16S rRNA vs. Metagenomics: A Researcher's Guide to Choosing the Right Microbiome Profiling Tool
This article provides a comprehensive comparison of 16S rRNA gene sequencing and shotgun metagenomics for researchers and drug development professionals.
16S rRNA vs. Metagenomics: A Researcher's Guide to Choosing the Right Microbiome Profiling Tool
Abstract
This article provides a comprehensive comparison of 16S rRNA gene sequencing and shotgun metagenomics for researchers and drug development professionals. It covers the foundational principles of each method, explores their specific applications and methodological workflows, and offers practical guidance for troubleshooting and optimizing study designs. A detailed validation and comparative analysis synthesizes empirical data on taxonomic resolution, functional insights, and cost-effectiveness, empowering scientists to make informed decisions for their specific biomedical and clinical research objectives.
Core Principles: Understanding 16S rRNA and Metagenomic Sequencing
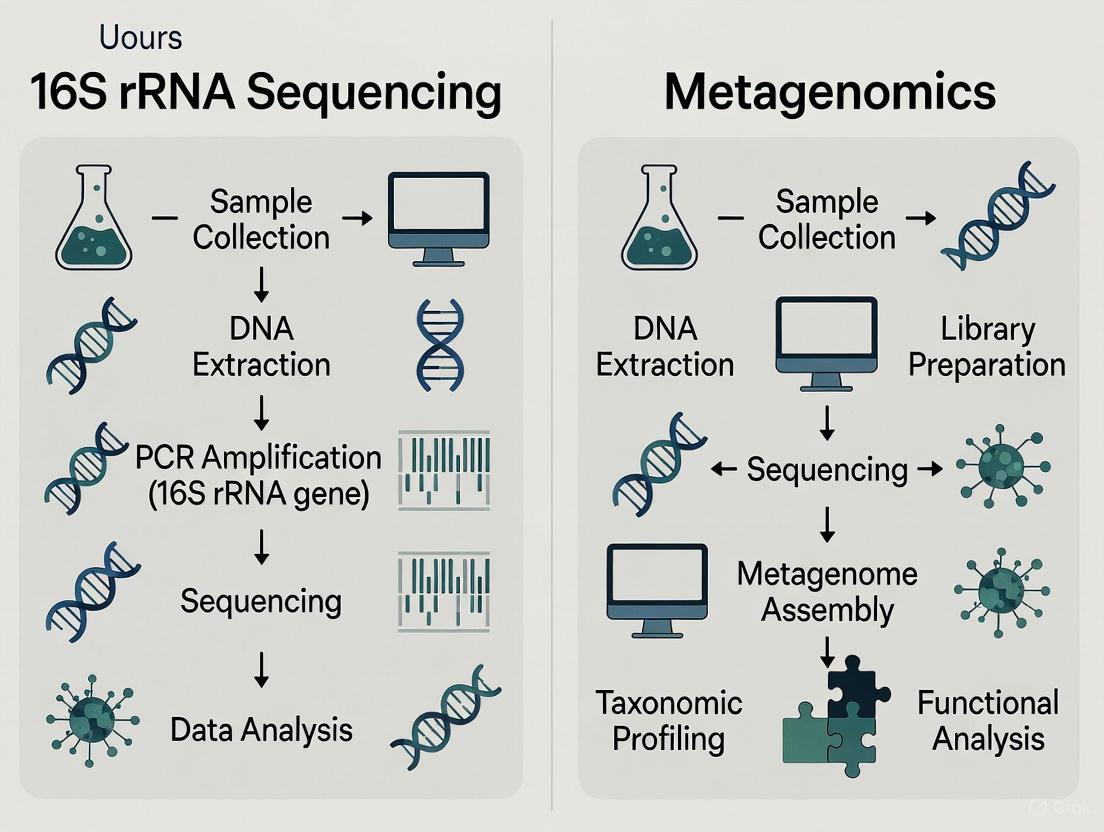
In the field of microbial genomics, two powerful sequencing techniques enable researchers to profile complex microbial communities: targeted amplicon sequencing and whole-genome shotgun metagenomic sequencing. The choice between these methods represents a critical decision point in research design, balancing factors such as cost, depth of information, and analytical resolution. Targeted amplicon sequencing, primarily focusing on the 16S rRNA gene for bacterial communities, uses polymerase chain reaction (PCR) to amplify specific genomic regions for taxonomic identification [1]. In contrast, shotgun metagenomic sequencing takes an untargeted approach by randomly fragmenting and sequencing all DNA present in a sample, enabling comprehensive taxonomic and functional analysis [2]. This guide provides an objective comparison of these techniques, supported by experimental data, to inform researchers and drug development professionals in selecting the appropriate method for their specific research context.
Technical Principles and Workflows
Targeted Amplicon Sequencing
Targeted amplicon sequencing employs PCR to amplify specific, taxonomically informative genomic regions before sequencing [1]. For bacterial identification, the 16S rRNA gene serves as the primary target due to its presence in all bacteria and archaea and its combination of highly conserved and variable regions [3]. The experimental workflow begins with DNA extraction, followed by PCR amplification using primers designed for specific hypervariable regions (V1-V9) of the 16S rRNA gene [1]. After purification and size selection, the amplified DNA fragments are sequenced using next-generation sequencing platforms [1]. Bioinformatic processing then clusters sequences into operational taxonomic units (OTUs) or amplicon sequence variants (ASVs) which are compared against reference databases such as SILVA or Greengenes for taxonomic classification [4].
Whole-Genome Shotgun Sequencing
Shotgun metagenomic sequencing fragments all DNA in a sample without target-specific amplification [2]. The workflow initiates with DNA extraction, followed by random fragmentation through mechanical shearing or enzymatic tagmentation [1]. After adapter ligation and library preparation, all DNA fragments undergo next-generation sequencing [2]. The resulting sequences are either assembled into contigs or aligned directly to reference databases for taxonomic assignment and functional annotation [5]. This method captures genomic information from all domains of life—bacteria, archaea, fungi, viruses, and eukaryotes—present in the sample [3]. Advanced bioinformatic pipelines such as MetaPhlAn and MEGAHIT enable species-level identification and functional pathway analysis [1].
Figure 1: Comparative workflows of targeted amplicon and whole-genome shotgun sequencing approaches.
Comparative Performance Analysis
Taxonomic Resolution and Coverage
The resolution and breadth of taxonomic identification differ substantially between these methods. Targeted 16S rRNA sequencing typically identifies microorganisms at the genus level, with limited capability for species-level discrimination [1] [3]. In contrast, shotgun metagenomic sequencing provides species-level resolution and can distinguish between strains in some cases [5] [1]. This enhanced resolution comes from shotgun sequencing's ability to access the entire genome rather than being restricted to a single gene region.
Regarding taxonomic coverage, 16S sequencing is limited to bacteria and archaea [1]. Shotgun metagenomics enables simultaneous detection of bacteria, fungi, viruses, and various other microorganisms from multiple kingdoms [2] [3]. Experimental data from a comparative study of gut microbiota demonstrated that shotgun sequencing detects a broader range of taxa, with 16S sequencing identifying only part of the microbial community revealed by shotgun methods [6].
Table 1: Taxonomic Profiling Capabilities of Sequencing Techniques
| Parameter | 16S Amplicon Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Primary Target | 16S rRNA gene (bacteria/archaea) | All genomic DNA in sample |
| Taxonomic Resolution | Genus level (sometimes species) | Species level (sometimes strains) |
| Kingdom Coverage | Bacteria and Archaea only | Bacteria, Archaea, Fungi, Viruses, Eukaryotes |
| Detection of Novel Taxa | Limited by primer specificity | Enhanced through assembly-based approaches |
| Reference Database | SILVA, Greengenes, RDP | NCBI RefSeq, GTDB, UHGG |
Functional Profiling Capabilities
A fundamental distinction between these methods lies in their capacity for functional analysis. 16S rRNA sequencing provides only taxonomic information, with functional potential inferred indirectly using tools like PICRUSt that predict gene functions based on taxonomic assignments [1]. These predictions are limited to known gene functions associated with reference taxa.
Shotgun metagenomics directly characterizes functional genes and metabolic pathways by sequencing all genomic material [5]. This enables comprehensive analysis of biological processes, including identification of antimicrobial resistance genes, carbohydrate-active enzymes, and virulence factors [7]. For example, a metagenomic study of goat gastrointestinal microbiota successfully annotated 6,095,352 predicted genes against KEGG and CAZy databases to understand functional potential and antimicrobial resistance traits [7].
Sensitivity and Detection Limitations
The sensitivity of each method varies significantly based on sample type and microbial abundance. In wastewater surveillance studies, untargeted shotgun sequencing without enrichment failed to generate sufficient genome coverage of human pathogenic viruses for robust genomic epidemiology, with samples dominated by bacterial sequences [8]. However, targeted hybrid-capture enrichment of shotgun libraries significantly increased genome coverage for respiratory viruses, making it viable for simultaneous genomic epidemiology of multiple viral pathogens [8].
16S rRNA sequencing demonstrates greater sensitivity in samples with high host DNA contamination, as the PCR amplification step specifically enriches for bacterial DNA [1]. Shotgun metagenomics is more susceptible to host DNA interference, particularly in samples with low microbial biomass, where host DNA can dominate sequencing output and reduce detection sensitivity for microbial taxa [3].
Table 2: Quantitative Performance Comparison from Experimental Studies
| Performance Metric | 16S Amplicon Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Typical Sequencing Depth | ~50,000 reads/sample [9] | Millions of reads/sample [8] |
| Alpha Diversity | Lower observed diversity [4] | Higher observed diversity [6] |
| Cost Per Sample | ~$50 USD [1] | Starting at ~$150 USD [1] |
| Host DNA Interference | Low (PCR enriches targets) [3] | High (requires mitigation strategies) [3] |
| Detection of Rare Taxa | Limited by amplification bias | Enhanced with sufficient sequencing depth |
Experimental Design Considerations
Sample Type and Preparation
The appropriate sequencing method depends heavily on sample characteristics. 16S rRNA sequencing performs well with samples containing low microbial biomass or high host DNA content, such as skin swabs, tissue biopsies, or environmental swabs [3]. The PCR amplification step enriches target sequences despite potential host DNA contamination.
Shotgun metagenomics is ideally suited for samples with high microbial biomass and low host DNA content, such as stool samples [3]. In samples with significant host contamination, DNA removal techniques or increased sequencing depth may be necessary to achieve sufficient microbial coverage [1]. For example, in a study comparing both techniques on human stool samples, shotgun sequencing required careful host DNA filtration using Bowtie2 against the human genome GRCh38 to improve microbial detection [4].
Sequencing Depth Requirements
The optimal sequencing depth varies between methods and sample types. 16S rRNA sequencing typically requires approximately 50,000 reads per sample to maximize identification of rare taxa [9]. Shotgun metagenomics demands significantly greater sequencing depth—millions of reads per sample—to adequately cover the diverse genomic content [8] [9].
Research on pediatric gut microbiomes has demonstrated that shallower shotgun sequencing depths may be sufficient for children under 30 months due to their lower gut microbial diversity compared to adults [9]. This "shallow shotgun" approach bridges the cost gap while providing >97% of the compositional and functional data obtained through deep sequencing [1].
Bioinformatics and Data Analysis
The complexity of bioinformatic analysis differs substantially between methods. 16S rRNA sequencing data typically undergoes processing through established pipelines such as QIIME2, MOTHUR, or DADA2 [1] [4]. These pipelines perform quality filtering, denoising, chimera removal, and taxonomic classification using reference databases [4].
Shotgun metagenomic data requires more complex bioinformatics approaches, involving either assembly-based methods (MEGAHIT) or read-based alignment (MetaPhlAn) [1]. The computational resources, expertise, and time investment are significantly greater for shotgun data analysis. Furthermore, shotgun taxonomic profiling depends heavily on reference databases, which can present challenges for identifying novel microbes without computationally expensive assembly [9].
Application-Based Method Selection
Comparative Experimental Evidence
Direct comparisons between these sequencing techniques reveal important performance differences. A 2021 study comparing 16S and shotgun sequencing for chicken gut microbiota characterization found that shotgun sequencing identified a statistically significant higher number of taxa when sufficient reads were available, corresponding to less abundant genera [6]. Similarly, a 2024 study on colorectal cancer microbiota found that 16S detects only part of the gut microbiota community revealed by shotgun sequencing, with 16S abundance data being sparser and exhibiting lower alpha diversity [4].
In wastewater surveillance, untargeted shotgun sequencing was unsuitable for genomic monitoring of low virus concentrations, while targeted hybrid-capture enrichment of shotgun libraries enabled simultaneous genomic epidemiology of multiple viral pathogens [8]. Tiled-PCR amplification (a targeted approach) provided optimal genome coverage for individual viruses with minimum sequencing depth [8].
Figure 2: Decision framework for selecting appropriate sequencing method based on research requirements.
Recommended Applications
16S rRNA Sequencing Applications: Large-scale microbiome studies with numerous samples [5], initial exploratory studies of bacterial communities [3], projects with limited budget or bioinformatics capabilities [1], and samples with high host DNA contamination or low microbial biomass [3].
Shotgun Metagenomic Sequencing Applications: Studies requiring species or strain-level resolution [5] [1], research investigating functional gene content or metabolic pathways [5], investigations spanning multiple microbial kingdoms [2], and deeply characterized sample sets where comprehensive genomic analysis is prioritized over sample number [5].
The Researcher's Toolkit: Essential Reagents and Materials
Table 3: Essential Research Reagents and Materials for Sequencing Approaches
| Reagent/Material | Function | Application in 16S | Application in Shotgun |
|---|---|---|---|
| DNA Extraction Kit (e.g., NucleoSpin Soil Kit, DNeasy PowerLyzer) | Isolation of high-quality genomic DNA from samples | Required [4] | Required [4] |
| PCR Primers (e.g., 16S V3-V4 primers) | Amplification of target regions | Essential for target enrichment [1] | Not typically used |
| Tagmentation Enzymes | Simultaneous fragmentation and adapter tagging | Not used | Essential for library prep [1] |
| Size Selection Beads | Selection of appropriately sized DNA fragments | Recommended [1] | Critical for library quality |
| Library Prep Kit (e.g., TruSeq Nano DNA LT, VAHTS Universal Plus) | Preparation of sequencing libraries | Required [7] | Required [7] |
| Sequenceing Platform (e.g., Illumina MiSeq/NovaSeq) | High-throughput sequencing | Standard implementation [7] | Standard implementation [7] |
| Bioinformatics Tools (e.g., QIIME2, MEGAHIT, MetaPhlAn) | Data processing and analysis | Essential [1] [4] | Essential [1] |
| Methyl clerodermate | Clerodermic Acid Methyl Ester | Clerodermic acid methyl ester is a natural diterpene for research. This product is For Research Use Only, not for human or veterinary use. | Bench Chemicals |
| Diacetamide | Diacetamide | High Purity Reagent for Research | High-purity Diacetamide for organic synthesis & biochemical research. For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. | Bench Chemicals |
Targeted amplicon and whole-genome shotgun sequencing offer complementary approaches for microbial community analysis, each with distinct advantages and limitations. 16S rRNA sequencing provides a cost-effective method for comprehensive taxonomic profiling of bacterial and archaeal communities, particularly valuable for large-scale studies and samples with challenging composition. Shotgun metagenomics delivers superior taxonomic resolution and direct functional insights across all microbial kingdoms, albeit at higher cost and computational requirements. The decision between these techniques should be guided by research objectives, sample characteristics, available resources, and analytical requirements. As sequencing technologies continue to advance and costs decrease, hybrid approaches such as shallow shotgun sequencing and targeted enrichment are emerging to bridge the gap between these powerful methodological paradigms.
In the field of microbial genomics, two powerful methods have emerged as cornerstones for profiling microbial populations: 16S ribosomal RNA (rRNA) gene sequencing and shotgun metagenomic sequencing [10]. Both techniques allow researchers to identify microorganisms from complex samples without the need for culturing, but they are founded on different principles and offer distinct insights. The 16S rRNA gene serves as a universal genetic barcode for bacteria and archaea, enabling targeted identification through its variable regions. In contrast, shotgun metagenomics takes a comprehensive approach by sequencing all the DNA in a sample [10]. This guide provides an objective comparison of these methodologies, supported by experimental data, to help researchers select the optimal approach for their specific scientific questions.
Core Methodologies and Workflows
16S rRNA Gene Sequencing: A Targeted Approach
The 16S rRNA gene is a conserved genetic marker approximately 1500 base pairs long, containing nine hypervariable regions (V1-V9) interspersed between conserved areas [11]. These variable regions provide the phylogenetic resolution necessary for taxonomic classification, while the conserved regions enable the design of universal PCR primers [10].
Key Experimental Protocol:
- DNA Extraction: Genomic DNA is isolated from the sample.
- Primer Selection & PCR Amplification: Universal primers target conserved areas surrounding specific variable regions (e.g., V3-V4 or V4 alone). The polymerase chain reaction (PCR) amplifies these target regions [10] [12].
- Library Preparation: Amplified DNA (amplicons) is cleaned and adapters are added for sequencing [10].
- Sequencing: High-throughput sequencers, such as those from Illumina or Oxford Nanopore Technologies (ONT), read the amplicons [13] [12].
- Bioinformatics Analysis: Tools like QIIME2 or MOTHUR process the reads, cluster them into Operational Taxonomic Units (OTUs) or Amplicon Sequence Variants (ASVs), and compare them to reference databases (e.g., SILVA, Greengenes) for taxonomic identification [10] [14].
Shotgun Metagenomic Sequencing: A Comprehensive View
Shotgun metagenomics bypasses targeted amplification and instead sequences all genomic DNA present in a sample, including from bacteria, archaea, viruses, and fungi [10].
Key Experimental Protocol:
- DNA Extraction: Total genomic DNA is extracted from the sample, ideally as high-molecular-weight DNA to improve assembly quality [15].
- Library Preparation: DNA is randomly fragmented, and sequencing adapters are ligated to the fragments [10].
- Sequencing: High-throughput shotgun sequencing is performed using short-read (e.g., Illumina) or long-read (e.g., ONT) platforms [12] [15].
- Assembly and Annotation: Sequencing reads are assembled into longer contigs, often binned into Metagenome-Assembled Genomes (MAGs). Functional annotation is performed using tools like HUMAnN or MG-RAST and databases such as KEGG and CARD [10] [15].
The following workflow diagram illustrates the key steps and differences between these two approaches.
Performance Comparison: Key Differences
The choice between 16S rRNA sequencing and metagenomics has significant implications for taxonomic resolution, functional insight, cost, and data complexity. The table below summarizes the core differences.
| Feature | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Taxonomic Resolution | Genus level, sometimes species [10] | Species and strain level [10] |
| Functional Insights | Limited to taxonomic inference [10] | Reveals metabolic pathways, AMR genes, and virulence factors [10] [16] |
| Organisms Detected | Primarily bacteria and archaea [10] | All domains: bacteria, archaea, viruses, fungi [10] |
| Amplification Bias | High (PCR introduces biases) [10] [14] | Low (no targeted PCR) [10] |
| Cost & Data Volume | Lower cost, smaller datasets [10] | Higher cost, very large datasets [10] |
| Bioinformatics Complexity | Less complex, user-friendly pipelines [10] | More complex, requires robust computational infrastructure [10] |
| Primary Application | Microbial diversity surveys, large-scale screening [10] | Functional potential analysis, pathogen discovery [10] [17] |
Supporting Experimental Data
Clinical Diagnostic Performance
A 2025 study compared Sanger sequencing with Next-Generation Sequencing (NGS) of the 16S rRNA gene for diagnosing infections in culture-negative clinical samples [13].
- Experimental Protocol: 101 clinical samples positive in 16S rRNA PCR were subjected to both Sanger and Oxford Nanopore Technologies (ONT) sequencing. ONT data were processed using the EPI2ME Fastq 16S workflow [13].
- Key Results:
- The positivity rate for clinically relevant pathogens was 72% for ONT versus 59% for Sanger sequencing.
- ONT detected more samples with polymicrobial presence (13 vs. 5).
- In one case, ONT identified the rare pathogen Borrelia bissettiiae in a joint fluid sample, which was missed by Sanger sequencing [13].
This demonstrates that NGS-based 16S sequencing can improve detection of both monobacterial and polymicrobial infections in a clinical setting.
Primer and Platform Selection Impact
A 2025 benchmarking study on mouse gut microbiota highlighted how methodological choices influence 16S rRNA sequencing results [12].
- Experimental Protocol: Mouse fecal samples were analyzed using different 16S rRNA primer combinations and sequencing platforms (Illumina and ONT). Metagenome sequencing (MS) was also performed for comparison [12].
- Key Results:
- Primer selection critically influenced results, with different combinations detecting unique taxa.
- ONT 16S sequencing captured a broader range of taxa compared to Illumina 16S.
- Metagenome sequencing on both Illumina and ONT platforms showed a high correlation and provided superior taxonomic resolution than 16S sequencing [12].
Algorithm Performance in 16S Analysis
The accuracy of 16S rRNA analysis is also affected by the bioinformatic algorithms used to process the data. A 2025 benchmarking study evaluated eight different clustering (OTU) and denoising (ASV) algorithms using a complex mock microbial community [14].
- Experimental Protocol: The performance of algorithms including DADA2, Deblur, UPARSE, and MED was tested on the most complex mock community available (227 bacterial strains). Metrics included error rates, over-splitting, over-merging, and resemblance to the expected community [14].
- Key Results:
- ASV algorithms (e.g., DADA2) produced a consistent output but suffered from over-splitting a single biological sequence into multiple variants.
- OTU algorithms (e.g., UPARSE) achieved clusters with lower error rates but with more over-merging of distinct sequences.
- UPARSE and DADA2 showed the closest resemblance to the intended microbial community structure [14].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of a microbial community study requires careful selection of reagents and materials. The following table details key solutions used in the featured experiments.
| Item | Function | Example Use-Case |
|---|---|---|
| Universal 16S Primers | Amplify target variable regions (e.g., V3-V4) for sequencing [10] [11] | Microbial diversity profiling in gut or environmental samples [12] |
| High-Fidelity DNA Polymerase | Perform accurate PCR amplification with minimal error introduction [13] | Preparation of 16S amplicon libraries for NGS [13] |
| Metagenomic DNA Extraction Kits | Isolate high-quality, high-molecular-weight DNA from complex samples [15] | Shotgun metagenomic library preparation for assembly of MAGs [15] |
| SILVA / Greengenes Database | Reference databases for taxonomic classification of 16S rRNA sequences [10] | Assigning taxonomy to OTUs or ASVs in bioinformatics pipelines [14] |
| KEGG / CARD Database | Databases for functional annotation of genes [10] [16] | Predicting metabolic pathways and antimicrobial resistance genes from metagenomic data [10] [17] |
| Bioinformatics Pipelines (QIIME2, MG-RAST) | Integrated tools for processing, analyzing, and interpreting sequencing data [10] | From raw sequencing reads to statistical analysis and visualization [10] [14] |
| Fodipir | Fodipir (MnDPDP) | Research Grade | Supplier | Fodipir (MnDPDP) is a manganese-based MRI contrast agent for research. For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
| 2,6-Dimethoxyphenol | 2,6-Dimethoxyphenol | High-Purity Reagent | RUO | High-purity 2,6-Dimethoxyphenol for lignin & polymer research. For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
The choice between 16S rRNA sequencing and shotgun metagenomics is not a matter of one being superior to the other, but rather which is best suited to the specific research goals and constraints.
- Choose 16S rRNA sequencing when your primary interest is in comparing microbial diversity and composition across a large number of samples in a cost-effective manner, and when taxonomic identification at the genus level is sufficient [10] [12]. It is an excellent tool for initial exploratory studies and large-scale screening.
- Choose shotgun metagenomics when your research question requires species- or strain-level resolution, or when you need to investigate the functional potential of the community, such as uncovering metabolic pathways, antimicrobial resistance genes, or virulence factors [10] [16] [17]. This method is indispensable for linking taxonomy to function.
As sequencing technologies and bioinformatics tools continue to advance, hybrid approaches that leverage the strengths of both methods are becoming more feasible. For instance, using 16S sequencing for broad surveillance and following up with metagenomics on key samples of interest can provide a powerful, comprehensive strategy for understanding complex microbial ecosystems [12].
In the captivating world of microbiology, researchers have two primary methods to study the composition, structure, and function of microbial communities: 16S ribosomal RNA (rRNA) gene sequencing and shotgun metagenomics [3]. While 16S rRNA sequencing employs a targeted approach to amplify and sequence a specific phylogenetic marker gene, shotgun metagenomics takes an untargeted approach by sequencing all the genetic material in a sample randomly [3] [1]. This fundamental difference in methodology creates a significant divergence in the type and scope of data generated, making each technique uniquely suited for specific research applications. The choice between these methods carries substantial implications for taxonomic resolution, functional insight, and overall experimental design, particularly for researchers in drug development and pharmaceutical sciences who require precise, actionable data from microbial communities. This guide provides a comprehensive, evidence-based comparison to inform these critical methodological decisions.
Fundamental Methodological Differences
The core distinction between these techniques lies in their basic approach to genetic analysis. 16S rRNA gene sequencing is a form of amplicon sequencing that targets, amplifies, and sequences specific hypervariable regions (V1-V9) of the 16S rRNA gene, which is found in all Bacteria and Archaea [1]. This process involves PCR amplification using primers designed for conserved regions flanking these variable areas, followed by sequencing of the amplified products [3]. The variation within these sequenced regions then allows for taxonomic differentiation between microbial organisms.
In contrast, shotgun metagenomic sequencing takes a comprehensive approach by fragmenting all DNA in a sample into numerous small pieces—akin to a shotgun blast—sequencing these fragments randomly, and then using bioinformatics to reconstruct the genetic content [1]. This method sequences the entire genomic content without targeting specific genes, enabling identification of all microorganisms—bacteria, fungi, viruses, and protists—simultaneously, while also providing direct access to the functional gene repertoire of the community [3] [18].
Figure 1: Comparative Workflows of 16S rRNA Sequencing and Shotgun Metagenomics. The 16S pathway (blue) is a targeted approach focusing on a specific marker gene, while shotgun metagenomics (red) comprehensively sequences all DNA, enabling more detailed functional and taxonomic analysis including Metagenome-Assembled Genomes (MAGs).
Technical Comparison: Capabilities and Limitations
The methodological differences between 16S rRNA sequencing and shotgun metagenomics translate directly into distinct technical capabilities, limitations, and appropriate applications for each approach.
Table 1: Technical Comparison of 16S rRNA Sequencing and Shotgun Metagenomics
| Feature | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Taxonomic Resolution | Family & Genus level (species level possible but with high false-positive rate) [3] | Species and strain-level resolution of multi-kingdom taxa [3] |
| Functional Profiling | Indirect inference only (e.g., PICRUSt); not direct functional data [3] | Direct characterization of functional genes and pathways [3] [18] |
| Taxonomic Coverage | Bacteria and Archaea only [3] | Bacteria, Fungi, Viruses, Protists (multi-kingdom) [3] [1] |
| Host DNA Interference | Low (PCR targets specific gene) [3] | High (requires mitigation strategies) [3] |
| Cost per Sample | ~$50 USD [1] | Starting at ~$150 (varies with depth) [1] |
| Minimum DNA Input | Low (successful with <1 ng DNA) [3] | Higher (typically minimum 1ng/μL) [3] |
| Bioinformatics Complexity | Beginner to intermediate [1] | Intermediate to advanced [1] |
| Recommended Sample Type | All types, especially low microbial biomass/high host DNA [3] | All types, best with high microbial biomass (e.g., stool) [3] |
Key Advantages and Trade-offs
16S rRNA sequencing offers significant cost advantages, particularly for large-scale studies where budget constraints are paramount [1]. Its lower sensitivity to host DNA contamination makes it particularly suitable for samples with low microbial biomass or high host DNA content, such as skin swabs, tissue biopsies, or blood samples [3]. The simpler bioinformatics pipeline and established, well-curated databases also make 16S sequencing more accessible to researchers without extensive computational expertise or resources [1].
Shotgun metagenomics provides superior taxonomic resolution, enabling discrimination not just at the species level but often at the strain level, which can be critical for understanding functional differences between closely related organisms [3] [18]. The ability to simultaneously profile multiple microbial kingdoms (bacteria, fungi, viruses, protists) without methodological adjustments offers a truly comprehensive view of microbial communities [3]. Most significantly, shotgun sequencing directly reveals the functional potential of microbial communities by cataloging metabolic pathways, virulence factors, and antibiotic resistance genes, moving beyond mere taxonomic census to functional understanding [18] [1].
A recent innovation in the field, shallow shotgun sequencing, has emerged as a cost-optimized intermediate approach. By sequencing at lower depths, this method reduces costs to levels comparable with 16S sequencing while still providing species-level resolution and multi-kingdom coverage, though with less robust data than deep shotgun sequencing [3].
Experimental Evidence and Performance Validation
Detection Sensitivity and Taxonomic Resolution
Comparative studies consistently demonstrate the enhanced detection capability of shotgun metagenomics. In a 2021 study comparing both methods on chicken gut microbiota, shotgun sequencing identified a statistically significant higher number of taxa compared to 16S sequencing when sufficient read depth was available (>500,000 reads) [6]. The less abundant genera detected exclusively by shotgun sequencing were biologically meaningful and able to discriminate between experimental conditions as effectively as the more abundant genera detected by both methods [6].
In clinical diagnostics, a 2025 study of 101 culture-negative samples found that Next-Generation Sequencing (NGS) of the 16S rRNA gene using Oxford Nanopore Technologies (ONT) showed a higher positivity rate for clinically relevant pathogens compared to Sanger sequencing (72% vs. 59%) [13]. Importantly, ONT detected more samples with polymicrobial presence compared to Sanger sequencing (13 vs. 5), and identified pathogens like Borrelia bissettiiae in a joint fluid sample that was missed by Sanger sequencing [13]. This demonstrates the improved detection of both monobacterial and multiple bacterial species with modern sequencing approaches.
Table 2: Experimental Performance Comparison from Clinical Studies
| Performance Metric | 16S Sanger Sequencing | 16S NGS (ONT) | Shotgun Metagenomics |
|---|---|---|---|
| Positivity Rate (Clinical Samples) | 59% [13] | 72% [13] | Complementary role [19] |
| Polymicrobial Detection | 5/101 samples [13] | 13/101 samples [13] | Superior for complex communities [18] |
| Species-Level Resolution | Limited [3] | Improved with full-length sequencing [20] | High resolution [3] [18] |
| Functional Insight | Not available | Not available | Comprehensive [18] [1] |
| Diagnostic Concordance | 80% with NGS methods [13] | Reference standard emerging | 70% sensitive vs. 16S [19] |
Methodological Protocols
16S rRNA Gene Sequencing Protocol (based on Illumina platform):
- DNA Extraction: Use commercial kits suitable for sample type (soil, stool, tissue)
- PCR Amplification: Target hypervariable regions (e.g., V3-V4) with barcoded primers
- Library Preparation: Clean up amplified DNA, size select, and pool samples in equal proportions
- Sequencing: Illumina MiSeq or HiSeq platforms (2×250 bp or 2×300 bp)
- Bioinformatics: QIIME2, MOTHUR, or USEARCH-UPARSE for OTU/ASV picking, taxonomy assignment with SILVA or Greengenes databases [1] [21]
Shotgun Metagenomic Sequencing Protocol:
- DNA Extraction: Use kits that maximize yield and minimize bias (critical for functional analysis)
- Library Preparation: Fragmentation and adapter ligation (tagmentation) using kits like Illumina Nextera
- Sequencing: Illumina NextSeq, HiSeq, or NovaSeq platforms (2×150 bp typical)
- Bioinformatics:
Genome-Resolved Metagenomics, an advanced shotgun approach, involves reconstructing microbial genomes directly from whole-metagenome sequencing data through a process of assembly and binning [18]. This enables researchers to study "microbial dark matter"—uncultured species that have not been previously characterized—by creating Metagenome-Assembled Genomes (MAGs) that provide insights into the genetic makeup and functional capabilities of novel microorganisms [18].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Computational Tools for Metagenomic Analysis
| Category | Item/Reagent | Function/Application |
|---|---|---|
| Wet Lab Materials | ZymoBIOMICS Gut Microbiome Standard (D6331) | Mock community control for method validation [22] |
| Quick-DNA Fecal/Soil Microbe Microprep Kit | DNA extraction optimized for difficult samples [20] | |
| Micro-Dx Kit with SelectNA plus | Selective lysis for human DNA depletion in clinical samples [13] | |
| SMRTbell Prep Kit 3.0 | Library preparation for PacBio long-read sequencing [20] | |
| Sequencing Platforms | Illumina MiSeq/NovaSeq | Short-read sequencing (16S and shotgun) [1] |
| Oxford Nanopore Technologies (ONT) | Long-read sequencing (full-length 16S and metagenomics) [13] [20] | |
| PacBio Sequel IIe | High-fidelity long-read sequencing [20] | |
| Bioinformatics Tools | QIIME2, MOTHUR | 16S rRNA data analysis pipelines [1] [21] |
| MetaPhlAn, HUMAnN | Taxonomic and functional profiling of shotgun data [18] [1] | |
| metaSPAdes, MEGAHIT | Metagenome assembly from sequencing reads [18] | |
| CheckM2, MaxBin | MAG quality assessment and binning [18] | |
| MetaDAVis, MicrobiomeAnalyst | Interactive data analysis and visualization [21] | |
| Antho-rwamide II | Antho-rwamide II | Neuropeptide Research Compound | RUO | Antho-rwamide II is a bioactive sea anemone neuropeptide for neuroscience research, modulating ion channels. For Research Use Only. |
| 1-Benzoylpiperazine | 1-Benzoylpiperazine, CAS:13754-38-6, MF:C11H14N2O, MW:190.24 g/mol | Chemical Reagent |
The choice between 16S rRNA sequencing and shotgun metagenomics depends fundamentally on research goals, sample type, and available resources. 16S rRNA sequencing remains a powerful, cost-effective tool for large-scale bacterial profiling studies, particularly when targeting specific bacterial questions or working with samples containing high host DNA or low microbial biomass. Its established protocols and simpler analysis pipelines make it accessible for researchers entering microbiome studies or those with limited bioinformatics support.
Shotgun metagenomics provides a comprehensive solution for studies requiring species- or strain-level resolution, multi-kingdom taxonomic profiling, or direct assessment of functional potential. The ability to reconstruct MAGs and explore functional gene content makes it particularly valuable for drug discovery efforts aimed at identifying novel microbial therapeutic targets, understanding mechanisms of action, or discovering bioactive metabolites.
For researchers and drug development professionals, the emerging paradigm suggests a complementary approach: using 16S sequencing for initial large-scale screening to identify candidate samples of interest, followed by deep shotgun metagenomics for detailed functional characterization of selected samples. This tiered strategy maximizes both statistical power and mechanistic insight while optimizing resource allocation. As sequencing costs continue to decline and analytical methods become more sophisticated, shotgun metagenomics is increasingly becoming the gold standard for comprehensive microbiome analysis, though 16S sequencing maintains its important role in the researcher's molecular toolkit for specific applications.
Key Historical Developments and the Evolution of Culture-Independent Microbiology
The field of microbiology has undergone a profound transformation, shifting from a reliance on culture-dependent techniques to the adoption of sophisticated, culture-independent sequencing methods. For over a century, the isolation and growth of microorganisms in pure culture on artificial media represented the cornerstone of microbial investigation. While foundational, this approach was intrinsically biased, as it could only detect a small fraction of microbial life—estimates suggest less than 2% of environmental bacteria are readily cultivable. This limitation obscured the true diversity and functional capacity of microbial communities, including those associated with the human body.
The development of culture-independent methods, primarily through the sequencing of genetic material directly from samples, has revolutionized the field. This paradigm shift began with the targeted sequencing of the 16S ribosomal RNA (rRNA) gene and has rapidly advanced towards comprehensive shotgun metagenomic sequencing and genome-resolved metagenomics. These technologies have enabled researchers to bypass the cultivation bottleneck, offering an unbiased view of microbial taxonomy, diversity, and function. This guide objectively compares the two most prevalent sequencing strategies—16S rRNA sequencing and shotgun metagenomics—within the context of their historical development, providing supporting experimental data and methodologies to inform researchers and drug development professionals.
Key Historical Developments in Sequencing Technologies
The evolution of culture-independent microbiology is inextricably linked to advancements in DNA sequencing technology. The journey began in the 1970s with the pioneering work of Carl Woese and George Fox, who utilized 16S rRNA sequencing to establish the phylogenetic structure of the prokaryotic world, ultimately leading to the discovery of the third domain of life, Archaea. This established the 16S rRNA gene as a powerful molecular chronometer due to its universal distribution in bacteria and archaea, its essential functional role, and the presence of both highly conserved and variable regions.
The subsequent development of the Polymerase Chain Reaction (PCR) in the 1980s, coupled with Sanger sequencing, allowed for the first detailed, sequence-based explorations of microbial communities. However, the true revolution began in the mid-2000s with the advent of Next-Generation Sequencing (NGS) platforms. NGS provided the high-throughput, cost-effective scalability required to deeply and broadly sample complex microbial ecosystems.
The Human Microbiome Project (HMP), launched in 2007, was a pivotal milestone that standardized and popularized these approaches for studying the human-associated microbiota. While the first phase of the HMP utilized both 16S and whole-metagenome sequencing (WMS), it highlighted the limitations of 16S data and catalyzed a shift towards WMS in its second phase to enable functional insights [18]. This trajectory continues today with the rise of genome-resolved metagenomics, a technique that reconstructs individual microbial genomes directly from metagenomic data, moving beyond community-level profiling to strain-level resolution and the exploration of "microbial dark matter" [18].
Table: Major Milestones in Culture-Independent Microbiology
| Time Period | Key Technological Development | Impact on Microbiology |
|---|---|---|
| 1970s | 16S rRNA Gene Sequencing (Sanger) | Enabled phylogenetic studies; discovery of Archaea. |
| 1980s | Polymerase Chain Reaction (PCR) | Allowed targeted amplification of 16S genes from complex samples. |
| Mid-2000s | Next-Generation Sequencing (NGS) | Enabled high-throughput, deep profiling of microbial communities. |
| 2007-2012 | Human Microbiome Project (Phase 1) | Catalyzed large-scale, standardized study of human microbiota using 16S and WMS. |
| 2013-Present | Advanced Shotgun Metagenomics & Genome-Resolved Metagenomics | Provided strain-level resolution and functional profiling; reconstruction of Metagenome-Assembled Genomes (MAGs). |
Comparative Analysis: 16S rRNA Sequencing vs. Shotgun Metagenomics
The choice between 16S rRNA sequencing and shotgun metagenomic sequencing is fundamental to experimental design, with each method offering distinct advantages and trade-offs regarding taxonomic resolution, functional insight, and cost.
Methodological Principles and Workflows
The core difference lies in their scope and approach. 16S rRNA sequencing is an amplicon-based method that uses PCR to amplify one or more hypervariable regions (V1-V9) of the bacterial and archaeal 16S rRNA gene prior to sequencing [1]. In contrast, shotgun metagenomic sequencing is a whole-genome approach that involves fragmenting all DNA in a sample—microbial and host—into short fragments that are sequenced randomly [6] [1]. The resulting reads are then computationally assembled and mapped to reference databases.
Performance and Experimental Data Comparison
Direct comparisons of these methods reveal critical differences in their performance. A 2021 study in Scientific Reports directly compared the two strategies using chicken gut microbiota samples. The research demonstrated that when sufficient sequencing depth is achieved (>500,000 reads), shotgun sequencing identified a statistically significant higher number of taxa, particularly among less abundant genera that were missed by 16S sequencing [6]. Furthermore, in differential abundance analysis, shotgun sequencing detected 152 significant changes in genera abundance between gut compartments that 16S sequencing failed to identify, whereas 16S found only 4 changes missed by shotgun sequencing [6].
Conversely, a 2024 clinical study on body fluid samples found that 16S rDNA Sanger sequencing remained a valuable diagnostic tool. In this setting, clinical metagenomics (CMg) using shotgun sequencing had a sensitivity of 70.1% compared to 16S sequencing, suggesting it may be best positioned as a complementary, rather than replacement, technique in certain clinical diagnostics [23].
Table: Comparative Analysis of 16S rRNA and Shotgun Metagenomic Sequencing
| Factor | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Principle | Targeted amplicon sequencing of the 16S gene [1]. | Whole-genome, untargeted sequencing of all DNA [1]. |
| Taxonomic Resolution | Genus-level (sometimes species) [1] [24]. | Species-level and strain-level (with sufficient depth) [1] [24]. |
| Taxonomic Coverage | Bacteria and Archaea only [1]. | All domains: Bacteria, Archaea, Fungi, Viruses [1]. |
| Functional Profiling | No direct functional data; requires prediction (e.g., PICRUSt) [1] [18]. | Yes, direct identification of microbial genes and pathways [1] [18]. |
| Cost per Sample (Approx.) | ~$50 - $80 USD [1] [24]. | ~$150 - $200 USD (deep); ~$120 (shallow) [1] [24]. |
| Sensitivity to Host DNA | Low (due to targeted PCR amplification) [24]. | High (requires high sequencing depth to overcome host DNA background) [1] [25]. |
| Bioinformatics Complexity | Beginner to Intermediate [1]. | Intermediate to Advanced [1]. |
| Key Advantage | Cost-effective for compositional profiling; high sensitivity for bacteria/archaea. | Comprehensive taxonomic and functional profiling. |
| Key Limitation | Limited resolution and functional inference; primer bias [9] [18]. | Higher cost; computationally intensive; host DNA interference [1]. |
Evolution to Genome-Resolved Metagenomics
The latest evolutionary step in this field is genome-resolved metagenomics, which moves beyond simply profiling taxonomic abundance from short reads. This method involves the de novo assembly of individual microbial genomes directly from metagenomic sequencing data, resulting in Metagenome-Assembled Genomes (MAGs) [18].
This approach is a "game changer" because it allows researchers to study the genetic makeup of uncultured microorganisms at a previously impossible resolution. It enables the study of within-species genetic diversity, including single nucleotide variants (SNVs) and structural variants (SVs), which can be associated with host phenotypes [18]. Furthermore, it facilitates the discovery of novel metagenome protein families and allows for genome-scale metabolic modeling of uncultured species, directly linking genetic capacity to ecosystem function [18]. This has been instrumental in illuminating the vast "microbial dark matter" that was previously inaccessible to researchers.
The Scientist's Toolkit: Essential Reagents and Materials
Successful culture-independent studies rely on a suite of carefully selected reagents and kits. The choice of kit can significantly impact DNA yield, shearing, and the representation of microbial communities.
Table: Essential Research Reagents for Culture-Independent Microbiology
| Reagent / Kit Name | Function / Application | Key Consideration |
|---|---|---|
| OMNIgene Gut OMR-200 Tube | Stool sample collection and stabilization at room temperature [9]. | Preserves microbial community structure during transport; critical for longitudinal studies. |
| Qiagen DNeasy PowerSoil Kit | DNA extraction from complex, hard-to-lyse samples (e.g., soil, stool). | Effective lysis of difficult-to-break Gram-positive bacteria; removes PCR inhibitors. |
| VAHTS Free-Circulating DNA Maxi Kit | Extraction of microbial cell-free DNA (cfDNA) from plasma or body fluid supernatants [25]. | Essential for liquid biopsy and sepsis diagnostics; targets DNA released by dying microbes. |
| ZymoBIOMICS Microbial Community Standard | Mock microbial community with fully sequenced genomes. | Serves as a positive control and benchmark for evaluating sequencing and bioinformatics accuracy. |
| HostZERO Microbial DNA Kit | Depletion of host DNA from samples (e.g., blood, tissue) [24]. | Increases the proportion of microbial reads in host-heavy samples, improving detection sensitivity. |
| VAHTS Universal Pro DNA Library Prep Kit | Preparation of sequencing libraries for Illumina platforms from low-input DNA [25]. | Facilitates the construction of sequencing-ready libraries from the nanogram DNA quantities typical of metagenomic samples. |
| Tridecanoyl chloride | Tridecanoyl chloride, CAS:17746-06-4, MF:C13H25ClO, MW:232.79 g/mol | Chemical Reagent |
| Tiemonium Iodide | Tiemonium Iodide | Tiemonium iodide is an anticholinergic research compound. It is a muscarinic receptor antagonist for research use only (RUO). Not for human consumption. |
Detailed Experimental Protocols from Key Studies
To ensure reproducibility and provide a clear framework for researchers, below are detailed methodologies from pivotal comparative studies.
This protocol is adapted from the 2021 Scientific Reports study that quantitatively compared taxonomic results from 16S and shotgun sequencing.
- Sample Collection and DNA Extraction: Gastrointestinal tracts (crop and caeca) from chickens were dissected. Total DNA was extracted from the contents using a commercial kit.
- 16S rRNA Library Preparation: The V3-V5 hypervariable regions of the 16S rRNA gene were amplified using primers 357F (5'-CCTACGGGAGGCAGCAG-3') and 926R (5'-CCGTCAATTCMTTTRAGT-3'). Amplification was performed with a touchdown PCR protocol. The resulting amplicons were purified, quantified, and pooled in equimolar ratios for sequencing.
- Shotgun Metagenomic Library Preparation: Total DNA was mechanically sheared, and libraries were prepared using a standard Illumina protocol with adapter ligation and PCR amplification. Libraries were quantified and pooled.
- Sequencing: Both 16S and shotgun libraries were sequenced on an Illumina MiSeq platform. A minimum of 500,000 reads per sample was set as a quality threshold for shotgun data based on rarefaction analysis.
- Bioinformatic Analysis:
- 16S Data: Raw reads were processed using the DADA2 pipeline within QIIME2 to infer Amplicon Sequence Variants (ASVs). Taxonomy was assigned against the SILVA database.
- Shotgun Data: Quality-filtered reads were analyzed for taxonomy using the MetaPhlAn tool, which relies on clade-specific marker genes.
This protocol is adapted from a 2025 clinical study comparing whole-cell DNA (wcDNA) mNGS, cell-free DNA (cfDNA) mNGS, and 16S rRNA NGS for pathogen identification.
- Sample Processing: Clinical body fluid samples (pleural, ascites, CSF) were centrifuged at 20,000 × g for 15 minutes to separate the cellular fraction from the supernatant.
- Dual DNA Extraction:
- cfDNA Extraction: Cell-free DNA was extracted from 400 μL of supernatant using the VAHTS Free-Circulating DNA Maxi Kit.
- wcDNA Extraction: The retained precipitate was subjected to mechanical bead-beating lysis. Whole-cell DNA was then extracted from the lysate using the Qiagen DNA Mini Kit.
- Library Preparation and Sequencing:
- mNGS (wcDNA and cfDNA): DNA libraries were prepared with the VAHTS Universal Pro DNA Library Prep Kit and sequenced on an Illumina NovaSeq platform to a depth of ~8 GB (26 million reads) per sample.
- 16S rRNA NGS: The 16S rRNA gene was amplified and prepared for sequencing on an Illumina NovaSeq platform, generating ~50,000 reads per sample.
- Bioinformatic and Reporting Criteria: For mNGS, a species was reported if its read count was >100, it mapped to ≥5 distinct genomic regions, and its z-score was threefold higher than in negative controls. For 16S NGS, a species was reported if reads were >100 and the z-score was threefold above the negative control.
The evolution from culture-dependent techniques to 16S rRNA sequencing and subsequently to shotgun and genome-resolved metagenomics represents a series of paradigm shifts in microbiology. Each technology has its place in the modern researcher's arsenal. 16S rRNA sequencing remains a powerful, cost-effective tool for large-scale, hypothesis-generating studies focused on bacterial and archaeal community composition. In contrast, shotgun metagenomics provides a deeper, more comprehensive view of the entire microbial community, delivering species-level taxonomy and critical insights into functional potential. The emerging frontier of genome-resolved metagenomics is now pushing the boundaries further, enabling the exploration of strain-level variation and the genetic repertoire of uncultured organisms. The choice among these methods is not a matter of which is universally superior, but rather which is most fit-for-purpose, balancing experimental goals, budget, and bioinformatic capabilities to advance both basic science and applied drug development.
In the field of microbial ecology, the choice of sequencing strategy fundamentally shapes the resolution and type of biological insights one can achieve. Two principal methodologies dominate: 16S rRNA gene amplicon sequencing (metataxonomics) and whole-genome shotgun metagenomic sequencing (metagenomics). The former typically yields fine-scale sequence variants like Amplicon Sequence Variants (ASVs), while the latter produces assembled genomic fragments known as contigs, from which genes can be predicted. These different outputs—ASVs versus contigs and genes—provide distinct yet complementary perspectives on microbial community structure and function. Amplicon sequencing, focusing on the highly conserved 16S rRNA gene, offers a cost-effective method for taxonomic profiling, but its resolution is inherently limited by the information within a single gene [6]. In contrast, shotgun metagenomics sequences all the DNA in a sample, enabling not only more precise taxonomic assignment but also direct access to the functional gene repertoire of the community [26]. This guide provides an objective comparison of these approaches, detailing their performance, supported by experimental data and methodologies relevant to researchers and drug development professionals.
Core Outputs Defined: ASVs, Contigs, and Genes
Amplicon Sequence Variants (ASVs)
ASVs represent unique, error-corrected sequences from high-throughput amplicon data. Unlike older Operational Taxonomic Unit (OTU) methods that cluster sequences at an arbitrary similarity threshold (e.g., 97%), ASV algorithms like DADA2 provide single-nucleotide resolution by distinguishing true biological variation from sequencing errors [27]. ASVs are highly reproducible exact sequences, facilitating direct comparison across different studies. However, as they are derived from a single marker gene (typically the 16S rRNA), their taxonomic resolution is often limited to the genus level and can be influenced by primer choice and intragenomic copy number variation [28] [4].
Contigs and Genes from Shotgun Metagenomics
Shotgun metagenomics involves randomly fragmenting and sequencing all DNA from a sample. These short reads are then assembled into longer, contiguous sequences called contigs [26]. This process of de novo assembly reconstructs stretches of microbial genomes without relying on reference databases. From these contigs, open reading frames (ORFs) and other genomic features can be predicted and annotated as genes. This allows for the direct identification of functional elements within the metagenome, providing insights into the metabolic capabilities, virulence factors, and antibiotic resistance genes present in the microbial community [26]. The analysis is dependent on comprehensive reference databases, and its effectiveness can be limited in environments with many novel, uncharacterized organisms.
Table 1: Fundamental Characteristics of Primary Outputs
| Feature | Amplicon Sequence Variants (ASVs) | Contigs & Genes (Shotgun) |
|---|---|---|
| Definition | Unique, error-corrected marker gene sequences | Assembled genomic fragments from whole DNA |
| Data Origin | Amplified 16S rRNA hypervariable regions | Randomly sheared total genomic DNA |
| Primary Use | Taxonomic profiling & diversity analysis | Taxonomic profiling & functional potential analysis |
| Resolution | Single-nucleotide (within the amplicon) | Varies with sequencing depth and assembly quality |
| Key Advantage | High reproducibility, cost-effective for taxonomy | Access to full genomic content, strain-level resolution |
Performance Comparison: Resolution, Accuracy, and Utility
Taxonomic Characterization and Detection Power
Comparative studies consistently demonstrate that shotgun sequencing detects a broader range of taxa, particularly those at low abundance. A 2021 study on chicken gut microbiota found that when a sufficient sequencing depth was achieved (>500,000 reads per sample), shotgun sequencing identified a statistically significant higher number of taxa compared to 16S sequencing [6]. The less abundant genera detected exclusively by shotgun sequencing were biologically meaningful and able to discriminate between experimental conditions. Similarly, a 2024 study on human colorectal cancer and healthy gut microbiota confirmed that 16S detects only part of the community revealed by shotgun, with the latter providing a more complete snapshot in both depth and breadth [4].
The ability to distinguish between experimental conditions also varies. In the chicken gut model, when comparing genera abundances between two gastrointestinal compartments, shotgun sequencing identified 256 statistically significant differences, whereas 16S sequencing identified only 108 [6]. This suggests that shotgun metagenomics has greater power to uncover biologically relevant taxonomic shifts.
Table 2: Taxonomic Profiling Performance: 16S vs. Shotgun
| Performance Metric | 16S rRNA Sequencing (ASVs) | Shotgun Metagenomics | Supporting Evidence |
|---|---|---|---|
| Detected Genera | Lower number; part of the community | Higher number; more comprehensive community view | [6] [4] |
| Low-Abundance Taxa | Limited detection power | Powerful detection with sufficient sequencing depth | [6] |
| Species/Strain Resolution | Limited, often to genus level | Possible, enables strain-level tracking | [28] [26] |
| Quantitative Accuracy | Biased by rRNA copy number and primer choice | More accurate, can be normalized by genome size | [29] |
| Differential Abundance Power | Lower | Higher (e.g., 256 vs. 108 significant genera found) | [6] |
Technical and Analytical Biases
Each method is susceptible to distinct technical biases. 16S rRNA sequencing relies on PCR amplification using primers targeting specific hypervariable regions (e.g., V3-V4, V4). The choice of primers can introduce significant bias, as no single region can adequately distinguish all species, leading to inconsistent representation of certain taxa like Proteobacteria or Actinobacteria [9] [28]. Furthermore, variability in the number of 16S rRNA gene copies between different bacteria can skew abundance estimates [4].
Shotgun metagenomics avoids PCR amplification biases related to a single gene but is strongly dependent on reference genome databases for taxonomic assignment and functional annotation [4]. This can make it challenging to identify novel microbes without computationally expensive assembly. Additionally, shotgun data from low-biomass samples or those with high host DNA (e.g., tissue biopsies) can suffer from a noisy signal and require deeper, more costly sequencing or host DNA depletion protocols [26] [4].
Experimental Protocols for Comparison
To ensure robust and comparable results in studies that benchmark these methods, consistent and well-documented experimental protocols are essential.
Sample Collection and DNA Extraction
The initial steps are critical for data quality. In a comparative study of infant gut microbiomes, stool samples were collected by parents in OMR-200 tubes (OMNIgene GUT, DNA Genotek), stored on ice, and transferred to the lab within 24 hours for immediate freezing at -80°C [9]. For the colorectal cancer study, fecal DNA for shotgun analysis was extracted using the NucleoSpin Soil Kit (Macherey-Nagel), while DNA for 16S sequencing was extracted using the Dneasy PowerLyzer Powersoil kit (Qiagen) from the same sample set [4]. The use of bead-beating in extraction protocols is crucial to ensure efficient lysis of Gram-positive bacteria.
Sequencing and Bioinformatics
For 16S rRNA Sequencing: The common approach is to amplify and sequence hypervariable regions. The colorectal cancer study used the V3-V4 region, with processing and analysis conducted with the DADA2 pipeline (v1.22.0) in R to infer ASVs [4]. Taxonomy was assigned using the SILVA 16S rRNA database (v138.1). To improve species-level classification, an additional taxonomic assignment was performed using a custom BLASTN database and k-mer based classification with Kraken2 and Bracken2 against the NCBI RefSeq Targeted Loci Project database [4].
For Shotgun Metagenomics: The same study sequenced libraries on an Illumina platform. Human sequence reads were filtered out by aligning to the human genome (GRCh38) using Bowtie2 [4]. For taxonomic profiling of non-host reads, tools like Kraken with custom databases built from complete bacterial, viral, fungal, and archaeal genomes are widely used [30].
Figure 1: Experimental workflow for comparative microbiome studies.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Reagents and Materials for Metagenomic Studies
| Item | Function/Application | Example Products & Kits |
|---|---|---|
| Sample Collection & Stabilization | Preserves microbial community integrity at point of collection | OMNIgene GUT OMR-200 tubes [9] |
| DNA Extraction Kit | Isolates high-quality, unbiased microbial DNA from complex samples | NucleoSpin Soil Kit (Macherey-Nagel), Dneasy PowerLyzer Powersoil (Qiagen) [4] |
| 16S rRNA PCR Primers | Amplifies specific hypervariable regions for sequencing | Primers for V3-V4, V4 regions [28] [4] |
| Sequencing Platform | Generates high-throughput sequence data | Illumina MiSeq/NovaSeq (short-read); PacBio Sequel, Oxford Nanopore (long-read) [26] |
| Reference Databases | Essential for taxonomic classification and functional annotation | SILVA, Greengenes (16S); NCBI RefSeq, GTDB, UHGG (Shotgun) [28] [4] |
| Bioinformatics Tools | Processes raw data into biological insights | DADA2, QIIME 2 (16S); Bowtie2, Kraken2, MEGAHIT (Shotgun) [4] [30] |
| Salipurpin | Apigenin 5-O-beta-D-glucopyranoside|28757-27-9 | |
| Bibapcitide | Bibapcitide, CAS:153507-46-1, MF:C112H162N36O43S10, MW:3021.4 g/mol | Chemical Reagent |
The choice between 16S rRNA sequencing and shotgun metagenomics is not a matter of which is universally superior, but which is the most appropriate tool for the specific research question, sample type, and budget [4].
- 16S rRNA sequencing (ASVs) is a powerful, cost-effective choice for large-scale studies focused on comparing taxonomic composition and diversity across a large number of samples, particularly when the research question resides at the genus level or above. It is also more suitable for samples with low microbial biomass or high host DNA contamination, such as tissue samples [26] [4].
- Shotgun metagenomics (Contigs & Genes) is the necessary approach when the research demands species- or strain-level resolution, an accurate quantification of taxa without marker gene biases, or direct insight into the functional potential of the microbial community [6] [26]. It is the preferred method for in-depth analysis of stool samples and for linking community structure to function.
As sequencing costs continue to decrease and analytical methods improve, shotgun metagenomics is becoming increasingly accessible. However, both techniques provide valuable, albeit different, lenses for examining microbial communities, and their combined application can offer a particularly powerful and comprehensive understanding.
Methodological Deep Dive: Workflows, Strengths, and Ideal Use Cases
In microbial ecology and clinical diagnostics, researchers primarily rely on two powerful sequencing approaches to characterize microbial communities: 16S rRNA sequencing and metagenomic sequencing [2] [10]. While both methods provide insights into microbial composition, they differ fundamentally in their workflow, data output, and applications. 16S rRNA sequencing is a targeted amplicon approach that amplifies and sequences the bacterial 16S ribosomal RNA gene, providing primarily taxonomic identification of bacteria and archaea [31] [32]. In contrast, metagenomic sequencing (often called shotgun metagenomics) randomly sequences all DNA fragments in a sample, enabling comprehensive taxonomic profiling across all microbial domains (bacteria, archaea, viruses, fungi) and functional potential analysis [2] [33]. This guide provides a detailed, step-by-step comparison of these methodologies from initial DNA extraction through final data analysis, equipping researchers with the knowledge to select the appropriate approach for their specific research questions.
Fundamental Workflow Comparison
The following diagram illustrates the core procedural differences between 16S rRNA sequencing and shotgun metagenomic sequencing workflows, highlighting the divergent paths each method takes from sample preparation to data interpretation:
Detailed Methodological Comparison
Sample Preparation and DNA Extraction
Both methods begin with careful sample collection and DNA extraction, but requirements differ significantly:
Shared Considerations:
- Sample Sterility: Containers must be sterile to prevent contamination from environmental microbes [32].
- Temperature Control: Samples are typically frozen at -20°C or -80°C immediately after collection to preserve microbial integrity [32].
- Rapid Processing: Minimize time between collection and freezing; temporary storage at 4°C or preservation buffers can be used when immediate freezing isn't possible [32].
Method-Specific Requirements:
- 16S rRNA Sequencing: Requires high-quality DNA but relatively small quantities (nanograms) due to subsequent PCR amplification [32].
- Shotgun Metagenomics: Demands larger DNA quantities (micrograms) and higher purity, especially for mate-pair libraries [33]. Host DNA removal is critical when sequencing host-associated microbiomes to prevent overwhelming microbial signals [33].
Library Preparation and Sequencing
This stage represents the most significant methodological divergence between the two approaches:
16S rRNA Sequencing Workflow:
- Targeted PCR Amplification: Universal primers target conserved regions surrounding hypervariable regions (V1-V9) of the 16S rRNA gene [31] [32].
- Primer Selection: Different hypervariable regions offer varying taxonomic resolution; common choices include V3-V4 (∼428 bp) for Illumina MiSeq or full-length 16S (∼1500 bp) for Pacific Bioscience [31] [34].
- Barcoding: Molecular barcodes are added to each sample during PCR for multiplexing [32].
- Library Cleaning: Magnetic bead-based purification removes impurities and size-selects amplified fragments [32].
Shotgun Metagenomics Workflow:
- Random Fragmentation: Mechanical shearing breaks all DNA in the sample into small fragments (typically 300-800 bp) [2] [33].
- Library Construction: Fragments are ligated with sequencing adapters without target-specific amplification [33] [10].
- No PCR Bias: Avoids amplification biases but requires sufficient starting material [33].
Table 1: Key Differences in Library Preparation and Sequencing
| Parameter | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Target Region | Specific hypervariable regions of 16S rRNA gene [31] | Entire genomic DNA of all organisms [2] |
| Amplification Required | Yes (PCR with universal primers) [32] | Optional (depending on DNA input) [33] |
| Common Sequencing Platforms | Illumina MiSeq, Roche 454, PacBio [31] | Illumina HiSeq, NovaSeq, PacBio [33] |
| Typical Read Length | 250-1500 bp [31] | 150-300 bp (Illumina), >10 kb (long-read) [33] |
| Multiplexing Capacity | High (hundreds of samples per run) | Moderate (typically 12-96 samples) |
Data Analysis Pipelines
Bioinformatic processing differs substantially between the two methods:
16S rRNA Sequencing Analysis:
- Quality Control: Tools like FastQC assess sequence quality [35].
- Denoising & Clustering: DADA2 or deblur generates amplicon sequence variants (ASVs), or VSEARCH clusters reads into operational taxonomic units (OTUs) [34].
- Taxonomic Assignment: Comparison against reference databases (SILVA, Greengenes, RDP) using classifiers like Naive Bayes [34].
- Diversity Analysis: Alpha diversity (within-sample) and beta diversity (between-sample) metrics calculated using tools like QIIME2 or mothur [34].
Shotgun Metagenomics Analysis:
- Quality Control & Host Removal: Trimmomatic or FastP removes low-quality reads; BBMAP filters host contamination [35].
- Assembly: Megahit or metaSPAdes performs de novo assembly of reads into contigs [33] [35].
- Taxonomic Profiling: Kraken2 or MetaPhlAn classifies reads against comprehensive databases [35].
- Functional Annotation: PROKKA or HUMAnN2 predicts genes and assigns functional categories using KEGG, COG, or Pfam databases [35].
- Binning: MetaBAT2 groups contigs into metagenome-assembled genomes (MAGs) based on sequence composition and coverage [35].
Table 2: Computational Requirements and Output
| Aspect | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Primary Analysis Tools | QIIME2, mothur, DADA2 [34] [32] | MetaPhlAn, HUMAnN2, MG-RAST, Kraken2 [10] [35] |
| Reference Databases | SILVA, Greengenes, RDP [31] [34] | NR, KEGG, CARD, RefSeq [10] [35] |
| Data Volume per Sample | 10-100 MB [10] | 1-100 GB [33] [35] |
| Computational Intensity | Low to Moderate [10] | High (requires 16+ cores, 100+ GB RAM) [35] |
| Technical Expertise Required | Beginner to Intermediate [10] | Advanced bioinformatics skills [10] [35] |
Performance Comparison and Experimental Data
Taxonomic Resolution and Coverage
The methods differ significantly in their taxonomic precision and range of detectable organisms:
Table 3: Taxonomic Profiling Capabilities
| Parameter | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Domains Detected | Bacteria and Archaea only [2] [32] | Bacteria, Archaea, Viruses, Fungi, Eukaryotes [2] [10] |
| Typical Resolution | Genus-level (sometimes species) [2] [36] | Species to strain-level [10] |
| Novel Organism Detection | Limited to divergent 16S sequences | Can reconstruct full genomes of novel organisms [2] [35] |
| Quantitative Accuracy | Affected by PCR bias, copy number variation | More directly quantitative [33] |
| Strain-Level Differentiation | Generally not possible [32] | Possible with sufficient coverage [10] |
Experimental data from a clinical study comparing these methods in 50 patients with suspected bacterial infections but negative cultures revealed complementary performance [19]. 16S rRNA Sanger sequencing identified clinically relevant bacteria in 27% of samples, while clinical metagenomics (shotgun approach) demonstrated 70% sensitivity compared to 16S but identified additional clinically relevant bacteria in 19% of samples that were negative by 16S sequencing [19]. This supports positioning the methods as complementary rather than mutually exclusive for diagnostic applications.
Functional Insights and Additional Applications
Beyond taxonomic classification, shotgun metagenomics provides extensive functional information absent from 16S data:
Functional Capabilities of Shotgun Metagenomics:
- Metabolic Pathway Analysis: Identification of complete metabolic pathways (e.g., nitrogen fixation, antibiotic synthesis) [33] [10].
- Antibiotic Resistance Genes: Comprehensive profiling of resistome using databases like CARD [10].
- Virulence Factors: Detection of genes associated with pathogenicity [10].
- Biosynthetic Gene Clusters: Identification of genes encoding bioactive compound synthesis [33].
16S rRNA Sequencing Applications:
- Community Ecology: Population structure analysis in environmental samples [32].
- Microbiome Dynamics: Tracking community changes over time or between conditions [34].
- Rapid Pathogen Screening: Initial identification of bacterial pathogens in clinical samples [36] [19].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of either method requires specific laboratory and computational resources:
Table 4: Essential Research Reagents and Solutions
| Reagent/Resource | Function | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|---|
| DNA Extraction Kits | Cell lysis and DNA purification | Standard bacterial kits [32] | Kits optimized for diverse organisms & high molecular weight DNA [33] |
| PCR Master Mix | Amplification of target regions | Required (with universal 16S primers) [32] | Not required for standard protocol |
| Size Selection Beads | Fragment size selection | Critical for removing primer dimers [32] | Optional for library normalization |
| Library Prep Kits | Sequencing library construction | Amplicon library kits [32] | Fragmentation and ligation kits [33] |
| Positive Controls | Protocol validation | Mock microbial communities [34] | Complex mock communities [35] |
| Negative Controls | Contamination assessment | Extraction and no-template controls [34] [32] | Extraction and no-template controls [35] |
| Reference Databases | Taxonomic classification | SILVA, Greengenes, RDP [31] [34] | NR, MetaPhlAn, KEGG [10] [35] |
| Bioinformatics Tools | Data processing and analysis | QIIME2, mothur [34] [32] | HUMAnN2, MetaPhlAn, Kraken2 [35] |
| Propyzamide | Propyzamide Herbicide | Propyzamide is a selective, systemic herbicide for agricultural and environmental research. This product is for Research Use Only (RUO). Not for personal use. | Bench Chemicals |
| Pukateine | Pukateine|CAS 81-67-4|RUO | Bench Chemicals |
Method Selection Guidelines
Choosing between 16S rRNA sequencing and shotgun metagenomics depends on research goals, budget, and computational resources:
Choose 16S rRNA Sequencing When:
- Research questions focus specifically on bacterial/archaeal community composition [2] [32].
- Studying large sample sizes with limited budget (16S is significantly more cost-effective) [10].
- Preliminary screening of microbial communities is needed [10].
- Laboratory and bioinformatics expertise is limited [10].
- Well-established reference databases exist for the studied environment [32].
Choose Shotgun Metagenomics When:
- Comprehensive taxonomic profiling across all domains is required [2] [10].
- Functional gene content, metabolic pathways, or antibiotic resistance genes are of interest [33] [10].
- Strain-level differentiation or genome reconstruction is needed [10] [35].
- Studying environments with limited prior characterization [2].
- Sufficient budget and computational resources are available [10] [35].
Future Perspectives
Emerging technologies are bridging the gap between these approaches:
- Full-length 16S sequencing with long-read technologies improves species-level resolution [31].
- Hybrid approaches combine 16S for taxonomic clarity with metagenomics for functional insights [10].
- Multi-omics integration correlates metagenomic data with metatranscriptomic, metaproteomic, and metabolomic data for comprehensive functional profiling [10].
- Standardized protocols and shared data repositories are addressing reproducibility challenges across studies [33] [35].
In conclusion, both 16S rRNA sequencing and shotgun metagenomics offer powerful but distinct approaches to microbial community analysis. 16S rRNA sequencing provides a cost-effective, targeted method for bacterial composition analysis, while shotgun metagenomics delivers comprehensive taxonomic and functional profiling across all domains of life. Understanding these workflow differences enables researchers to select the optimal method for their specific research objectives, budget constraints, and technical capabilities. As sequencing technologies continue to advance and costs decrease, shotgun metagenomics is likely to see increased adoption, though 16S rRNA sequencing will remain valuable for large-scale epidemiological studies and targeted bacterial analysis.
In the field of microbiome research, selecting the appropriate sequencing method is a critical first step in experimental design. For studies focused on characterizing bacterial diversity and taxonomy, 16S rRNA gene sequencing has long been the workhorse technology, prized for its cost-effectiveness and specialized capabilities. While metagenomic shotgun sequencing provides a broader view of all genetic material in a sample, 16S sequencing remains the gold standard for efficiently answering specific questions about bacterial composition [1]. This guide provides an objective comparison of these technologies, framed by experimental data to help researchers make evidence-based decisions for their specific research contexts.
Methodological Comparison: 16S rRNA vs. Shotgun Metagenomic Sequencing
The fundamental difference between these approaches lies in their scope and target. 16S rRNA sequencing is an amplicon-based method that uses polymerase chain reaction (PCR) to amplify specific hypervariable regions of the bacterial 16S rRNA gene, which is present in all bacteria and archaea [1]. In contrast, shotgun metagenomic sequencing is a comprehensive approach that fragments and sequences all DNA present in a sample—bacterial, archaeal, viral, fungal, and even host DNA—without targeting specific genes [1].
Table 1: Core Methodological Differences Between 16S rRNA and Metagenomic Sequencing
| Factor | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Target | Specific 16S rRNA gene regions | All genomic DNA in sample |
| Taxonomic Coverage | Bacteria and Archaea only | All taxa (Bacteria, Archaea, Viruses, Fungi) |
| Taxonomic Resolution | Genus-level (sometimes species) [1] [37] | Species-level and strain-level [1] |
| Functional Profiling | No (but prediction possible with tools like PICRUSt) [1] | Yes (reveals functional potential) [1] |
| Host DNA Sensitivity | Low [1] | High [1] |
| Bioinformatics Requirements | Beginner to Intermediate [1] | Intermediate to Advanced [1] |
| Approximate Cost per Sample | ~$50 USD [1] | Starting at ~$150 USD [1] |
Performance and Experimental Data Comparison
Taxonomic Profiling and Diversity Assessment
Multiple studies have directly compared the performance of 16S and shotgun sequencing for taxonomic profiling. A 2022 study on pediatric ulcerative colitis found that both methods yielded consistent patterns of gut microbiome signatures, with 16S rRNA data showing similar results to shotgun data in terms of alpha diversity (within-sample diversity) and beta diversity (between-sample diversity) [38]. Furthermore, the study demonstrated that 16S data could predict disease status with accuracy comparable to shotgun data (AUROC close to 0.90) [38].
However, the choice of 16S primer sets can influence results. A 2025 evaluation highlighted the critical influence of primer selection on 16S rRNA sequencing results, with certain primer combinations detecting unique taxa that others missed [12]. Despite this variability, the core finding—significant microbial differences between experimental groups—remained consistently detectable across primer choices [12].
Resolution and Identification Capabilities
The primary limitation of 16S sequencing is its resolution. It is generally limited to genus-level identification and sometimes species, whereas shotgun sequencing can achieve species and strain-level resolution [1]. This is because the 16S rRNA gene is highly conserved, and even with full-length sequencing, it may not reliably differentiate between closely related species that have nearly identical 16S sequences but different genomic profiles [37] [18].
Table 2: Experimental Performance Comparison Based on Peer-Reviewed Studies
| Study Focus | 16S rRNA Sequencing Performance | Shotgun Metagenomic Sequencing Performance | Citation |
|---|---|---|---|
| Pathogen ID in Body Fluids | 58.5% (24/41) consistency with culture results. | 70.7% (29/41) consistency with culture results. | [25] |
| Pediatric UC Prediction | AUROC ~0.90 for predicting UC status. | AUROC ~0.90 for predicting UC status. | [38] |
| Taxonomic Resolution | Low phylogenetic power at species level; cannot distinguish recently diverged species. [37] | Enables species and strain-level identification; can profile single nucleotide variants. [1] | [1] [37] |
| Platform Comparison | Captured a narrower range of taxa compared to ONT. | Illumina and ONT platforms showed a high degree of correlation. | [12] |
Experimental Protocols for 16S rRNA Sequencing
A standardized protocol for 16S rRNA sequencing, as used in contemporary studies, involves the following key steps [12] [38]:
- DNA Extraction: Microbial DNA is extracted from the sample (e.g., feces, soil, water) using commercial kits, such as the QIAamp Powerfecal DNA kit. This step often includes a mechanical lysis step to break open resilient microbial cell walls [12] [38].
- PCR Amplification: The extracted DNA is used as a template for PCR to amplify one or more selected hypervariable regions of the 16S rRNA gene (e.g., V4 region). Primers are tagged with unique molecular barcodes to allow for multiplexing of multiple samples in a single sequencing run [38].
- Library Preparation and Clean-up: The amplified DNA is cleaned to remove impurities and primers, and then size-selected to ensure the fragments are appropriate for sequencing [1].
- Pooling and Quantification: The barcoded samples are pooled together in equal proportions, and the final library pool is quantified to ensure optimal sequencing concentration [1].
- Sequencing: The pooled library is sequenced on a platform such as the Illumina MiSeq or iSeq, typically using 2x150bp or 2x250bp paired-end chemistry [1] [38].
The following workflow diagram illustrates this process:
The Scientist's Toolkit: Essential Reagents and Materials
Successful 16S rRNA sequencing relies on a suite of specialized reagents and tools. The following table details key solutions required for the experimental workflow.
Table 3: Essential Research Reagent Solutions for 16S rRNA Sequencing
| Reagent/Material | Function | Example Product |
|---|---|---|
| DNA Extraction Kit | To lyse microbial cells and purify high-quality genomic DNA from complex samples. | QIAamp Powerfecal DNA Kit (Qiagen) [38] |
| 16S-Targeted PCR Primers | To specifically amplify hypervariable regions of the bacterial 16S rRNA gene. | 515FB / 806RB (for V4 region) [38] |
| PCR Master Mix | Contains enzymes, buffers, and nucleotides necessary for the amplification of target DNA. | Not specified in results |
| Library Preparation Kit | For attaching sequencing adapters and sample-specific barcodes to amplified DNA. | Illumina Nextera XT (for shotgun, adaptable) [38] |
| Size Selection Beads | To clean up PCR products and select for the desired fragment size, removing impurities. | AMPure XP Beads (common practice) |
| Sequencing Platform | Instrumentation to perform high-throughput sequencing of the prepared library. | Illumina MiSeq System [38] |
| DRI-C21041 (DIEA) | DRI-C21041 (DIEA), MF:C38H40N4O7S, MW:696.8 g/mol | Chemical Reagent |
| Diethyl succinate-13C4 | Diethyl succinate-13C4, MF:C8H14O4, MW:178.17 g/mol | Chemical Reagent |
16S rRNA sequencing remains a powerful, cost-effective tool for profiling bacterial communities, especially in large-scale studies where budget constraints are a primary concern. It is the recommended method when the research question is focused specifically on bacterial taxonomy, diversity, and community structure. However, for projects requiring species-level resolution, functional gene analysis, or detection of non-bacterial kingdoms, shotgun metagenomic sequencing, despite its higher cost and bioinformatic complexity, is the superior choice [1] [18]. The decision between these technologies should be guided by the specific research objectives, desired resolution, and available resources.
The study of microbial communities has been revolutionized by culture-independent sequencing methods, primarily 16S rRNA gene amplicon sequencing and shotgun metagenomics [26]. While 16S rRNA sequencing (16S) targets a single, highly conserved bacterial gene to reveal taxonomic composition, shotgun metagenomics (MG) sequences all the DNA in a sample, enabling comprehensive insights into both the taxonomic identity and the functional potential of a microbial community [9]. Each method presents distinct trade-offs between cost, resolution, and informational output. This guide objectively compares their performance in key application areas—assessing functional potential, characterizing antibiotic resistance genes (ARGs), and conducting cross-kingdom analysis—by synthesizing experimental data and protocols from recent research.
Methodological Comparison and Performance Metrics
Technical and Performance Characteristics
The core difference between the methods lies in their approach: 16S is a targeted analysis, while MG is an untargeted, comprehensive genomic survey [26]. This fundamental distinction leads to varied performance in resolution, required resources, and data output.
Table 1: Core Methodological Comparison of 16S rRNA Sequencing vs. Shotgun Metagenomics
| Characteristic | 16S rRNA Amplicon Sequencing | Shotgun Metagenomics |
|---|---|---|
| Target | Amplified 16S rRNA gene regions [9] | Total genomic DNA from all organisms [26] |
| Taxonomic Resolution | Typically genus-level, sometimes species [9] | Species-level and strain-level [26] |
| Functional Insight | Indirectly inferred from taxonomy [9] | Direct assessment of genes and metabolic pathways [26] |
| Host DNA Depletion | Less critical (targeted amplification) | Critical for low-biomass/high-host-DNA samples [26] |
| Sequencing Depth | ~50,000 reads/sample often sufficient [9] | Millions to billions of reads required; deeper for complex communities [26] |
| Cost | Lower cost per sample [9] | Higher cost per sample (sequencing, computation) [9] |
Experimental Validation in Clinical and Environmental Studies
Quantitative comparisons across studies demonstrate the practical implications of these methodological differences. In clinical diagnostics, a 2025 study of 41 body fluid samples found that whole-cell DNA metagenomics (wcDNA mNGS) showed 70.7% consistency with culture results for bacterial detection, outperforming 16S rRNA NGS at 58.54% [25]. However, this increased sensitivity can come at the cost of specificity, which was compromised for wcDNA mNGS (56.34%), necessitating careful interpretation of results [25].
For broader biodiversity assessments, a pediatric gut microbiome study revealed that 16S profiling identified a larger number of genera than shotgun metagenomics, with each method detecting unique genera missed by the other [9]. This suggests that for primary taxonomic profiling, especially in lower-diversity environments like the infant gut, 16S remains a potent and cost-effective tool.
Table 2: Experimental Performance Comparison from Recent Studies
| Study Context | 16S rRNA Sequencing Performance | Shotgun Metagenomics Performance | Reference |
|---|---|---|---|
| Clinical Diagnosis (Body Fluids) | 58.54% consistency with culture (bacterial detection) [25] | 70.7% consistency with culture (wcDNA mNGS); higher sensitivity [25] | [25] |
| Bacterial Diagnosis (Culture-Negative Samples) | 27% of samples had clinically relevant results (reference method) [19] | 70% sensitivity vs. 16S; found additional clinically-relevant bacteria in 19% of 16S-negative samples [19] | [19] |
| Pediatric Gut Microbiome | Identified a larger number of genera; some genera missed by MG [9] | Identified some genera missed by 16S; provides functional data and higher taxonomic resolution [9] | [9] |
Assessing Functional Potential and Antibiotic Resistance Genes
Revealing Metabolic Capabilities
A primary advantage of metagenomics is its direct access to the functional gene repertoire of a community. For example, a 2025 study of eutrophic urban lakes used MG to analyze microbial vitamin B12 (VB12) synthesis pathways. The research quantified key functional genes like hemL and cobA (involved in the precorrin-2 synthesis pathway) and revealed that eutrophication can enhance specific VB12 synthesis routes while inhibiting others [39]. This level of mechanistic insight into metabolic pathways is beyond the reach of 16S sequencing.
Investigating the Antibiotic Resistome
MG is indispensable for comprehensively characterizing the diversity and mechanisms of antibiotic resistance genes (ARGs), a field known as resistome research [40]. Two metagenomic approaches are prevalent:
- Sequence-based metagenomics: Identifies known ARGs by comparing sequenced reads to reference databases [40].
- Functional metagenomics: Involves cloning metagenomic DNA into a surrogate host (e.g., E. coli) and selecting for antibiotic-resistant clones. This phenotype-driven approach can discover completely novel ARGs without prior sequence knowledge [40] [41].
A key application of functional metagenomics is exploring environmental resistomes. A study of agricultural soils in China identified 45 clones conferring resistance to eight different antibiotics. Notably, over 60% of the discovered ARGs had low similarity (<60%) to known sequences at the amino acid level, highlighting the vast, unexplored reservoir of novel resistance mechanisms in nature [41]. The same study found that soils with manure application contained approximately 70% of the identified ARGs, demonstrating how agricultural practices can expand the environmental resistome [41].
Fig 1. Workflow for Metagenomic Antibiotic Resistome Analysis. Two primary approaches are used to identify antibiotic resistance genes (ARGs) from environmental samples. Sequence-based analysis maps reads to databases to profile known ARGs, while functional screening clones DNA into surrogate hosts to select for resistant phenotypes and discover novel ARGs.
Cross-Kingdom Community Analysis
Limitations of 16S and Advantages of MG
The 16S rRNA gene is exclusive to bacteria and archaea, making 16S sequencing unsuitable for studying eukaryotic microbes like fungi, protozoa, and other micro-eukaryotes [9]. Shotgun metagenomics overcomes this limitation by sequencing all DNA, enabling simultaneous, cross-kingdom analysis of prokaryotic and eukaryotic communities [42]. This provides a more holistic view of the entire microbial ecosystem and its complex interactions.
Case Study: Landfill Microbiome Profiling
A 2025 study of landfill leachates across China exemplifies the power of cross-kingdom metagenomics. Researchers developed a "Microbial Panorama Profiling Program (MP3)" method that used MG data, spike-in standards, and gene copy number correction to quantify the absolute cell abundance of both prokaryotes and eukaryotes [42]. This approach revealed:
- Landfills clustered into two distinct groups based on microbial composition, aligning with landfill age.
- A shift in metabolic processes: Younger landfills (<10 years) were characterized by fermentation and multi-pathway methanogenesis, while older landfills (≥10 years) were dominated by aerobic heterotrophs and synergistic degradation of recalcitrant organic matter by bacteria and fungi.
- Eukaryotic predators (protozoa and metazoa) decreased the stability of the microbial community in older landfills in a "top-down" manner [42].
These insights into the synergistic and predatory interactions between kingdoms would not be possible with 16S sequencing alone and underscore the value of MG for studying complex, multi-kingdom ecosystems.
Fig 2. Cross-Kingdom Analysis via Shotgun Metagenomics. Metagenomic data enables simultaneous profiling of prokaryotic and eukaryotic microorganisms. Integrating taxonomic and functional data from both kingdoms, along with absolute abundance quantification, allows researchers to model complex inter-kingdom interactions such as synergism and predation.
Essential Protocols and Research Toolkit
Key Experimental Workflows
Protocol 1: Standard Metagenomic Analysis for Taxonomy and Function [7] [39]
- DNA Extraction: Use kits designed for complex samples (e.g., soil, stool). For body fluids, separate whole-cell DNA (from pellet) and cell-free DNA (from supernatant) [25].
- Library Preparation & Sequencing: Use Illumina NovaSeq or similar platforms for PE150 sequencing. Generate ~8+ Gb of data per sample for complex communities [7] [25].
- Bioinformatic Processing:
- Quality Control: Use Fastp or Trimmomatic to remove low-quality reads and adapters [7] [39].
- Assembly: Assemble quality-filtered reads into contigs using MEGAHIT [7] [39].
- Gene Prediction & Annotation: Predict open reading frames with Prodigal or MetaGeneMark. Annotate against functional databases (KEGG, CAZy) and taxonomic databases (NR) [7] [39].
Protocol 2: Functional Metagenomics for Novel ARG Discovery [40] [41]
- Metagenomic Library Construction: Extract environmental DNA and shotgun clone it into a vector (e.g., fosmid, BAC) to create a gene library in a surrogate host like E. coli.
- Phenotypic Selection: Plate the library clones on media containing antibiotics at a concentration that would kill the host without a resistance gene.
- Identification of Resistant Clones: Isolate and sequence the DNA inserts from resistant colonies to identify the novel ARG responsible for the resistance phenotype.
The Scientist's Toolkit
Table 3: Essential Reagents and Solutions for Metagenomic Workflows
| Research Reagent / Tool | Primary Function | Example Use Case |
|---|---|---|
| DNeasy PowerWater Kit / PowerSoil Kit (QIAGEN) | Extraction of high-quality DNA from low-biomass water or complex soil/stool samples. | Standardized DNA extraction for reproducibility in community studies [39]. |
| VAHTS Universal Pro DNA Library Prep Kit (Vazyme) | Preparation of Illumina-compatible sequencing libraries from metagenomic DNA. | Preparing whole-cell DNA libraries from clinical body fluid samples [25]. |
| Fastp / Trimmomatic | Quality control and adapter trimming of raw sequencing reads. | Pre-processing raw metagenomic data to remove low-quality sequences prior to assembly [7] [39]. |
| MEGAHIT | De novo assembly of metagenomic reads into longer contigs. | Assembling contiguous genomic fragments from complex environmental communities [7] [39]. |
| Prodigal | Prediction of protein-coding genes in assembled metagenomic contigs. | Identifying open reading frames (ORFs) for subsequent functional annotation [39]. |
| MetaPhlAn4 | Profiling taxonomic abundance from metagenomic reads using unique clade-specific markers. | Determining bacterial species composition and relative abundance in clinical samples [19]. |
| (2,4,6-Trichlorophenoxy)acetic acid-13C6 | (2,4,6-Trichlorophenoxy)acetic acid-13C6, MF:C8H5Cl3O3, MW:261.43 g/mol | Chemical Reagent |
The choice between 16S rRNA sequencing and shotgun metagenomics is not a matter of one being universally superior, but rather of selecting the right tool for the research question and budget [26]. 16S rRNA sequencing remains a powerful, cost-effective method for high-throughput taxonomic profiling, especially when tracking community changes over time or across many samples. In contrast, shotgun metagenomics is indispensable for applications requiring high taxonomic resolution, direct assessment of functional potential (like vitamin synthesis pathways or novel ARG discovery), and integrated cross-kingdom analysis. As evidenced by recent studies, MG can uncover novel resistance mechanisms [41] and elucidate complex multi-kingdom interactions [42] that are invisible to targeted approaches. For a comprehensive understanding of microbial ecosystems, many studies optimally employ both methods in a complementary fashion, using 16S for broad surveillance and MG for deep functional and cross-kingdom insights [19] [9].
In microbiome research, the choice between 16S rRNA gene sequencing and shotgun metagenomics is fundamental. While shotgun metagenomics offers broader functional insights and higher taxonomic resolution, 16S rRNA sequencing retains critical advantages that make it indispensable for many studies. This guide objectively examines the experimental data supporting the key practical benefits of 16S sequencing: significantly lower cost, reduced sequencing depth requirements, and a more streamlined analysis workflow.
Quantitative Comparison: 16S rRNA Sequencing vs. Shotgun Metagenomics
The following table summarizes the direct comparative metrics between the two approaches, highlighting the practical advantages of 16S sequencing.
Table 1: Direct comparison of key performance metrics between 16S rRNA and shotgun metagenomic sequencing.
| Factor | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Approximate Cost per Sample | ~$50 - $80 USD [1] [43] | Starting at ~$150 - $200 USD [1] [43] |
| Typical Sequencing Depth Required | ~50,000 reads/sample [9] [1] | Millions of reads/sample [9] |
| Bioinformatics Requirements | Beginner to intermediate expertise [1] [44] | Intermediate to advanced expertise [1] [44] |
| Minimum DNA Input | Femtograms; as low as 10 copies of the 16S gene [43] | 1 ng (may require host DNA depletion) [43] |
| Sensitivity to Host DNA | Low (PCR targets microbial gene) [1] [44] | High (sequences all DNA, including host) [1] [43] |
| Functional Profiling | No (but predicted profiling is possible with tools like PICRUSt) [1] [43] | Yes (direct measurement of functional genes) [1] [44] |
| Taxonomic Resolution | Genus-level (sometimes species) [1] [9] | Species-level and sometimes strain-level [1] [44] |
Experimental Evidence and Protocols
The advantages of 16S rRNA sequencing are not merely theoretical but are supported by robust experimental evidence. The following sections detail key studies and the methodologies that validate its cost-effectiveness and efficiency.
Protocol Optimization for Cost and Time Savings
A 2023 study systematically investigated two rate-limiting steps in 16S rRNA library preparation to improve efficiency without compromising data quality [45].
- Experimental Design: Researchers used nasal samples from healthy human participants and a serially diluted mock microbial community (ZymoBIOMICS Microbial Community DNA Standard II). They compared:
- PCR Pooling: Conducting PCR amplification in triplicate, duplicate, or as a single reaction per sample.
- Mastermix Preparation: Using a manually prepared mastermix versus a commercially available premixed mastermix.
- Key Findings: The study found no significant difference in high-quality read counts, alpha diversity, or beta diversity between single and pooled PCR reactions, or between the two mastermix preparation methods [45].
- Conclusion and Implication: Eliminating the need for PCR pooling and using premixed mastermixes reduces manual handling time, reagent costs, and the risk of contamination. This enables more efficient scaling of 16S sequencing for large-scale studies [45].
Lower Sequencing Depth Requirements
Research has directly quantified the adequate sequencing depth for 16S sequencing, particularly when compared to shotgun metagenomics.
- Experimental Design: A 2021 study compared paired 16S rRNA and shotgun metagenomic datasets from 338 pediatric gut samples across three age groups. They investigated the effect of sequencing depth on diversity measurements [9].
- Key Findings: The research demonstrated that 16S sequencing requires a relatively low number of sequenced reads (~50,000) per sample to maximize the identification of rare taxa. In contrast, shotgun metagenomics "typically requires more sequenced reads per sample to find unique taxonomic identifiers," carrying a higher cost [9].
- Conclusion and Implication: For studies focused on taxonomic profiling, especially of bacterial communities, 16S sequencing provides a cost-effective and efficient path to saturated diversity analysis without the need for deep, expensive sequencing runs [9].
Workflow Visualization
The simplified workflow of 16S rRNA sequencing, from sample to result, is a key contributor to its lower cost and streamlined analysis. The following diagram illustrates this process.
Diagram 1: 16S rRNA sequencing workflow.
Research Reagent Solutions
The table below lists essential reagents and kits used in a typical 16S rRNA gene sequencing experiment, as referenced in the cited studies.
Table 2: Key research reagents and their functions in 16S rRNA sequencing protocols.
| Reagent / Kit | Function | Example Use Case |
|---|---|---|
| DNA Extraction Kit (e.g., MPure Bacterial DNA kit) [45] | Isolation of total genomic DNA from complex samples. | Standardized cell lysis and DNA purification from fecal, soil, or swab samples. |
| High-Fidelity DNA Polymerase (e.g., Q5 Hot Start from NEB) [45] | PCR amplification of target 16S hypervariable regions. | Accurate amplification with minimal bias for library preparation, using primers like 27F/1492R. |
| Premixed PCR Mastermix [45] | A ready-to-use solution containing polymerase, dNTPs, and buffer. | Reduces manual handling steps, liquid transfer errors, and contamination risk during scaling. |
| AMPure XP Beads (Beckman Coulter) [45] | Solid-phase reversible immobilization (SPRI) for DNA clean-up and size selection. | Purification of PCR amplicons and library preparation to remove impurities and primers. |
| Mock Microbial Community (e.g., ZymoBIOMICS Standard) [45] [43] | A defined control with known microbial composition. | Validating entire workflow performance, from DNA extraction to bioinformatics, and detecting contamination. |
| 16S rRNA Reference Database (e.g., SILVA, Greengenes) [1] [9] | Curated collection of 16S gene sequences. | Taxonomic classification of sequencing reads into biological identities. |
Empirical evidence firmly supports 16S rRNA sequencing as a powerful and efficient method for microbiome taxonomic profiling. Its significantly lower cost per sample, minimal sequencing depth requirements, and less computationally intensive analysis pipeline make it an ideal choice for large-scale studies, projects with budget constraints, or initial surveys of microbial communities. While shotgun metagenomics excels in functional insight and high taxonomic resolution, the streamlined and cost-effective nature of 16S sequencing ensures its continued vital role in the microbiome researcher's toolkit.
In the field of microbial ecology, the choice of sequencing methodology fundamentally shapes the depth and scope of biological insights. While 16S rRNA gene sequencing has long been a staple for profiling microbial communities, shotgun metagenomics provides a more powerful, comprehensive lens for exploration. This guide objectively compares these approaches, demonstrating how metagenomics delivers superior resolution by enabling strain-level taxonomic classification, direct gene discovery, and detailed pathway analysis—capabilities that remain largely inaccessible with 16S methods. For researchers and drug development professionals, these advantages are critical for unlocking the functional potential of microbiomes in health, disease, and biotechnological application.
Technical Comparison: 16S rRNA Sequencing vs. Shotgun Metagenomics
The core difference between these techniques lies in their scope. 16S rRNA sequencing uses PCR to amplify a single, conserved gene region, using its variation to identify taxa [3]. In contrast, shotgun metagenomics involves randomly sequencing all DNA fragments in a sample, which are then computationally reconstructed into genomic information [3] [46].
Table 1: Fundamental Technical Differences
| Feature | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Target | Specific hypervariable regions of the 16S rRNA gene [3] | Total genomic DNA from all organisms in a sample [3] |
| Method | PCR amplification with targeted primers [3] | Random fragmentation and whole-genome sequencing [3] |
| Primary Output | Amplicon Sequence Variants (ASVs) or Operational Taxonomic Units (OTUs) [6] | Short reads assembled into contigs and genes [47] |
| Multikingdom Coverage | Primarily bacteria and archaea [3] | Bacteria, archaea, viruses, fungi, and protists [48] [3] |
This fundamental distinction in methodology underlies the significant differences in data output and analytical power, as detailed in the following sections.
Advantage 1: Strain-Level Resolution and Enhanced Taxonomic Profiling
Metagenomics provides a much finer-grained view of microbial communities, allowing identification down to the species and strain level, whereas 16S sequencing is typically limited to genus-level classification [3].
Experimental Evidence from Comparative Studies
A 2024 study on colorectal cancer gut microbiota directly compared taxonomic results from the same stool samples sequenced with both 16S and shotgun methods. The research concluded that "16S detects only part of the gut microbiota community revealed by shotgun," with the latter providing a more detailed snapshot in both depth and breadth [4]. Similarly, a comparison in chicken gut microbiota found that when a sufficient number of reads is available, shotgun sequencing has more power to identify less abundant taxa than 16S sequencing [6].
This superior resolution is not merely about detecting more taxa; it enables the tracking of specific bacterial strains within a community. For example, a longitudinal study of the pregnancy microbiome used metagenomic assembly to bin sequences into 97 draft quality genomes and recover co-occurring Gardnerella vaginalis strains with predicted distinct functions from the same subject [47]. Such strain-level dynamics are invisible to 16S analysis.
Key Methodological Protocols for Strain Resolution
Achieving strain-level resolution requires specific analytical approaches:
- High-Qepth Sequencing & Assembly: Deep sequencing is performed (e.g., 5-20 Gb per sample) to ensure sufficient coverage of genomes. Non-human reads are assembled into scaffolds using tools like MEGAHIT or metaSPAdes [47].
- Binning and Genome Reconstruction: Assembled scaffolds are binned into Metagenome-Assembled Genomes (MAGs) based on composition and abundance. This process involves tools such as MaxBin or MetaBAT, resulting in draft genomes [47] [46].
- Variant Analysis: Within a specific species, single-nucleotide polymorphisms (SNPs) and structural variants are identified to distinguish co-occurring strains [47].
Table 2: Comparative Taxonomic Power
| Taxonomic Level | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Phylum/Class | Reliable identification [6] | Reliable identification |
| Genus | Primary level of reliable identification [3] [4] | Reliable identification |
| Species | High rate of false positives; often unreliable [3] | Reliable identification [4] |
| Strain | Not possible | Possible via MAGs and variant analysis [47] [3] |
Figure: The workflow for achieving strain-level resolution from a complex sample using shotgun metagenomics, culminating in the ability to track individual strains.
Advantage 2: Direct Gene Discovery and Functional Potential
Unlike 16S sequencing, which only infers function from taxonomy, metagenomics directly characterizes the entire genetic repertoire of a microbial community, enabling true gene discovery and functional profiling [48] [3].
Quantifying Functional Capacity
A direct comparison of the two methods highlighted that 16S sequencing provides only taxonomic information, and any functional profile is an inference based on the presumed functions of the identified taxa. In contrast, shotgun sequencing directly provides information on the functional genes and pathways present in the microbial community, capturing both known and novel genes [3]. This direct approach avoids the inaccuracies of inference, which can be misleading as functional capacity can vary significantly between closely related strains.
Protocol for Functional Annotation
The standard workflow for functional characterization from metagenomic data involves:
- Gene Calling: Assembled contigs are processed with gene prediction tools (e.g., Prodigal) to identify open reading frames (ORFs) [47].
- Annotation against Databases: Predicted genes are compared against functional databases (e.g., KEGG, COG, EggNOG) using tools like BLASTP or DIAMOND to assign putative functions [47] [49].
- Pathway Reconstruction: Annotated genes are mapped to metabolic pathways to reconstruct the functional potential of the community [49].
Application in Pharmaceutical Development
This direct gene discovery capability is pivotal for pharmaceutical development. Metagenomics allows researchers to hunt for novel bacterial species and their biosynthetic gene clusters (BGCs) from environmental samples, which are a promising source for new therapeutics [17] [50]. For instance, the novel antibiotic teixobactin was discovered by identifying a previously undescribed soil bacterium and isolating its bioactive compound, a process powered by metagenomic insights [17].
Table 3: Key Reagents and Tools for Functional Metagenomics
| Research Reagent / Tool | Function in Analysis |
|---|---|
| Prodigal | Gene prediction software for identifying open reading frames in metagenomic assemblies [47]. |
| KEGG Database | Reference database for annotating predicted genes and reconstructing metabolic pathways [49]. |
| AntiSMASH | A bioinformatic pipeline for the identification and analysis of biosynthetic gene clusters (BGCs) from metagenomic data [50]. |
| Heterologous Host (e.g., E. coli) | A model organism used to express DNA clones from metagenomic libraries to screen for bioactive compounds [50]. |
Advantage 3: Comprehensive Pathway Analysis and Novel Compound Discovery
Building on functional gene annotation, metagenomics enables the reconstruction of complete metabolic pathways and the discovery of novel enzymes and small molecules, moving beyond description to functional prediction and validation.
Sequence-Based Discovery of Biosynthetic Pathways
The precipitous reduction in DNA sequencing costs has transformed natural product discovery [50]. Researchers can use shotgun sequencing of complex environmental DNA (eDNA) followed by computational tools like AntiSMASH to scan assembled contigs for biosynthetic gene clusters (BGCs) encoding pathways for compounds like polyketides and non-ribosomal peptides [50]. This sequence-based approach allows for the in-silico characterization of the biosynthetic potential of uncultured organisms before any laboratory isolation is attempted.
Protocol for Pathway-Centric Metagenomics
The workflow for pathway analysis and enzyme discovery often includes:
- Deep Metagenomic Sequencing: High-depth sequencing of environmental DNA (e.g., from soil or marine sponges) to adequately cover diverse and rare genomes [50].
- Biosynthetic Gene Cluster (BGC) Prediction: Process assembled contigs with AntiSMASH or similar software to identify BGCs and predict their encoded natural products [50] [51].
- Heterologous Expression: Clone candidate BGCs into culturable host bacteria (e.g., Streptomyces) to express and produce the predicted novel compound [50].
Figure: A sequence-based metagenomic workflow for the discovery of novel biosynthetic pathways and compounds, from environmental DNA to functional validation.
The choice between 16S rRNA sequencing and shotgun metagenomics is not a matter of one being universally "better" than the other, but rather of selecting the right tool for the research objective [4].
- 16S rRNA sequencing remains a powerful, cost-effective choice for large-scale studies focused on answering "Who is there?" at the genus level, especially when analyzing hundreds to thousands of samples or working with low-biomass samples where host DNA contamination can be problematic [3] [4].
- Shotgun metagenomics is the unequivocal choice when the research question demands "What can they do?" It is essential for studies requiring strain-level tracking, direct gene discovery, comprehensive pathway analysis, and multi-kingdom profiling [47] [48] [3].
For researchers and drug development professionals, the advanced capabilities of metagenomics in providing strain-level resolution, direct access to genetic material for novel gene discovery, and detailed pathway analysis make it an indispensable technology for understanding microbial function and driving innovation in therapeutics and biotechnology.
The study of gastrointestinal (GI) microbiota development represents a critical frontier in understanding host health, physiological functions, and disease pathogenesis. The complex successional patterns of microbial communities during early development require sophisticated analytical approaches to unravel both compositional and functional dynamics [7]. Two principal molecular methodologies have emerged as cornerstone techniques in this field: 16S ribosomal RNA (rRNA) gene sequencing and shotgun metagenomic sequencing. Each approach offers distinct advantages and limitations for characterizing microbial ecosystems during crucial developmental windows [3] [52].
This case study objectively compares the application of these techniques for investigating GI microbiota development, with a specific focus on the perinatal period in ruminant models—a critical phase for fetal organ maturation and microbial community establishment [7]. We present experimental data, detailed methodologies, and comparative analyses to guide researchers in selecting appropriate methods for specific research questions related to GI microbiota development.
Fundamental Principles and Methodologies
16S rRNA gene sequencing is a targeted amplicon sequencing approach that isolates and sequences specific variable regions (V4, V9, etc.) of the bacterial 16S rRNA gene [3]. This gene contains both highly conserved regions (enabling broad PCR amplification across bacterial taxa) and variable regions (providing taxonomic discriminatory power) [28]. The methodology involves DNA extraction, PCR amplification of target regions, library preparation, and sequencing, typically on Illumina platforms [1]. Following sequencing, bioinformatic processing clusters sequences into operational taxonomic units (OTUs) or amplicon sequence variants (ASVs) for taxonomic classification against reference databases [52].
Shotgun metagenomic sequencing employs an untargeted approach wherein total DNA is extracted from samples and randomly fragmented prior to sequencing [3]. This technique sequences all genomic content, enabling comprehensive characterization of all microorganisms—bacteria, archaea, viruses, and fungi—without prior targeting [3] [1]. The resulting sequences can be analyzed through either assembly-based approaches (reconstructing partial or complete genomes) or read-based approaches (aligning to marker gene databases) [1].
Key Technical Differences
Table 1: Comparative Analysis of 16S rRNA Sequencing and Shotgun Metagenomics
| Factor | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Taxonomic Resolution | Family/Genus level (species level possible but with high false positive rate) [3] | Species and strain-level resolution [3] |
| Taxonomic Coverage | Bacteria and Archaea only [3] | Multi-kingdom (Bacteria, Fungi, Virus, Protist) [3] |
| Functional Profiling | Indirect prediction via tools like PICRUSt2 (limited accuracy) [3] [53] | Direct detection of functional genes and pathways [3] |
| Recommended Sample Types | All types, particularly advantageous for low microbial biomass samples [3] | All types, best for samples with high microbial biomass (e.g., stool) [3] |
| Host DNA Interference | Minimal (PCR amplifies only target gene) [3] | Significant (requires host DNA removal or increased sequencing depth) [3] |
| Cost per Sample | Lower (~$50 USD) [1] | Higher (starting at ~$150 USD; varies with sequencing depth) [3] [1] |
| Minimum DNA Input | Low (successful with <1 ng DNA) [3] | Higher (typically minimum 1ng/μL) [3] |
| Bioinformatics Complexity | Beginner to intermediate [1] | Intermediate to advanced [1] |
Experimental Design and Methodologies
Case Study: GI Microbiota Development in Perinatal Goats
A recent investigation characterized early microbiota dynamics in Hunan local goats (Hutianshi Goat) during the perinatal period using both 16S rRNA sequencing and metagenomics [7]. This experimental design provides an ideal framework for comparing the two methodologies in the context of GI microbiota development.
Sample Collection and Processing
Biological Samples:
- Fetal goats (90 ± 10 gestational days): Contents from reticulum, rumen, small intestine, and large intestine (13 total samples) [7]
- 7-day-old goat kids: Contents from four stomachs (rumen, omasum, abomasum, reticulum), large intestine (cecum, colon, rectum), and small intestine (ileum, jejunum) (27 total samples) [7]
Ethical Considerations: All procedures were approved by the Animal Experimental Ethics Committee of Hunan Agricultural University (protocol number: HAU ACC 2022120) [7].
DNA Extraction: The TGuide S96 magnetic bead-based soil/fecal genomic DNA extraction kit was used for all samples [7].
Sequencing Protocols
16S rRNA Gene Sequencing:
- Library Preparation: TruSeq Nano DNA LT Library Prep Kit (Illumina)
- Sequencing Platform: Illumina MiSeq/NovaSeq
- Target Region: Variable regions of 16S rRNA gene
- Bioinformatic Processing: QIIME2 pipeline with DADA2 for quality control, denoising, joining, and chimera removal [7]
Shotgun Metagenomic Sequencing:
- Library Preparation: VAHTS Universal Plus DNA Library Prep Kit for Illumina
- Sequencing Platform: Illumina NovaSeq 6000 with PE150 strategy
- Bioinformatic Processing:
- Quality control: fastp and bowtie2
- Assembly: MEGAHIT with contigs <300 bp filtered out
- Gene prediction: MetaGeneMark
- Annotation: Multi-database (NR, KEGG, CAZy) [7]
Figure 1: Experimental workflow for comparative analysis of GI microbiota using 16S rRNA sequencing and shotgun metagenomics
Comparative Performance Assessment
Taxonomic Profiling Capabilities
Studies directly comparing taxonomic results from 16S rRNA sequencing and shotgun metagenomics in GI microbiota reveal significant differences in detection sensitivity and resolution [6]. When sufficient sequencing depth is achieved (>500,000 reads), shotgun sequencing identifies a statistically significant higher number of taxa compared to 16S sequencing, particularly among less abundant genera [6].
Table 2: Taxonomic Comparison in Gastrointestinal Microbiota Studies
| Parameter | 16S rRNA Sequencing | Shotgun Metagenomics | Experimental Evidence |
|---|---|---|---|
| Genera Detection | 288 genera detected | 288+ genera detected (additional rare taxa) | Analysis of chicken GI tract showed shotgun sequencing detects more rare taxa [6] |
| Differential Abundance | 108 significant differences (caeca vs. crop) | 256 significant differences (caeca vs. crop) | 152 changes detected only by shotgun; 4 only by 16S [6] |
| Species-Level Resolution | Limited with short reads | Reliable species and strain-level identification | Full-length 16S improves resolution but not equivalent to shotgun [28] [54] |
| Multi-Kingdom Coverage | Restricted to Bacteria/Archaea | Comprehensive (Bacteria, Fungi, Viruses, Eukaryotes) | Shotgun enables viral and eukaryotic profiling [3] [1] |
In one poultry study comparing the two approaches for characterizing gut microbiota, shotgun sequencing identified 152 statistically significant changes in genera abundance between GI compartments that 16S sequencing failed to detect, while 16S found only 4 changes that shotgun sequencing did not identify [6]. This demonstrates the enhanced sensitivity of shotgun approaches for detecting subtle microbial community changes during development.
Functional Profiling Capabilities
A critical advantage of shotgun metagenomics is direct access to functional genetic elements, including metabolic pathways and antibiotic resistance genes (ARGs) [3] [55]. In the duck gut microbiota, integrated 16S rRNA and metagenomic sequencing revealed distinct functional specialization along intestinal segments: foregut microbes were enriched for genetic information processing (transcription, translation, replication), while hindgut microbes showed greater capacity for biosynthesis of secondary metabolites and diverse metabolic pathways [55].
Tools such as PICRUSt2, Tax4Fun2, and PanFP attempt to predict functional profiles from 16S rRNA data, but systematic evaluations demonstrate they "generally do not have the necessary sensitivity to delineate health-related functional changes in the microbiome" [53]. These inference tools show particularly poor performance for detecting subtle functional shifts associated with human health conditions [53].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for GI Microbiota Studies
| Item | Function | Application Notes |
|---|---|---|
| TGuide S96 Magnetic Bead-based DNA Extraction Kit | DNA extraction from complex samples | Optimal for soil/fecal samples; effective for difficult-to-lyse microorganisms [7] |
| TruSeq Nano DNA LT Library Prep Kit (Illumina) | Library preparation for 16S sequencing | Compatible with low DNA input; ideal for amplicon sequencing [7] |
| VAHTS Universal Plus DNA Library Prep Kit | Library preparation for shotgun sequencing | Includes tagmentation for efficient fragmentation and adapter addition [7] |
| QIIME2 Bioinformatics Pipeline | Analysis of 16S rRNA sequencing data | Incorporates DADA2 for quality control and denoising [7] |
| MEGAHIT Assembly Software | Metagenomic assembly from shotgun reads | Efficient for complex microbial communities; maintains strain diversity [7] |
| MetaGeneMark | Gene prediction from metagenomic assemblies | Identifies coding regions in assembled contigs [7] |
| Greengenes/SILVA Databases | Taxonomic classification reference | Curated 16S rRNA databases for bacterial taxonomy assignment [7] [52] |
Advanced Considerations in Method Selection
Impact of Sequencing Depth and Coverage
The reliability of both 16S rRNA and shotgun metagenomic sequencing is highly dependent on sequencing depth. Research demonstrates that shotgun samples with fewer than 500,000 reads exhibit rarefaction curves that fail to reach a plateau, indicating insufficient sampling depth [6]. This is particularly relevant for developmental studies where microbial biomass may be limited, such as in fetal or early postnatal samples [7].
For 16S rRNA sequencing, the selection of variable regions significantly impacts taxonomic resolution. Computational experiments reveal that the V4 region performs poorly, with 56% of in-silico amplicons failing to confidently match their sequence of origin at the species level, while full-length 16S sequences correctly classify nearly all sequences [28]. Different variable regions also exhibit taxonomic biases; for example, V1-V2 performs poorly for Proteobacteria, while V3-V5 struggles with Actinobacteria classification [28].
Emerging Technologies: Full-Length 16S Sequencing
Third-generation sequencing platforms (PacBio SMRT, Oxford Nanopore) enable full-length 16S rRNA gene sequencing (≈1,500 bp), significantly improving taxonomic resolution compared to short-read approaches [28] [54]. Recent evaluations demonstrate that PacBio Sequel II sequencing assigns a higher proportion of reads to the species level (74.14%) compared to Illumina V3-V4 sequencing (55.23%) [54].
While samples sequenced using both Illumina and PacBio technologies show comparable clustering by microbial niche, the improved species-level discrimination of full-length 16S sequencing makes it particularly valuable for studying genera containing multiple species with highly similar 16S rRNA gene sequences (e.g., Streptococcus, Escherichia/Shigella) [54]. This enhanced resolution is crucial for developmental studies where specific species successional patterns may have important functional implications.
Figure 2: Decision framework for selecting appropriate sequencing methodology in GI microbiota development studies
The selection between 16S rRNA sequencing and shotgun metagenomics for studying gastrointestinal microbiota development depends on specific research questions, resources, and required resolution. 16S rRNA sequencing remains a cost-effective approach for comprehensive taxonomic profiling at genus level, particularly advantageous for low-biomass samples or studies requiring high sample throughput [3] [1]. Shotgun metagenomics provides superior species and strain-level resolution, multi-kingdom taxonomic coverage, and direct access to functional genetic elements, making it ideal for investigating functional potential and subtle taxonomic shifts during developmental transitions [3] [6].
For comprehensive investigations of GI microbiota development, a hybrid approach—using 16S rRNA sequencing for broad sampling across multiple subjects and timepoints, complemented by targeted shotgun metagenomics on key samples—often provides the most balanced strategy [1]. This approach leverages the cost-effectiveness and sensitivity of 16S sequencing for detecting compositional changes while utilizing shotgun metagenomics to gain deeper functional insights and higher taxonomic resolution for critical developmental transitions.
As sequencing technologies continue to advance and costs decrease, full-length 16S sequencing and shallow shotgun approaches are emerging as compelling alternatives that bridge the gap between these traditional methods, offering improved resolution at intermediate costs [3] [54]. The optimal methodology should be selected based on explicit research questions, recognizing that each approach contributes unique insights into the complex process of gastrointestinal microbiota development.
Navigating Challenges: Technical Limitations and Strategic Decision-Making
16S ribosomal RNA (rRNA) gene sequencing has served as a cornerstone technique in microbial ecology for decades, providing invaluable insights into the composition of bacterial and archaeal communities across diverse environments. Its cost-effectiveness and standardized workflows have made it particularly popular for large-scale microbiome studies. However, as research questions have evolved to demand greater taxonomic precision and functional insights, significant limitations inherent to 16S rRNA sequencing have become increasingly apparent. This guide objectively details three fundamental constraints—primer bias, limited taxonomic resolution, and the lack of direct functional data—by comparing 16S rRNA sequencing results directly to those obtained from shotgun metagenomic sequencing, the latter often serving as a more comprehensive reference point.
Primer Bias in 16S rRNA Sequencing
The Impact of Variable Region Selection
A core vulnerability of 16S rRNA sequencing lies in its reliance on PCR amplification of specific hypervariable regions (V1-V9) of the 16S rRNA gene. The choice of which region(s) to amplify is not neutral; different primer sets exhibit distinct amplification efficiencies due to sequence mismatches, leading to skewed representations of the actual microbial community [56]. This primer bias means that the same sample processed with different primer pairs can yield dramatically different taxonomic profiles.
Table 1: Impact of Primer Choice on Microbial Community Profiling
| Targeted V-Region | Example Primer Pairs | Key Documented Biases and Impacts |
|---|---|---|
| V1-V2 | 27F-338R [56] | Lower off-target amplification of human DNA in biopsy samples; can miss certain taxa like Fusobacteriota without primer modification [57]. |
| V3-V4 | 341F-785R [56] | Susceptible to off-target amplification of human DNA (e.g., mitochondrial DNA), leading to significant data loss in host-dominated samples [57]. |
| V4 | 515F-806R [56] | A widely used region, yet it may miss specific bacterial groups entirely (e.g., Bacteroidetes with 515F-944R primers) and is highly prone to human DNA co-amplification [56] [57]. |
| V4-V5 | 515F-944R [56] | Can completely miss entire phyla such as Bacteroidetes, drastically altering perceived community structure [56]. |
| V6-V8 | 939F-1378R [56] | Performance and biases are sample-dependent; independent validation is required for reliable profiling [56]. |
Experimental evidence underscores this critical limitation. In a systematic evaluation, human stool samples and mock communities sequenced with seven different primer pairs clustered primarily by primer choice rather than by sample origin [56]. Furthermore, the use of unsuitable primers can lead to the complete failure to detect specific, important taxa. For instance, the phylum Bacteroidetes was missed when using the 515F-944R primer pair targeting the V4-V5 region [56]. The problem of "off-target amplification" is particularly acute in clinical samples with high host-DNA content, such as biopsies from the gastrointestinal tract. One study found that primers targeting the V4 region produced sequencing data where an average of 70% of amplicon sequence variants (ASVs) were derived from the human genome, rendering the majority of the data useless for microbiome analysis [57]. In contrast, a modified V1-V2 primer set reduced this off-target amplification to nearly zero [57].
Figure 1: Workflow illustrating how primer choice introduces bias. The same sample, when amplified with different primer sets targeting different variable regions (V1-V2, V3-V4, V4), can yield fundamentally different and non-comparable taxonomic profiles.
Experimental Protocol: Assessing Primer Bias
To evaluate primer bias in a study, researchers often employ a combination of mock communities and comparative amplification.
- Mock Community Design: Create a mock microbial community with a known, defined composition of bacterial strains. The true abundance of each member is precisely known.
- Parallel Amplification and Sequencing: Extract DNA from the mock community and split it into multiple aliquots. Perform 16S rRNA amplification on each aliquot using a different primer set targeting various variable regions (e.g., V1-V2, V3-V4, V4). Sequence all libraries on the same platform.
- Bioinformatic Processing: Process the raw sequencing data through the same bioinformatics pipeline (e.g., QIIME2 with DADA2 for ASV inference) to generate taxonomic profiles for each primer set [7].
- Bias Quantification: Compare the observed abundance of each taxon in the sequencing data to its known abundance in the mock community. Calculate metrics like fold-change differences to identify which taxa are systematically over- or under-represented by each primer set [56].
Limited Taxonomic Resolution of 16S rRNA Sequencing
Genus-Level vs. Species-Level Identification
While shotgun metagenomics can frequently achieve species-level and sometimes even strain-level resolution, 16S rRNA sequencing is often limited to genus-level assignments [10] [1]. This cap stems from the fact that short reads from a single hypervariable region simply do not contain enough nucleotide variability to distinguish between closely related species [9]. Consequently, 16S rRNA sequencing provides a blurred picture of the microbial community, missing critical details that are often biologically and clinically relevant.
Table 2: Taxonomic Resolution: 16S rRNA vs. Shotgun Metagenomics
| Metric | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Typical Resolution | Genus level (sometimes species) [1] | Species level (sometimes strains and single nucleotide variants) [1] |
| Detectable Taxa | Bacteria and Archaea only [1] | All domains: Bacteria, Archaea, Viruses, Fungi, and other micro-eukaryotes [1] |
| Community Depth | Detects only part of the community, weighted toward dominant members [6] [4] | Reveals a broader and deeper spectrum of taxa, including low-abundance organisms [6] |
| Quantitative Correlation | Lower correlation with shotgun data at finer taxonomic levels (species) due to database and resolution disagreements [4] | Serves as a reference for species-level abundance; higher correlation with 16S at coarser levels (genus, family) [4] |
Direct comparative studies highlight this resolution gap. A study on the chicken gut microbiome found that shotgun sequencing identified a statistically significant higher number of taxa than 16S sequencing, with the additional taxa primarily belonging to less abundant genera [6]. Similarly, a large study on human colorectal cancer found that while the two methods showed general agreement at the genus level, they "highly differed" at the species level, partly due to disagreements in reference databases [4]. The limitations of 16S sequencing are not just about missing rare taxa; they also impact quantitative accuracy. The data from 16S sequencing is typically sparser and exhibits lower alpha diversity compared to shotgun data [4]. This is compounded by variation in 16S rRNA gene copy numbers between different bacterial taxa, which can confound abundance estimates—a problem that shotgun metagenomics avoids [53].
The Lack of Functional Data
Inference vs. Direct Measurement
The 16S rRNA gene is a phylogenetic marker; its sequence reveals "who is there" but provides no direct information about the functional capabilities of the community (the "what are they doing"). While tools like PICRUSt2 and Tax4Fun2 have been developed to infer gene families and metabolic pathways from 16S rRNA data, these predictions are indirect, relying on the assumption that closely related taxa have similar functional gene repertoires [53]. In contrast, shotgun metagenomics directly sequences all genes in a sample, allowing for a comprehensive and direct profile of the community's functional potential, including metabolic pathways, virulence factors, and antibiotic resistance genes [1].
The table below summarizes the key differences in functional profiling capability.
Table 3: Functional Profiling Capabilities: Inference vs. Shotgun Sequencing
| Aspect | 16S rRNA with Predictive Tools (e.g., PICRUSt2) | Shotgun Metagenomic Sequencing |
|---|---|---|
| Data Type | Inferred / Predicted functional potential [53] | Direct measurement of gene content (functional potential) [1] |
| Basis | Uses taxonomic data and reference genomes to predict gene families [53] | Sequences all DNA; identifies microbial genes via alignment to functional databases (e.g., KEGG, CAZy) [7] [1] |
| Sensitivity to Health-Related Changes | Generally lacks the necessary sensitivity to delineate subtle, health-related functional changes in the microbiome [53] | Capable of identifying specific functional genes and pathways associated with health and disease states [58] [4] |
| Key Limitation | Predictions are limited by the quality and completeness of reference genomes and can be misled by horizontal gene transfer [53] | Analysis is dependent on functional reference databases, which may be incomplete for novel genes [1] |
A systematic benchmark study raised serious concerns about the reliability of functional inference from 16S rRNA data, particularly for identifying subtle changes linked to human health. The study concluded that 16S rRNA gene-based functional inference tools generally do not have the necessary sensitivity to delineate health-related functional changes in the microbiome and should thus be used with care [53]. Furthermore, while these tools might show high correlation in gene abundance with metagenomic data, this correlation can persist even when sample labels are permuted, indicating that correlation alone is a poor metric for assessing prediction accuracy [53]. For discovering disease-specific biomarkers, the direct approach of shotgun sequencing is superior. For example, a study on colorectal cancer used Nanopore sequencing of the full-length 16S rRNA gene (V1-V9) to identify specific bacterial biomarkers like Parvimonas micra and Fusobacterium nucleatum at species level, which was a significant improvement over standard short-read 16S approaches [58].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Kits for 16S rRNA and Metagenomic Studies
| Item | Function / Application | Example Use Case |
|---|---|---|
| OMNIgene GUT OMR-200 Tube | Non-invasive stool collection and ambient stabilization of microbial DNA [9]. | Population-scale gut microbiome studies where immediate freezing is impractical. |
| NucleoSpin Soil Kit / DNeasy PowerLyzer PowerSoil Kit | Efficient DNA extraction from complex biological samples (stool, soil) while inhibiting humic acids and other PCR inhibitors [4]. | Standardized DNA extraction for both 16S and shotgun sequencing from fecal samples [4]. |
| TruSeq Nano DNA LT Library Prep Kit | Preparation of sequencing libraries for Illumina platforms from low-input amplicon DNA [7]. | 16S rRNA amplicon sequencing on Illumina MiSeq/NovaSeq platforms [7]. |
| VAHTS Universal Plus DNA Library Prep Kit | Preparation of whole-genome shotgun sequencing libraries for Illumina platforms [7]. | Shotgun metagenomic sequencing on Illumina NovaSeq 6000 [7]. |
| SILVA Database | A comprehensive, curated database of aligned ribosomal RNA sequences for taxonomic classification [56] [7]. | Assigning taxonomy to 16S rRNA ASVs or OTUs [7] [4]. |
| Integrated Gene Catalog (IGC) / UHGG | A curated database of non-redundant human gut microbial genes for functional annotation. | Functional annotation of metagenomic sequencing reads from human stool samples. |
The limitations of 16S rRNA sequencing—significant primer bias, a cap on taxonomic resolution at the genus level, and an inability to provide direct functional insights—are well-documented and supported by extensive comparative data. Primer choice can fundamentally alter the perceived microbial community structure, while the technique's inherent resolution fails to capture the species-level diversity and functional capacity revealed by shotgun metagenomics. For studies where the research question demands accurate quantification beyond dominant genera, species-level identification, or direct knowledge of community function, shotgun metagenomic sequencing is the more powerful and appropriate tool. The choice between them should be a deliberate one, guided by the specific scientific objectives, acknowledging that 16S rRNA sequencing provides a useful but often incomplete picture of the microbial world.
In the field of microbial ecology, researchers primarily rely on two powerful sequencing technologies to profile complex microbial communities: 16S rRNA gene sequencing and shotgun metagenomic sequencing. The choice between these methods involves critical trade-offs concerning data resolution, cost, and analytical complexity. While 16S sequencing provides a cost-effective way to profile bacterial and archaeal composition, shotgun metagenomics offers superior taxonomic resolution and direct functional insights but is often hindered by significant limitations. This guide provides an objective comparison of these techniques, focusing on three major challenges in shotgun metagenomics: host DNA contamination, high operational costs, and complex bioinformatics requirements. We summarize experimental data to help researchers and drug development professionals select the most appropriate methodology for their projects.
Technical Comparison: 16S rRNA vs. Shotgun Metagenomic Sequencing
The fundamental difference between these techniques lies in their scope. 16S rRNA sequencing is an amplicon-based method that targets and amplifies a specific region of the 16S rRNA gene found in all bacteria and archaea, which is then sequenced to identify the types of microbes present [1] [59]. In contrast, shotgun metagenomic sequencing is a comprehensive approach that fragments and sequences all the DNA in a sample—microbial and host—which is then reconstructed bioinformatically to identify species and genes [1].
Table: Head-to-Head Comparison of 16S rRNA and Shotgun Metagenomic Sequencing
| Factor | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Cost per Sample | ~$50 USD [1] | Starting at ~$150; price depends on sequencing depth [1] |
| Taxonomic Resolution | Genus-level (sometimes species); dependent on targeted region(s) [1] | Species-level (sometimes strains and single nucleotide variants) [1] [6] |
| Taxonomic Coverage | Bacteria and Archaea only [1] | All domains of life (Bacteria, Archaea, Fungi, Viruses) [1] |
| Functional Profiling | No (only predicted via tools like PICRUSt) [1] | Yes (direct profiling of microbial genes and functional potential) [1] |
| Bioinformatics Requirements | Beginner to Intermediate [1] | Intermediate to Advanced [1] |
| Sensitivity to Host DNA | Low (PCR specifically targets the 16S gene) [1] | High (sequences all DNA; varies with sample type) [1] [60] |
| Experimental Bias | Medium to High (dependent on primer choice and targeted variable region) [1] | Lower ("untargeted," though biases can be introduced) [1] |
Experimental Analysis of Key Limitations
Host DNA Contamination
Host DNA contamination is a major constraint in shotgun metagenomics, particularly in samples with low microbial biomass. This contamination consumes sequencing depth, reducing the sensitivity for detecting low-abundance pathogens or community members.
Experimental Evidence: A 2025 clinical study of body fluid samples directly compared host DNA proportions in whole-cell DNA (wcDNA) versus cell-free DNA (cfDNA) metagenomic sequencing. The mean host DNA proportion in wcDNA mNGS was 84%, which was significantly lower than the 95% observed in cfDNA mNGS (p < 0.05). This higher host proportion in cfDNA mNGS corresponded with a lower concordance rate with culture results (46.67%) compared to wcDNA mNGS (63.33%) [25]. This demonstrates that the sample type and DNA extraction method critically influence the severity of host contamination.
Impact on Sensitivity: High host DNA content directly impairs detection sensitivity. A study using a synthetic bacterial community found that with high host DNA (99%), reliance on marker-gene-based tools like MetaPhlAn2 led to nine species becoming undetectable. However, switching to a more sensitive read-binning tool (Kraken 2 with Bracken) allowed all 20 expected organisms to be detected even with 99% host DNA, though off-target "contaminant" reads then exceeded the counts of many target genera [60]. This highlights the interplay between host DNA, sequencing depth, and bioinformatic tool selection.
High Cost and Sequencing Depth Requirements
The comprehensive nature of shotgun metagenomics makes it substantially more expensive than 16S rRNA sequencing. While 16S sequencing costs approximately $50 per sample, shotgun sequencing starts at around $150 per sample, with the final price heavily dependent on the required sequencing depth [1]. This cost disparity is a critical factor in designing studies, especially those with large sample sizes.
Sequencing Depth vs. Identification Power: The relationship between sequencing depth and taxonomic identification in shotgun sequencing was demonstrated in a 2021 chicken gut microbiome study. Researchers classified shotgun samples into two groups: a "low-depth" group with <500,000 reads and a "high-depth" group with >500,000 reads. The low-depth group showed highly skewed relative species abundance distributions and did not reach a plateau in rarefaction curves, indicating poor sampling of the microbial diversity. In contrast, the high-depth group showed more symmetrical distributions and better sampling [6]. This confirms that insufficient depth truncates the observable diversity, particularly for low-abundance taxa.
The Shallow Shotgun Approach: To bridge the cost-resolution gap, "shallow" shotgun metagenomics has emerged. This approach combines more samples in a single run and uses a modified protocol with fewer reagents, providing over 97% of the compositional and functional data of deep sequencing at a cost similar to 16S rRNA gene sequencing. However, it is currently best suited for samples with a high microbial-to-host DNA ratio, such as fecal samples [1].
Table: Cost and Performance Comparison Across Sequencing Strategies
| Sequencing Strategy | Estimated Cost per Sample | Typical Sequencing Depth | Key Advantages | Key Limitations |
|---|---|---|---|---|
| 16S rRNA Sequencing | ~$50 [1] | 50,000 reads [9] | Highly cost-effective for large-scale studies; simpler analysis [1] | Limited to bacteria/archaea; genus-level resolution; no direct functional data [1] |
| Shallow Shotgun | Similar to 16S [1] | Lower than deep shotgun | Good compositional data at lower cost; reveals functional potential [1] | Best for high-microbial-biomass samples; may miss rare species [1] |
| Deep Shotgun | Starting at ~$150 [1] | Millions of reads (e.g., 8 GB data) [25] | Species/strain-level resolution; profiles all microbial domains; direct functional analysis [1] [6] | High cost; requires advanced computing resources; complex data analysis [1] |
Complex Bioinformatics and Data Challenges
The richness of shotgun metagenomic data comes with a significant computational burden. The analysis requires more powerful computers, time, and expertise compared to 16S rRNA data [1]. A key challenge is the reliance on reference databases, which are still growing and can lead to false positives or impede the discovery of novel microbes if they are not comprehensive [9] [6].
Bioinformatic Pipelines and Tool Selection: The choice of bioinformatic tool significantly impacts results, especially in challenging scenarios. As noted in the host DNA contamination section, a tool like Kraken 2/Bracken (a read-binning approach) demonstrated superior sensitivity for detecting all expected organisms in a high-host-DNA background, whereas a marker-gene-based tool like MetaPhlAn2 failed to detect several low-abundance species under the same conditions [60]. This shows that tool selection is not trivial and must be tailored to the specific experimental conditions.
The Contamination Challenge: Sensitive tools are a double-edged sword. The same study found that with 99% host DNA, over 10% of microbial reads were "off-target," and the counts for these contaminant genera exceeded the counts of 13 out of 17 target genera. However, applying a contaminant detection tool (Decontam) successfully removed 61% of off-target species and 79% of the off-target reads [60]. This underscores the necessity of incorporating robust contamination screening into metagenomic workflows, particularly for low-biomass samples.
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key reagents and materials used in a typical shotgun metagenomics workflow, based on protocols cited in the search results [60] [25].
Table: Key Research Reagent Solutions for Metagenomic Sequencing
| Item | Function/Description | Example Product |
|---|---|---|
| DNA Extraction Kit | Extracts total genomic DNA (host and microbial) from sample precipitates for whole-cell DNA metagenomics. | Qiagen DNA Mini Kit [25] |
| Cell-Free DNA Extraction Kit | Specifically extracts microbial cell-free DNA from sample supernatants; used for cfDNA mNGS. | VAHTS Free-Circulating DNA Maxi Kit [25] |
| DNA Library Prep Kit | Prepares sequencing libraries from extracted DNA by fragmenting, end-repairing, and adding adapters/indexes. | VAHTS Universal Pro DNA Library Prep Kit for Illumina [25] |
| Magnetic Beads | Used in various cleanup and size-selection steps during library preparation to purify DNA. | Included in VAHTS Kits [25] |
| Proteinase K | An enzyme used during DNA extraction to digest proteins and degrade nucleases, facilitating more efficient DNA release. | Included in VAHTS Kits [25] |
| Negative Control Reagents | Sterile water or buffer processed alongside samples to identify reagent or environmental contaminants. | Nuclease-Free Water [60] |
The choice between 16S rRNA and shotgun metagenomic sequencing is not a matter of which is universally better, but which is more appropriate for the specific research question, budget, and analytical capacity.
- Opt for 16S rRNA sequencing when the study involves a large number of samples, the primary goal is to profile bacterial and archaeal communities at the genus level, and when budget or bioinformatics expertise is limited [1]. Its lower sensitivity to host DNA also makes it a robust choice for samples like skin or oral swabs [1].
- Choose shotgun metagenomic sequencing when the research requires species- or strain-level resolution, comprehensive functional profiling, or the detection of non-bacterial microorganisms (e.g., fungi, viruses) [1] [6]. This method is powerful for linking microbial communities to functional traits, such as in drug discovery or detailed mechanistic studies.
To mitigate the limitations of shotgun metagenomics, researchers can adopt strategies such as host DNA depletion protocols, the use of shallow sequencing for large-scale studies, leveraging more sensitive and contamination-aware bioinformatics tools like Kraken 2 with Decontam, and ensuring sufficient sequencing depth (typically >500,000 reads per sample) to adequately capture community diversity [1] [60] [6]. By understanding these trade-offs and limitations, scientists can make informed decisions that optimize their research outcomes in microbiome science and drug development.
Database Dependencies and Their Impact on Taxonomic Assignment Accuracy
In microbiome research, the choice of sequencing method—16S rRNA gene sequencing or shotgun metagenomics—defines the landscape of available bioinformatics tools and reference databases. This selection is not merely technical but fundamentally shapes the taxonomic composition and functional interpretation derived from a dataset. While much attention is given to sequencing platforms and bioinformatics pipelines, the impact of the reference database itself is a critical, yet sometimes overlooked, factor. The database acts as the lens through which raw sequencing data is interpreted; its comprehensiveness, curation quality, and taxonomic framework directly determine the accuracy, resolution, and very validity of taxonomic assignments. This guide objectively compares how database choice differentially impacts 16S rRNA and shotgun metagenomic sequencing, providing researchers with a data-driven framework for selecting the most appropriate methods and resources for their specific study systems and hypotheses.
Fundamental Differences in Database Architecture and Application
The core distinction in database usage between 16S rRNA and shotgun metagenomic sequencing stems from the fundamental nature of the data each method generates.
16S rRNA Gene Sequencing Databases (e.g., SILVA, Greengenes, RDP) contain curated collections of the 16S rRNA gene sequence from identified bacteria and archaea. Taxonomic assignment works by comparing sequenced amplicons (typically of one or more hypervariable regions) to this reference collection [36] [4]. The resolution is inherently limited by the degree of sequence variation within the 16S gene itself, often capping at the genus level and sometimes preventing discrimination between closely related species [1] [18]. Furthermore, the specific hypervariable region sequenced (e.g., V1-V2, V3-V4) significantly influences the taxonomic profile obtained, as different regions possess varying degrees of discriminative power [61].
Shotgun Metagenomic Sequencing Databases (e.g., RefSeq, GTDB, UHGG) are composed of whole-genome sequences. Classifiers map short reads to these genomes or to a set of clade-specific marker genes, allowing for identification down to the species and sometimes even strain level [1] [18]. This method also enables the detection of all domains of life—bacteria, archaea, fungi, and viruses—from a single dataset, provided the DNA extraction method is comprehensive [1]. However, its effectiveness is heavily dependent on the presence of a high-quality, representative genome in the database for the microbe in question.
Table 1: Core Characteristics of Database Types for 16S vs. Shotgun Sequencing
| Feature | 16S rRNA Databases | Shotgun Metagenomic Databases |
|---|---|---|
| Primary Content | Curated 16S rRNA gene sequences | Whole microbial genomes |
| Taxonomic Resolution | Genus-level (sometimes species) | Species-level, strain-level (with deep sequencing) |
| Taxonomic Coverage | Bacteria and Archaea | All domains of life (Bacteria, Archaea, Fungi, Viruses) |
| Key Databases | SILVA, Greengenes, RDP | RefSeq, GTDB, UHGG |
| Inherent Limitation | Limited by 16S gene variation; primer/probe bias | Dependent on genome quality and representativeness |
Experimental Evidence of Database-Dependent Performance
Impact on Shotgun Metagenomic Classification Accuracy
A seminal 2022 study directly quantified the impact of reference database choice on taxonomic classification accuracy using shotgun metagenomic data [62]. Researchers created a simulated metagenomic dataset from known rumen microbial genomes and classified it using Kraken 2 against multiple custom databases.
Table 2: Database Performance in Metagenomic Read Classification [62]
| Database | Description | Key Finding on Classification |
|---|---|---|
| RefSeq | Standard public database of complete genomes | Poor classification rate for a rumen microbiome sample |
| Hungate | Custom database of 460 cultured rumen microbes | Improved classification rate vs. RefSeq alone |
| RUG | Database of Metagenome-Assembled Genomes (MAGs) from rumen | Greatest improvement in classification rate and accuracy |
| RefSeq + Hungate | Combined standard and cultured genomes | Enhanced performance over RefSeq |
| RefSeq + RUG | Combined standard and MAGs | Substantial improvement in classification accuracy |
The study demonstrated that classification accuracy varies significantly between databases and taxonomic levels. Crucially, it highlighted that the addition of Metagenome-Assembled Genomes (MAGs), which represent uncultivated microbes from the specific environment under study, substantially improved read classification accuracy. This underscores that accurate classification requires the reference database to be environmentally representative, not just large [62].
Comparative Performance in Clinical and Gut Microbiome Studies
A 2024 prospective clinical study compared shotgun metagenomics to Sanger 16S sequencing for bacterial identification in culture-negative samples [63]. Shotgun metagenomics demonstrated significantly better performance at the species level, identifying a bacterial etiology in 46.3% of cases versus 38.8% with Sanger 16S. The difference was more pronounced when comparing species-level identifications (28/67 vs. 13/67) [63]. This enhanced resolution is a direct benefit of shotgun sequencing's reliance on whole-genome databases, which contain the genetic variation necessary for species-level discrimination.
Furthermore, a comprehensive 2024 study of 156 human stool samples sequenced by both 16S and shotgun methods found that while the two techniques could reveal common patterns, "16S will tend to show only part of the picture, giving greater weight to dominant bacteria in a sample" [4]. The study noted that abundance data from 16S was sparser and exhibited lower alpha diversity. Discrepancies were particularly large at lower taxonomic ranks, which the authors attributed partially to "disagreement in reference databases" used for the two methods [4].
Methodological Considerations and Best Practices
Detailed Experimental Protocol for Database Comparison
To systematically evaluate database choice in a microbiome study, researchers can adopt the following validated experimental workflow [62] [4]:
Sample Preparation & Sequencing: Extract total genomic DNA from the sample set (e.g., human stool, environmental sample). For a balanced comparison, split each sample aliquot for both 16S rRNA amplicon sequencing (targeting an appropriate hypervariable region like V3-V4 or V1-V2) and whole-genome shotgun sequencing on a platform such as Illumina.
Bioinformatic Processing:
- 16S Data: Process raw sequences through a pipeline like DADA2 or QIIME2 to infer Amplicon Sequence Variants (ASVs). Perform taxonomic assignment against major 16S databases (SILVA, Greengenes).
- Shotgun Data: Perform quality trimming and filter out host DNA (if applicable). Use a read classifier like Kraken 2 for taxonomic profiling. Conduct parallel classifications against multiple databases, including:
- A standard database (e.g., RefSeq).
- A specialized database for the sample type (e.g., Hungate for rumen).
- A custom database augmented with relevant MAGs.
Comparative Analysis:
- Taxonomic Composition: Compare the relative abundances of major taxa and the number of taxa identified at different ranks (genus, species) across different database results.
- Alpha and Beta Diversity: Calculate and compare diversity indices (e.g., Shannon, Chao1) and beta-diversity ordinations (e.g., PCoA) derived from the different taxonomic profiles.
- Accuracy Assessment (if possible): Use a mock microbial community with a known composition as a positive control to benchmark the accuracy of each database and sequencing method.
Table 3: Key Reagents and Resources for Microbiome Database Studies
| Item/Category | Function | Example Products/Tools |
|---|---|---|
| DNA Extraction Kits | Isolation of high-quality, unbiased genomic DNA from samples. | NucleoSpin Soil Kit, DNeasy PowerLyzer PowerSoil Kit |
| 16S rRNA Primers | Amplification of specific hypervariable regions for targeted sequencing. | 341F/806R (V3-V4), 27F/338R (V1-V2) |
| Sequencing Platforms | Generation of high-throughput sequencing data. | Illumina MiSeq/NovaSeq, PacBio Sequel (for full-length 16S) |
| Bioinformatics Pipelines | Processing raw sequences into taxonomic and functional profiles. | QIIME2, DADA2 (16S); Kraken 2, MetaPhlAn (Shotgun) |
| Reference Databases | Essential resources for assigning taxonomy to sequence reads. | SILVA, Greengenes (16S); RefSeq, GTDB (Shotgun) |
| Mock Community | Control standard with known composition to benchmark accuracy. | ZymoBIOMICS Microbial Community Standard |
Integrated Workflow and Decision Framework
The following diagram summarizes the experimental workflow and the pivotal role of database choice in determining taxonomic assignment outcomes for the two main sequencing approaches.
The decision to use 16S rRNA or shotgun metagenomic sequencing, and consequently their associated databases, should be guided by the study's primary objective, sample type, and resources.
Choose 16S rRNA Sequencing When: The research question focuses on broad taxonomic composition (community ecology) or shifts in dominant bacterial/archaeal populations at the genus level [1] [4]. It is also the preferred choice for studies with limited budget, large sample sizes, or when analyzing samples with high host DNA contamination (e.g., tissue, skin swabs), as the targeted approach minimizes host read generation [1].
Choose Shotgun Metagenomic Sequencing When: The goal requires species- or strain-level identification, profiling of non-bacterial microbes (fungi, viruses), or analysis of the functional potential (genes and pathways) of the community [1] [63] [18]. It is highly recommended for stool samples and in-depth analyses where comprehensive characterization is the priority [4]. To maximize its potential, researchers should invest in building or selecting environmentally relevant reference databases, including the use of MAGs where possible [62] [18].
The dependence of taxonomic assignment on reference databases is a fundamental reality in microbiome research, but this dependency manifests differently for 16S rRNA and shotgun metagenomic sequencing. 16S rRNA analysis offers a cost-effective, focused view of the bacterial and archaeal community but is constrained by the resolution of the 16S gene and the primers used. In contrast, shotgun metagenomics provides a comprehensive, high-resolution census of the entire microbial community but demands robust computational resources and, most critically, high-quality, representative reference databases for accurate interpretation.
The experimental data clearly shows that database choice is not a neutral parameter but a primary driver of taxonomic results. Inaccurate or incomplete databases lead directly to misclassification and a loss of biological insight. Therefore, the most advanced sequencing technology and powerful bioinformatics pipeline cannot compensate for an inadequate database. For researchers, this means that database selection—and increasingly, the curation of custom, study-specific databases enhanced with MAGs—must be considered a primary, deliberate step in experimental design, one that is as crucial as the choice of sequencing technology itself.
For researchers navigating the complex landscape of microbiome analysis, the choice between 16S rRNA gene sequencing and shotgun metagenomic sequencing represents a fundamental trade-off between cost, resolution, and analytical depth. This guide provides an objective comparison of these foundational methods to inform strategic experimental design in pharmaceutical and biomedical research.
Core Methodological Comparison: 16S rRNA vs. Shotgun Metagenomics
The two primary culture-independent methods for microbial community analysis differ fundamentally in their targets and capabilities [9] [44].
16S rRNA sequencing employs PCR to amplify and sequence specific variable regions of the bacterial 16S ribosomal RNA gene, a conserved genetic marker present in all bacteria and archaea [64] [10]. The approach leverages the fact that this gene contains both highly conserved regions (ideal for primer binding) and variable regions (providing taxonomic differentiation) [64].
In contrast, shotgun metagenomic sequencing takes an untargeted approach by fragmenting and sequencing all DNA present in a sample, enabling simultaneous identification of bacteria, archaea, viruses, fungi, and other microorganisms without prior amplification or selection [18] [64] [44].
Table 1: Fundamental Characteristics of 16S rRNA and Metagenomic Sequencing
| Feature | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Target | 16S rRNA gene (specific regions) [64] | Entire metagenome (all DNA) [64] [44] |
| Taxonomic Resolution | Genus-level (sometimes species) [64] [44] | Species- and strain-level [18] [44] |
| Functional Insights | Limited to prediction algorithms [18] | Direct assessment of genes and pathways [18] [64] |
| Organisms Detected | Bacteria and Archaea only [64] | Bacteria, Archaea, Viruses, Fungi, Eukaryotes [64] [44] |
| Bioinformatics Complexity | Low to Moderate [64] [10] | High [64] [10] |
| Reference Databases | Well-established (SILVA, Greengenes) [64] [10] | Evolving (RefSeq, KEGG, CARD) [18] [10] |
Quantitative Performance and Cost Analysis
Sequencing Depth and Diversity Capture
The relationship between sequencing depth and microbial diversity assessment varies significantly between methods and sample types. A 2021 comparative analysis of 338 pediatric fecal samples revealed that 16S rRNA profiling identified a larger number of genera across multiple age groups compared to standard metagenomic sequencing [9]. However, each method detected unique genera missed by the other, highlighting complementary strengths [9].
For shotgun metagenomics, the required sequencing depth depends heavily on sample complexity. Infant gut samples with lower microbial diversity may be adequately characterized at lower sequencing depths, while adult samples with higher diversity require deeper sequencing to capture rare taxa [9]. One core facility offers tiered metagenomic sequencing services at 5 million reads for basic characterization versus 30 million reads for comprehensive analysis [65].
Cost Considerations and Budget Impact
The financial implications of method selection are substantial, particularly for large-scale studies:
Table 2: Cost and Practical Considerations for Microbial Sequencing
| Parameter | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Cost per Sample | ~$55 (sequencing) + $20 (bioinformatics) [65] | ~$90 (5M reads) to $190 (30M reads) [65] |
| Additional Costs | $15/sample (DNA extraction) [65] | $15/sample (DNA extraction) [65] |
| Data Volume | ~50,000 reads/sample often sufficient [9] | Millions of reads per sample required [9] |
| Infrastructure Requirements | Standard computing resources [10] | High-performance computing essential [10] |
| Hands-on Time | Moderate (PCR amplification required) [10] | Moderate (library preparation) [10] |
The global market data reflects the economic dynamics, with the metagenomics sector projected to grow from USD 2.68 billion in 2025 to USD 8.39 billion by 2034, driven by expanding applications in drug discovery and clinical diagnostics [66].
Experimental Design and Workflow Considerations
Method Selection Framework
Choosing between these methods requires alignment with specific research objectives:
Choose 16S rRNA sequencing when:
- Conducting large-scale taxonomic surveys with limited budget [64] [44]
- Focusing exclusively on bacterial and archaeal communities [64]
- Performing exploratory studies to map microbial diversity [64]
- Working with samples potentially contaminated with host DNA [44]
Choose shotgun metagenomics when:
- Investigating functional potential, metabolic pathways, or resistance genes [18] [44]
- Detection of viruses, fungi, or eukaryotic microbes is required [64] [44]
- Strain-level differentiation is critical [18] [44]
- Studying novel or poorly characterized microbial communities [18]
Workflow Visualization and Experimental Protocols
The methodological workflows differ significantly in their core processes:
Diagram 1: Comparative Workflows for Microbial Community Analysis
Research Reagent Solutions and Essential Materials
Successful implementation requires careful selection of laboratory reagents and computational resources:
Table 3: Essential Research Reagents and Resources for Microbial Sequencing
| Item Category | Specific Examples | Function/Purpose |
|---|---|---|
| DNA Extraction Kits | OMNIgene·GUT collection tubes [9] | Sample stabilization and DNA preservation |
| Amplification Reagents | 16S V3-V4 primers [44] [10] | Target-specific amplification for 16S sequencing |
| Library Prep Kits | Commercial NGS library preparation systems | Fragment processing and adapter ligation |
| Sequencing Platforms | Illumina, Oxford Nanopore, PacBio [66] | High-throughput DNA sequencing |
| Bioinformatics Tools | QIIME2, Mothur (16S) [10]MetaPhlAn, HUMAnN (Metagenomics) [10] | Data processing, taxonomic assignment, functional analysis |
| Reference Databases | SILVA, Greengenes (16S) [64] [10]KEGG, CARD, RefSeq (Metagenomics) [18] [10] | Taxonomic classification and functional annotation |
Emerging Innovations and Clinical Applications
Technological Advancements
The field is rapidly evolving with several key innovations enhancing both approaches:
Long-read sequencing technologies from Oxford Nanopore and PacBio now enable full-length 16S rRNA sequencing, improving species-level discrimination [67]. Clinical validation studies demonstrate that long-read 16S sequencing provides higher taxonomic resolution at approximately one-third the cost of Sanger sequencing ($25.30 vs. $74 per test) [67].
Genome-resolved metagenomics represents a transformative advancement, allowing reconstruction of metagenome-assembled genomes (MAGs) directly from complex samples [18]. This approach facilitates strain-level tracking, functional characterization, and discovery of novel microbial dark matter [18].
Clinical Translation and Diagnostic Applications
Both methods show increasing utility in clinical diagnostics and therapeutic development:
16S rRNA sequencing provides rapid pathogen detection in cases of culture-negative infections, with demonstrated utility in bone and joint infections where it improved diagnostic yield by approximately 18% over culture alone [68].
Shotgun metagenomics enables comprehensive pathogen detection and antimicrobial resistance profiling in complex clinical scenarios. In critically ill patients with sepsis, metagenomic sequencing identified pathogens up to 30 hours earlier than traditional cultures while simultaneously detecting resistance genes [68]. The method has also proven valuable for monitoring strain engraftment following fecal microbiota transplantation (FMT) and guiding personalized microbiome therapies [68].
Strategic Implementation Framework
Decision Algorithm for Method Selection
Diagram 2: Method Selection Decision Framework
Integrated Approaches and Future Directions
For comprehensive studies, researchers increasingly employ hybrid strategies that leverage the complementary strengths of both methods [44]. This may involve using 16S rRNA sequencing for large-scale screening followed by targeted metagenomic analysis of select samples, or implementing shallow shotgun sequencing as a cost-effective intermediate approach [44].
The growing integration of artificial intelligence and machine learning with metagenomic data analysis is enhancing pattern recognition, anomaly detection, and predictive modeling of microbial community dynamics [66]. These computational advances are particularly valuable for identifying microbial signatures associated with disease states and treatment outcomes in pharmaceutical development [68] [66].
Selecting the optimal sequencing depth and method requires careful consideration of research goals, sample characteristics, and resource constraints. While 16S rRNA sequencing remains the most cost-effective approach for comprehensive taxonomic profiling of bacterial communities, shotgun metagenomics provides unparalleled resolution and functional insights across all microbial domains. The emerging paradigm favors method selection based on specific research questions rather than one-size-fits-all approaches, with integrated multi-method frameworks offering the most comprehensive path forward for advanced microbiome research in drug development and clinical applications.
In the field of microbial analysis, two powerful sequencing methods have become foundational: 16S rRNA gene sequencing and shotgun metagenomic sequencing. While both techniques profile microbial populations from complex samples, their objectives, methodologies, resolutions, and outputs are distinctly different. This guide provides an objective, data-driven comparison to help researchers, scientists, and drug development professionals select the optimal tool for their specific research scenario.
The table below summarizes the core differences between these two technologies, providing a high-level overview for initial orientation.
| Feature | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Core Principle | Targets & amplifies a single, conserved gene [10] | Sequences all DNA in a sample indiscriminately [10] |
| Target Organisms | Bacteria and Archaea [10] | All domains: Bacteria, Archaea, Viruses, Fungi [10] |
| Typical Taxonomic Resolution | Genus-level (species-level with full-length sequencing) [10] [18] | Species-level and strain-level [10] [18] |
| Functional Insights | Limited to prediction from taxonomy (e.g., PICRUSt) [18] | Direct revelation of metabolic pathways, virulence factors, and AMR genes [7] [10] |
| Primary Workflow | PCR amplification of 16S gene regions → Sequencing [10] | Direct sequencing of fragmented total DNA (shotgun approach) [10] |
| Key Limitations | PCR bias, primer selection bias, limited resolution [10] [18] | High host DNA contamination, high cost, complex data analysis [10] [25] |
| Typical Cost | Lower [10] | Higher [10] |
| Data Output Size | Smaller, more manageable datasets [10] | Very large, computationally intensive datasets [10] |
Technical Performance and Experimental Data
Choosing a method requires understanding its real-world performance. The following tables consolidate quantitative data from peer-reviewed studies comparing these two approaches across different applications.
Table 1: Comparative Diagnostic Performance in Clinical Settings
Data from clinical studies highlight the trade-offs between sensitivity and specificity in pathogen identification.
| Study Context / Metric | 16S rRNA Sequencing | Shotgun Metagenomics | Citation |
|---|---|---|---|
| 73 culture-negative clinical samples (Sanger 16S vs. CMg) | 27% (20/73) clinically relevant detection rate | 70% sensitivity vs. 16S; detected clinically relevant bacteria in 19% (10/53) of 16S-negative samples | [19] |
| 41 body fluid samples (vs. culture as reference) | 58.54% (24/41) consistency with culture | 74.07% sensitivity; 70.7% (29/41) consistency with culture | [25] |
| 30 body fluid samples (wcDNA mNGS vs. cfDNA mNGS) | Not Applicable | wcDNA mNGS: 63.33% (19/30) concordance with culturecfDNA mNGS: 46.67% (14/30) concordance with culture | [25] |
| Host DNA Proportion | Not Applicable | Mean host DNA in wcDNA mNGS: 84%Mean host DNA in cfDNA mNGS: 95% (p < 0.05) | [25] |
Table 2: Taxonomic and Functional Profiling Capabilities
This comparison delves into the fundamental output differences between the two methods, from a goat microbiome study and technical reviews.
| Profiling Aspect | 16S rRNA Sequencing | Metagenomic Sequencing | Citation |
|---|---|---|---|
| Typical Data Output (per sample) | 852,694 high-quality reads (e.g., from fetal goat GI tract) | 1.08 billion final valid reads (e.g., from 7-day-old goat kid) [7] | [7] |
| Assembly Output | Not Applicable | 8.56 million contigs; 6.10 million predicted genes [7] | [7] |
| Functional Annotation | Limited to prediction (e.g., PICRUSt) [18] | Direct annotation via multiple databases (KEGG, CAZy, NR) revealing functional potential and AMR traits [7] | [7] [18] |
| Impact of Primer Selection | High; different primers detect unique taxa, though group differences remain detectable [12] | Not Applicable | [12] |
| Platform Comparison (Illumina vs. ONT) | ONT captures a broader range of taxa compared to Illumina [12] | High correlation between Illumina and ONT platforms for taxonomic diversity [12] | [12] |
Methodological Deep Dive: Experimental Protocols
To ensure reproducibility and informed decision-making, here are the detailed experimental protocols as cited in the research.
Protocol 1: Standard 16S rRNA Gene Sequencing Workflow
This protocol is adapted from the goat gastrointestinal microbiota study and comparative evaluations [7] [12].
- DNA Extraction: Extract genomic DNA from the sample (e.g., using a magnetic bead-based soil/fecal DNA kit) [7].
- PCR Amplification: Use universal primers targeting specific hypervariable regions (e.g., V3-V4) of the 16S rRNA gene. The choice of primer pair is critical and can significantly influence results [12].
- Library Preparation: Prepare the sequencing library from the amplified PCR products (amplicons) using a kit such as the Illumina TruSeq Nano DNA LT Library Prep Kit [7].
- Sequencing: Perform high-throughput paired-end sequencing on a platform like the Illumina MiSeq or NovaSeq [7].
- Bioinformatic Analysis:
- Quality Control & Denoising: Process raw sequences using a pipeline like QIIME2. This includes trimming primers, quality filtering, denoising, joining paired-end reads, and removing chimeras with DADA2 [7].
- Taxonomic Classification: Assign taxonomy to the resulting Amplicon Sequence Variants (ASVs) using a pre-trained classifier (e.g., Naive Bayes) against a reference database such as Greengenes or SILVA [7].
Protocol 2: Shotgun Metagenomic Sequencing Workflow
This protocol is outlined in the goat microbiome and clinical metagenomics studies [7] [25].
- DNA Extraction: Extract total genomic DNA from the sample, aiming for high yield and quality. The extraction protocol can bias the representation of certain taxa (e.g., Gram-positive bacteria with tough cell walls) [12].
- Library Preparation: Fragment the extracted DNA and prepare the sequencing library without a targeted amplification step. Kits such as the VAHTS Universal Plus DNA Library Prep Kit for Illumina are commonly used [7].
- Sequencing: Conduct shotgun sequencing on a high-output platform like the Illumina NovaSeq 6000 with a PE150 strategy, generating billions of reads [7].
- Bioinformatic Analysis:
- Quality Control & Host Depletion: Trim adapters and remove low-quality reads using tools like fastp. Subtract reads that align to the host genome (if applicable) using Bowtie2 [7] [25].
- Assembly & Binning: De novo assemble the clean reads into contigs using assemblers like MEGAHIT. Reconstruct individual microbial genomes from the assembled contigs through a process called "binning" to generate Metagenome-Assembled Genomes (MAGs) [7] [18].
- Gene Prediction & Annotation: Predict coding sequences on contigs or within MAGs using tools like MetaGeneMark. Functionally and taxonomically annotate the non-redundant gene catalog against databases like NR, KEGG, and CAZy [7].
Visualizing the Workflows
The diagram below illustrates the core procedural differences between the 16S rRNA and metagenomic sequencing workflows, highlighting the key steps where their methodologies diverge.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of either sequencing method relies on a suite of trusted reagents, kits, and software tools.
| Item Name | Function/Benefit | Applicable Field |
|---|---|---|
| TGuide S96 Magnetic Bead DNA Kit | Efficient genomic DNA extraction from complex samples like soil and feces [7]. | 16S & Metagenomics |
| VAHTS Universal Plus DNA Library Prep Kit | Prepares high-quality sequencing libraries from fragmented DNA for Illumina platforms [7]. | Metagenomics |
| TruSeq Nano DNA LT Library Prep Kit | Prepares sequencing libraries from amplified 16S rRNA amplicons [7]. | 16S Sequencing |
| QIIME 2 (Bioinformatics Platform) | A powerful, user-friendly platform for processing and analyzing 16S rRNA sequencing data, from denoising to taxonomic classification [7]. | 16S Sequencing |
| MEGAHIT | An efficient and fast assembler for de novo assembly of large and complex metagenomic datasets [7]. | Metagenomics |
| MetaPhlAn | A profiling tool that uses unique clade-specific marker genes to provide species-level taxonomic resolution from metagenomic data [19] [10]. | Metagenomics |
| Greengenes & SILVA Databases | Curated databases of 16S rRNA gene sequences used as references for taxonomic classification [7]. | 16S Sequencing |
| KEGG & CAZy Databases | Functional databases used for annotating genes and pathways identified in metagenomic studies (e.g., metabolic pathways, carbohydrate-active enzymes) [7]. | Metagenomics |
The Decision Matrix: Choosing Your Tool
To synthesize this information into an actionable guide, use the following matrix to align your research goals with the appropriate technology.
| Research Goal | Recommended Tool | Rationale and Considerations |
|---|---|---|
| Large-scale microbial community screening (e.g., environmental samples, cohort studies) | 16S rRNA Sequencing | Cost-effective for processing hundreds of samples. Provides robust community composition (alpha/beta diversity) at the genus level [10]. |
| Strain-level tracking (e.g., outbreak investigation) | Shotgun Metagenomics | Provides the resolution needed to distinguish between closely related bacterial strains and track transmission events [10] [18]. |
| Functional potential analysis (e.g., discovering novel enzymes, AMR genes) | Shotgun Metagenomics | Directly sequences and annotates functional genes, revealing metabolic pathways, virulence factors, and resistance genes without inference [7] [10] [51]. |
| Infectious disease diagnosis (culture-negative, polymicrobial, or rare pathogens) | Shotgun Metagenomics (with caveats) | Offers unbiased detection of all pathogens (bacterial, viral, fungal) in a single run. Superior sensitivity to 16S rRNA Sanger sequencing, though requires careful interpretation to manage background noise and ensure specificity [19] [25]. |
| Preliminary mapping of a new microbiome (e.g., gut, skin, oral) | 16S rRNA Sequencing | An excellent first step to economically characterize the overall structure, diversity, and key genera of an unknown microbial community [10]. |
| Microbial "dark matter" exploration (uncultured organisms) | Shotgun Metagenomics | Genome-resolved metagenomics (binning) can reconstruct genomes of novel, uncultured microbes, moving beyond database-dependent identification [18] [51]. |
Future Outlook and Emerging Trends
The field of microbiome research is rapidly evolving. Key trends include the rise of long-read sequencing (e.g., Oxford Nanopore Technologies, PacBio), which improves the assembly of complete microbial genomes from metagenomes [12] [10]. Furthermore, the future lies in multi-omics integration, where metagenomic data is combined with metatranscriptomics, metabolomics, and proteomics to gain a dynamic, systems-level understanding of microbial ecosystem function and its interaction with the host [10]. For the most challenging diagnostic and research questions, a hybrid approach that leverages both 16S and metagenomic sequencing on a subset of samples may provide the most comprehensive insights [10].
The study of low-biomass microbial environments, such as those potentially found in fetal tissues, the placenta, and blood, represents one of the most technically challenging frontiers in microbiome research. These environments harbor minimal microbial DNA, often approaching the detection limits of standard sequencing approaches [69]. The fascination with these niches is undeniable, as they promise to reveal previously unknown host-microbe interactions from the earliest stages of human development. However, this promise is tempered by significant technical challenges, most notably the profound risk of contamination from exogenous DNA sources that can readily distort results and lead to spurious biological conclusions [69] [70].
The field was sobered by the reevaluation of the "placental microbiome," once thought to harbor a resident microbial community but later shown to likely represent contamination introduced during sampling and laboratory processing [69] [70]. This example underscores a critical lesson: findings in low-biomass systems require extraordinary rigor in validation. The relative scarcity of authentic microbial signal DNA in these samples means that even minute amounts of contaminating DNA, which would be negligible in high-biomass samples like stool, can dominate the sequencing results and create the illusion of a microbial community where none exists [69]. This Consensus Statement will synthesize best practices derived from fetal microbiome studies and other low-biomass environments, providing a framework for conducting robust and reproducible research.
Methodological Comparison: 16S rRNA Sequencing vs. Shotgun Metagenomics
The choice between targeted 16S rRNA gene sequencing and whole-genome shotgun metagenomics is fundamental to any microbiome study design. Each method offers distinct advantages and suffers from particular vulnerabilities when applied to low-biomass samples. Understanding these trade-offs is essential for selecting the appropriate tool and correctly interpreting the resulting data.
Table 1: Core Methodological Differences Between 16S and Metagenomic Sequencing
| Feature | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Target | Amplification of specific 16S rRNA hypervariable regions [3] | All genomic DNA in a sample [3] |
| Taxonomic Resolution | Genus level (sometimes species); high false-positive rate at species level [3] [18] | Species and strain level for multiple kingdoms [3] [71] |
| Functional Profiling | Indirect prediction only (e.g., PICRUSt) [3] [18] | Direct detection of functional genes and pathways [3] [1] |
| Coverage | Bacteria and Archaea only [3] [1] | Bacteria, Archaea, Viruses, Fungi, Protists [3] |
| Host DNA Interference | Low (PCR amplifies only the target bacterial gene) [3] [1] | High (host DNA consumes sequencing reads) [3] [70] |
| Cost per Sample | Lower [3] [1] | Higher (especially deep sequencing) [3] [1] |
16S rRNA Gene Sequencing: A Targeted, Yet Limited, Approach
16S rRNA sequencing involves PCR amplification of specific hypervariable regions of the bacterial 16S ribosomal RNA gene, which is then sequenced [3] [1]. This targeted approach makes it less susceptible to host DNA interference, as the PCR step selectively enriches for bacterial sequences. This can be advantageous for samples like placental or fetal tissue, where human DNA is abundant [3]. However, this method has significant limitations. Its taxonomic resolution is generally restricted to the genus level, with a high rate of false positives when attempting species-level identification [3] [18]. Furthermore, it cannot identify non-bacterial members of the community (e.g., viruses, fungi) and provides no direct information about the functional potential of the microbiota, relying instead on inference from taxonomic data [3] [18] [1].
Shotgun Metagenomic Sequencing: A Comprehensive but Demanding Technique
Shotgun metagenomics sequences all the DNA fragments in a sample, providing a much broader view of the microbial community [3] [71]. Its primary advantages are superior taxonomic resolution down to the species and strain level for multiple kingdoms, and the direct identification of functional genes [3] [71] [18]. The major drawback for low-biomass applications is its vulnerability to host DNA. In samples where microbial DNA is extremely rare, the vast majority of sequences will be from the host, requiring deep sequencing to obtain sufficient microbial data and potentially making it a less cost-effective option [3] [70]. "Shallow shotgun" sequencing has emerged as a cost-compromise, offering many of the advantages of full metagenomics at a cost closer to 16S sequencing, but it may still be less suitable for very low-biomass, high-host-DNA samples [3].
Foundational Experimental Protocols for Contamination Control
Robust low-biomass microbiome science requires meticulous experimental design from the moment of sample collection. The following protocols, distilled from consensus guidelines, are non-negotiable for generating credible data [69] [70].
Pre-Sampling Preparation and Decontamination
Prior to sample collection, researchers must identify and mitigate all potential contamination sources. This involves using single-use, DNA-free collection equipment where possible [69]. When re-usable tools are necessary, a two-step decontamination process is recommended: first, using a solution like 80% ethanol to kill contaminating organisms, followed by a nucleic acid-degrading solution (e.g., sodium hypochlorite/bleach, commercial DNA removal kits) to remove residual environmental DNA that can persist even on sterilized surfaces [69]. All plasticware and glassware for sample storage should be pre-sterilized by autoclaving and/or UV-C irradiation and remain sealed until the moment of use [69].
Sampling Procedure and Personal Protective Equipment (PPE)
During sampling, minimizing contact between the sample and potential contaminants is critical. Personnel should wear extensive PPE—including gloves, masks, goggles, coveralls or cleansuits, and shoe covers—to prevent contamination from skin, hair, aerosols, and clothing [69]. The sample itself should be handled as little as possible. For clinical procedures like cesarean sections for fetal sample collection, swabbing the maternal skin before the procedure and exposing a swab to the operating theatre air can serve as vital procedural controls [69].
Incorporation of Comprehensive Control Samples
The inclusion of various control samples throughout the experimental workflow is perhaps the most critical component for identifying contamination sources post-sequencing [69] [70] [72]. These controls should be processed alongside the biological samples through every step (DNA extraction, library preparation, sequencing).
Table 2: Essential Control Samples for Low-Biomass Studies
| Control Type | Description | Purpose |
|---|---|---|
| Negative Extraction Control | A blank sample (e.g., water) taken through the DNA extraction kit [70] [72]. | Identifies contaminants introduced from DNA extraction reagents and kits [69] [72]. |
| No-Template Control (NTC) | A water sample included during the PCR amplification or library preparation step [70]. | Detects contamination from PCR/librar y preparation reagents [70]. |
| Sample Collection Control | An empty collection vessel or a swab exposed to the air during sampling [69]. | Reveals contaminants from the collection tubes, swabs, or the sampling environment [69]. |
| Mock Community | A defined mixture of known microorganisms [72]. | Serves as a positive control to evaluate the fidelity of the entire wet-lab and bioinformatic workflow [72]. |
Laboratory Logistics to Minimate Cross-Contamination
A often-overlooked source of contamination is "well-to-well leakage" or "cross-contamination," where DNA from one sample leaches into an adjacent well on a plate during laboratory processing [69] [70]. To mitigate this, researchers should avoid arranging high-biomass samples (even if from other studies) next to precious low-biomass samples on the same plate. If possible, leaving empty wells between low-biomass samples can provide a physical buffer [70].
Computational Decontamination Strategies
Even with impeccable laboratory practices, some contamination is inevitable. Computational decontamination tools are therefore essential for identifying and removing suspect signals from sequencing data before biological interpretation.
Several software packages have been developed to tackle contamination in microbiome data. Their approaches generally fall into three categories: (1) Control-based methods that subtract sequences or abundances found in negative controls (e.g., SCRuB, microDecon) [73]; (2) Sample-based methods that identify contaminants based on their behavior across samples, such as an inverse correlation with DNA concentration (e.g., Decontam) [72]; and (3) Blocklist methods that remove taxa previously identified as common contaminants [73]. A key consideration is whether a tool removes entire microbial taxa (features) identified as contaminants or attempts to subtract only the proportion of reads attributed to contamination, with the latter being more nuanced for low-biomass studies [73].
Implementing a Decontamination Pipeline with micRoclean
The micRoclean R package provides a structured framework for decontaminating 16S rRNA data, offering two distinct pipelines based on the research goal [73].
- The "Original Composition Estimation" Pipeline: This pipeline is ideal when the goal is to estimate the true microbial composition of the sample as closely as possible. It leverages the SCRuB algorithm, which is particularly effective at accounting for well-to-well leakage if well-location information is provided. It works by partially removing reads attributed to contamination rather than discarding entire taxa [73].
- The "Biomarker Identification" Pipeline: This pipeline is better suited for studies aiming to find microbial biomarkers associated with a disease or condition. It takes a more conservative, "better safe than sorry" approach by aggressively removing entire taxa identified as contaminants across multiple batches of samples, thereby minimizing false positive associations [73].
A critical feature of micRoclean is the calculation of a Filtering Loss (FL) statistic. This value quantifies the impact of decontamination on the overall covariance structure of the dataset. An FL value close to 1 indicates that the removed features contributed heavily to the sample distributions, which could be a warning sign of over-filtering and the potential loss of true biological signal [73].
The following diagram illustrates the logical workflow for selecting and applying these decontamination strategies.
The Researcher's Toolkit: Essential Reagents and Materials
Successful low-biomass research relies on a suite of specialized reagents and materials designed to minimize and monitor DNA contamination.
Table 3: Essential Research Reagent Solutions for Low-Biomass Studies
| Item | Function | Example & Notes |
|---|---|---|
| DNA Decontamination Solution | To degrade contaminating DNA on surfaces and reusable equipment. | Sodium hypochlorite (bleach), hydrogen peroxide, or commercial solutions (e.g., DNA-ExitusPlus) [69]. |
| DNA-Free Nucleic Acid Extraction Kits | To isolate microbial DNA while minimizing reagent-derived contaminant DNA. | Kits specifically designed for low-biomass (e.g., MoBio PowerSoil, QIAamp PowerFecal Pro). Verify lot-to-lot contaminant profiles [71] [74]. |
| Ultra-Pure Water | Serves as a negative control and a solvent for molecular biology reactions. | Must be certified nuclease-free and DNA-free. Used for no-template controls (NTCs) [72]. |
| Mock Microbial Community | A defined mix of microbial cells or DNA from known species. | ZymoBIOMICS Microbial Community Standard is widely used. Serves as a positive control for the entire workflow [72] [74]. |
| DNA-Free Collection Supplies | Sterile swabs, tubes, and containers for sample acquisition and storage. | Single-use, sterilized, and certified DNA-free. Pre-treat with UV light if possible [69]. |
The investigation of low-biomass environments like the fetal microbiome demands a paradigm shift from standard microbiome research. It is no longer sufficient to simply sequence and report; researchers must build a fortress of evidence around their findings to distinguish true biological signal from technical artifact. This entails a holistic strategy that integrates meticulous sample collection with comprehensive controls, informed choice of sequencing technology, and robust computational decontamination. By adopting the rigorous, multi-layered best practices outlined in this guide—from the use of extensive PPE and DNA-degrading solutions to the careful implementation of tools like micRoclean—the research community can advance the field confidently. The goal is to ensure that future discoveries about the microscopic inhabitants of our most pristine tissues are built upon an unshakable foundation of methodological rigor.
Head-to-Head Validation: Empirical Comparisons and Performance Metrics
The accurate characterization of microbial communities is a cornerstone of modern microbiome research, with 16S rRNA gene sequencing and shotgun metagenomics serving as the two predominant techniques [3]. While both methods are widely used to determine the taxonomic composition of samples, their resolution, biases, and resulting microbial profiles can differ significantly [6] [4]. This guide provides a direct objective comparison of these technologies, focusing on their concordance and discrepancies in genus and species-level identification. Understanding these differences is critical for researchers, scientists, and drug development professionals to select the appropriate methodology, interpret results accurately, and integrate findings from studies utilizing different techniques. We synthesize evidence from multiple comparative studies to offer a data-driven perspective on the performance of each method.
Core Technological Differences and Their Impact on Taxonomy
The fundamental difference between the two methods lies in their scope and approach. 16S rRNA gene sequencing is a targeted amplicon-based method that involves PCR amplification and sequencing of specific hypervariable regions (e.g., V3-V4, V4) of the bacterial and archaeal 16S rRNA gene [3] [1]. In contrast, shotgun metagenomic sequencing is an untargeted approach that sequences all genomic DNA from a sample, followed by computational analysis to assign taxonomy based on whole-genome or marker-gene data [3] [75].
This core distinction leads to several key implications for taxonomic profiling:
- Scope of Detection: 16S sequencing is generally restricted to bacteria and archaea, whereas shotgun sequencing can simultaneously profile bacteria, archaea, viruses, fungi, and other microorganisms [1] [75].
- Reference Databases: 16S analysis relies on 16S-specific databases (e.g., SILVA, Greengenes), while shotgun analysis uses whole-genome databases (e.g., NCBI RefSeq, GTDB) [4]. Discrepancies between these databases can lead to different taxonomic assignments [4].
- PCR Bias: The PCR amplification step in 16S sequencing can introduce artifacts and amplification biases, potentially over- or under-representing certain taxa [76].
- Host DNA Interference: Shotgun sequencing is more susceptible to host DNA contamination, which can reduce the effective microbial sequencing depth unless removal techniques are employed [3] [75].
The following diagram illustrates the foundational workflows of each method and their primary outputs.
Figure 1: Comparative Workflows of 16S rRNA Gene and Shotgun Metagenomic Sequencing. ASV, Amplicon Sequence Variant; OTU, Operational Taxonomic Unit.
Quantitative Comparison of Taxonomic Profiling
Detection Sensitivity and Community Richness
Multiple studies have consistently demonstrated that shotgun metagenomic sequencing detects a greater number of taxa and provides a more comprehensive view of microbial community richness compared to 16S sequencing.
Table 1: Comparison of Taxon Detection and Diversity Between Sequencing Methods
| Study & Sample Type | 16S rRNA Sequencing Findings | Shotgun Metagenomic Findings | Key Comparative Result |
|---|---|---|---|
| Human Gut (24 samples) [77] | 616 filtered bacterial features | 1,041 filtered bacterial features | sFL16S classified 69% more bacterial features than V3V4. |
| Chicken Gut (50 samples) [6] | Lower alpha-diversity (genus level) | Higher alpha-diversity (genus level) | Shotgun RSA distributions were more symmetrical, indicating better sampling of rare taxa. |
| Pediatric Gut (338 samples) [76] | Identified a larger number of genera | Detected genera missed by 16S | Each method uniquely identified some genera; 16S detected more overall in this cohort. |
| Human Colorectal Cancer (156 samples) [4] | Sparse abundance data; lower alpha-diversity | Less sparse data; higher alpha-diversity | 16S revealed only part of the community, giving greater weight to dominant bacteria. |
A study on human gut microbiota found that a full-length 16S method (sFL16S) identified 1,041 bacterial features, significantly more than the 616 features identified by standard V3-V4 short-read sequencing [77]. Similarly, in a chicken gut model, shotgun sequencing revealed a statistically significant higher number of taxa, particularly among less abundant genera, when sequencing depth was sufficient (>500,000 reads per sample) [6]. The analysis of Relative Species Abundance (RSA) distributions in that study showed that shotgun samples exhibited skewness closer to zero, indicating more symmetrical distributions and better sampling of low-abundance species, whereas 16S samples were often left-skewed, a potential artifact of insufficient sampling [6].
However, the relationship is not absolute. One study on pediatric gut microbiomes found that 16S rRNA profiling identified a larger number of genera than shotgun metagenomic sequencing [76]. This highlights that the performance can be influenced by factors such as the specific bioinformatics pipelines, reference databases, and the age-related complexity of the microbiome under investigation.
Taxonomic Resolution: Genus and Species-Level Concordance
The resolution of taxonomic classification is a critical differentiator between the two methods. While 16S sequencing can often provide reliable genus-level assignments, its ability to resolve species is limited. Shotgun sequencing, with its access to genomic information beyond a single gene, typically achieves higher resolution.
Table 2: Resolution and Concordance at Different Taxonomic Levels
| Taxonomic Level | 16S rRNA Sequencing | Shotgun Metagenomics | Concordance Notes |
|---|---|---|---|
| Genus Level | Reliable for most genera [1]. | Reliable for most genera [1]. | High correlation (Average r = 0.69) in relative abundances of shared genera [6]. |
| Species Level | Limited resolution; high false positive rate [3]. Possible with full-length 16S [77]. | Species and sometimes strain-level resolution [3] [75]. | High disagreement; 16S misses or misidentifies species with high 16S sequence similarity [77] [4]. |
| Differential Abundance | Fewer significant findings (e.g., 108 genera in caeca vs crop) [6]. | More significant findings (e.g., 256 genera in caeca vs crop) [6]. | Shotgun detects more subtle, significant changes, often in less abundant taxa. |
A comparison of gut microbiota in chickens found a strong positive correlation (average Pearson's r = 0.69) in the relative abundances of genera common to both 16S and shotgun sequencing [6]. However, the agreement deteriorates at the species level. A study on human gut samples concluded that 16S sequencing is limited in classifying strains with high similarity at the species level [77]. This is because short reads from a single hypervariable region lack the information to distinguish between species with highly similar 16S rRNA genes.
Furthermore, in differential analysis, shotgun sequencing demonstrates superior power to identify taxa with statistically significant abundance changes between experimental conditions. In the chicken gut study, shotgun sequencing identified 256 genera with significant differences between caeca and crop compartments, compared to only 108 genera identified by 16S sequencing [6]. The 152 additional significant genera found only by shotgun were biologically meaningful and able to discriminate between the experimental conditions [6].
Experimental Protocols from Key Comparative Studies
To ensure reproducibility and provide context for the data presented, this section outlines the experimental methodologies from several key studies cited in this guide.
Protocol: Human Gut Microbiota Comparison (sFL16S vs. V3V4)
- Sample Preparation: Human fecal samples were collected from three healthy adults. Each sample was divided into eight identical tubes, totaling 24 specimens. Integrity of extracted microbial genomic DNA was confirmed based on concentration, purity, and lack of degradation [77].
- Library Preparation & Sequencing:
- sFL16S: The LoopSeq 16S Microbiome Kit (Loop Genomics) was used. This technology employs a unique molecule barcoding strategy to assemble short Illumina reads into synthetic long-reads covering the full-length 16S rRNA gene (V1-V9) [77].
- V3V4: The V3-V4 hypervariable region was amplified and sequenced on the Illumina MiSeq platform, following standard amplicon sequencing procedures [77].
- Bioinformatics & Analysis: Amplicon Sequence Variants (ASVs) were generated for both methods. Taxonomic classification was performed using the SILVA reference database with a >70% confidence threshold. Alpha-diversity indices (ObservedOTUs, Chao1, Shannon, Simpson, Pieloue) were calculated and compared [77].
Protocol: Chicken Gut Microbiota Comparison
- Sample Preparation: The same DNA samples extracted from chicken crop and caeca in a previous study were used for both sequencing methods [6].
- Library Preparation & Sequencing:
- Shotgun Metagenomics: Standard whole-genome shotgun sequencing libraries were prepared and sequenced.
- 16S rRNA Sequencing: Targeted 16S rRNA gene sequencing was performed on the same DNA samples [6].
- Bioinformatics & Analysis: For a balanced comparison, shotgun samples with less than 500,000 reads were discarded, as their rarefaction curves did not plateau. Corresponding 16S samples were also removed. Relative Species Abundance (RSA) distributions and their skewness were analyzed. Differential abundance analysis was performed using DESeq2, and Pearson's correlation was calculated for genus abundances common to both methods [6].
Protocol: Colorectal Cancer Gut Microbiota Comparison
- Sample Collection: 156 human stool samples were collected from controls, patients with high-risk colorectal lesions (HRL), and colorectal cancer (CRC) cases. Samples were stored at -20°C by participants and then at -80°C upon delivery [4].
- DNA Extraction & Sequencing:
- Bioinformatics & Analysis:
- 16S Data: Processed using DADA2 for quality filtering, denoising, and merging. Taxonomy was assigned using the SILVA database. An additional classification step using BLASTN against a custom SILVA database and k-mer based classification (Kraken2/Bracken2) was added to improve species-level assignment [4].
- Shotgun Data: Processed to filter out human reads using Bowtie2 against the GRCh38 human genome. Taxonomic profiling was performed using the MGnify pipeline [4].
Table 3: Key Research Reagent Solutions for Method Comparison
| Item | Function | Example Products / Tools (from cited studies) |
|---|---|---|
| DNA Extraction Kits | To isolate high-quality microbial DNA from complex samples. | NucleoSpin Soil Kit [4], Dneasy PowerLyzer Powersoil Kit [4], DNeasy Blood and Tissue Kit [78] |
| 16S Sequencing Kits | For amplification and library preparation of 16S rRNA gene regions. | LoopSeq 16S Microbiome Kit (for sFL16S) [77], Various primers for V3-V4 [77] [4] |
| Shotgun Library Prep Kits | For random fragmentation, adapter ligation, and library preparation for whole-genome sequencing. | Not specified in results, but Illumina kits (e.g., Nextera) are industry standard. |
| Reference Databases | Essential for accurate taxonomic classification of sequencing reads. | 16S: SILVA [77] [4], GreengenesShotgun: NCBI RefSeq [4], GTDB |
| Bioinformatics Pipelines | Software for processing raw sequencing data, from quality control to taxonomic assignment. | 16S: DADA2 [4], QIIME, MOTHURShotgun: MGnify [4], MetaPhlAn, Kraken2, HUMAnN |
| Mock Microbial Communities | Controls to assess accuracy, sensitivity, and false positive rates of the entire workflow. | ZymoBIOMICS Microbial Community Standard [78] [75] |
Both 16S rRNA gene sequencing and shotgun metagenomics provide valuable insights into microbial community composition, but they offer different lenses through which to view the microbiome [4]. The choice between them should be guided by the specific research questions, available resources, and sample types.
For 16S rRNA Gene Sequencing: This method is a cost-effective choice for large-scale studies focused primarily on bacterial composition and diversity at the genus level. It is particularly useful for samples with low microbial biomass or high host DNA content, and when budget or bioinformatics expertise is limited [1] [75]. However, researchers must be cautious about its limitations in species-level resolution and functional inference.
For Shotgun Metagenomic Sequencing: This is the preferred method when the research demands species- or strain-level resolution, comprehensive functional profiling, or detection of non-bacterial community members (e.g., viruses, fungi) [6] [1]. It is highly recommended for stool samples where depth and breadth of analysis are paramount [4]. The higher cost and computational demands are traded for a much more detailed and comprehensive snapshot of the microbial community.
In summary, while shotgun metagenomics generally provides a more detailed and broader view of the microbiome, 16S sequencing remains a powerful tool for targeted bacterial profiling. Researchers should be aware that results from these two methods are not directly interchangeable, and consistent methodology is key for comparative studies.
In microbiome research, accurately quantifying microbial diversity is fundamental to understanding community structure, stability, and its relationship with the host or environment. Two principal sequencing methods—16S rRNA gene amplicon sequencing and shotgun metagenomic sequencing—are widely employed for this purpose, each with distinct technical principles and implications for diversity measurement [3]. Diversity is typically assessed through two lenses: alpha diversity, which measures the richness and evenness of species within a single sample, and beta diversity, which quantifies the differences in microbial composition between samples [79]. The choice of sequencing method profoundly influences the resulting taxonomic resolution, detection sensitivity, and consequently, the ecological conclusions drawn. This guide objectively compares how these two techniques measure alpha and beta diversity, supported by experimental data and detailed methodologies from recent studies.
Fundamental Methodological Differences
At their core, 16S and shotgun metagenomics differ in what parts of the genomic material are sequenced.
- 16S rRNA Gene Sequencing: This is an amplicon-based, targeted approach. It uses polymerase chain reaction (PCR) to amplify one or more hypervariable regions (e.g., V3-V4, V4, V6-V8) of the bacterial and archaeal 16S rRNA gene. The resulting sequences are clustered into Operational Taxonomic Units (OTUs) or, with higher resolution, Amplicon Sequence Variants (ASVs) for taxonomic classification [34]. This method focuses on a single, highly conserved gene, which acts as a barcode for identifying microbial taxa [3].
- Shotgun Metagenomic Sequencing: This is a whole-genome approach. It involves randomly fragmenting and sequencing all the DNA present in a sample—bacterial, viral, fungal, and host. These sequences can then be aligned to reference databases for taxonomic assignment at the species or even strain level, or assembled into genomes, providing a comprehensive view of the entire genetic content [6] [3].
These foundational differences lead to variations in taxonomic resolution, multi-kingdom coverage, and functional insight, which directly impact diversity assessments. The experimental workflow for each method is summarized in the diagram below.
Comparative Analysis of Alpha Diversity Measurement
Alpha diversity summarizes the microbial community structure within a single sample, incorporating concepts of richness (number of different taxa) and evenness (distribution of their abundances) [79]. Common metrics include the Chao1 and ACE indices (which estimate richness), and the Shannon and Simpson indices (which combine richness and evenness) [79].
Table 1: Impact of Sequencing Method on Alpha Diversity Metrics
| Alpha Diversity Aspect | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Typical Observed Richness | Generally lower, as it detects only a portion of the community [6] [4]. | Statistically significantly higher, especially for less abundant taxa, given sufficient read depth [6]. |
| Data Sparsity | Higher; abundance data is sparser, with more zero counts [4]. | Lower; provides a more complete and symmetrical relative abundance distribution [6]. |
| Skewness of Abundance Distribution | More left-skewed (positively skewed) at the genus level, an artifact of smaller effective sample size [6]. | More symmetrical (skewness closer to zero), indicating better sampling of low-abundance species [6]. |
| Dependence on Sequencing Depth | Reaches saturation with fewer reads (e.g., ~50,000 reads/sample) [9]. | Requires a high number of reads (>500,000) to reach a stable plateau and avoid skewness [6]. |
| Correlation Between Methods | Moderate correlation with shotgun-derived alpha diversity measures [4]. | Considered the more comprehensive benchmark for true community diversity [6] [4]. |
Experimental data consistently shows that shotgun sequencing reveals a greater alpha diversity compared to 16S sequencing. A 2021 study on chicken gut microbiota demonstrated that when shotgun sequencing achieves sufficient depth (>500,000 reads per sample), it identifies a statistically significant higher number of taxa, corresponding to the less abundant members of the community that 16S sequencing misses [6]. This results in a more symmetrical relative species abundance distribution for shotgun data, whereas 16S data tends to be left-skewed—a pattern indicative of an undersampled community [6]. A 2024 study on colorectal cancer confirmed these findings, noting that 16S abundance data is sparser and exhibits lower alpha diversity [4].
Comparative Analysis of Beta Diversity Measurement
Beta diversity measures the compositional dissimilarity between microbial communities from different samples. It is typically visualized using ordination plots (e.g., PCoA, NMDS) based on distance metrics like Bray-Curtis (abundance-weighted), Jaccard (presence-absence), and UniFrac (phylogenetic).
Table 2: Impact of Sequencing Method on Beta Diversity Analysis
| Beta Diversity Aspect | 16S rRNA Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Overall Pattern Consistency | Beta diversity projections often show moderate correlation with shotgun results and can cluster samples by condition similarly [4] [38]. | Considered the more accurate reflection of true biological variation; patterns are more distinct and robust [6]. |
| Power to Discriminate Conditions | Can distinguish between major experimental conditions (e.g., disease vs. healthy) but with fewer significant taxa [6] [38]. | Identifies a much larger number of statistically significant taxa that differentiate conditions, providing finer resolution [6]. |
| Representation of Community Variation | In disease states (e.g., ulcerative colitis), shows higher beta diversity within cases compared to controls [38]. | Confirms higher within-group variation in disease states and provides a deeper understanding of the drivers of this variation [38]. |
| Key Differentiating Metrics | Bray-Curtis and Jaccard dissimilarities are standard [34]. Unweighted UniFrac is also commonly used. | Same metrics (Bray-Curtis, Jaccard, UniFrac) can be applied, but are calculated from a more comprehensive species-level profile. |
While both methods can reveal consistent ecological patterns, shotgun sequencing generally provides greater discriminatory power. In the chicken gut study, shotgun sequencing identified 256 genera with significantly different abundances between gut compartments, compared to only 108 by 16S sequencing [6]. Importantly, the less abundant genera detected exclusively by shotgun sequencing were biologically meaningful and able to discriminate between experimental conditions as effectively as the more abundant genera [6]. Studies in human health, such as one on pediatric ulcerative colitis, have found that both methods can produce similar beta diversity patterns and prediction accuracy for disease status, demonstrating that 16S can be sufficient for identifying broad compositional shifts [38].
Experimental Protocols for Method Comparison
To ensure a fair and objective comparison between 16S and shotgun sequencing, researchers must follow a rigorous paired design. The following protocol, synthesized from multiple studies, outlines the key steps.
Key Experimental Steps:
- Sample Splitting and DNA Extraction: The most critical step is splitting a single, homogenized sample aliquot for parallel processing. Using the same DNA extract for both methods is ideal but not always feasible due to different extraction kit optimizations (e.g., Dneasy PowerLyzer for 16S vs. NucleoSpin Soil for shotgun) [4]. Including negative controls and mock microbial communities (e.g., ZymoBIOMICS Standard) is mandatory to monitor contamination and PCR biases [45] [34].
- Library Preparation and Sequencing:
- 16S Protocol: Amplify the target hypervariable region (e.g., V4 using 515F/806R primers) [38]. Recent optimizations show that pooling multiple PCR replicates per sample may be unnecessary, streamlining the protocol [45]. Sequence on an Illumina MiSeq to achieve ~50,000-100,000 reads per sample.
- Shotgun Protocol: Use a kit like Nextera XT for library preparation without target amplification [38]. Sequence on an Illumina NextSeq or HiSeq platform. For stool samples, "shallow shotgun" (reduced sequencing depth) can be a cost-effective compromise [9] [3]. For host-rich samples (e.g., tissue), implement host DNA depletion protocols.
- Bioinformatics Processing:
- 16S Analysis: Process demultiplexed reads through a pipeline like QIIME2 with DADA2 for quality filtering, denoising, and Amplicon Sequence Variant (ASV) calling [34]. Assign taxonomy using reference databases such as SILVA or Greengenes.
- Shotgun Analysis: Quality-trim reads with tools like Trim Galore! Remove host-derived reads using KneadData (with a human reference genome) [38]. Perform taxonomic profiling with tools like MetaPhlAn or Kraken2 against curated genome databases.
- Statistical Comparison: Calculate alpha diversity (Shannon, Chao1) and beta diversity (Bray-Curtis) metrics for both datasets. Use PERMANOVA to test for group differences in beta diversity and tools like DESeq2 to identify differentially abundant taxa, comparing the number and concordance of significant findings between the two methods [6] [4].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for 16S and Shotgun Sequencing Studies
| Item | Function/Description | Example Products/Kits |
|---|---|---|
| Sample Collection Kit | Standardizes sample preservation at the point of collection to prevent microbial community shifts. | OMNIgene GUT (feces), RNAlater (tissue) [9]. |
| DNA Extraction Kit | Isolates high-quality, high-molecular-weight genomic DNA from complex samples. | QIAamp PowerFecal Pro DNA Kit, DNeasy PowerLyzer Powersoil Kit (16S), NucleoSpin Soil Kit (shotgun) [4] [38]. |
| 16S PCR Primers | Target-specific primers that amplify hypervariable regions of the 16S rRNA gene for sequencing. | 515FB/806RB (V4 region) [38], 27F/338R (V1-V2), 338F/806R (V3-V4). |
| PCR Master Mix | A pre-mixed solution containing enzymes, dNTPs, and buffers for efficient and consistent PCR amplification. | Q5 Hot Start High-Fidelity Master Mix (premixed reduces handling) [45]. |
| Library Prep Kit (Shotgun) | Prepares DNA fragments for sequencing by adding adapters and indexing barcodes. | Illumina Nextera XT DNA Library Preparation Kit [38]. |
| Mock Community | A defined mix of microbial strains with known abundance, used as a positive control to assess accuracy and bias in sequencing and bioinformatics. | ZymoBIOMICS Microbial Community DNA Standard [45]. |
| Bioinformatics Databases | Curated collections of reference sequences used for taxonomic classification. | 16S: SILVA, Greengenes. Shotgun: NCBI RefSeq, GTDB, UHGG [4]. |
The choice between 16S rRNA and shotgun metagenomic sequencing for quantifying diversity involves a clear trade-off between cost and resolution. 16S sequencing is a powerful, cost-effective tool for revealing broad-scale ecological patterns, such as shifts in alpha and beta diversity associated with disease states or environmental perturbations. When the research question focuses on dominant bacterial community members and the budget is constrained, 16S remains a robust choice [38] [3].
In contrast, shotgun metagenomic sequencing provides a more comprehensive and detailed census of the microbial community. It delivers higher resolution taxonomic profiling, capturing rare and less abundant taxa that are often missed by 16S, leading to more powerful differential analyses and a truer representation of community structure [6] [4]. For studies requiring species-level discrimination, functional insights, or characterization of non-bacterial kingdoms, shotgun is the unequivocally superior method. As sequencing costs continue to decrease, shotgun metagenomics, including the cost-effective "shallow shotgun" approach for stool samples, is becoming an increasingly accessible and informative standard for in-depth microbiome diversity analysis [9] [3].
The microbial "rare biosphere," composed of low-abundance taxa, represents a vast reservoir of genetic diversity and functional potential within microbiomes. The accurate detection and characterization of these rare species are crucial for understanding ecosystem resilience, host-microbe interactions, and uncovering novel biosynthetic genes. This review objectively compares the performance of 16S rRNA gene sequencing and whole-genome shotgun metagenomics in revealing this hidden dimension of microbial life. We synthesize experimental data demonstrating that shotgun sequencing, by providing broader genomic coverage and higher resolution, consistently detects a greater proportion of low-abundance taxa and achieves superior strain-level differentiation compared to the targeted approach of 16S sequencing. Supported by detailed methodologies and quantitative comparisons, we conclude that shotgun metagenomics is the more powerful tool for comprehensively studying the rare biosphere, despite higher computational costs and DNA input requirements.
Microbial communities are predominantly composed of a vast number of low-abundance species, a collective now known as the "rare biosphere" [80]. This component is not merely a statistical artifact; it serves as a reservoir of genetic diversity that can bolster ecosystem resistance and resilience, shape host-associated microbiomes, and be a source of novel genes with industrial or clinical applications [80]. The accurate identification of these rare species is, therefore, a priority in microbial ecology and clinical diagnostics. However, the study of these taxa is fraught with technical challenges, principally because their signal can be easily obscured by sequencing errors, contamination, or methodological biases [81].
The two foremost culture-independent techniques used to profile microbial communities are 16S rRNA gene amplicon sequencing (16S sequencing) and whole-genome shotgun metagenomic sequencing (shotgun sequencing). The former is a targeted approach that amplifies and sequences a specific hypervariable region of the bacterial and archaeal 16S rRNA gene, providing a cost-effective means for taxonomic census [11]. The latter sequences all the DNA fragments in a sample in an untargeted manner, enabling not only taxonomic profiling of all domains of life but also functional gene analysis [3].
A critical and recurring question is: which of these methods is more capable of detecting and accurately identifying the members of the rare biosphere? This guide directly addresses this question by synthesizing evidence from recent comparative studies, presenting structured experimental data, and outlining the underlying methodologies. The consensus emerging from the literature is that while 16S sequencing offers an accessible entry point, shotgun sequencing provides a more powerful and detailed lens for observing the rare biosphere [6] [4].
Technical Comparison: 16S rRNA vs. Shotgun Sequencing
The fundamental differences in the underlying technology of 16S and shotgun sequencing directly impact their ability to detect low-abundance taxa.
Table 1: Core Technical Differences Between 16S and Shotgun Sequencing
| Feature | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Target | Specific hypervariable regions of the 16S rRNA gene [11] | All genomic DNA in a sample (whole-genome) [3] |
| Taxonomic Resolution | Typically genus-level; species-level is challenging and often unreliable [28] [3] | Species and strain-level resolution is achievable [81] [3] |
| Kingdom Coverage | Primarily Bacteria and Archaea [11] | Multi-kingdom: Bacteria, Archaea, Viruses, Fungi, Protists [3] |
| Functional Insight | Indirect inference based on taxonomy [3] | Direct characterization of functional genes and pathways [6] [3] |
| Host DNA Contamination | Minimal; PCR amplification enriches for the target gene [3] | Can be significant; requires deeper sequencing or host DNA depletion [3] |
| Relative Cost & DNA Input | Lower cost and minimal DNA input (can be <1 ng) [3] | Higher cost and higher DNA input required (e.g., >1 ng/μL) [3] |
The process of 16S sequencing relies on PCR amplification using primers that bind to conserved regions flanking one or more variable regions (e.g., V4, V3-V5). This targeted amplification is both its strength and its key limitation for rare biosphere studies. Primer bias can lead to the under-representation or complete omission of certain taxa, as no single primer pair can perfectly amplify all 16S genes [4] [28]. Furthermore, the short read lengths (typically 250-300 bp on Illumina platforms) from a single variable region contain limited phylogenetic information, which often precludes confident species-level assignment [28].
In contrast, shotgun sequencing avoids PCR amplification of a specific gene, thereby circumventing primer bias. Its ability to sequence random fragments from across the entire genome provides a richer dataset for taxonomic assignment. The analysis relies on mapping reads to comprehensive genomic databases, allowing for discrimination based on unique genomic signatures beyond the 16S gene. This is particularly critical for differentiating between closely related strains and for detecting organisms whose 16S genes are highly similar but whose genomes have diverged, such as Escherichia coli and Shigella spp. [81].
Experimental Data: Quantitative Performance in Detecting Low-Abundance Taxa
Direct comparative studies consistently demonstrate the enhanced sensitivity of shotgun sequencing for detecting less abundant community members.
Comparative Studies on Real-World Samples
A comprehensive 2021 study comparing 16S and shotgun sequencing of the chicken gut microbiota found that shotgun sequencing identified a statistically significant higher number of taxa [6]. The researchers showed that the genera detected exclusively by shotgun sequencing were biologically meaningful and able to discriminate between different experimental conditions (e.g., different gastrointestinal tract compartments) as effectively as the more abundant genera detected by both methods. This indicates that the low-abundance taxa revealed by shotgun are not merely noise but represent ecologically relevant signals.
Similarly, a 2024 study on human colorectal cancer and healthy gut microbiota revealed that 16S sequencing detects only part of the gut microbiota community revealed by shotgun sequencing [4]. The abundance data from 16S was sparser and exhibited lower alpha diversity. The disagreement was more pronounced at lower taxonomic ranks (species), partially due to differences in reference databases, but also because 16S lacks the resolution to confidently assign species based on short, single-gene fragments.
Detection Thresholds and Methodological Limitations
The definition of the "rare biosphere" itself is a methodological challenge. Many studies use arbitrary relative abundance thresholds (e.g., 0.1% or 0.01%) [80]. However, this approach is flawed for cross-method comparisons because 16S and shotgun data have abundance scores in different orders of magnitude. A 0.1% threshold applied to a 16S dataset will capture a different biological reality than the same threshold applied to a shotgun dataset from the same sample [80].
To address this, novel computational tools like the Unsupervised Learning based Definition of the Rare Biosphere (ulrb) have been developed. This method uses unsupervised machine learning (k-medoids clustering) to classify taxa into "rare," "intermediate," and "abundant" categories based solely on their abundance distribution within a sample, providing a more consistent and user-independent definition [80].
Furthermore, specialized algorithms for analyzing shotgun data, such as the Rare Species Identifier (raspir), have been shown to differentiate rare species with genome coverages of less than 0.2% and successfully distinguish between genetically similar species like E. coli and Shigella spp. with low false discovery (1.3%) and omission rates (13%) [81]. Another advanced method, Latent Strain Analysis (LSA), can deconvolve complex communities and enable the assembly of partial genomes for bacterial taxa present at relative abundances as low as 0.00001%, and can even separate reads from different strains of the same species [82].
Table 2: Quantitative Comparison of Detection Capabilities from Key Studies
| Study (Context) | Shotgun Sequencing Performance | 16S Sequencing Performance |
|---|---|---|
| Cassotta et al., 2021 (Chicken Gut) [6] | Identified significantly more genera; low-abundance genera were biologically meaningful. | Failed to detect 152 genera that shotgun found to be statistically significant. |
| Pust et al., 2021 (Mock Community) [81] | Detected rare species with <0.2% genome coverage; differentiated E. coli and Shigella. | Not directly tested, but methods based on full genomes are inherently more specific. |
| Truong et al., 2015 (Spiked HMP Data) [82] | Assembled genomes from taxa at 0.00001% abundance; separated closely related strains. | Not applicable to this specific methodology (LSA is for shotgun data). |
| BenÃtez-Páez et al., 2024 (Human Gut) [4] | Revealed a broader and more diverse community; higher alpha diversity. | Detected only a portion of the community; sparser data and lower alpha diversity. |
Detailed Experimental Protocols
To ensure the reproducibility of the results cited, this section outlines the core methodologies employed in the featured comparative studies.
Protocol 1: Standard 16S rRNA Gene Amplicon Sequencing
- DNA Extraction: Genomic DNA is extracted from the biospecimen (e.g., stool, tissue) using commercial kits, such as the Dneasy PowerLyzer Powersoil kit (Qiagen) [4].
- Library Preparation (PCR Amplification): The hypervariable regions of the 16S rRNA gene (e.g., V3-V4) are amplified using universal primer sets. This step enriches for the target gene and attaches sequencing adapters [83] [4].
- Sequencing: Amplified libraries are purified, normalized, and pooled. Sequencing is typically performed on short-read platforms like the Illumina MiSeq or HiSeq systems, generating paired-end reads [83] [4].
- Bioinformatic Analysis:
- Quality Filtering & Denoising: Raw reads are trimmed and filtered based on quality. Tools like DADA2 are used to correct errors and infer exact Amplicon Sequence Variants (ASVs) [4].
- Taxonomic Assignment: ASV sequences are classified by comparison to curated 16S databases (e.g., SILVA, Greengenes) [4].
Protocol 2: Whole-Genome Shotgun Metagenomic Sequencing
- DNA Extraction: Total DNA is extracted from the sample using kits like the NucleoSpin Soil Kit (Macherey-Nagel). For samples with high host DNA, additional depletion steps may be incorporated [4].
- Library Preparation (Random Fragmentation): Extracted DNA is randomly sheared (by mechanical or enzymatic means) into short fragments, to which sequencing adapters are ligated. This is a critical difference: no target-specific PCR is used [3].
- Sequencing: Libraries are sequenced on platforms such as the Illumina HiSeq or NovaSeq, generating tens of millions of short reads per sample. "Shallow" shotgun sequencing at lower depth is a cost-effective alternative for high-microbial-biomass samples like stool [3].
- Bioinformatic Analysis:
Diagram 1: A comparative workflow of 16S rRNA amplicon sequencing and whole-genome shotgun metagenomic sequencing, highlighting the key methodological divergence at the library preparation stage.
Essential Research Reagent Solutions
The following table details key reagents and kits used in the protocols derived from the cited studies, which are essential for conducting this type of research.
Table 3: Key Research Reagents and Kits for Microbiome Sequencing
| Product Name / Type | Function in Workflow | Specific Application |
|---|---|---|
| NucleoSpin Soil Kit (Macherey-Nagel) [4] | DNA extraction from complex samples. | Optimal for stool and environmental samples in shotgun metagenomics. |
| Dneasy PowerLyzer Powersoil Kit (Qiagen) [4] | DNA extraction with bead-beating for lysis. | Used for 16S sequencing from various sample types. |
| Illumina DNA Prep Kit [11] | Library preparation for NGS. | Used for preparing sequencing libraries from fragmented DNA. |
| Universal 16S Primers (e.g., V3-V4) [4] | PCR amplification of target gene. | Essential for creating 16S amplicon libraries. |
| SILVA Database [4] | Reference for 16S taxonomic assignment. | A curated database for classifying 16S rRNA sequences. |
| GreenGenes Database [11] | Reference for 16S taxonomic assignment. | A commonly used taxonomic database for 16S data. |
| NCBI RefSeq Database | Reference for shotgun read alignment. | A comprehensive genome database for taxonomic and functional profiling in shotgun analysis. |
The collective evidence from methodological comparisons and experimental data strongly supports the superior capability of shotgun metagenomic sequencing for revealing the rare biosphere. Its key advantages include:
- Higher Taxonomic Resolution: Ability to detect low-abundance taxa and differentiate at the species and strain level [6] [81].
- Reduced Primer Bias: Untargeted nature avoids the amplification biases inherent in 16S PCR, allowing for a more representative profile [4] [3].
- Functional Insights: Provides direct access to the functional gene content of the community, including the genetic potential of rare members [6] [3].
However, 16S rRNA sequencing remains a valuable and powerful tool, particularly for large-scale cohort studies where cost is a primary constraint, or for samples with very low microbial biomass where its PCR-based amplification is advantageous [3].
Recommendations for researchers:
- For in-depth rare biosphere discovery, strain-level tracking, or functional analysis, shotgun sequencing is the recommended method. If budget is a concern, shallow shotgun sequencing provides a compelling middle ground for high-biomass samples [3].
- For large-scale, hypothesis-generating studies focused on dominant bacterial community shifts at the genus level, 16S sequencing remains a robust and cost-effective choice.
- Regardless of the chosen method, researchers should employ modern bioinformatic tools (e.g.,
ulrbfor defining rarity,raspirfor validating rare species) to maximize the robustness of their findings concerning the rare biosphere [81] [80].
The decision between these two powerful techniques should be guided by the specific research question, the sample type, and the available resources, with a clear understanding that shotgun metagenomics offers a more comprehensive and detailed window into the hidden world of low-abundance microbial taxa.
A fundamental goal in microbiome research is to identify microorganisms that differ in abundance between conditions, such as health versus disease. The choice of sequencing technology—16S rRNA gene amplicon sequencing or whole-genome shotgun metagenomics—is a critical initial decision that directly influences the statistical power and biological conclusions of a study [6] [76]. While 16S sequencing is a cost-effective method for profiling bacterial composition, shotgun sequencing provides a more comprehensive view of the entire microbial community, including bacteria, archaea, viruses, and fungi, and allows for direct functional profiling [76] [4]. This guide objectively compares the performance of these two sequencing strategies in the context of differential abundance (DA) analysis, synthesizing evidence from recent, direct comparative studies to inform researchers and drug development professionals.
Technical Comparison: 16S rRNA vs. Shotgun Metagenomic Sequencing
The two techniques differ fundamentally in their approach, which leads to distinct trade-offs.
- 16S rRNA Gene Sequencing: This is a targeted amplicon sequencing approach. It uses polymerase chain reaction (PCR) to amplify specific hypervariable regions of the bacterial and archaeal 16S rRNA gene before sequencing [76]. This method is widely used due to its lower cost and computational requirements. However, it is limited to bacteria and archaea, offers lower taxonomic resolution (typically genus-level), and its reliance on PCR can introduce amplification biases. Furthermore, it does not provide direct information on the functional potential of the community [76] [4].
- Shotgun Metagenomic Sequencing: This is an untargeted approach that sequences all the DNA fragments in a sample. It does not require a PCR amplification step and provides significantly higher taxonomic resolution, often enabling species- and strain-level identification. Its primary advantage is the ability to profile all domains of life and to directly identify functional genes and metabolic pathways [6] [4]. The main drawbacks are higher cost per sample and greater computational complexity for data analysis [76].
Table 1: Core Technical Characteristics of 16S rRNA and Shotgun Metagenomic Sequencing
| Feature | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Target | Specific hypervariable regions of the 16S rRNA gene | All genomic DNA in a sample |
| Taxonomic Scope | Primarily Bacteria and Archaea | Bacteria, Archaea, Viruses, Fungi, other Micro-eukaryotes |
| Taxonomic Resolution | Typically genus-level, sometimes species | Species-level and strain-level |
| Functional Profiling | Indirect inference only (e.g., via PICRUSt2) | Direct assessment of genes and pathways |
| PCR Amplification Bias | Yes | No |
| Cost & Computational Demand | Lower | Higher |
| Reference Database Dependency | Moderate (e.g., SILVA, Greengenes) | High (e.g., NCBI RefSeq, GTDB) |
Comparative Performance in Differential Abundance Analysis
Direct comparisons using matched samples demonstrate that the choice of sequencing method significantly impacts the outcome of DA testing.
Detection Depth and Sparsity
A consistent finding across studies is that shotgun sequencing detects a broader and less abundant fraction of the microbial community than 16S sequencing. In a study of the chicken gut microbiome, shotgun sequencing, when provided with a sufficient depth (>500,000 reads), identified a statistically significant higher number of taxa, particularly among less abundant genera [6]. Similarly, in a human colorectal cancer (CRC) study, 16S sequencing detected only a part of the community revealed by shotgun, with 16S data being sparser and exhibiting lower alpha diversity [4].
This difference arises because shotgun sequencing surveys the entire genome, providing more sequence information per organism for taxonomic assignment, whereas 16S is limited to a single gene. The relative species abundance distributions at the genus level are more symmetrical in shotgun data, indicating better sampling of rare taxa, whereas 16S data often show a left-skewed distribution, an artifact of undersampling [6].
Statistical Power to Detect Differences
The enhanced detection of low-abundance taxa with shotgun sequencing translates directly into greater statistical power to identify condition-specific biomarkers.
In the chicken gut study, when comparing microbial communities between two gastrointestinal compartments (caeca vs. crop), shotgun sequencing identified 256 genera with statistically significant abundance differences, while 16S sequencing identified only 108 [6]. Notably, shotgun sequencing found 152 significant changes that 16S missed, whereas 16S found only 4 changes not identified by shotgun. This represents a more than two-fold increase in sensitivity for shotgun sequencing in this experimental model [6].
Table 2: Quantitative Comparison of Differential Abundance Findings from Direct Comparative Studies
| Study & Experimental Condition | Number of Significant Genera (Shotgun) | Number of Significant Genera (16S) | Key Finding |
|---|---|---|---|
| Chicken GI Tract (Caeca vs. Crop) [6] | 256 | 108 | Shotgun detected 152 significant genera missed by 16S, demonstrating superior power for less abundant taxa. |
| Chicken GI Tract (14th vs. 35th Day) [6] | 75 | 58 | Shotgun consistently identified more temporally dynamic taxa. |
| Human Colorectal Cancer [4] | Unique microbial signature | Unique microbial signature | Shotgun provided a more detailed snapshot in depth and breadth; both methods identified taxa like Parvimonas micra. |
Despite differences in detection power, both methods can identify major, well-established microbial patterns. For example, in CRC studies, both 16S and shotgun sequencing have revealed microbial signatures that include known CRC-associated bacteria such as Fusobacterium and Parvimonas micra [4]. The high-level trends in alpha- and beta-diversity with factors like age or disease state also often appear similar between the two methods [76] [4].
However, the biological interpretation becomes more nuanced and potentially divergent when focusing on specific, less abundant taxa. The genera detected exclusively by shotgun sequencing have been shown to be biologically meaningful and capable of discriminating between experimental conditions just as well as the more abundant genera detected by both methods [6]. Therefore, relying solely on 16S rRNA sequencing risks overlooking a significant portion of the biologically relevant signal residing in the rare biosphere.
Experimental Protocols for Method Comparison
To ensure a fair and robust comparison between 16S and shotgun sequencing, the following experimental design and protocols, derived from the cited literature, are recommended.
Sample Collection and DNA Extraction
- Sample Type: The comparisons are most valid when performed on the same sample aliquot. Stool samples are commonly used for gut microbiome studies [4] [84].
- DNA Extraction: The same extraction kit and protocol should be used for both sequencing methods from a single sample homogenate. However, some studies optimize the protocol for each technique (e.g., using the NucleoSpin Soil Kit for shotgun and the Dneasy PowerLyzer Powersoil kit for 16S from the same sample) [4].
Sequencing Protocols
16S rRNA Gene Sequencing:
- Target Region: The V3-V4 or V4-V5 hypervariable regions are frequently used [76] [84].
- PCR Amplification: Amplify the target region with primers (e.g., 338F/806R for V3-V4) [84].
- Sequencing Platform: Illumina MiSeq or NovaSeq platforms are standard [84].
- Sequencing Depth: A minimum of 50,000 reads per sample is often targeted to maximize the identification of rare taxa [76].
Shotgun Metagenomic Sequencing:
- Library Preparation: Prepare sequencing libraries without target-specific amplification [4].
- Sequencing Platform: Illumina NovaSeq or HiSeq X Ten platforms are typical [84].
- Sequencing Depth: Depth is critical. A minimum of 5-10 million reads per sample is often recommended for complex gut samples, with deeper sequencing providing better resolution for low-abundance members [6] [76].
Bioinformatic Analysis
16S rRNA Data Processing:
Shotgun Data Processing:
Differential Abundance Analysis:
- Tool Selection: Use robust DA tools that account for compositionality and sparsity, such as ALDEx2 or ANCOM-II, which show more consistent results across studies [85].
- Consensus Approach: Given the high variability in results produced by different DA tools, applying a consensus approach based on multiple methods is recommended for robust biological interpretation [85].
The following workflow summarizes the key experimental and analytical steps for a comparative study.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for 16S and Shotgun Metagenomic Studies
| Item | Function | Example Products/Catalogs |
|---|---|---|
| Stool Collection Kit | Standardized sample stabilization at room temperature for transport. | OMNIgene GUT (OMR-200) [76] |
| DNA Extraction Kit | Isolation of high-quality, inhibitor-free microbial DNA from complex samples. | NucleoSpin Soil Kit (for shotgun), Dneasy PowerLyzer Powersoil Kit (for 16S) [4] |
| 16S PCR Primers | Amplification of specific hypervariable regions of the 16S rRNA gene. | 338F / 806R (for V3-V4 region) [84] |
| Sequencing Kits | Generation of sequence-ready libraries. | Illumina MiSeq Reagent Kit v3 (16S), Illumina NovaSeq 6000 S-Plex (Shotgun) |
| Bioinformatics Tools | Processing raw data into taxonomic and functional profiles. | DADA2 [4], QIIME 2 [84], MEGAHIT [84], MetaPhlAn, HUMAnN3 [53] |
| Reference Databases | Taxonomic classification of sequence data. | SILVA [4] (16S), NCBI RefSeq [4], GTDB (Shotgun) |
The choice between 16S rRNA and shotgun metagenomic sequencing has a profound impact on the statistical power and biological conclusions of a microbiome study. 16S sequencing is a cost-effective choice for large-scale, cross-sectional studies focused on dominant bacterial taxa. However, shotgun metagenomics is unequivocally superior for detecting less abundant taxa, providing higher taxonomic resolution, and uncovering condition-specific biomarkers that would otherwise be missed. For research and drug development programs where discovering a comprehensive microbial signature is critical, shotgun sequencing, despite its higher initial cost, provides a much richer and more powerful dataset, ultimately leading to more robust and translatable biological insights.
In the study of complex microbial communities, researchers primarily rely on two powerful sequencing technologies: 16S rRNA gene sequencing and shotgun metagenomic sequencing. While 16S rRNA sequencing provides a cost-effective method for taxonomic classification of bacteria and archaea, it offers limited functional insights. In contrast, shotgun metagenomics sequences all the DNA in a sample, enabling comprehensive analysis of both taxonomic composition and functional potential, including antibiotic resistance genes and metabolic pathways [10]. This capability makes metagenomics an indispensable tool for researchers and drug development professionals investigating the functional dynamics of microbiomes in health, disease, and environmental settings.
The selection between these methods involves important trade-offs. 16S rRNA sequencing targets specific variable regions of the conserved 16S ribosomal RNA gene, typically providing taxonomic resolution to the genus level, though species-level identification is often limited by sequence similarity [10] [76]. Metagenomic sequencing adopts an unbiased approach, fragmenting and sequencing all DNA, which allows for species- and sometimes strain-level identification, while simultaneously revealing the functional gene repertoire of the microbial community [10] [86]. This direct access to functional genes provides unique value for applications requiring insights into antibiotic resistance profiles, metabolic capabilities, and virulence factors.
Comparative Performance: Key Experimental Findings
Antibiotic Resistance Gene Profiling
Direct comparisons of 16S rRNA sequencing and metagenomics for antibiotic resistance gene (ARG) monitoring reveal significant differences in detection capabilities and application value. A 2025 study quantitatively comparing high-throughput qPCR (HT-qPCR based on 16S rRNA) and metagenomics for ARG profiling in aquaculture environments demonstrated that 28 out of 31 targeted ARGs were detected by HT-qPCR/16S rRNA, while metagenomics identified 18 of these ARGs [87]. Despite these methodological differences, both approaches effectively captured variations in ARG profiles across different environments, with metagenomics excelling in comprehensive diversity profiling and HT-qPCR demonstrating strength in absolute quantification [87].
Metagenomics provides transformative advantages for ARG surveillance by enabling the detection of mobile genetic elements (MGEs) such as integrons, transposons, plasmids, and bacteriophages, which play critical roles in horizontal gene transfer across diverse environments and clinical settings [88]. This capability is crucial for understanding the dissemination mechanisms of resistance determinants. Furthermore, metagenomic analysis of wastewater treatment systems has identified specific host-pathogen relationships, revealing that Pseudomonas serves as both a primary nitrogen-metabolizing genus and a major ARG host, carrying up to 16 different ARGs in some operational systems [89].
Table 1: Antibiotic Resistance Gene Profiling Capabilities
| Profiling Aspect | 16S rRNA-Based Approaches | Metagenomic Sequencing |
|---|---|---|
| ARG Detection Method | Indirect (via co-occurrence analysis) or HT-qPCR | Direct detection and identification |
| Typical ARGs Detected | Limited, targeted panels (e.g., 31 in one study) | Comprehensive, untargeted profiling |
| Host Identification | Indirect inference via correlation | Direct linkage via assembly |
| Mobile Element Context | Not available | Comprehensive analysis possible |
| Quantification Strength | Absolute quantification (HT-qPCR) | Relative abundance |
| Key Finding | Identified high-risk ARGs: mexF, ereA, sul2, aadA, floR [87] | Confirmed high-risk ARGs and identified Pseudomonas as key host [89] |
Metabolic Pathway Analysis
Metagenomics provides unparalleled access to the functional potential of microbial communities, enabling comprehensive metabolic profiling that far exceeds the capabilities of 16S rRNA sequencing. Research on duck gut microbiota demonstrated that metagenomic sequencing could identify distinct functional specializations along different intestinal segments, with foregut microbial functions primarily related to genetic information processing (transcription, translation, replication), while hindgut functions were predominantly associated with the biosynthesis of secondary metabolites and various metabolic pathways [55].
The ability to directly sequence and analyze metabolic genes allows researchers to reconstruct complete metabolic pathways and understand how microbial communities contribute to system-level functions. For instance, in wastewater treatment research, metagenomics revealed that organic nitrogen degradation and synthesis genes had the highest abundance among nitrogen-metabolizing genes, followed by denitrification, dissimilatory nitrate reduction, and nitrification genes [89]. This level of functional resolution is simply not possible with 16S rRNA sequencing alone, which is limited to taxonomic classification without direct functional insights.
Table 2: Metabolic Profiling Capabilities Across Environments
| Environment | 16S rRNA Sequencing Limitations | Metagenomic Sequencing Advantages |
|---|---|---|
| Animal Gut Microbiome | No functional data on regional specialization | Revealed foregut/hindgut functional differences [55] |
| Wastewater Treatment | Cannot link taxonomy to nitrogen cycling functions | Identified 180 high-quality MAGs of N-metabolizing microbes with ARGs [89] |
| Clinical Diagnostics | Limited to pathogen identification | Enables AMR profiling and therapy guidance [68] |
| Environmental Systems | Biodiversity assessment only | Reveals functional potential for bioremediation, nutrient cycling [10] |
Experimental Protocols and Workflows
Methodological Framework
The fundamental difference between 16S rRNA sequencing and metagenomic sequencing begins at the experimental design stage and extends through all phases of workflow and analysis. The 16S rRNA workflow employs universal primers to target conserved regions surrounding variable portions of the 16S gene, followed by PCR amplification, library preparation, and sequencing of these amplicons [10]. This targeted approach provides sensitivity for bacterial detection but introduces potential PCR biases and primer mismatches that can affect quantitative representation of community structure [10].
In contrast, the metagenomic sequencing workflow begins with total DNA extraction from all organisms in the sample (bacteria, viruses, fungi, hosts), followed by random fragmentation and library preparation without target-specific amplification [10]. This comprehensive approach eliminates PCR amplification biases but generates vastly more complex data requiring sophisticated bioinformatic processing for assembly, annotation, and analysis [10]. The computational demands for metagenomics are substantially higher, requiring robust infrastructure for data storage and processing, often involving tools like MEGAHIT for assembly and MetaGeneMark for gene prediction [7].
Workflow Diagram: 16S rRNA vs. Metagenomic Sequencing
Key Research Reagents and Solutions
Successful implementation of either sequencing approach requires specific research reagents and computational tools optimized for each method's unique requirements.
Table 3: Essential Research Reagents and Solutions
| Category | Specific Tool/Reagent | Function/Purpose |
|---|---|---|
| Wet Lab Reagents | TGuide S96 DNA Extraction Kit [7] | Magnetic bead-based genomic DNA extraction from soil/feces |
| TruSeq Nano DNA LT Library Prep Kit [7] | Illumina library preparation for 16S rRNA sequencing | |
| VAHTS Universal Plus DNA Library Prep Kit [7] | Library preparation for Illumina metagenomic sequencing | |
| Primer Sets | 16S rRNA Variable Region Primers (e.g., V1-V3, V3-V4, V4-V5) [86] [90] | Target amplification of specific 16S rRNA regions |
| Bioinformatics Tools | QIIME2, DADA2 [10] [7] | 16S rRNA data processing, denoising, and analysis |
| MEGAHIT [7] | Metagenomic assembly from short reads | |
| MetaGeneMark [7] | Gene prediction in metagenomic assemblies | |
| MetaPhlAn, HUMAnN [10] | Taxonomic profiling and metabolic reconstruction | |
| Reference Databases | SILVA, Greengenes [10] [7] | 16S rRNA taxonomy classification |
| KEGG, CARD [10] | Functional annotation of metagenomic genes |
Analysis of a Key Experiment: ARG Risk Assessment in Aquaculture
Experimental Design and Methodology
A 2025 study provides an excellent comparative framework for understanding the relative strengths of HT-qPCR/16S rRNA and metagenomic approaches for antibiotic resistance assessment [87]. The researchers monitored ARG profiles in aquaculture environments using both methods, employing a novel risk assessment model that integrated absolute abundance, detection frequency, mobility, and host pathogenicity. The experimental protocol involved:
- Sample Collection: Environmental samples from aquaculture water and sediment.
- Parallel Processing: Simultaneous analysis using HT-qPCR targeting 31 specific ARGs and metagenomic sequencing.
- Host Identification: Co-occurrence analysis of HT-qPCR data with 16S rRNA sequencing to identify potential ARG hosts, compared to direct host identification via metagenomic assembly.
- Risk Assessment: Application of a unified model to identify high-risk ARGs using outputs from both methods.
This experimental design allowed for direct comparison of methodological performance in a real-world environmental monitoring scenario, highlighting the complementary nature of these approaches while identifying specific advantages unique to each method.
Results and Interpretation
The study demonstrated that 28 ARGs were detected via HT-qPCR/16S rRNA, while metagenomics identified 18 overlapping ARGs, confirming that both methods effectively captured spatial variations in ARG profiles across different aquaculture environments [87]. The co-occurrence analysis with HT-qPCR and 16S rRNA identified Pseudomonas and Acinetobacter as dominant potential ARG hosts, results that overlapped significantly with metagenomic findings [87].
Both methodologies consistently identified the same high-risk ARGs: mexF, ereA, sul2, aadA, and floR, providing confidence in the risk assessment outcomes regardless of technical approach [87]. The integrated risk assessment revealed that ARGs in aquaculture water posed higher potential risk than those in sediment, demonstrating the practical environmental insights generated by this comparative approach.
Application in Clinical and Environmental Settings
Clinical Diagnostic Applications
Metagenomics has emerged as a transformative tool in clinical diagnostics, enabling culture-independent pathogen detection and comprehensive antimicrobial resistance profiling. In clinical practice, metagenomic sequencing has demonstrated exceptional value for diagnosing complex infections where traditional culture-based methods fail, particularly in cases of culture-negative infections and polymicrobial diseases [68]. The ability to simultaneously identify pathogens and their resistance markers directly from clinical specimens represents a significant advancement for infectious disease management.
Research has shown that metagenomic approaches can achieve diagnostic sensitivity of 96.6% for lower respiratory infections compared to culture, with the additional capability of real-time AMR gene identification enabling early, tailored therapy adjustments [68]. In critical care settings, shotgun metagenomics applied directly to blood samples from septic patients has identified pathogens up to 30 hours earlier than traditional cultures, while simultaneously detecting resistance genes to guide appropriate antimicrobial therapy [68]. This rapid, comprehensive diagnostic capability supports antimicrobial stewardship by reducing reliance on empirical broad-spectrum antibiotics.
Environmental Monitoring Applications
In environmental contexts, metagenomics provides unparalleled insights into the spread and evolution of antibiotic resistance across diverse ecosystems. Studies of wastewater treatment systems, recognized as hotspots for ARG dissemination, have utilized metagenomics to identify significant positive correlations between various types of nitrogen-metabolizing genes and ARGs, revealing fundamental linkages between nutrient cycling and resistance proliferation [89]. This approach has enabled researchers to assemble 1,492 high-quality metagenome-assembled genomes from wastewater systems, with 12.06% identified as major nitrogen-metabolizing microbes carrying ARGs [89].
The functional insights provided by metagenomics extend beyond resistance profiling to encompass comprehensive metabolic pathway analysis. Unlike 16S rRNA sequencing, which is limited to taxonomic assessment, metagenomics can elucidate functional potential for pollution degradation, carbon cycling, and nitrogen fixation [10]. This capability is particularly valuable for environmental engineering and bioremediation applications, where understanding microbial metabolic potential is essential for optimizing treatment processes and monitoring ecosystem health.
The choice between 16S rRNA sequencing and metagenomics depends fundamentally on research objectives, budgetary constraints, and analytical requirements. 16S rRNA sequencing remains the preferred choice for large-scale taxonomic surveys, rapid pathogen screening, and studies requiring cost-effective analysis of many samples [10]. Its advantages include lower sequencing costs, simpler bioinformatic analysis, and established standardized protocols.
In contrast, metagenomic sequencing provides unique value for applications requiring functional insights, including antibiotic resistance profiling, metabolic pathway analysis, and comprehensive characterization of microbial communities beyond bacteria alone [10] [68]. While more expensive and computationally demanding, metagenomics delivers superior taxonomic resolution and direct access to functional genetic elements that define microbial activities in clinical, environmental, and host-associated ecosystems.
For researchers investigating antibiotic resistance or metabolic profiling, metagenomics offers irreplaceable advantages that justify its additional resource requirements. The ability to directly identify resistance genes, elucidate their genomic context including mobile genetic elements, and simultaneously profile metabolic potential makes metagenomics an essential tool for addressing the complex challenges of antimicrobial resistance and microbial functional ecology.
The choice between 16S rRNA gene sequencing and shotgun metagenomics is a fundamental decision in microbiome research, with significant implications for project budget, data depth, and experimental outcomes. This guide provides an objective comparison of these technologies, focusing on their cost-benefit ratio, taxonomic resolution, and functional insights. While 16S sequencing offers a cost-effective solution for high-throughput bacterial profiling, shotgun metagenomics provides unparalleled resolution and functional gene analysis at a higher cost. Recent advancements in "shallow shotgun" sequencing are now bridging this gap, offering a compelling middle ground for large-scale studies.
16S rRNA Gene Sequencing
16S rRNA gene sequencing is a targeted amplicon sequencing approach that amplifies and sequences specific hypervariable regions (e.g., V4, V9) of the bacterial and archaeal 16S rRNA gene. This gene is ideal for phylogenetic differentiation due to its highly conserved nature and taxonomically informative variable regions [3] [91]. The process involves DNA extraction, PCR amplification of the target region, library preparation, and sequencing. The resulting data provides a profile of the microbial community composition, but is generally limited to genus-level taxonomic identification for bacteria and archaea only [1].
Shotgun Metagenomic Sequencing
Shotgun metagenomics takes an untargeted approach by sequencing all genomic DNA present in a sample after random fragmentation. This method sequences the entirety of the genetic material, enabling strain-level multi-kingdom taxonomic classification (bacteria, viruses, fungi, protists) and allowing for functional profile characterization by identifying microbial genes and pathways [3]. The process includes DNA extraction, random fragmentation, library preparation without targeted PCR amplification, and deep sequencing [3] [1].
Table 1: Core Technological Differences
| Feature | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Target | Specific hypervariable regions of the 16S rRNA gene [3] | All genomic DNA in a sample [3] |
| Primary Method | PCR amplification of target region [3] | Random fragmentation and sequencing [3] |
| Taxonomic Coverage | Bacteria and Archaea only [3] [1] | Multi-kingdom: Bacteria, Archaea, Viruses, Fungi [3] |
| Functional Profiling | Indirect prediction only (e.g., PICRUSt) [3] [1] | Direct characterization of genes and pathways [3] |
Figure 1: Comparative Workflows of 16S rRNA Sequencing and Shotgun Metagenomics.
Quantitative Cost-Benefit Analysis
Financial and Operational Comparison
Table 2: Financial and Operational Cost-Benefit Analysis
| Factor | 16S rRNA Sequencing | Shotgun Metagenomics | Shallow Shotgun |
|---|---|---|---|
| Approximate Cost per Sample | ~$50 USD [1] | Starting at ~$150 USD [1] (Deeper sequencing costs more) | Close to 16S costs [3] |
| Taxonomic Resolution | Family & Genus level; Species level possible but with high false-positive rate [3] | Species and Strain-level resolution [3] | Species and Strain-level (but less robust than deep shotgun) [3] |
| Functional Profiling | No direct functional data; requires prediction algorithms [3] [1] | Direct functional gene and pathway characterization [3] | Enables functional profiling [3] |
| Handling of Host DNA | Low interference (PCR amplifies only the 16S gene) [3] | High interference; requires host DNA removal or increased sequencing depth [3] | Varies with sample type |
| Minimum DNA Input | Low (can be <1 ng due to PCR amplification) [3] | Higher (typically ≥1 ng/μL) [3] | Similar to shotgun |
| Bioinformatics Complexity | Beginner to Intermediate [1] | Intermediate to Advanced [1] | Intermediate |
Information Yield and Data Quality
Comparative studies consistently demonstrate that shotgun sequencing provides a more comprehensive view of microbial communities. A 2021 study comparing both methods on chicken gut samples found that shotgun sequencing identified a statistically significant higher number of taxa, particularly among less abundant genera that were missed by 16S sequencing [6]. Similarly, a 2024 study on human gut microbiota in colorectal cancer confirmed that 16S detects only part of the gut microbiota community revealed by shotgun, with 16S data being sparser and exhibiting lower alpha diversity [4].
The superiority of shotgun sequencing for detecting meaningful biological differences is evident in differential abundance testing. In the chicken gut study, when comparing microbial communities between different gastrointestinal compartments, shotgun sequencing identified 256 statistically significant differences in genera abundance, while 16S sequencing identified only 108 [6]. This enhanced detection power enables researchers to identify subtle but biologically relevant microbial shifts.
Experimental Design and Protocol Considerations
Sample Type and DNA Extraction
The optimal sequencing method depends heavily on sample type and DNA quality:
- High-host DNA samples (skin swabs, tissue biopsies): 16S sequencing is often more suitable because PCR amplification specifically targets the 16S gene, minimizing host DNA interference [3] [1]. Shotgun sequencing these samples requires host DNA removal techniques or substantial sequencing depth to obtain sufficient microbial data [3].
- High-microbial biomass samples (stool, environmental samples): Shotgun sequencing is preferred, with shallow shotgun offering a cost-effective alternative [3]. These samples typically contain enough microbial DNA to support untargeted sequencing without excessive host contamination.
- Low-biomass samples: 16S sequencing may be more successful due to its PCR-based approach, which can amplify targets from minimal starting material [3]. However, caution is needed to avoid contamination artifacts.
DNA extraction methods must be tailored to both the sample type and sequencing technology. For 16S sequencing, standard commercial kits like the DNeasy PowerSoil Pro Kit are commonly used [4]. For shotgun sequencing, methods that maximize yield and minimize bias are critical, with some studies using the NucleoSpin Soil Kit for optimal results [4]. A high-throughput DNA extraction method using AMPure XP magnetic beads has been developed specifically for plant root microbiota studies, demonstrating comparable performance to traditional kit-based methods [92].
Bioinformatics and Data Analysis
The bioinformatic requirements differ substantially between the two methods:
16S rRNA Sequencing Analysis:
- Typical pipelines: QIIME2, MOTHUR, DADA2 [1]
- Key steps: Quality filtering, denoising (DADA2) or OTU clustering, chimera removal, taxonomic assignment using reference databases (SILVA, Greengenes) [7] [4]
- Output: Taxonomic relative abundances, alpha and beta diversity metrics
Shotgun Metagenomic Analysis:
- Typical pipelines: MetaPhlAn, HUMAnN, MEGAHIT [1]
- Key steps: Quality control, host DNA removal (Bowtie2), taxonomic profiling, functional annotation (KEGG, CAZy), assembly, and gene prediction [7] [4]
- Output: Species/strain-level taxonomy, functional gene abundances, pathway analyses
Table 3: Essential Research Reagent Solutions
| Reagent/Kit | Function | Application Context |
|---|---|---|
| DNeasy PowerSoil Pro Kit (QIAGEN) | DNA extraction from soil, stool, and other complex samples | Standardized DNA extraction for both 16S and shotgun; effective for difficult-to-lyse organisms [4] |
| NucleoSpin Soil Kit (Macherey-Nagel) | DNA extraction for metagenomic analysis | Used in shotgun metagenomic studies of human stool samples [4] |
| 16S Barcoding Kit (Oxford Nanopore) | Library preparation for long-read 16S sequencing | Enables full-length 16S gene sequencing for improved taxonomic resolution [93] |
| AMPure XP Magnetic Beads (Beckman Coulter) | DNA purification and size selection | High-throughput DNA extraction and PCR clean-up; reduces sample handling time [92] |
| ZymoBIOMICS Microbial Community Standard | Mock community control for method validation | Quality control for both 16S and shotgun sequencing; assesses accuracy and bias [92] |
Decision Framework and Recommendations
Method Selection Guide
The choice between 16S and shotgun sequencing should be guided by research objectives, budget constraints, and sample characteristics:
Figure 2: Decision Framework for Selecting the Appropriate Sequencing Method.
Emerging Trends and Future Directions
Long-read 16S sequencing using Oxford Nanopore Technologies (ONT) is emerging as a way to enhance the resolution of amplicon sequencing. A 2025 study demonstrated that full-length (~1,500 bp) 16S rRNA gene sequencing with ONT provided higher taxonomic resolution at the genus level compared to traditional ~500 bp Sanger sequencing, at a significantly lower cost per test (~$25.30 vs $74) [93]. This approach bridges the gap between short-read 16S and shotgun sequencing by providing more precise taxonomic assignment while remaining cost-effective.
Integrated approaches that combine both methods are also gaining traction. A study on duck gut microbiota used 16S sequencing for broad microbial composition analysis alongside metagenomics to identify and track antibiotic resistance genes (ARGs), demonstrating how the techniques can be complementary [55]. This dual approach maximizes the strengths of each method - using 16S for cost-effective community profiling while employing metagenomics for specific functional gene analysis.
The decision between 16S rRNA gene sequencing and shotgun metagenomics represents a fundamental trade-off between cost and information depth. 16S sequencing remains the most cost-effective choice for large-scale bacterial profiling studies where genus-level resolution is sufficient and functional gene data is not required. Shotgun metagenomics provides superior resolution and functional insights but at a significantly higher cost, making it ideal for hypothesis-driven research requiring species- or strain-level discrimination or functional potential assessment.
The emergence of shallow shotgun sequencing and long-read 16S technologies is creating new opportunities to optimize this cost-benefit ratio. Researchers should carefully consider their specific project goals, sample types, and analytical capabilities when selecting a sequencing strategy, as the optimal choice is highly context-dependent. As sequencing costs continue to decline and bioinformatic tools become more accessible, shotgun metagenomics is likely to become increasingly prevalent, though 16S sequencing will maintain its utility for targeted bacterial analyses and large-scale epidemiological studies.
Conclusion
The choice between 16S rRNA sequencing and shotgun metagenomics is not a question of which is superior, but which is most appropriate for the specific research question and resources. 16S rRNA remains a powerful, cost-effective tool for high-throughput studies focused on bacterial community structure and composition. In contrast, shotgun metagenomics provides a deeper, more comprehensive view, delivering strain-level taxonomy, functional gene content, and cross-kingdom insights at a higher cost and computational burden. Future directions in clinical and biomedical research will likely involve hybrid approaches, using 16S for initial surveillance and metagenomics for deep mechanistic investigation. As databases expand and sequencing costs decline, metagenomics is poised to become the gold standard for pathogen discovery, antibiotic resistance tracking, and personalized medicine, fundamentally advancing our understanding of host-microbe interactions in health and disease.
References
- 1. 16S rRNA Gene Sequencing vs. Shotgun Metagenomic ... [https://blog.microbiomeinsights.com/16s-rrna-sequencing-vs-shotgun-metagenomic-sequencing]
- 2. Unravel Your Metagenomic Sample - Shotgun Sequencing ... [https://cegat.com/unravel-your-metagenomic-sample-shotgun-sequencing-vs-16s-sequencing/]
- 3. 16S vs Metagenomics: What's the Difference? [https://www.cosmosid.com/blog/16s-vs-metagenomics-whats-the-difference/]
- 4. Comparison between 16S rRNA and shotgun sequencing in ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10621-7]
- 5. Amplicon-Based Next-Generation Sequencing vs. ... [https://www.cd-genomics.com/microbioseq/amplicon-based-next-generation-sequencing-vs-metagenomic-shotgun-sequencing.html]
- 6. Comparison between 16S rRNA and shotgun sequencing ... [https://www.nature.com/articles/s41598-021-82726-y]
- 7. 16S rRNA and Metagenomic Datasets of Gastrointestinal ... [https://www.nature.com/articles/s41597-025-05555-y]
- 8. Comparison of metagenomic and targeted methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10746264/]
- 9. Comparative Analysis of 16S rRNA Gene and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8320171/]
- 10. Difference Between 16S rRNA and Metagenomic Sequencing [https://uncoded.in/difference-between-16s-rrna-and-metagenomic-sequencing/]
- 11. 16S and ITS rRNA Sequencing | Identify bacteria & fungi ... [https://www.illumina.com/areas-of-interest/microbiology/microbial-sequencing-methods/16s-rrna-sequencing.html]
- 12. Comparative evaluation of sequencing technologies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12130029/]
- 13. Next Generation Sequencing Improves Diagnostic 16S ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12418051/]
- 14. The unresolved struggle of 16S rRNA amplicon sequencing [https://environmentalmicrobiome.biomedcentral.com/articles/10.1186/s40793-025-00705-6]
- 15. Metagenome-Assembled Genomes (MAGs): Advances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114606/]
- 16. Potential applications and future prospects of ... [https://www.sciencedirect.com/science/article/abs/pii/S0378111925005098]
- 17. Uses of Metagenomics in Pharmaceutical Development [https://www.psomagen.com/blog/metagenomics-pharmaceutical-development]
- 18. Genome-resolved metagenomics: a game changer for ... [https://www.nature.com/articles/s12276-024-01262-7]
- 19. Comparison of clinical metagenomics with 16S rDNA ... [https://www.sciencedirect.com/science/article/pii/S1438422125000062]
- 20. Comparative evaluation of sequencing platforms: Pacific ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1633360/full]
- 21. MetaDAVis: An R shiny application for metagenomic data ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0319949]
- 22. Unveiling the impact of 16S rRNA gene intergenomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12081361/]
- 23. Comparison of clinical metagenomics with 16S rDNA ... [https://www.medrxiv.org/content/10.1101/2024.06.18.24309080v1.full-text]
- 24. 16S Sequencing vs Shotgun Metagenomic Sequencing [https://www.zymoresearch.com/blogs/blog/16s-sequencing-vs-shotgun-metagenomic-sequencing?srsltid=AfmBOoqb9DMgEvHfrzu2JAH9p0zoVBBcz10BNKILNFjgzyfp0LX4QQT2]
- 25. Comparative analysis of metagenomic next-generation ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-025-03887-8]
- 26. Metagenomic approaches in microbial ecology: an update ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7641418/]
- 27. OTU vs. ASV in Metagenomic Analysis [https://www.seqme.eu/en/blog/article/otu-vs-asv-in-metagenomic-analysis]
- 28. Evaluation of 16S rRNA gene sequencing for species and ... [https://www.nature.com/articles/s41467-019-13036-1]
- 29. High-resolution microbiome analysis enabled by linking of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8715429/]
- 30. Impact of sequencing depth on the characterization ... [https://www.nature.com/articles/s41598-018-24280-8]
- 31. Introduce to 16S rRNA and 16S rRNA Sequencing [https://www.cd-genomics.com/blog/introduce-to-16s-rrna-and-16s-rrna-sequencing/]
- 32. 16S rRNA Sequencing Guide [https://blog.microbiomeinsights.com/16s-rrna-sequencing-guide]
- 33. Metagenomics - a guide from sampling to data analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3351745/]
- 34. 16S rRNA Methodology and Analysis Guide [https://cores.emory.edu/eicc/_includes/documents/sections/resources/16s_methodology_2024-01-24.html]
- 35. GitHub - grimmlab/MicrobiomeBestPracticeReview: Current Challenges... [https://github.com/grimmlab/MicrobiomeBestPracticeReview]
- 36. Impact of 16S rRNA Gene Sequence Analysis for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC523561/]
- 37. 16S rRNA Gene Sequencing for Bacterial Identification in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2045242/]
- 38. 16S rRNA and metagenomic shotgun sequencing data ... [https://www.nature.com/articles/s41598-022-07995-7]
- 39. Metagenomic insights into resistance trends related to ... [https://www.nature.com/articles/s41598-025-06125-3]
- 40. Functional metagenomics for the investigation of antibiotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3979872/]
- 41. Functional metagenomic characterization of antibiotic ... [https://www.sciencedirect.com/science/article/abs/pii/S0160412013002997]
- 42. Spatiotemporal profiling and succession of microbial ... [https://www.sciencedirect.com/science/article/abs/pii/S0043135425002489]
- 43. 16S Sequencing vs Shotgun Metagenomic Sequencing [https://www.zymoresearch.com/blogs/blog/16s-sequencing-vs-shotgun-metagenomic-sequencing?srsltid=AfmBOorTuEEbXI5ChExl6pD5DaoK0YxoFU2JXjwZi2xPQ51a7KK-RAE6]
- 44. 16S rRNA Sequencing vs. Metagenome Sequencing [https://www.cd-genomics.com/resource-16s-rRNA-sequencing-vs-metagenome-sequencing.html]
- 45. Optimization of high-throughput 16S rRNA gene amplicon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10634443/]
- 46. Metagenome vs Genome: The Differences [https://www.cosmosid.com/blog/metagenome-vs-genome-the-differences/]
- 47. Metagenomic analysis with strain-level resolution reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6169887/]
- 48. What can metagenomics do for you? [https://blog.microbiomeinsights.com/what-can-metagenomics-do-for-you]
- 49. Metagenomics Sequencing Overview [https://www.cd-genomics.com/microbioseq/resource-metagenomics-sequencing-overview.html]
- 50. Metagenomic small molecule discovery methods - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4135586/]
- 51. Metagenomic applications in exploration and development ... [https://www.sciencedirect.com/science/article/pii/S1687157X23005607]
- 52. Characterization of the Gut Microbiome Using 16S or ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4837688/]
- 53. On the limits of 16S rRNA gene-based metagenome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10926695/]
- 54. Full-length 16S rRNA gene sequencing by PacBio improves ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10213-5]
- 55. Integrated 16S rRNA and metagenomic sequencing ... [https://www.sciencedirect.com/science/article/pii/S0032579125004481]
- 56. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8544895/]
- 57. 16S rRNA gene primer choice impacts off-target ... [https://www.nature.com/articles/s41598-023-39575-8]
- 58. Nanopore full length 16S rRNA gene sequencing ... [https://www.nature.com/articles/s41598-025-10999-8]
- 59. 16S rRNA Sequencing VS Shotgun ... [https://www.cd-genomics.com/microbioseq/resource-16s-rrna-sequencing-vs-shotgun-sequencing-for-microbial-research]
- 60. Sensitivity of shotgun metagenomics to host DNA [https://pmc.ncbi.nlm.nih.gov/articles/PMC7523627/]
- 61. Determining the most accurate 16S rRNA hypervariable ... [https://www.nature.com/articles/s41598-023-30764-z]
- 62. Investigating the impact of database choice on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9673341/]
- 63. Prospective Comparison Between Shotgun Metagenomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9020828/]
- 64. 16S rRNA vs. Metagenomic Sequencing [https://live-biotherapeutic.creative-biolabs.com/16s-rrna-vs-metagenomic-sequencing-which-is-best-for-microbial-identification.htm]
- 65. Pricing - Forsyth Microbiome Core [https://microbiome.forsyth.org/pricing.php]
- 66. Metagenomics Market Size Booms at 13.55% CAGR ... [https://finance.yahoo.com/news/metagenomics-market-size-booms-13-160000902.html]
- 67. Evaluation of long-read 16S rRNA next-generation ... [https://pubmed.ncbi.nlm.nih.gov/40261041/]
- 68. Gut microbiome metagenomics in clinical practice [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562794/]
- 69. Guidelines for preventing and reporting contamination in ... [https://www.nature.com/articles/s41564-025-02035-2]
- 70. Planning and analyzing a low-biomass microbiome study [https://pmc.ncbi.nlm.nih.gov/articles/PMC12445924/]
- 71. Analysis of the microbiome: Advantages of whole genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4830092/]
- 72. Controlling for Contaminants in Low-Biomass 16S rRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6550369/]
- 73. an R package for decontaminating low-biomass 16S-rRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12095030/]
- 74. Full-length 16S rRNA Sequencing Reveals Gut Microbiome ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-025-03849-0]
- 75. 16S Sequencing vs Shotgun Metagenomic Sequencing [https://www.zymoresearch.com/blogs/blog/16s-sequencing-vs-shotgun-metagenomic-sequencing?srsltid=AfmBOortCRCcvBiBIuzcXb7EiLq3QCVjxAAYH0sANFOHPjWcDbjVPyI1]
- 76. Comparative Analysis of 16S rRNA Gene and ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.670336/full]
- 77. The effect of taxonomic classification by full-length 16S ... [https://www.nature.com/articles/s41598-020-80826-9]
- 78. A Comparison of Three Different Bioinformatics Analyses ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2019.00620/full]
- 79. The Use and Types of Alpha-Diversity Metrics in Microbial ... [https://www.cd-genomics.com/microbioseq/the-use-and-types-of-alpha-diversity-metrics-in-microbial-ngs.html]
- 80. Definition of the microbial rare biosphere through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11965487/]
- 81. Identification of core and rare species in metagenome ... [https://www.nature.com/articles/s43705-021-00010-6]
- 82. Detection of low-abundance bacterial strains in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4720164/]
- 83. Advantages and Limitations of 16S rRNA Next-Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7602188/]
- 84. Integrated 16S rRNA sequencing and metagenomics ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1375804/full]
- 85. Microbiome differential abundance methods produce ... [https://www.nature.com/articles/s41467-022-28034-z]
- 86. Comparative evaluation of sequencing technologies and ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1584359/full]
- 87. Quantitative comparison of HT-qPCR/16S rRNA ... [https://pubmed.ncbi.nlm.nih.gov/40972162/]
- 88. Metagenomics as a Transformative Tool for Antibiotic ... [https://www.mdpi.com/2079-6382/14/3/296]
- 89. Antibiotic resistance profile of nitrogen-metabolizing ... [https://www.nature.com/articles/s41545-025-00464-4]
- 90. Refining microbiome diversity analysis by concatenating ... [https://www.nature.com/articles/s41522-025-00686-x]
- 91. Research progress on the application of 16S rRNA gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10869621/]
- 92. High throughput method of 16S rRNA gene sequencing ... [https://www.nature.com/articles/s41598-022-23943-x]
- 93. Evaluation of long-read 16S rRNA next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12077174/]