring method. dPCR provides absolute quantification without a standard curve and minimizes host DNA amplification bias [12]. |
| Contaminant sequences appear in data | Contaminating DNA from reagents or the environment is amplified when the true microbial load of the sample is very low [12]. | Include negative control extractions (e.g., with no sample) in the sequencing run. Filter out any taxa found in these controls from your experimental data [12]. |
| Misinterpretation of a taxon's response to diet | Relative abundance analysis cannot distinguish if a taxon increased, decreased, or stayed the same in absolute terms [12]. | Use absolute abundance data to determine the true direction and magnitude of change for individual taxa, as relative data can create false positives [12]. |
Frequently Asked Questions (FAQs)
Q: What is the core limitation of using only relative abundance data in a diet study? Relative abundance measurements are compositionally constrained; an increase in one taxon's relative abundance forces an artificial decrease in all others. This can lead to high false-positive rates in identifying differentially abundant taxa and makes it impossible to determine if an individual taxon's population truly expanded or contracted in absolute terms [12].
Q: When should I use absolute abundance quantification in my research? Absolute quantification is critical when your research question involves changes in total microbial load, the true magnitude of change of specific taxa, or when analyzing samples with vastly different microbial densities (e.g., stool vs. small intestine mucosa). It is also superior for estimating the time since deposition in forensic samples [12] [83].
Q: What are the main methodological approaches for absolute quantification? The primary anchoring methods are:
- Spiked Standards: Adding a known quantity of exogenous DNA to the sample [12].
- Digital PCR (dPCR): Precisely counting 16S rRNA gene copies to establish a total load anchor, which is then used to transform relative sequencing data into absolute abundances [12].
- Flow Cytometry: Directly counting total bacterial cells [12].
- Total DNA Quantification: Effective only for samples free of host DNA [12].
Q: How does a ketogenic diet affect the gut microbiome, and why is it a good model for this method? The ketogenic diet induces substantial and rapid compositional changes in the gut microbiota [12]. In a murine model, absolute quantification revealed that the diet not changed the balance of taxa but also decreased the total microbial load. This critical finding, which directly impacts physiological interpretation, is invisible to relative abundance analysis alone [12].
Q: How do I calculate the absolute abundance of a specific taxon from sequencing data?
After using dPCR to determine the total number of 16S rRNA gene copies in your sample, you multiply this total by the relative abundance of your taxon of interest (obtained from 16S rRNA amplicon sequencing).
Formula: Absolute Abundance of Taxon A = Total 16S rRNA Gene Copies (from dPCR) × Relative Abundance of Taxon A (from sequencing)
Experimental Protocol: dPCR Anchoring for Absolute Abundance
This protocol details the framework for quantifying absolute abundances using dPCR anchoring, as applied in the murine ketogenic diet study [12].
The following diagram illustrates the complete experimental workflow from sample preparation to data integration.
Step-by-Step Methodology
Sample Collection and Storage:
- Collect luminal and mucosal samples from the gastrointestinal tract (e.g., small intestine, cecum, colon) and stool.
- Immediately freeze samples at -80°C to preserve nucleic acid integrity.
DNA Extraction with Validated Efficiency:
- Use a standardized kit-based DNA extraction protocol (e.g., 20-µg column-based kits).
- Critical: Prior to the experiment, validate DNA extraction efficiency by spiking a defined microbial community into germ-free mouse samples across a dilution series (e.g., from 1.4 × 109 to 1.4 × 105 CFU/mL). Confirm near-complete recovery via dPCR [12].
- Adhere to maximum input masses: 200 mg for stool/cecal contents and 8 mg for mucosal samples to prevent column over-saturation by host DNA [12].
Digital PCR (dPCR) for Total Load Quantification:
- Perform dPCR with primers targeting the 16S rRNA gene.
- The dPCR reaction is partitioned into thousands of nanoliter-sized droplets. After amplification, count the positive droplets to obtain an absolute count of 16S rRNA gene copies in the sample without relying on a standard curve [12].
- This value represents the "Total Microbial Load."
16S rRNA Gene Amplicon Sequencing for Relative Abundance:
- Prepare sequencing libraries using improved "universal" 16S rRNA primers and a validated protocol [12].
- Critical: Monitor amplification reactions with real-time qPCR and stop cycles in the late exponential phase to limit chimera formation and over-amplification biases [12].
- Sequence the libraries on a high-throughput platform. Process the data through a standard bioinformatics pipeline (quality filtering, OTU clustering, taxonomy assignment) to generate "Relative Abundance" profiles for each taxon.
Data Integration and Absolute Abundance Calculation:
- For each sample, multiply the relative abundance of each taxon by the total 16S rRNA gene copies measured by dPCR.
- Formula:
Absolute Abundance of Taxon A = Total 16S rRNA Gene Copies (from dPCR) × Relative Abundance of Taxon A (from sequencing)
Table 2: Key Quantitative Findings from the Murine Ketogenic Diet Study [12]
| Measurement | Result | Experimental Context & Significance |
|---|---|---|
| DNA Extraction Efficiency | ~2x accuracy (near complete recovery) | Observed when spiking a defined microbial community into germ-free mouse samples. Efficiency was consistent across 5 orders of magnitude (1.4×105 to 1.4×109 CFU/mL) and different sample matrices (cecum, stool, SI mucosa) [12]. |
| Lower Limit of Quantification (LLOQ) per Gram | 4.2×105 16S copies (stool/cecum) 1.0×107 16S copies (mucosa) | Dictated by the maximum sample mass that can be loaded without over-saturating the extraction column. Mucosal samples have a higher LLOQ due to high host DNA content [12]. |
| Minimum 16S Input for Reliable Sequencing | >1×104 gene copies | Library preparation with input below this level led to taxon "dropouts" (loss of lowest abundance taxa) and the appearance of contaminant sequences [12]. |
| Ketogenic Diet Effect on Total Load | Decrease in total microbial load | Revealed only by absolute abundance analysis. Relative abundance data alone could not detect this overall collapse in the microbial community [12]. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Methods for Absolute Quantification
| Item | Function in the Protocol |
|---|---|
| Digital PCR (dPCR) System | Provides absolute quantification of total 16S rRNA gene copies in a DNA sample without a standard curve, serving as the anchor for converting relative to absolute data [12]. |
| Validated DNA Extraction Kit (20-µg column) | Standardizes microbial lysis and DNA purification. Must be validated for efficiency and evenness across Gram-positive and Gram-negative bacteria and different sample types [12]. |
| 16S rRNA Gene Primers (Improved) | "Universal" primers for amplicon sequencing that provide comprehensive coverage while minimizing amplification bias [12]. |
| Defined Microbial Community | A mixture of known bacterial strains used as a spike-in control to empirically validate DNA extraction efficiency and evenness across the expected microbial load range [12]. |
| Germ-Free Mouse Samples | Tissues or stool from germ-free animals used as a blank matrix for spike-in control experiments, ensuring no background microbial DNA interferes with efficiency calculations [12]. |
Assessing Reproducibility and Accuracy Using Mock Microbial Communities
In the field of microbial ecology, data derived from high-throughput sequencing is inherently compositional. This means that traditional analyses report the relative abundance of microbial taxa, where an increase in one taxon necessarily leads to an apparent decrease in others. This compositional nature can obscure true biological changes, such as whether a taxon is genuinely increasing or if other community members are decreasing. The measurement of absolute abundance is therefore critical, as it quantifies the actual number of microbial cells or gene copies in a sample, providing a direct and unambiguous view of microbial load and dynamics [11] [4].
Mock microbial communities—synthetic mixes of known microorganisms with defined compositions—serve as essential ground-truth controls. By using these communities, researchers can identify technical biases, optimize workflows, and ultimately ensure that their data reflects biological reality rather than methodological artefacts. This guide provides a practical framework for leveraging mock communities to assess and improve the reproducibility and accuracy of your microbiome studies, with a focus on absolute abundance measurement.
FAQs on Mock Communities and Absolute Abundance
1. What is the key difference between relative and absolute abundance, and why does it matter?
- Relative Abundance measures the proportion of each microbial species within a sample, summing to 100%. It does not provide information about the actual quantity of microbes.
- Absolute Abundance quantifies the actual number of microbial cells or gene copies in a given sample volume [11].
The distinction is critical for correct interpretation. For example, two samples can both contain 50% of a bacterial species, but if one sample has a total of 2 million cells and the other has 20 million, the absolute abundance of that species is ten times higher in the second sample. Relying solely on relative data can lead to misinterpretations; a decrease in one taxon's relative abundance might simply be a dilution effect caused by the bloom of another, rather than a true decrease in its absolute numbers [84] [4].
2. How can mock communities improve the reproducibility of my microbiome data?
Mock communities are a powerful tool for identifying technical biases introduced at various stages of your workflow [85] [86]:
- DNA Extraction: Cells with different wall strengths (e.g., Gram-positive vs. Gram-negative) lyse with varying efficiency. A mock community containing both types reveals this bias.
- PCR Amplification: Variations in 16S rRNA gene copy number (GCN) and genomic GC-content can cause uneven amplification.
- Sequencing and Bioinformatics: Platform-specific errors and database limitations can affect taxonomic assignments.
By including a mock community in every run as a positive control and comparing your observed results to the expected composition, you can quantify the technical variability and bias in your data, which is the first step toward improving reproducibility [87] [88].
3. What methods are available for measuring absolute abundance?
Several methods can be used to convert relative data into absolute quantities, each with its own strengths:
| Method | Principle | Key Advantages | Key Limitations |
|---|---|---|---|
| Spike-In Controls [11] | Adding a known quantity of foreign cells or DNA to the sample before processing. | Accounts for biases in DNA extraction and sequencing; precise. | Requires unique species not found in native samples. |
| Flow Cytometry [84] [89] | Directly counting total bacterial cells in a sample using fluorescent staining. | Direct cell count; no amplification bias. | Requires specialized equipment; staining can be biased by cell physiology. |
| Quantitative PCR (qPCR) [11] | Quantifying 16S rRNA gene copies using a standard curve. | Cost-effective; uses same DNA for sequencing and qPCR. | Affected by DNA extraction efficiency and variable 16S GCN. |
| Digital PCR (dPCR) [4] | Partitioning a PCR reaction into thousands of nanoliter droplets for absolute nucleic acid counting. | Highly precise; does not require a standard curve. | Lower throughput; requires specialized equipment. |
Troubleshooting Guide: Common Issues and Solutions
This guide helps diagnose and correct common problems identified through the use of mock communities.
Problem 1: Inaccurate Representation of Gram-Positive vs. Gram-Negative Bacteria
- Observed Issue: In your mock community data, Gram-negative bacteria are overrepresented while Gram-positive bacteria are underrepresented compared to the expected ratios [86].
- Underlying Cause: Inefficient lysis of Gram-positive bacterial cells due to their thicker, more resilient cell walls.
- Solutions:
- Optimize Bead-Beating: Increase the intensity or duration of mechanical bead-beating during DNA extraction [87].
- Enzymatic Treatment: Incorporate additional enzymatic lysis steps (e.g., lysozyme) to help break down Gram-positive cell walls.
- Validate with Standards: Use a cellular mock community like the ZymoBIOMICS Microbial Community Standard, which includes both Gram-positive and Gram-negative species, to optimize your lysis protocol [87] [85].
Problem 2: Over- or Under-Estimation of Taxa with High or Low GC Content
- Observed Issue: Taxa with exceptionally high or low genomic Guanine-Cytosine (GC) content deviate from their expected abundance.
- Underlying Cause: PCR amplification bias. Polymerases can have difficulty amplifying high-GC templates, leading to their under-representation [88].
- Solutions:
Problem 3: High Variability in Absolute Abundance Measurements Between Samples
- Observed Issue: The total microbial load, as measured by spike-in controls or flow cytometry, varies unexpectedly between technically similar samples.
- Underlying Cause: Inconsistent sample handling, DNA extraction efficiency, or inaccurate quantification of the internal standard.
- Solutions:
- Standardize Protocols: Use the same lot of reagents and process all samples for a project in the same batch to minimize batch effects [87].
- Correct Spike-in Workflow: Ensure the spike-in material is added at the very beginning of the DNA extraction process to control for all downstream losses [11].
- Use Appropriate Controls: For low-biomass samples, use a spike-in control designed for low microbial loads to improve quantification accuracy [86].
Essential Experimental Protocols
Protocol 1: Implementing Spike-In Controls for Absolute Abundance
This protocol allows you to calculate the absolute abundance of each taxon in your samples.
- Step 1: Select a Spike-In Control. Choose a synthetic DNA sequence or a whole-cell preparation from an organism not present in your native samples (e.g., Aliivibrio fischeri) [86].
- Step 2: Add a Known Quantity. Spike a precise, known amount of the control material into each sample before DNA extraction. This is critical, as it accounts for losses during extraction [11].
- Step 3: Proceed with DNA Extraction and Sequencing.
- Step 4: Calculate Absolute Abundance. Use the following formula for each taxon i in each sample:
Absolute Abundance (Taxon i) = (Relative Abundance of Taxon i / Relative Abundance of Spike-in) * Known Quantity of Spike-inThis calculation transforms your relative sequencing data into absolute counts [11].
Protocol 2: Using a Mock Community to Calculate a Measurement Integrity Quotient (MIQ) Score
The MIQ score provides a simple, standardized metric (0-100) to quantify the accuracy of your entire workflow [85].
- Step 1: Sequence a Mock Community. Include a well-characterized mock community (e.g., ZymoBIOMICS Microbial Community Standard) in your sequencing run.
- Step 2: Compare Observed vs. Expected. Bioinformatically determine the observed relative abundance of each species in the mock community.
- Step 3: Account for Manufacturing Tolerance. The expected abundance of each species has a tolerance range (e.g., ±15%). Observed values within this range are considered accurate.
- Step 4: Calculate the MIQ Score. The score is calculated by measuring the Root Mean Square Error (RMSE) of observed abundances that fall outside the manufacturing tolerance band. The formula is:
MIQ Score = 100 - RMSE. A score above 90 is considered excellent, while a score below 80 indicates significant technical bias that requires investigation [85].
The Scientist's Toolkit: Key Research Reagent Solutions
The following table lists essential materials and their specific functions in quality control for microbiome studies.
| Reagent or Tool | Type | Primary Function and Utility |
|---|---|---|
| ZymoBIOMICS Microbial Community Standard [85] [86] | Cellular Mock Community | Serves as a positive control for the entire workflow, from cell lysis to sequencing. Ideal for optimizing lysis methods due to its mix of Gram-positive, Gram-negative, and yeast cells with varying cell wall toughness. |
| ZymoBIOMICS Spike-in Controls I & II [86] | Spike-in Control | Added directly to native samples to enable absolute abundance quantification and act as an internal control for each sample. Control I is for high-biomass samples (e.g., stool), and Control II is for low-biomass samples (e.g., sputum). |
| ZymoBIOMICS Microbial Community DNA Standard [86] | DNA Mock Community | Used to optimize library preparation and bioinformatics pipelines, as it bypasses the DNA extraction step. Helps identify biases in amplification and taxonomic classification. |
| ZymoBIOMICS Fecal Reference [86] | True Diversity Reference | Provides a stable, complex natural microbiome profile for assessing run-to-run consistency, challenging bioinformatic pipelines, and enabling inter-laboratory comparisons. |
| Microbial Cytometric Mock Community (mCMC) [89] | Flow Cytometry Standard | A defined mix of cells used to validate accurate cell treatment, test cytometer alignment, and ensure proper use of flow cytometry bioinformatics pipelines for quantitative analysis. |
| Digital PCR (dPCR) [4] | Quantification Technology | Provides an ultrasensitive and precise method for the absolute quantification of 16S rRNA gene copies without a standard curve, useful for samples with low microbial load or high host DNA background. |
The Impact on Absolute Quantification on Differential Abundance Testing
Frequently Asked Questions
Q1: Why does my differential abundance analysis (DAA) produce misleading results when using relative data?
Relative abundance data is compositional, meaning all measurements are interdependent. An increase in one taxon's relative abundance necessarily causes an apparent decrease in others. This leads to compositional bias, where the observed log fold change between groups is contaminated by an additive bias term that depends on the ratio of total microbial content across groups, not just the taxon of interest [90]. Consequently, you may identify taxa that appear to change significantly due to the expansion or contraction of the rest of the community, rather than a true biological change in the taxon itself [10] [91].
Q2: What are the primary methods for obtaining absolute microbial abundance data?
The main methods are digital PCR (dPCR), flow cytometry, and spike-in standards. dPCR provides absolute quantification of 16S rRNA gene copies without a standard curve by partitioning samples into thousands of nanoliter reactions [4]. Flow cytometry directly counts bacterial cells in a sample [10]. Spike-in methods add a known quantity of exogenous cells or DNA to the sample prior to DNA extraction, providing an internal standard for calculating absolute abundances [10] [4].
Q3: My absolute abundance measurements show different trends for the same taxa compared to relative data. Is this normal?
Yes, this is a common and critical finding. Relative and absolute profiling can reveal opposing successional trends for major microbial phyla [91]. For example, a study on carcass decomposition found that Pseudomonadota displayed a decreasing trend in tissue based on relative abundance, while absolute quantification revealed an increasing trend [91]. Similarly, in antibiotic treatment studies, flow cytometry-based absolute counting revealed decreased abundances of specific bacterial families that were not detectable by standard relative analysis [10].
Q4: What are the key advantages of group-wise normalization methods like G-RLE and FTSS?
Traditional normalization methods (RLE, TMM) perform sample-to-sample comparisons, which can struggle with false discovery rate control when compositional bias is large. Group-wise normalization methods like G-RLE and FTSS reduce bias by re-conceptualizing normalization as a group-level task [90]. They achieve higher statistical power for identifying differentially abundant taxa and maintain better false discovery rate control in challenging scenarios, especially when used with DAA methods like MetagenomeSeq [90].
Troubleshooting Guides
Issue 1: Inconsistent or Misleading Differential Abundance Results
Problem: Your DAA identifies taxa that appear differentially abundant, but the results don't align with biological expectations or other experimental data.
Solution:
- Root Cause: This often stems from using relative abundance data, which is subject to compositional effects.
- Actionable Steps:
- Transition to Absolute Quantification: Implement a method to obtain absolute abundance data. For example, use dPCR to anchor your 16S rRNA gene sequencing data [4].
- Apply Group-wise Normalization: If you must use relative data, employ novel group-wise normalization methods like Group-wise Relative Log Expression (G-RLE) or Fold-Truncated Sum Scaling (FTSS). These methods calculate normalization factors using group-level summary statistics, which better corrects for compositional bias [90].
- Validate with a Second Method: Confirm key findings using an orthogonal absolute quantification method, such as flow cytometry or ddPCR [10].
Issue 2: Low Accuracy in Predicting Absolute Prokaryotic Load
Problem: Your model for predicting absolute microbial abundance (e.g., from DNA concentration) shows limited prediction accuracy on external validation cohorts.
Solution:
- Root Cause: The predictive model may be oversimplified or trained on a dataset with a small dynamic range.
- Actionable Steps:
- Incorporate Multiple Predictors: While DNA concentration is strongly correlated with absolute prokaryotic abundance (Spearman's rho = 0.92), a model including additional features (host read fraction, prokaryotic alpha diversity, etc.) can significantly improve performance [5].
- Ensure Sufficient Dynamic Range: Train your model on datasets that encompass a large dynamic range of absolute abundance, spanning multiple orders of magnitude, as this improves model robustness [5].
- Model Selection: Use a robust algorithm like random forest. One study showed a "DNA-only" random forest model achieved a Spearman correlation of 0.89, which improved to 0.91 with a full model including additional features [5].
Issue 3: Technical Challenges in Digital PCR (dPCR) Experiments
Problem: You encounter issues such as incorrect dye setup, concentration errors (NaN results), or problems with plate loading during dPCR runs.
Solution:
- Root Cause: Protocol deviations or software setup errors.
- Actionable Steps:
- Dye Setup: Confirm dye channels are correctly selected before starting the run. Unused channels can be turned off after the run in the software setup, but data will not be collected for channels turned off before the run [92].
- Concentration Readout (NaN): If the software displays "NaN" (not a number), it indicates an issue during the analysis of array images. Restart the software and instrument. If the problem persists, contact technical support [92].
- Plate Loading: Adhere to loading precautions to ensure accuracy.
- Do not let the pipette tip contact the well bottom or puncture the membrane.
- Avoid introducing bubbles.
- Do not mix the isolation buffer and sample.
- Load exactly 9 µL of sample along with 15 µL of isolation buffer per well. Deviations will affect microchamber filling and results [92].
Experimental Protocols for Absolute Quantification
Protocol 1: Absolute Quantification Framework Using dPCR Anchoring
This protocol provides a rigorous method for absolute abundance measurement across diverse sample types, from microbe-rich stool to host-rich mucosal samples [4].
Workflow Diagram:
Detailed Steps:
- Sample Collection and DNA Extraction:
- Homogenize samples and extract DNA using a standardized protocol.
- Assess extraction efficiency by spiking a defined microbial community into germ-free samples across a dilution series. Recovery should be near-equal over 5 orders of magnitude [4].
- Digital PCR (dPCR) for Total Load:
- Perform dPCR targeting the 16S rRNA gene to obtain an absolute count of gene copies per gram of sample.
- The lower limit of quantification (LLOQ) is approximately 4.2 × 10ⵠcopies/gram for stool and 1 × 10ⷠcopies/gram for mucosa due to high host DNA [4].
- 16S rRNA Gene Amplicon Sequencing:
- Prepare sequencing libraries, monitoring amplification with qPCR and stopping in the late exponential phase to limit chimera formation [4].
- Data Integration:
- For each taxon j in sample i, calculate its absolute abundance using the formula:
Absolute Abundanceᵢⱼ = (Relative Abundanceᵢⱼ from Sequencing) × (Total 16S rRNA Gene Copiesᵢ from dPCR).
- For each taxon j in sample i, calculate its absolute abundance using the formula:
Protocol 2: Flow Cytometry for Quantitative Microbiome Profiling (QMP)
This method quantifies total bacterial cell counts to convert relative sequencing data to absolute abundances [10].
Workflow Diagram:
Detailed Steps:
- Sample Preparation:
- Suspend a known mass of feces in phosphate-buffered saline (PBS) and homogenize thoroughly.
- Filtration and Staining:
- Filter the suspension to remove large particles.
- Stain with a fluorescent DNA dye (e.g., SYBR Green I).
- Flow Cytometry:
- Analyze the stained samples on a flow cytometer to obtain absolute bacterial cell counts per gram of sample.
- Data Integration:
- Multiply the relative abundance from 16S sequencing by the total bacterial cell count to obtain absolute abundances for each taxon.
Comparative Data: Relative vs. Absolute Abundance Analysis
Table 1: Impact of Absolute Quantification on Differential Abundance Findings in Selected Studies
| Study Context | Findings Based on Relative Abundance | Findings Based on Absolute Abundance | Implication |
|---|---|---|---|
| Antibiotic Study (Pigs) [10] | Limited detection of antibiotic effects. | Flow cytometry identified decreased absolute abundances of 5 families and 10 genera post-tylosin. | Absolute quantification reveals a broader and more significant impact of interventions. |
| Carcass Decomposition [91] | Pseudomonadota showed a decreasing trend. | Pseudomonadota showed an increasing trend. | Relative and absolute methods can show opposing ecological trends, changing biological interpretation. |
| Ketogenic Diet (Mice) [4] | Standard relative analysis. | Revealed a decrease in total microbial load and enabled accurate determination of the magnitude and direction of change for each taxon. | Resolves ambiguity in interpreting taxon ratios; clarifies true diet effect. |
Table 2: Performance of Machine Learning Models for Predicting Absolute Abundance
| Model Predictors | Spearman's rho | Key Performance Insight |
|---|---|---|
| DNA Concentration Only [5] | 0.89 | Strong correlation exists but model is sub-optimal. |
| Full Model (DNA concentration, host reads, alpha diversity, etc.) [5] | 0.91 | Incorporating multiple features significantly improves prediction accuracy. |
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for Absolute Quantification
| Item | Function | Example & Specification |
|---|---|---|
| Digital PCR System | Absolute quantification of target genes (e.g., 16S rRNA) without a standard curve. | QuantStudio Absolute Q System; uses MAP16 plates with 20,000 microchambers per sample [92]. |
| Validated Master Mix | Ensures optimal performance and quantification in dPCR reactions. | Absolute Q DNA Digital PCR Master Mix (5X) or 1-step RT-dPCR Master Mix (4X); designed for specific systems [92]. |
| Flow Cytometer | Direct enumeration of bacterial cells in a sample for total load calculation. | Used for Quantitative Microbiome Profiling (QMP) to obtain cells/gram [10]. |
| Spike-in Standards | Exogenous cells or DNA added to sample pre-extraction as an internal control for quantification. | Synthetic 16S rRNA genes or defined microbial communities of known concentration [10] [4]. |
| Universal 16S rRNA Primers | Amplification of the bacterial 16S gene for both sequencing and dPCR quantification. | Primers covering hypervariable regions (e.g., V4); validated for even amplification across taxa [4]. |
| DNA Binding Dye | Staining of microbial DNA for cell counting via flow cytometry. | SYBR Green I; must account for potential bias from varying nucleic acid content [10]. |
Evaluating Scalability, Cost, and Throughput for Large-Scale Clinical Studies
Frequently Asked Questions (FAQs)
FAQ 1: What are the primary cost drivers in a large-scale clinical trial, and how can they be managed? Clinical trial costs are influenced by multiple factors, with expenses escalating significantly with each phase. Key drivers include patient recruitment and retention, regulatory compliance, data collection and management, and clinical supplies [93]. Effective management strategies include designing efficient protocols to avoid unnecessary procedures, utilizing decentralized trials with remote monitoring to reduce site-related costs, and leveraging technology like electronic health records (EHRs) and AI-driven recruitment to streamline processes [93].
FAQ 2: How can I assess the scalability of a clinical study protocol before large-scale implementation? Scalability assessment should be integrated early in the study design process [94]. Utilize structured scalability assessment tools that examine key dimensions such as innovation attributes, implementer capabilities, adopting community characteristics, socio-political context, and scale-up strategy [95]. A study protocol complexity scoring model can help evaluate parameters like study arms, enrollment feasibility, investigational product administration, data collection complexity, and follow-up requirements [96]. Engaging potential clinical sites for feedback during protocol development can significantly improve feasibility and scalability [96].
FAQ 3: What methodological considerations are crucial for measuring absolute abundance in microbial communities within clinical studies? For absolute abundance quantification in microbial communities, digital PCR (dPCR) offers significant advantages over relative methods. dPCR provides absolute quantification without relying on standard curves or Ct values, is less sensitive to PCR inhibitors in crude samples, and enables more precise quantification of specific bacteria in complex communities [97]. When combining different methods, ensure computational adjustments are made to account for methodological differences in quantification approaches.
FAQ 4: How does high-throughput sequencing technology choice affect data quality and cost in large microbiome studies? The choice of sequencing technology creates a direct trade-off between resolution and cost. 16S rRNA amplicon sequencing is cost-effective for determining bacterial components at genus level but offers limited taxonomic resolution [97] [98]. Whole Genome Shotgun (WGS) sequencing provides higher resolution species/strain-level information and functional potential but at significantly higher cost [97]. Shallow WGS sequencing offers a middle ground, providing higher taxonomic depth than 16S sequencing at lower cost than full WGS, though with reduced ability to assess low-abundance community members [97].
Table 1: Comparison of High-Throughput Sequencing Methods for Microbial Community Analysis
| Method | Taxonomic Resolution | Functional Information | Relative Cost | Best Use Cases |
|---|---|---|---|---|
| 16S rRNA Amplicon Sequencing | Genus/Species level | No | $ | Large cohort studies, initial community profiling |
| Shallow WGS Sequencing | Species/Strain level for abundant members | Limited | $$ | Studies requiring higher resolution than 16S but with budget constraints |
| Whole Genome Shotgun (WGS) Sequencing | Species/Strain level | Yes (enzymatic pathways, gene families) | $$$ | Mechanistic studies, functional potential assessment |
| RNA Sequencing (Meta-transcriptomics) | Species/Strain level | Active gene expression | $$$$ | Studies of community functional activity |
FAQ 5: What strategies can optimize throughput without compromising data integrity in large clinical studies? Implement high-throughput process development (HTPD) approaches that use automation, robotic systems, and advanced liquid handling platforms to minimize manual intervention and reduce human error [99]. Optimize experimental layouts using standardized formats like 96-well plates to enhance throughput, accuracy, and reproducibility [99]. Integrate advanced analytical tools that enable rapid characterization of multiple samples simultaneously while maintaining data quality [99].
Troubleshooting Guides
Issue 1: Unexpected Cost Overruns in Clinical Trial Implementation
Symptoms: Trial expenses exceeding projected budget, particularly in patient recruitment, site management, or data collection phases.
Diagnosis and Resolution:
- Conduct Protocol Complexity Assessment: Use a standardized complexity scoring model to identify overly complex protocol elements that increase costs [96]. Evaluate parameters such as number of study arms, enrollment feasibility, subject registration process, and data collection requirements.
- Implement Cost Management Strategies:
- Optimize Protocol Design: Eliminate unnecessary procedures or endpoints not critical to primary objectives [93] [96].
- Utilize Centralized Monitoring: Reduce site monitoring costs through risk-based monitoring approaches [96].
- Leverage Technology: Implement electronic data capture (EDC) systems and consider decentralized trial elements to reduce site visits [93].
Prevention: Engage clinical sites early for feasibility feedback, employ adaptive trial designs that allow modifications based on interim results, and establish rigorous budget contingency planning based on phase-specific cost benchmarks [93] [96].
Issue 2: Inadequate Throughput in Microbial Sample Processing
Symptoms: Processing bottlenecks, sample backlog, delayed results, or compromised sample quality due to processing delays.
Diagnosis and Resolution:
- Automate Repetitive Processes: Implement robotic liquid handling systems for consistent, high-volume sample processing [99].
- Optimize Workflow Layout: Use 96-well plate formats strategically to maximize parallel processing while minimizing cross-contamination risks [99].
- Implement Quality Control Checkpoints: Integrate automated quality assessment for nucleic acid quantification and sample integrity verification [97].
- Utilize High-Throughput Analytical Platforms: Employ systems like the Fluidigm Access Array for targeted high-throughput qPCR or digital PCR systems for absolute quantification [97].
Prevention: Establish standardized operating procedures with throughput capacity planning, implement regular maintenance schedules for automated equipment, and maintain adequate reagent inventories to avoid processing delays.
Issue 3: Challenges in Scaling Microbial Community Analysis Methods
Symptoms: Method performance degradation with increased sample size, inconsistent results across batches, inability to maintain data quality at scale.
Diagnosis and Resolution:
- Standardize Sample Processing: Implement batch-effect minimization strategies through randomized processing and control samples in each batch [98].
- Validate Scalability of Quantification Methods: Transition from relative to absolute quantification methods like digital PCR for more robust scaling [97].
- Implement Computational Corrections: Apply batch-effect correction algorithms and normalization methods appropriate for the specific quantification technology [98].
- Establish Reference Materials: Use characterized control samples across batches to monitor technical variability [97].
Prevention: Conduct pilot studies at projected scale, validate all methods across expected sample size ranges, and establish quality thresholds before full study implementation.
Experimental Protocols
Protocol 1: Absolute Quantification of Microbial Abundance Using Digital PCR
Purpose: To precisely quantify absolute abundance of specific bacterial taxa in complex communities without reliance on standard curves.
Materials:
- Extracted genomic DNA from microbial samples
- Target-specific primers and probes
- Digital PCR reaction mix
- Digital PCR chip or droplet generator
- Thermal cycler compatible with digital PCR
- Quantification software
Procedure:
- Assay Design: Design and validate primers/probes for target taxonomic markers (e.g., specific 16S rRNA variable regions, functional genes).
- Reaction Setup: Prepare digital PCR reactions according to manufacturer specifications, partitioning each sample into thousands of individual reactions [97].
- Amplification: Run thermal cycling with optimized conditions for target amplification.
- Quantification Analysis: Count positive and negative partitions after amplification. Calculate absolute copy number using Poisson distribution statistics without reference to standards or controls [97].
- Data Normalization: Normalize to sample input mass or volume for cross-sample comparisons.
Troubleshooting Notes:
- If partition failure occurs, verify reaction mix composition and partitioning efficiency.
- For low abundance targets, increase sample input concentration while maintaining proper reaction chemistry.
- If high false positive rates occur, re-optimize primer specificity and annealing temperature.
Protocol 2: Scalability Assessment for Clinical Study Protocols
Purpose: To systematically evaluate and optimize clinical study protocols for large-scale implementation potential.
Materials:
- Draft study protocol document
- Scalability assessment tool [95]
- Protocol complexity scoring model [96]
- Multidisciplinary stakeholder team
Procedure:
- Stakeholder Engagement: Assemble team including investigators, clinical site staff, methodologists, and potential end-users [96].
- Scalability Dimension Assessment: Evaluate the innovation across five key dimensions [94] [95]:
- Innovation attributes (relative advantage, compatibility, complexity)
- Implementer characteristics (capacity, motivation)
- Adopting community attributes (needs, values)
- Socio-political context (policy alignment, regulatory environment)
- Proposed scale-up strategy (resource planning, monitoring)
- Protocol Complexity Scoring: Apply standardized complexity scoring to 10 key parameters [96]:
- Study arms/groups
- Informed consent process complexity
- Enrollment feasibility and population specificity
- Subject registration and randomization process
- Investigational product administration complexity
- Treatment phase duration and variability
- Study team multidisciplinary requirements
- Data collection and reporting requirements
- Follow-up phase duration and intensity
- Ancillary study requirements
- Feasibility Analysis: Identify potential implementation barriers and resource constraints.
- Protocol Optimization: Iteratively refine protocol based on assessment findings to enhance scalability potential.
Troubleshooting Notes:
- If scalability assessment identifies major barriers, consider protocol simplification or phased implementation approach.
- For high complexity scores in specific domains, develop targeted mitigation strategies before scale-up.
- If stakeholder feedback reveals significant concerns, conduct additional feasibility testing before proceeding.
Table 2: Clinical Trial Cost Benchmarks by Phase and Geographic Location
| Trial Phase | Participant Range | U.S. Cost Range (million $) | Western Europe Cost Range (million $) | Key Cost Drivers |
|---|---|---|---|---|
| Phase I | 20-100 | $1-4 | ~20-30% lower than U.S. [93] | Investigator fees, safety monitoring, specialized testing |
| Phase II | 100-500 | $7-20 | ~20-30% lower than U.S. [93] | Increased participant numbers, longer duration, detailed endpoint analyses |
| Phase III | 1,000+ | $20-100+ | ~20-30% lower than U.S. [93] | Large-scale recruitment, multiple sites, comprehensive data collection |
| Phase IV | Variable | $1-50+ | ~20-30% lower than U.S. [93] | Long-term follow-up, extensive safety monitoring |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for Microbial Community Analysis
| Resource | Function/Application | Key Considerations |
|---|---|---|
| 16S rRNA Primers (V3-V4 region) | Amplification of bacterial taxonomic markers for community profiling [97] | Region selection affects taxonomic resolution; V3-V4 provides balance of length and discrimination |
| Digital PCR Reagents | Absolute quantification of specific taxa without standard curves [97] | Provides precise quantification but requires specific equipment; less affected by inhibitors |
| Whole Genome Shotgun Library Prep Kits | Preparation of metagenomic sequencing libraries for functional profiling [97] | Higher cost than 16S but provides strain-level resolution and functional information |
| Metagenomic DNA Extraction Kits | Isolation of high-quality DNA from complex microbial communities [97] | Efficiency varies by sample type; critical for representative community analysis |
| Fluidigm Access Array | High-throughput qPCR platform for multiple targets across many samples [97] | Enables large-scale targeted quantification with minimal sample volume |
| Reference Strain Collections | Well-characterized microbial strains for method validation and controls [97] | Essential for quantification accuracy and cross-study comparisons |
Workflow Diagrams
Conclusion
The adoption of absolute abundance measurement is not merely a technical refinement but a fundamental shift essential for robust and reproducible microbiome science. This synthesis demonstrates that moving beyond relative data overcomes inherent compositional biases, reveals true biological effect sizes—such as distinguishing between an actual increase in a taxon versus a decrease in others—and provides a more accurate map of microbial ecology in health and disease. Methodologically, no single technique is universally superior; the choice between flow cytometry, spike-in standards, or digital PCR depends on the specific research question, sample type, and available resources. Looking forward, the integration of absolute quantification is poised to revolutionize clinical applications, from personalizing pharmaceutical treatments by accurately assessing their microbial impact to establishing definitive microbial biomarkers for disease. The future of microbiome research lies in embracing these quantitative frameworks, ensuring that our interpretations are grounded in biological reality rather than compositional illusion.
References
- 1. Microbiome Datasets Are Compositional: And This Is Not ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2017.02224/full]
- 2. Relative abundance data can misrepresent heritability of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10561453/]
- 3. a review of normalization and differential abundance analysis [https://www.nature.com/articles/s41522-020-00160-w]
- 4. A quantitative sequencing framework for absolute ... [https://www.nature.com/articles/s41467-020-16224-6]
- 5. Accurate prediction of absolute prokaryotic abundance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12146642/]
- 6. Troubleshooting Cell Culture Contamination: A Practical ... [https://www.yeasenbio.com/blogs/cell/troubleshooting-cell-culture-contamination-a-practical-solution-guide]
- 7. Absolute Abundance vs Relative Abundance in the ... [https://www.cd-genomics.com/resource-absolute-abundance-relative-abundance.html]
- 8. Microbiome differential abundance methods produce ... [https://www.nature.com/articles/s41467-022-28034-z]
- 9. A quantitative approach to measure and predict microbiome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11423569/]
- 10. Absolute abundance calculation enhances the significance ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1481197/full]
- 11. Absolute Abundance Microbiome Testing Services [https://cmbio.io/omics-sequencing-services/absolute-abundance]
- 12. A quantitative sequencing framework for absolute ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7244552/]
- 13. Assessment of absolute abundance in mother-infant gut ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12367464/]
- 14. Fecal microbial load is a major determinant of gut ... [https://www.sciencedirect.com/science/article/pii/S0092867424012042]
- 15. Tools for Analysis of the Microbiome - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7598837/]
- 16. MicrobiomeAnalyst [https://www.microbiomeanalyst.ca/]
- 17. A quantitative sequencing framework for absolute ... abundance [https://www.nature.com/articles/s41467-020-16224-6?error=cookies_not_supported&code=b8b85400-becf-4eb4-a436-e0f3807abe33]
- 18. Development of a quantitative PMA-16S rRNA gene sequencing... [https://environmentalmicrobiome.biomedcentral.com/articles/10.1186/s40793-025-00741-2]
- 19. Viability Differentiation Enhances Microbial ... [https://pubmed.ncbi.nlm.nih.gov/41211753/]
- 20. Flow Cytometry Total Cell Counts: A Field Study Assessing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3684093/]
- 21. A Flow Cytometry Method for Rapid Detection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC91970/]
- 22. Optimization of the flow cytometry method of detection, ... [https://www.sciencedirect.com/science/article/pii/S0308814624022568]
- 23. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOopOI6ItgZgpqJUw0xTd0D3l0BHQqfPk-1BWXAR4PeVw7JGcMnPs]
- 24. Microbiological Analysis Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/cell-analysis-support-center/microbiological-analysis-support/microbiological-analysis-support-troubleshooting.html]
- 25. How to Identify Bad Flow Cytometry Data – Bad Data Part 1 [https://voices.uchicago.edu/ucflow/2020/09/30/how-to-identify-bad-flow-cytometry-data-part-1/]
- 26. Identifying and fixing errors in flow data [https://www.colibri-cytometry.com/post/identifying-and-fixing-errors-in-flow-data]
- 27. synDNA—a Synthetic DNA Spike-in Method for Absolute ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9765022/]
- 28. Use of Recombinant Bacteria with Unique Tags as Spike-in ... [https://www.atcc.org/resources/application-notes/use-of-recombinant-bacteria-with-unique-tags-as-spike-in-controls-for-microbiome-studies]
- 29. Troubleshooting guidelines for CUT&RUN [https://support.epicypher.com/docs/troubleshooting-cut-and-run]
- 30. SNAP Spike-in Controls [https://www.epicypher.com/technologies/snap-spike-in-controls/]
- 31. Nucleic Acid Quantification Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/nucleic-acid-purification-analysis-support-center/nucleic-acid-quantification-support/nucleic-acid-quantification-support-troubleshooting.html]
- 32. Troubleshooting qPCR: Interpreting Amplification Curves [https://blog.biosearchtech.com/thebiosearchtechblog/bid/63622/qpcr-troubleshooting]
- 33. Troubleshooting Your qPCR Data and Amplification Curves [https://eu.idtdna.com/page/support-and-education/decoded-plus/a-quick-guide-for-troubleshooting-qpcr-data/]
- 34. Analytical techniques for nucleic acid and protein detection ... [https://www.nature.com/articles/s12276-025-01453-w]
- 35. Highly accurate and sensitive absolute quantification of ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-024-01881-2]
- 36. Digital PCR: from early developments to its future application ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00055f]
- 37. Recent advancements and emerging techniques in nucleic ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993625000408]
- 38. The accuracy of absolute differential abundance analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9302745/]
- 39. A Statistical Perspective on the Challenges in Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9667415/]
- 40. Field-of-view subsampling: A novel 'exotic marker' method for ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0320887]
- 41. A toolbox of machine learning software to support microbiome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10704913/]
- 42. NatureååˆŠï¼šæ•™ä½ é›¶åŸºç¡€å¼€å±•å¾®ç”Ÿç‰©ç»„æ•°æ®åˆ†æžå’Œå¯è§†åŒ–原创 [https://blog.csdn.net/qazplm12_3/article/details/141954155]
- 43. Predicting microbial community structure and temporal ... [https://www.nature.com/articles/s41467-025-64175-7]
- 44. Evaluation of variability in cell-free DNA extraction ... [https://www.nature.com/articles/s41598-025-06563-z]
- 45. Development of a quantitative PMA-16S rRNA gene ... [https://pubmed.ncbi.nlm.nih.gov/40597447/]
- 46. A Sample Extraction Method for Faster, More Sensitive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5707197/]
- 47. Evaluating the impact of sample storage, handling, and ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0270659]
- 48. Microbial Diversity Biased Estimation Caused by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10231250/]
- 49. Correcting for 16S rRNA gene copy numbers in microbiome ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-018-0420-9]
- 50. Accounting for 16S rRNA copy number prediction ... [https://www.nature.com/articles/s43705-023-00266-0]
- 51. Accounting for 16S rRNA copy number prediction ... [https://pubmed.ncbi.nlm.nih.gov/37301942/]
- 52. 16Stimator: statistical estimation of ribosomal gene copy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4796925/]
- 53. QMD: A new method to quantify microbial absolute ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10989753/]
- 54. Optimization of the 16S rRNA sequencing analysis pipeline ... [https://www.sciencedirect.com/science/article/pii/S2589004222001778]
- 55. Unveiling the impact of 16S rRNA gene intergenomic ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1573920/full]
- 56. Creating unity: linking 16S rRNA gene sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570451/]
- 57. Limit of Blank, Limit of Detection and Limit of Quantitation [https://pmc.ncbi.nlm.nih.gov/articles/PMC2556583/]
- 58. Current Applications of Absolute Bacterial Quantification in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8467167/]
- 59. The limits of detection (LOD) and quantification (LOQ) ... [https://www.mtc-usa.com/kb-article/aa-03949]
- 60. Limit of detection and Limit of quantification [https://www.chromforum.org/viewtopic.php?t=99414]
- 61. Troubleshooting & FAQs [https://www.cygnustechnologies.com/resources/guidelines?srsltid=AfmBOor4Xy3Wm3H_m9PhehYhI-M0J4qucll8UtMc6waKxiQVGlHQ6K6C]
- 62. Methods to determine limit of detection and ... [https://www.sciencedirect.com/science/article/pii/S2214753516300286]
- 63. Optimizing Small RNA-Seq with Spike-In Controls. [https://www.revvity.com/blog/optimizing-small-rna-seq-spike-controls]
- 64. ZymoBIOMICS Spike-in Control I (High Microbial Load) [https://www.zymoresearch.com/products/zymobiomics-spike-in-control-i-high-microbial-load?srsltid=AfmBOoorUihOFIwjhfK4KO9S5azkkX0WlB6p2-OPcR6Z2Yx4afsU30m8]
- 65. Synthetic spike-in standards for RNA-seq experiments - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3166838/]
- 66. Spike and Recovery and Linearity of Dilution Assessment [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/spike-recovery-linearity-assessment.html]
- 67. DspikeIn: Estimating Absolute Abundance from ... - bioc [https://bioc.r-universe.dev/DspikeIn]
- 68. How much PhiX spike in is recommended when ... [https://knowledge.illumina.com/instrumentation/general/instrumentation-general-reference_material-list/000001527]
- 69. Guidelines for preventing and reporting contamination in ... [https://www.nature.com/articles/s41564-025-02035-2]
- 70. Benefits and challenges of host depletion methods in ... [https://www.nature.com/articles/s41522-025-00762-2]
- 71. Sensitivity of shotgun metagenomics to host DNA [https://pmc.ncbi.nlm.nih.gov/articles/PMC7523627/]
- 72. Seven tips for avoiding DNA contamination in your PCR [https://www.takarabio.com/about/bioview-blog/tips-and-troubleshooting/avoid-dna-contamination-in-pcr?srsltid=AfmBOopHaIL7IUvsrYwxYz8IceKxgVtSJkFP8YE_sW_jyohMLIfrOm_u]
- 73. Host Cell DNA Quantification: Tips and Tricks [https://www.cygnustechnologies.com/host-cell-dna-quantification-tips-and-tricks?srsltid=AfmBOopiZnsFOWRTUB-XlqxntedB4WA5UH5bwdNYb98obOD0-55Va7vz]
- 74. Targeted decontamination of sequencing data with CLEAN [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288876/]
- 75. What should researchers know about host cell DNA ... [https://www.statnews.com/sponsor/2022/02/23/what-should-researchers-know-about-host-cell-dna-contamination-risks-in-cell-and-gene-therapies/]
- 76. Efficient Depletion of Host DNA Contamination in Malaria ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3592063/]
- 77. Fast quantification of gut bacterial species in cocultures ... [https://www.nature.com/articles/s43705-022-00123-6]
- 78. How to Count Our Microbes? The Effect of Different ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2020.00403/full]
- 79. The Power of Flow Cytometry in Biomarker-Driven Clinical ... [https://www.precisionformedicine.com/blog/harnessing-flow-cytometry-biomarker-driven-clinical-trials/]
- 80. A comparison of DNA methylation specific droplet digital ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4622657/]
- 81. Insights on Droplet Digital PCR–Based Cellular Kinetics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7925486/]
- 82. Application of Droplet Digital PCR Technology in Muscular ... [https://www.mdpi.com/1422-0067/23/9/4802]
- 83. Multi-kingdom microbiota dynamics in exposed saliva [https://pubmed.ncbi.nlm.nih.gov/41240731/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=0ylbq-RFuBigtUi7J-G4hkJERU9n9p7ydPX6iXUtWBX&fc=None&ff=20251120002007&v=2.18.0.post22+67771e2]
- 84. Absolute abundance calculation enhances the significance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11973300/]
- 85. Measure metagenomics accuracy with a simple 0-100 score [https://www.zymoresearch.com/blogs/blog/how-to-quantify-bias-with-mock-microbial-community-standards?srsltid=AfmBOopE5z15Ys5GPG5QPx1blVoDLreh4NEj9whiSL6R3sFQs2LGBwy_]
- 86. How To Choose A Microbiome Standard [https://blog.quartzy.com/how-to-choose-a-microbiome-standard]
- 87. Rigor and Reproducibility | Microbiome Core [https://www.med.unc.edu/microbiome/rigor-and-reproducibility/]
- 88. Characterization and Demonstration of Mock Communities as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8941912/]
- 89. Bacterial mock communities as standards for reproducible ... [http://flowrepository.org/id/FR-FCM-Z2CJ]
- 90. Group-wise normalization in differential abundance analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12308967/]
- 91. Quantifying the relative contributions of bacterial and ... [https://www.nature.com/articles/s41522-025-00842-3]
- 92. QuantStudioâ„¢ Absolute Qâ„¢ Digital PCR Starter Kit - FAQs [https://www.thermofisher.com/store/v3/products/faqs/A52732]
- 93. The Ultimate Guide to Clinical Trial Costs in 2025 [https://www.sofpromed.com/ultimate-guide-clinical-trial-costs]
- 94. Assessing scalability of an intervention: why, how and who? [https://pmc.ncbi.nlm.nih.gov/articles/PMC6788216/]
- 95. Tools for assessing the scalability of innovations in health [https://health-policy-systems.biomedcentral.com/articles/10.1186/s12961-022-00830-5]
- 96. Optimization of protocol design: a path to efficient, lower cost ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5137936/]
- 97. Services and Rates | Microbiome Core [https://www.med.unc.edu/microbiome/services-and-rates/]
- 98. High-Throughput Sequencing and Metagenomics [https://pmc.ncbi.nlm.nih.gov/articles/PMC3685257/]
- 99. Optimizations in High Throughput Process Development [https://www.halolabs.com/blog/high-throughput-process-development/]